

Abstract
17β-hydroxysteroid dehydrogenase (17β-HSD) type 1 is known as a critical target to block the final step of estrogen production in estrogen-dependent breast cancer. Recent confirmation of the role of dyhydroxytestosterone (DHT) in counteracting estrogen-induced cell growth prompted us to study the reductive 17β-HSD type 7 (17β-HSD7), which activates estrone while markedly inactivating DHT. The role of DHT in breast cancer cell proliferation is demonstrated by its independent suppression of cell growth in the presence of a physiological concentration of estradiol (E2). Moreover, an integral analysis of a large number of clinical samples in Oncomine datasets demonstrated the overexpression of 17β-HSD7 in breast carcinoma. Inhibition of 17β-HSD7 in breast cancer cells resulted in a lower level of E2 and a higher level of DHT, successively induced regulation of cyclinD1, p21, Bcl-2, and Bik, consequently arrested cell cycle in the G0/G1 phase, and triggered apoptosis and auto-downregulation feedback of the enzyme. Such inhibition led to significant shrinkage of xenograft tumors with decreased cancer cell density and reduced 17β-HSD7 expression. Decreased plasma E2 and elevated plasma DHT levels were also found. Thus, the dual functional 17β-HSD7 is proposed as a novel target for estrogen-dependent breast cancer by regulating the balance of E2 and DHT. This demonstrates a conceptual advance on the general belief that the major role of this enzyme is in cholesterol metabolism.
Keywords: 17β-hydroxysteroid dehydrogenase type 7, breast cancer, xenograft tumor, steroid enzyme inhibition
Introduction
Breast cancer (BC) is the most commonly diagnosed cancer and the second highest cause of cancer-related death in females worldwide (Jemal et al., 2011). A large proportion of BCs are initially estrogen-dependent (Russo and Russo, 1998): these account for 60% of BC cases in premenopausal women and 75% of cases in postmenopausal women (Jonat et al., 2006). In the latter, ovarian-derived estrogens are dramatically decreased and replaced by estrogens synthesized by steroidogenic enzymes from precursor steroids in an intracrine manner (Labrie, 1991, 2003). The most potent estrogen, estradiol (E2), which is formed by the reduction of estrone (E1) and/or by the aromatization of testosterone (T) (Payne and Hales, 2004), serves a dual role in BC as a hormone stimulating cell proliferation through binding to estrogen receptors (ERs) (Yager and Davidson, 2006), as well as a pro-carcinogen inducing genetic damage and mutations (Liehr, 2000). Consequently, blockage of E2 production with specific inhibitors of steroidogenic enzymes such as aromatase inhibitors (AIs) comprises one of the two currently available standard therapies. The other approach involves blocking the ER (Pearson et al., 1982) with selective estrogen receptor modulators (SERMs).
In contrast to E2, which predominantly contributes to breast carcinoma growth through ER, the non-aromatic androgen, dihydrotestosterone (DHT), has recently been reported to exert anti-proliferative effects via the androgen receptor (AR) by activating p21waf1/cip1 and/or inhibiting cyclinD1 (Greeve et al., 2004; Lanzino et al., 2010). Indeed, the AR is expressed in ∼80% of primary BCs, which is greater than the proportion expressing ER (70%) (Hu et al., 2011). Furthermore, nearly 80% of ER-positive (ER+) BCs were found to co-express AR (Niemeier et al., 2010). A systematic meta-analysis demonstrated that co-expression of ARs in female breast tumors is associated with a better prognosis and outcome (Vera-Badillo et al., 2014). Moreover, metabolites of DHT such as 5α-androstane-3β,17β-diol (3β-diol) displayed weak estrogenic potency toward MCF-7 cells (Adams et al., 1981) through binding to ER (Chen et al., 2013), which was thought to be a possible mechanism for inducing AI resistance (Sikora et al., 2009; Hanamura et al., 2013). Therefore, a new concept for the treatment of BC has emerged through the joint targeting of E2 (reduction) and DHT (restoration) (Aka et al., 2010; Lin et al., 2010).
The intracrine pathways for estrogen synthesis include: (i) the ‘aromatase pathway’, which transforms androgens into estrogens; (ii) the ‘sulfatase pathway’, which desulfates dehydroepiandrosterone-sulfate (DHEA-S) and estrone-sulfate (E1-S) followed by the reductive 17β-HSD1 and 17β-HSD7 that are integrated into both pathways converting E1 into potent E2. Therefore, aromatase, steroid sulfatase (STS), and reductive 17β-HSDs involved in the final steps of E2 biosynthesis are primary targets for blocking E2 production (Pasqualini, 2004; Lin et al., 2010). Aromatase inhibitors have been used in the clinic with favorable outcomes (Campos, 2004); however, resistance to AIs has become an inevitable occurrence (Miller and Larionov, 2012). Thus, approaches targeting other enzymes could be effective at reducing estrogen synthesis. 17β-HSD1 has been accepted as a critical therapeutic target to block E2 production (Lin et al., 2010). Structure-based studies suggested that 17β-HSD1 alternatively binds to and inactivates DHT (Gangloff et al., 2003), providing a basis for dual catalytic function by this long-held ‘estrogenic enzyme’. Another dual functional enzyme, 17β-HSD7, also converts E1 into E2 and catalyzes a marked inactivation of DHT into the weak estrogen, 3β-diol (Törn et al., 2003). This prompted us to investigate in vitro and in vivo whether 17β-HSD7 can be a potential therapeutic target for estrogen-dependent BC.
Expression of human 17β-HSD7 has been reported in the ovary, placenta, mammary gland, liver, and brain (Krazeisen et al., 1999). At present, it is generally accepted that 17β-HSD7 is primarily involved in cholesterol synthesis rather than in steroidogenesis (Marijanovic et al., 2003; Ohnesorg et al., 2006). This has had a marked effect on the direction of studies involving this enzyme and explains the limited number of studies addressing its function in steroid hormone biosynthesis and related diseases including BC. 17β-HSD7 was first detected as prolactin receptor-associated protein in rat (Duan et al., 1997). Detection of a high expression level in the corpus luteum of pregnant mice supported the assumption of its role in E2 synthesis (Nokelainen et al., 1998). The predominant involvement of 17β-HSD7 in cholesterol metabolism rather than in sex steroid synthesis, was further supported by the observation that although 17β-HSD7 knockout mice were fertile, they bred nonviable fetuses due to defective in situ cholesterol biosynthesis in the brain (Breitling et al., 2001; Shehu et al., 2008). In order to gain a better understanding of the role of 17β-HSD7 in BC, we re-initiated this functional study of 17β-HSD7 with an emphasis on clarifying its contribution to sex hormone biosynthesis and BC stimulation (Canadian Institutes of Health Research Project ‘Sulfatase and aromatase pathways for estradiol synthesis in human breast cancer cells, tissues and animal models: identifying a combinatory therapy’, since 2009).
In the present study, 17β-HSD7 in BC cells (ER+ cell lines MCF-7 and T47D; ER-negative (ER−) cell line BT-20) was inhibited with a selective inhibitor (Bellavance et al., 2009). The effects generated by 17β-HSD7 inhibition were carefully evaluated in terms of cell proliferation, cell cycle progression, and E2/DHT formation. An experimental therapeutic study was also performed on a murine xenograft model generated with wild-type MCF-7 cells. Moreover, the Oncomine dataset (Rhodes et al., 2004) with an extensive cancer microarray database was interrogated to confirm the overexpression status of 17β-HSD7 in various breast carcinomas. The critical involvement of 17β-HSD7 in steroid metabolism and stimulation of BC cells was demonstrated, and through in vitro and in vivo studies, 17β-HSD7 was characterized as a novel therapeutic target for postmenopausal ER+ BC.
Results
17β-HSD7. inhibitor at low concentrations suppressed cell proliferation and arrested cell cycle in the G0/G1 phase by inhibiting cyclin D1 and activating p21
With reference to the IC50 values of the inhibitors (INH7 or INH1) (Table 1), concentrations ranging from 0.2 to 2 µM (IC50 to 10 × IC50) were selected to investigate the anti-proliferative effect in response to specific enzyme inhibition. A significant dose-dependent reduction in DNA synthesis was observed in parallel to attenuated cell proliferation in MCF-7 (Figure 1A) and T47D cells (Supplementary Figure S1A). Treatment with 2 µM (10 × IC50) INH7 suppressed MCF-7 cell proliferation by 33% vs. 18% with INH1, and 1.2 µM INH7 reduced proliferation of T47D cells by 26% vs. 35% with INH1. However, neither INH7 nor INH1 displayed an anti-proliferative effect in ER− BT-20 cells (Supplementary Figure S2A). Cell viability at a low concentration range (0.2–2 µM) was tested with MTT (data not shown) and no cytotoxic effect was observed within this dose range. These results demonstrated that INH7 showed higher anti-proliferative efficacy than INH1 in MCF-7, whereas they showed similar efficacies in T47D cells with higher expression of 17β-HSD1 (Table 2).
Table 1.
Characteristics of 17β-HSD1 and 17β-HSD7 inhibitors used in this study.
| Compound | Targeting enzyme | Chemical structure | IC50a of E1∼E2 | IC50b of DHT∼3β-diol |
|---|---|---|---|---|
| E2B-Methoxy (INH1) | 17β-HSD type1 | 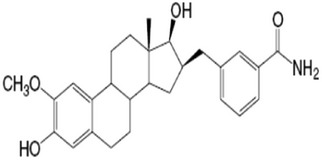 |
275 ± 5 nM | None |
| EB-357-030 (INH7) | 17β-HSD type7 | 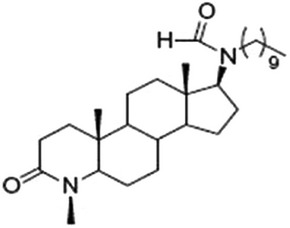 |
195 ± 18 nM | 230 ± 15 nM |
None, data not shown.
aIC50, concentration of the compound inhibiting 50% of E1 to E2 conversion in T47D cells (Laplante et al., 2008).
bIC50, concentration of the compound inhibiting 50% of DHT inactivation in HEK293 cells overexpressing 17β-HSD7 (Bellavance et al., 2009).
Figure 1.

Cytostatic effect of INH7/INH1 in MCF-7 cells. (A) Cell proliferation assay of MCF-7 treated with INH7 or INH1. Data are reported as % of DNA synthesis vs. control (100%). Each point represents the mean of experiments carried out in quadruplicate (mean ± SD). Statistical significance by Student's t-test: *P < 0.05 vs. control (Ctrl). (B) Cell cycle analysis of MCF-7 cells treated with INH7 or INH1. Data are reported as % of living cells (G0/G1, S, and G2/M cells = 100%). Each number represents the mean of experiments carried out in triplicate (mean ± SD). Statistical significance (P < 0.05) by Student's t-test is highlighted in bold box in table. (C) E2 and DHT formation in MCF-7 cells treated with INH7 or INH1. Each bar represents the mean of experiments carried out in quadruplicate (mean ± SD). Statistical significance by Student's t-test: *P < 0.05 vs. control (Ctrl); **P < 0.001 vs. Ctrl. (D) Cyclin D1, p21, and PCNA protein expression determined by western blot in MCF-7 cells treated with INH7. Data are reported as mean ± SD (n = 3) of the individual experiments. Statistical significance by Student's t-test: *P < 0.05 vs. control (Ctrl); **P < 0.001 vs. Ctrl. (E) 17β-HSD1/7 protein expression determined by western blot in MCF-7 cells treated with INH1 and INH7, respectively. Data are reported as mean ± SD (n = 3) of the individual experiments. Significant differences by Student's t-test: *P < 0.05 vs. control (Ctrl).
Table 2.
Expression of receptors and 17β-HSDs in different breast cancer cell lines.
| Cell lines | Receptors |
17β-HSDs mRNA level |
Tumor type | Gene cluster | ||
|---|---|---|---|---|---|---|
| ER | AR | 17β-HSD1 | 17β-HSD7 | |||
| MCF7 | 653 | 96 | 83 | 159 | IDC | Lu |
| T47D | 190 | 127 | 697 | 150 | IDC | Lu |
| BT20 | N | N | 294 | 26 | IDC | BaA |
qRT–PCR value shows mRNA copies/µg total RNA. N, negligible by q-RT–PCR or IHC (Neve et al., 2006; Laplante et al., 2009; Aka and Lin, 2012).
ER, estrogen receptor; AR, androgen receptor (Laplante et al., 2009; Aka and Lin, 2012).
IDC, invasive ductal carcinoma; Lu, luminal; BaA, Basal A (Neve et al., 2006).
In MCF-7 cells (Figure 1B), 2 µM INH7 arrested 8.2% of cells in the G0/G1 phase (76.9% ± 0.4% vs. 68.7% ± 0.9% of Ctrl), and the number of cells in S-phase decreased by 7.5% (23.4% ± 0.9% of Ctrl vs. 15.9% ± 0.4% of INH7). In contrast, INH1 displayed less potency with regard to G0/G1 arrest (3.7%) and S phase cell reduction (3.4%). In T47D cells (Supplementary Figure S1B), INH7 (2 µM) arrested 8.3% of cells in the G0/G1 phase and decreased the number of cells in the S phase by 3.7%, whereas INH1 arrested 4.6% cells in G0/G1 and decreased the number of cells in the S phase by 3.4%. However, in BT-20 cells (Supplementary Figure S2B), neither INH7 nor INH1 displayed any effect on cell cycle arrest. Results indicated that INH7 was more potent than INH1 with regard to cell cycle disruption in MCF-7 and T47D cells.
The G1/S cell cycle checkpoint is primarily monitored by the Cdk4–cyclin D1 complex as well as by cyclin-dependent kinase inhibitors such as p21waf1/cip1. Western blot analysis indicated that in MCF-7 cells (Figure 1D), INH7 (2 µM) induced a 2.2-fold (P < 0.001) downregulation of cyclin D1 and a 2.4-fold (P < 0.001) upregulation of p21waf1/cip1. INH7 suppressed expression of PCNA by 2.2-fold vs. Ctrl. These observations suggest that INH7 arrests cell cycle in G0/G1 by downregulation of cyclin D1 and upregulation of p21.
17β-HSD7. inhibitor blocked E2 formation and DHT degradation
In MCF-7 cells (Figure 1C), INH7 reduced the E2 level from 0.068 ± 0.006 nM to 0.011 ± 0.001 nM (Ctrl vs. 2 µM INH7), whereas INH1 reduced E2 from 0.067 ± 0.002 nM to 0.015 ± 0.002 nM (Ctrl vs. 2 µM INH1). In T47D cells (Supplementary Figure S1C), 2 µM INH7 decreased the E2 level from 0.098 ± 0.001 nM to 0.011 ± 0.003 nM, whereas INH1 reduced the E2 level from 0.089 ± 0.001 nM to 0.013 ± 0.003 nM. These results indicated that INH7 displayed similar potency in both MCF-7 and T47D cells with regard to the blockage of E2 formation. However, only INH7 displayed a significant inhibitory effect on DHT inactivation, thereby inducing DHT accumulation in cells: INH7 (2 µM) induced the accumulation of DHT from 0.01 ± 0.003 nM to 0.12 ± 0.002 nM (Ctrl vs. INH7) in MCF-7 cells (Figure 1C), and increased the DHT level in T47D cells from 0.01 ± 0.002 nM to 0.11 ± 0.002 nM (Supplementary Figure S1C). These results show that inhibition of 17β-HSD7 blocked E2 formation and restored DHT within cells, consequently regulating the cell cycle through cyclin D1 and p21 (see discussion).
17β-HSD7. inhibitor inhibited the expression of 17β-HSD7 in MCF-7 cells
Treatment with INH7 at 2 µM for 4 days inhibited the expression of 17β-HSD7 in MCF-7 cells. Western blot demonstrated that INH7 reduced 17β-HSD7 expression from 100% (Ctrl) to 58% ± 5% (INH7) (P < 0.05), whereas treatment with 0.1 nM E2 induced an increase in 17β-HSD7 expression of 115% ± 1.8% (P < 0.05) vs. Ctrl. However, the same dose of INH1 (2 µM for 4 days) had no effect on the expression of 17β-HSD1 in MCF-7 cells (Figure 1E).
A high concentration of 17β-HSD7 inhibitor induced apoptosis by suppressing anti-apoptosis protein Bcl-2 and inducing pro-apoptosis protein Bik
MTT assay indicated that in MCF-7 cells (Figure 2A), the number of viable cells was significantly decreased in response to treatment with INH7 compared with the control (100% of Ctrl vs. 26.7% ± 2.3% at 8 µM), whereas INH1 had a minor effect (100% vs. 87.8% ± 3.5%). In T47D cells (Supplementary Figure 1D), 8 µM INH7 decreased cell viability from 100% of Ctrl to 42.9% ± 3.4%, whereas INH1 at the same concentration decreased viability from 100% to 78.6% ± 1.5%. Therefore, INH7 demonstrated a more remarkable cytotoxic effect than INH1.
Figure 2.

Apoptotic effect of INH7/INH1 in MCF-7 cells. (A) Cell viability of MCF-7 treated with INH7 or INH1 by MTT assay. Each point represents the mean of experiments carried out in quadruplicate (mean ± SD). Statistical significance by Student's t-test: *P < 0.05 vs. Ctrl. **P < 0.001 vs. Ctrl. (B) Cell apoptosis of MCF-7 treated with INH7 or INH1. Each number represents the mean of experiments carried out in triplicate (mean ± SD). Statistical significance (P < 0.05) by Student's t-test is highlighted in bold box in table. Flow cytometry panels represent the distribution of each cell type after treatment. (C) Expression of Bcl-2, Bik, and pro-caspase 7 in INH7-treated MCF-7 cells. Data are reported as mean ± SD (n = 3) of the individual experiments. Statistical significance by Student's t-test: *P < 0.05 vs. Ctrl; **P < 0.001 vs. Ctrl.
Apoptosis analysis was carried out by flow cytometry. Treatment of MCF-7 cells with INH7 (8 µM) (Figure 2B) decreased the number of living cells from 83.4% ± 0.6% to 38.5% ± 2%, whereas the population of apoptotic cells increased from 9.3% ± 0.5% to 52.1% ± 1.5%; however, INH1 at the same concentration did not produce the same effect. In T47D cells, INH7 (8 µM) (Supplementary Figure S1E) increased the population of apoptotic cells from 5.5% ± 0.3% to 31.5% ± 1.3%, whereas the population of viable cells decreased from 90.2% ± 1.2% to 56.2% ± 0.1%. The INH7-induced statistically significant increase in apoptotic cells was evident from 5 µM in MCF-7 cells and T47D cells, which was in agreement with the MTT assay results. The results revealed that INH7 displayed a more significant apoptotic effect than INH1.
Western blot analysis was further carried out to detect the expression of the anti-apoptotic protein Bcl-2 and pro-apoptotic protein Bik (Figure 2C). The expression of Bcl-2 was downregulated 2.1-fold by INH7 (6 µM) vs. Ctrl (P < 0.001), whereas Bik was upregulated 1.5-fold by INH7 (6 µM) vs. Ctrl (P < 0.001). The expression of pro-caspase 7 was reduced 2.9-fold by INH7 (6 µM) vs. Ctrl (P < 0.001). These results suggest that INH7 induced apoptosis by suppressing Bcl-2 and inducing Bik, with subsequent activation of pro-caspase 7.
17β-HSD7. inhibitor induced MCF-7 xenograft tumor shrinkage
The effects of INH7 on tumor growth were investigated in a xenograft tumor model with MCF-7 cells in ovariectomized mice. The in vivo study was divided into three phases (Phase 1: from Day 0 to Day 21; Phase 2: from Day 21 to Day 28; Phase 3: from Day 28 to Day 45) (Figure 3A). Examination of the results from Phase 1 revealed that INH7 induced tumor shrinkage from 67 ± 8 mm3 to 26 ± 6 mm3 on Day 10 (P < 0.05 vs. Ctrl) and that the tumor volume continued to decrease to 18 ± 7 mm3 on Day 21 (P < 0.001 vs. Ctrl). The inhibitor was withdrawn from the INH7 group (E1 injection without administration of inhibitor) for 1 week during Phase 2 (from Day 21 to Day 28). Of interest was the observation that the tumor reduced by INH7 during Phase 1 regrew with inhibitor withdrawal and continued injection of E1. Tumors regrew to 38 ± 11 mm3 on Day 28, which approached the tumor volume in the Ctrl (E1 alone) group (48 ± 12 mm3). The regrown tumor from Phase 2 showed shrinkage after reinstatement of inhibitor injection during Phase 3. The inhibitor induced significant tumor reduction on Day 38 (23 ± 11 mm3, P < 0.05 vs. Ctrl), and until the termination of treatment on Day 45 (19 ± 9 mm3, P < 0.05). The average tumor weight decreased from 22 ± 3.4 g in the Ctrl group to 13.8 ± 4.3 g in the INH7-treated group, which was similar to that observed in the w/o group (10.5 ± 5.6 g). Mouse body weights were also recorded during the study period: body weights increased slightly over the 45-day treatment period in each group including the INH7 group (data not shown), indicating that there was no apparent toxicity by INH7 at 10 mg/kg/mouse/day (s.c. injection).
Figure 3.

In vivo study of INH7-treated xenograft MCF-7 breast tumor. (A) Regression of xenograft tumor growth in OVX nude mice by INH7. The growth regression curves represent the decrease in tumor volume in response to INH7 treatment. Black arrow represents the date of treatment interruption (Day 21). Blue arrow represents the treatment starting again on Day 28. Statistical significance by one-way ANOVA assay is shown by *P < 0.05. Tumor weight bar graph represents the shrinkage of tumor by INH7. Statistical significance by T-test is shown by *P < 0.05. (B) Pathohistological analysis of INH7-treated MCF-7 xenograft tumors. Representative images show histological tumor sections stained by hematoxylin–eosin–saffron (HES Stain) and Masson's Trichrome (MT Stain). The table reports the pathohistological scores of cancer cell organization and tumor stroma quantity. Bar graph represents cancer cell density within tissue. Statistical significance by Student's t-test: *P < 0.05. Scale bar, 40 µm. (C) Immunohistological analysis of 17β-HSD7 expression level within xenograft tumor tissue. Representative images of immune-reactivity by DAB (brown) reveal 17β-HSD7 expression within tumor tissue. Bar graph represents quantification results of immune-reactivity of 17β-HSD7 with ImageJ software. Statistical significance by Student's t-test: *P < 0.05. Scale bar, 100 µm. (D) The effect of INH7 on plasma E2 and DHT concentrations (left) and estrogen-responsive organ (uterus and vagina) weights (right) after treatment with INH7. Statistical significance by Student's t-test: *P < 0.05 vs. control (Ctrl).
17β-HSD7. inhibitor disrupted cancer cell organization and decreased cancer cell density within xenograft tissue
Cancer epithelial cells were typically organized in strands as shown in the E2 group (Figure 3B, HES stain) with less tumor stroma within tumor tissue (Figure 3B, MT stain). A similar histological organization was observed in the E1 injection group (Ctrl). However, in the INH7-treated group, the distribution of cancer cells was particularly dispersed with an abundance of tumor stroma, which were stained blue by MT stain. From a pathological viewpoint, ∼80% of cancer cells were organized in strands in Ctrl, whereas only 10% of cancer cells were organized as such in the INH7-treated group. Meanwhile, a large proportion of tumor cells (70%) in the INH7-treated group were sparsely distributed with a stromal reaction. The quantification of cancer cell density showed that INH7 treatment significantly decreased cancer cell density (density value 86 ± 7) within xenograft tissue compared with the Ctrl group (density value 108 ± 11) (P < 0.05).
17β-HSD7. inhibitor suppressed 17β-HSD7 expression within xenograft tumor tissue
Immunohistochemistry (IHC) was used to detect the expression of 17β-HSD7 enzyme within xenograft tissue (Figure 3C). Abundant expression of 17β-HSD7 was observed in the E2 group and Ctrl group, in contrast to the INH7-treated group and w/o group. The density of DAB staining was quantified with ImageJ software. The expression of 17β-HSD7 was significantly decreased within the INH7-treated group (DAB density value 83 ± 6) compared with Ctrl group (DAB density value 102 ± 4) (P < 0.05).
17β-HSD7. inhibitor modulated E2 and DHT levels in blood serum
E2 and DHT levels in murine blood serum were determined by ELISA (Figure 3D). The results indicated that the serum E2 level was 431.9 ± 7.7 pg/ml in the Ctrl group vs. 566.9 ± 8.1 pg/ml in the E2-supplemented group. However, INH7 significantly reduced the serum E2 level to 245 ± 11 pg/ml, which was comparable to that in the w/o group (255.6 ± 16 pg/ml). Meanwhile, INH7 significantly increased the serum DHT level to 458.8 ± 25 pg/ml from 288.9 ± 15 pg/ml in the Ctrl group. Estrogen-sensitive organs including the uterus and vagina were weighed (Figure 3D). The uterine and vaginal weights of the INH7-treated group were similar to the w/o group and markedly lower than those of the E2 group. However, there was no statistically significant difference between the Ctrl group and INH7 group. These results suggest that INH7 modulated circulating sex hormones without a systemic inhibitory effect on estrogen-sensitive organs under the experimental conditions using a physiological s.c. dose of E1 (see discussion).
Integrative datasets analysis: overexpression of 17β-HSD7 in invasive breast carcinoma
Integration of ‘TCGA (The Cancer Genome Atlas) breast cancer’ datasets that include 593 clinical samples from the Oncomine database (www.oncomine.org) confirmed significantly higher expression of 17β-HSD7 in various breast carcinomas compared with normal breast tissue (Figure 4A), with P-values and fold changes shown in Figure 4B. For example, comparison of invasive breast carcinoma (legend 3, 76 cases) vs. normal breast tissue showed that 17β-HSD7 was upregulated by 1.8-fold (P = 1.302 × 10−12). The expression of 17β-HSD7 was upregulated by 1.7-fold (P = 0.896 × 10−16) in invasive ductal breast carcinoma (legend 5, 392 cases) vs. normal breast tissue, and by 1.87-fold (P = 0.4 × 10−7) in invasive lobular breast carcinoma (legend 6, 36 cases) vs. normal breast tissue.
Figure 4.

Integrative analysis of 17β-HSD7 overexpression in neoplastic breast vs. normal breast type. (A) A box plot shows relative 17β-HSD7 expression in TCGA breast datasets grouped by cancer and normal tissue. The box reflects the interquartile range, the whiskers reflect the 10%–90% range, and the dots reflect the minimum and maximum values. Legend code indicates different cancer/normal type described in B. Horizontal axis units are normalized expression values (standard deviation above or below the medium per array). Data are normalized to facilitate inter-study comparison. The box plot was downloaded from ONCOMINE (Rhodes et al., 2004). (B) Comparison of 17β-HSD7 overexpression in different cancer types vs. normal breast tissue. P-value indicates the difference between two group comparisons. Fold change indicates the fold difference in 17β-HSD7 overexpression in cancer type vs. normal breast tissue.
Discussion
During the last half century, research and clinical efforts against breast cancer have resulted in two successful milestone therapies: the SERMs such as tamoxifen (Jordan, 2003) and AIs such as letrozole (Campos, 2004). Unfortunately, resistance to tamoxifen (Ring and Dowsett, 2004) and the associated potent induction of endometrial cancer (Wu et al., 2005) are common in patients with progressive cancer. Moreover, an increasing number of AI resistance have been observed in the clinical setting (Miller and Larionov, 2012), which has driven the urgent demand for new efficient drugs. However, currently there are no available inhibitors directed against the critical sulfatase pathway involved in E2 activation (Williams, 2013). Most recently, 667-Coumate, a promising steroid sulfatase inhibitor (Stanway et al., 2006), was investigated as an alternative approach for the treatment of hormone-dependent BC. Despite the fact that an associated Phase I clinical trial met with success, these results were not repeated in the Phase II trial (Williams, 2013). The principal enzymes in the sulfatase pathway (Aka et al., 2009) involved in the final step of E1 to E2 conversion are the reductive 17β-HSDs, including types 1, 7, and 12. The importance of targeting these enzymes has been recognized more recently as a potential therapeutic approach for hormone-dependent BC. However, as reported (Day et al., 2008), no involvement of 17β-HSD type 12 was found for ER+ BC in vitro or in vivo. In particular, the most significant efforts during recent years have centered on the design of 17β-HSD1 inhibitors. Progress has been made through rational inhibitor design based on the enzyme 3D-structure with the elimination of residual estrogenic activity in the inhibitors. Several inhibitors have been developed (Poirier, 2010), but no drugs have yet been approved due to the unavoidable estrogenic activity of inhibitors (Lin et al., 2013). Difficulties associated with the development of 17β-HSD1 inhibitors have prompted the search for alternative targets.
The multifunctional enzyme 17β-HSD7 involved in cholesterogenesis as well as steroidogenesis came to our attention. The recent description of the dual role of 17β-HSD1 in BC (Aka et al., 2010) further convinced us to study the function of 17β-HSD7 in BC in vitro and in vivo. During steroid biosynthesis, 17β-HSD7 converts E1 into E2, and to a significant extent, it inactivates DHT into the weak estrogen 3β-diol, thereby making it a dual intracrine regulator of the most potent estrogen and androgen (Törn et al., 2003). Although E2 and ERs are accepted as the predominant promoters in estrogen-dependent BC (Pearson et al., 1982), DHT can inhibit the proliferation of ER+ BC cells through the AR pathway (Greeve et al., 2004; Lanzino et al., 2010). Therefore, jointly targeting AR and ER pathways would generate a more favorable treatment outcome. Hence, 17β-HSD7 would constitute a novel endocrine therapeutic target in the blockade of E2 production with a concomitant increase of anti-proliferative efficacy induced by DHT.
By careful integration of 593 clinical microarrays by multiple centers and cohorts from the Oncomine database, we first confirmed the overexpression of 17β-HSD7 in various breast carcinomas. This confirmation further strengthens previous reports from BC biopsies (34 samples), that compared with 17β-HSD1, the expression of 17β-HSD7 was significantly increased by 3.5-fold in ER+ BC vs. normal tissue. Furthermore, such overexpression was correlated with an increased ratio of E2 to E1 in breast tumors compared with normal tissue (Haynes et al., 2010).
Moreover, the function of 17β-HSD7 in BC was carefully investigated by in vitro and in vivo models with a selective inhibitor (INH7). The in vitro study suggested that INH7 displayed dose-related cytostatic and cytotoxic effects toward ER+ BC cells similar to tamoxifen and AIs (Thiantanawat et al., 2003; Han et al., 2009). The cytostatic effect induced by anti-estrogen agents in MCF-7 cells was associated with cell cycle arrest in the G0/G1 phase due to estrogen blockage, resulting in a decrease in the relative proportion of cells synthesizing DNA during the S phase in ER+ (but not ER−) BC cells (Doisneau-Sixou et al., 2003; Dalvai and Bystricky, 2010). The G1/S cell cycle checkpoint is primarily monitored by the Cdk4–cyclin D1 complex as well as by cyclin-dependent kinase inhibitors such as p21waf1/cip1. CyclinD1, an estrogen-responsive gene, was established as an oncogene in ER+ BC (Arnold and Papanikolaou, 2005). CyclinD1 has recently been identified as a novel DHT response element by AR in ER+ BC cells (Lanzino et al., 2010). The cyclin-dependent kinase inhibitor p21, which was activated by DHT in MCF-7 cells, inhibits cell proliferation (Greeve et al., 2004). The interesting finding in the present study was that INH7 regulated both E2 and DHT; therefore, blockage of E2 induced a decrease in cyclin D1, and elevated DHT-stimulated p21 expression, producing a synergistic effect on cell cycle arrest in the G0/G1 phase. Another interesting observation was that INH7 downregulated the expression of 17β-HSD7 itself; however, INH1 had no effect on the expression of 17β-HSD1. Recently, Shehu et al. (2011) found that the expression of 17β-HSD7 is stimulated by E2 in a positive-feedback manner that ultimately promotes E2 biosynthesis within BC cells. Therefore, such downregulation may be generated by E2 blockage.
It was considered that the extent of cytostasis and/or apoptosis in vitro may depend on the drug concentration and the time course of drug administration (Rixe and Fojo, 2007). By regulating the Bcl-2 family, tamoxifen and/or AIs induced apoptosis in ER+ BC cells. The anti-apoptotic protein Bcl-2 is overexpressed in ER+ BC cells, protecting MCF-7 cells against apoptosis (Martin and Dowsett, 2013), while the pro-apoptotic Bik was induced by anti-estrogens in ER+ BC cells to promote apoptosis (Hur et al., 2004). INH7 displayed remarkable apoptotic effects by suppressing the anti-apoptotic protein Bcl-2 and elevating the pro-apoptotic protein Bik. However, caspase 3 was not expressed in MCF-7 cells, and activation of apoptosis was alternatively induced by caspase 7 (Jänicke, 2009). The results demonstrated that INH7 significantly decreased the expression level of pro-caspase 7 compared with Ctrl (Figure 2C). Results from the present study demonstrate that INH7 decreased the expression of Bcl-2 and induced the expression of Bik, thereby promoting apoptosis in MCF-7 cells.
The present in vivo xenograft model was established by supplying a physiological concentration of E1 according to the estrogen concentration in the circulation of postmenopausal women, instead of providing a hyper-physiological concentration of E1 as applied in other studies (Day et al., 2008; Ayan et al., 2012). This approach was adopted in order to better simulate physiological estrogen status in postmenopausal women with BC. Results from the in vivo study suggest that tumor volume was reduced by specific inhibition of 17β-HSD7, which correlated with decreased E2 and increased DHT levels in plasma. In addition, IHC analysis of 17β-HSD7 within tumor tissue showed that INH7 suppressed the expression of 17β-HSD7, in agreement with the in vitro study. Furthermore, uterine and vaginal weights were increased by E2 injection, but treatment with INH7 had no such effect although INH7 decreased tumor volume in the xenograft. Similar results were observed in a study with the 17β-HSD1 inhibitor (Day et al., 2008; Ayan et al., 2012). This may indicate that INH7 activity was directed against human 17β-HSD7 in the xenograft tumor rather than the murine enzyme present in the uterus. Thus, both in vitro and in vivo studies demonstrate that INH7 can block E2 production, restore DHT, and suppress enzyme expression, resulting in xenograft tumor shrinkage.
In conclusion, inhibition of 17β-HSD7 modulated sex hormones and produced a synergistic suppression of BC cell growth. Furthermore, blockage of E2 conversely attenuated the expression of 17β-HSD7 itself. These combined effects could magnify the inhibitory effect of 17β-HSD7 (Figure 5). Therefore, through in vitro and in vivo study with analysis of clinical datasets, we provide the first demonstration that 17β-HSD7 is a novel target for ER+ BC through the dual regulation of estrogen and androgen.
Figure 5.

Schematic mechanisms of INH7 toward ER+ breast cancer cells. Inhibition of 17β-HSD7 by INH7 blocks E2 formation from E1 and inhibits DHT conversion into the weak estrogen metabolite (3β-diol). Furthermore, blockage of E2 conversely attenuates the expression of 17β-HSD7 itself. A decrease in E2 weakens the estrogenic effect through ER and an increase in DHT strengthens androgenic effect through AR, synergistically leading to post-receptor gene transcription culminating in cytostatic and/or cytotoxic effects.
Materials and methods
Inhibitors and chemicals
The inhibitors E2B-Methoxy against 17β-HSD1 (INH1) (Laplante et al., 2008) and EB-357-030 against 17β-HSD7 (INH7) (Bellavance et al., 2009) were chemically synthesized and biologically selected. The chemical structures and enzyme specificities are shown in Table 1. Stock solutions were prepared by dissolving the compounds in ethanol, and these were diluted to the desired concentrations with culture medium for in vitro assays. Dilution of the stock solutions for in vivo assays was performed with 0.4% methylcellulose solution (e.g. 0.4% methylcellulose and ethanol; 92:8 v/v).
Cell culture
The ER+ BC cell lines MCF-7 and T47D, and ER− BC cell line BT-20, which all express 17β-HSD1 and 17β-HSD7 (Laplante et al., 2009; Aka and Lin, 2012) in addition to ER and AR (Neve et al., 2006) (Table 2), were obtained from the American Type Culture Collection (ATCC). MCF-7, T47D, and BT-20 cells were propagated according to a previous protocol (Aka and Lin, 2012). All culture media were phenol red-free. Medium containing 10% dextran-coated charcoal-stripped fetal bovine serum (FBS; PAA) was used as the protocol medium. Charcoal-stripped FBS was obtained as before (Aka and Lin, 2012). Cells were plated in protocol medium for 24 h before testing. After 24 h (experimental Day 0), the medium was replaced with fresh protocol medium containing the desired concentrations of test compounds and 0.1 nM E1 as the substrate. This concentration of E1 is close to the physiological intracellular concentration in BC cells (Pasqualini et al., 1996). Medium containing E1 without test compounds was set as treatment control (Ctrl); medium without E1 or E2 was set as the baseline control (w/o); and medium containing 0.1 nM E2 was set as the reference control (E2).
Cell proliferation assay
The most accurate parameter for analyzing cell proliferation is the measurement of DNA synthesis (Quent et al., 2010). CyQUANT cell proliferation assay kit (Molecular Probes, Invitrogen) was used to determine the anti-proliferative effects of INH7 or INH1. Cells were plated into 96-well plates with 1 × 103 cells/well, and were treated with compounds at concentrations ranging from 0.2 to 2 µM (IC50 to 10 × IC50 in Table 1) for 4 days. Medium was renewed every 2 days. Cell culture supernatants were pooled and stored frozen at −80°C for determining E2 and DHT concentrations. On Day 4, the plates were washed with PBS and frozen at −80°C. Cell proliferation in response to E1 supplementation (Ctrl) was fixed as 100%. Data were reported as the percentage (%) of DNA synthesis vs. Ctrl (100%). Each condition was performed in quadruplicate and experiments were repeated three times.
Cell cycle analysis by flow cytometry
Click-iTEdu assay kit and propidium iodide (PI) (Molecular Probes, Invitrogen) were used to perform cell cycle assay. Cells were seeded in 6-wells plates with 0.5 × 105 cells/well, and were treated with compounds at concentrations ranging from 0.2 to 2 µM for 4 days. Medium was renewed every 2 days. On Day 4, cells were treated according to manufacturer's instructions. Cells were analyzed by flow cytometry using a BD FACSCanto II instrument (BD Bioscience). The percentage of cells in the G0/G1, S, and G2/M phases was calculated using Multi Cycle AV software (Phoenix Flow Systems). Data were reported as the percentage of living cells (G0/G1, S, and G2/M = 100%). Each condition was performed in triplicate and experiments were repeated three times.
Determination of E2 and DHT levels
Collected cell culture supernatants or blood specimens from in vivo study were thawed and vortexed before testing. The levels of E2 and DHT in the culture supernatants were determined using the Estradiol EIA Kit (Cayman Chemical; Cross-reactivities toward E2 and E1 were 100% and 12%, respectively) and DHT ELISA Kit (Alpha Diagnostic International; Cross-reactivities toward DHT and E1 were 100% and <0.01%, respectively) according to the manufacturers' instructions. Data were reported as concentrations (nM) per well. Each condition was performed in quadruplicate and experiments were repeated three times.
Cell viability assay using MTT
Cell viability assays were performed with the MTT [3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide] kit (ATCC). Cells were plated into 96-wells plates with 3 × 103 cells/well, and were treated with compounds at concentrations ranging from 2 to 8 µM (10 × IC50 to 40 × IC50 in Table 1) for 4 days. Medium was renewed every 2 days. On Day 4, plates were processed according to the manufacturer's instructions. Cell viability in response to E1 supplementation (Ctrl) was fixed as 100%. Data were reported as percentage of viable cells vs. the Ctrl (100%). Each condition was performed in quadruplicate and experiments were repeated three times.
Analysis of cellular apoptosis by flow cytometry
Cellular apoptosis was evaluated by flow cytometry using Annexin V and PI dyes (Molecular Probes, Invitrogen). Cells were plated into 6-well plates with 1 × 105 cells/well and treated with compounds at concentrations ranging from 2 to 8 µM. Media were renewed every 2 days. On Day 4, the adherent and non-adherent cells were pooled, washed, and then incubated on ice for 30 min in 500 µl binding buffer containing annexin V (5 µl) and PI (1 µl). The samples were analyzed by flow cytometry on a BD FACS Canto II instrument (BD Bioscience). Data were reported as percentage of living cells, apoptotic cells, or late apoptotic/necrotic cells among total detected cells. Each condition was performed in triplicate and experiments were repeated three times.
Western blotting
MCF-7 cells were treated with INH7 or INH1 at 2 µM (10 × IC50) or 6 µM (30 × IC50) for 4 days. Total protein was extracted with RIPA buffer (Sigma) containing a protease inhibitor cocktail (Calbiochem). Equal amounts of protein (40 µg) were separated by 15% SDS–PAGE. The primary antibodies included anti-17β-HSD1 (ab27501), anti-17β-HSD7 (ab112006), anti-p21 (ab109199), anti-cyclin D1 (ab134175), anti-Bik (ab52182), anti-Bcl-2 (ab32124), anti-pro caspase 7 (ab32522), and anti-actin (ab3280; Abcam), as well as anti-proliferating cell nuclear antigen (PCNA) (sc-7907; Santa Cruz Biotechnology). The secondary antibodies used were goat-anti-rabbit IgG-HRP (sc-2004) and goat-anti-mouse IgG-HRP (sc-2005; Santa Cruz Biotechnology). Blots were visualized with enhanced Chemi-luminescence (ECL plus™ Western Blotting Detection Kit, PerkinElmer) and quantified using ImageJ software. The ratios between the proteins of interest and β-actin were calculated to determine the relative protein expression values. Each condition was performed in triplicate and experiments were repeated three times.
MCF-7 xenograft tumor studies
Animal experiments were approved by the local Institutional Animal Care Committee and were conducted in agreement with the guidelines of the Canadian Council for Animal Care. Female athymic nude mice (4 weeks) (FOxnnu, Harlan Laboratories, Montreal, Canada) were ovariectomized under isoflurane-induced anesthesia. Estradiol implants with an average release of 0.1 µg E2/day over a 30-day period were subcutaneously inserted at the time of ovariectomy. The E2 implants were prepared according to a previous protocol (Gutman et al., 1999). One week after ovariectomy, 1 × 106 MCF-7 cells suspended in a 100 µl mixture of 1:3 (v/v) Matrigel (BD) and PBS were administered by s.c. injection on one side of each mouse. The E2 implants were removed once the average tumor volume reached 100 mm3 (after 3 weeks), and were substituted by daily s.c. injection of E2 or E1 at 100 pg/mouse/day, which was equivalent to the physiological concentration of estrogen in postmenopausal women (Woolcott et al., 2010), for the remaining weeks with the exception of the w/o group (s.c injection of PBS), in order to maintain the xenograft tumor and to provide an optimized model that mimics the physiological hormonal status of BC in postmenopausal women. Mice were distributed into four groups after 1 week of E1 injection on the basis of similar average tumor volume. No statistical method was used to predetermine tumor size. Ten mice were assigned to each group (E2 group: vehicle and injection of 100 pg E2/mouse/day; Ctrl group: vehicle and injection of 100 pg E1/mouse/day; w/o group: vehicle and PBS injection; INH7 group: 10 mg/kg/mouse/day of INH7 with injection of 100 pg E1/mouse/day). On Day 0, INH7 or vehicle solution (0.4% methylcellulose and ethanol; 92:8 v/v) was administered by s.c. injection, and either E2 or E1, or PBS was injected into a separate s.c. site at the same time. Body weight and tumor volumes were determined twice weekly. Tumor volumes were determined by measuring the greatest longitudinal diameter (length) and the greatest transverse diameter (width) with external calipers and calculating with the modified ellipsoidal formula: Tumor volume =0.52 × (length × width2). Treatment with INH7 was interrupted on Day 21 for 1 week and recommenced on Day 28 until Day 45. The animals were euthanized on Day 45, and tumor specimens and blood samples were collected. The estrogen-sensitive organs (uterus and vagina) were removed and excised of fat and weighed. Tumor specimens were photographed and weighed before fixation for histological assessment.
Histopathological analysis of xenograft tumor tissue
Tumor specimens were fixed in 4% paraformaldehyde and were embedded in paraffin and then sections (4 µM) were processed. Tissue sections were stained with standard hematoxylin–eosin–saffron (HES) stain. The same section was processed with Masson's Trichrome (MT) stain to observe the density of cancer cells vs. tumor stroma. Two independent observers assessed the tumor section with regard to the formation of assembled (in strands) vs. sparsely distributed epithelial cancer cells within the tumor. Tumor stroma reactions around cancer cells were also assessed quantitatively. ImageJ software was used to quantify the cancer cell density vs. tumor stroma density, and results were reported as grey levels within the image based on MT-stained sections.
Immunohistological analysis of 17β-HSD7 expression level within xenograft tumor tissue
Immunohistological (IHC) analysis was performed according to the standard IHC-paraffin protocol from Abcam. The sections were then incubated with anti-17β-HSD7 (ab112006) at 4°C overnight. Immuno-reactivity was detected by the rabbit-specific HRP/DAB (ABC) Detection IHC Kit (ab64261) from Abcam. Hematoxylin stain was applied as counter stain. ImageJ software was used to quantify the density of immune reaction and data were reported as grey levels within the image.
Integrative analysis of clinical datasets
Oncomine database (https://www.oncomine.org), which is a cancer microarray database and web-based data-mining platform, was interrogated to validate the expression status of 17β-HSD7 in various types of BC. Filter indexes were set in Oncomine based on research interests. Primary filters were set as differential analysis (cancer vs. normal) and cancer type (breast cancer). Sample filters were set as demographic (female, postmenopausal). Dataset filters were set as data type (mRNA) and dataset size (>150 samples). Datasets were ordered by overexpression with P-value. Datasets were set by P-value (1E-6) and fold change (2+). The datasets were then sorted to analyze the expression of 17β-HSD7 associated with different cancer types vs. normal breast tissue.
Statistical analysis
Analysis by one-way ANOVA was used to determine the significance of differences observed in the in vivo data. Multiple comparisons were performed with the Student–Newman–Keuls method. The Student's t-test was applied when only two groups were compared. Data are presented as mean ± SD. P-values that were < 0.05 were considered as statistically different, and P < 0.001 was considered as significantly different.
Supplementary material
Supplementary Material is available at Journal of Molecular Cell Biology online.
Funding
This work was supported by operating grants from Canadian Institutes of Health Research (CIHR, MOP 97917 to S.-X.L., D.P., and C.J.D.; MOP 89851 to S.-X.L. and D.P.) and China Scholarship Council (PhD Fellowship, #2010621032 to X.Q.W.).
Conflict of interest: none declared.
Supplementary Material
Acknowledgements
The authors acknowledge Dr Muriel Kelly (Scientific Editing Service, Madbury, NH, USA) for manuscript editing.
References
- Adams J., Garcia M., Rochefort H. (1981). Estrogenic effects of physiological concentrations of 5-androstene-3 β, 17β-diol and its metabolism in MCF7 human breast cancer cells. Cancer Res. 41, 4720–4726. [PubMed] [Google Scholar]
- Aka J.A., Lin S.X. (2012). Comparison of functional proteomic analyses of human breast cancer cell lines T47D and MCF7. PLoS One 7, e31532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aka J.A., Mazumdar M., Lin S.X. (2009). Reductive 17β-hydroxysteroid dehydrogenases in the sulfatase pathway: critical in the cell proliferation of breast cancer. Mol. Cell. Endocrinol. 301, 183–190. [DOI] [PubMed] [Google Scholar]
- Aka J.A., Mazumdar M., Chen C.Q., et al. (2010). 17β hydroxysteroid dehydrogenase type 1 stimulates breast cancer by dihydrotestosterone inactivation in addition to estradiol production. Mol. Endocrinol. 24, 832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold A., Papanikolaou A. (2005). Cyclin D1 in breast cancer pathogenesis. J. Clin. Oncol. 23, 4215–4224. [DOI] [PubMed] [Google Scholar]
- Ayan D., Maltais R., Roy J., et al. (2012). A new nonestrogenic steroidal inhibitor of 17β-hydroxysteroid dehydrogenase type I blocks the estrogen-dependent breast cancer tumor growth induced by estrone. Mol. Cancer Ther. 11, 2096–2104. [DOI] [PubMed] [Google Scholar]
- Bellavance E., Luu-The V., Poirier D. (2009). Potent and selective steroidal inhibitors of 17β-hydroxysteroid dehydrogenase type 7, an enzyme that catalyzes the reduction of the key hormones estrone and dihydrotestosterone. J. Med. Chem. 52, 7488–7502. [DOI] [PubMed] [Google Scholar]
- Breitling R., Krazeisen G., Moller G., et al. (2001). 17β-hydroxysteroid dehydrogenase type 7- an ancient 3-ketosteroid reductase of cholesterogenesis. Mol. Cell. Endocrinol. 171, 199–204. [DOI] [PubMed] [Google Scholar]
- Campos S.M. (2004). Aromatase inhibitors for breast cancer in postmenopausal women. Oncologist 9, 126–136. [DOI] [PubMed] [Google Scholar]
- Chen J., Wang W.Q., Lin S.X. (2013). Interaction of Androst-5-ene-3β,17β-diol and 5α-androstane-3β,17β-diol with estrogen and androgen receptors: a combined binding and cell study. J. Steroid Biochem. Mol. Biol. 137, 316–321. [DOI] [PubMed] [Google Scholar]
- Dalvai M., Bystricky K. (2010). Cell cycle and anti-estrogen effects synergize to regulate cell proliferation and ER target gene expression. PLoS One 5, e11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day J.M., Foster P.A., Tutill H.J., et al. (2008). 17β-hydroxysteroid dehydrogenase Type 1, and not Type 12, is a target for endocrine therapy of hormone-dependent breast cancer. Int. J. Cancer 122, 1931–1940. [DOI] [PubMed] [Google Scholar]
- Doisneau-Sixou S.F., Sergio C.M., Carroll J.S., et al. (2003). Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr. Relat. Cancer 10, 179–186. [DOI] [PubMed] [Google Scholar]
- Duan W.R., Parmer T.G., Albarracin C.T., et al. (1997). PRAP, a prolactin receptor associated protein: its gene expression and regulation in the corpus luteum. Endocrinology 138, 3216–3221. [DOI] [PubMed] [Google Scholar]
- Gangloff A., Shi R., Nahoum V., et al. (2003). Pseudo-symmetry of C19 steroids, alternative binding orientations, and multispecificity in human estrogenic 17β-hydroxysteroid dehydrogenase. FASEB J. 17, 274–276. [DOI] [PubMed] [Google Scholar]
- Greeve M.A., Allan R.K., Harvey J.M., et al. (2004). Inhibition of MCF-7 breast cancer cell proliferation by 5α-dihydrotestosterone; a role for p21Cip1/Waf1. J. Mol. Endocrinol. 32, 793–810. [DOI] [PubMed] [Google Scholar]
- Gutman M., Couillard S., Labrie F., et al. (1999). Effects of the antiestrogen EM-800 (SCH 57050) and cyclophosphamide alone and in combination on growth of human ZR-75–1 breast cancer xenografts in nude mice. Cancer Res. 59, 5176–5180. [PubMed] [Google Scholar]
- Han P., Kang J.H., Li H.L., et al. (2009). Antiproliferation and apoptosis induced by tamoxifen in human bile duct carcinoma QBC939 cells via up regulated p53 expression. Biochem. Biophys. Res. Commun. 385, 251–256. [DOI] [PubMed] [Google Scholar]
- Hanamura T., Niwa T., Nishikawa S., et al. (2013). Androgen metabolite-dependent growth of hormone receptor-positive breast cancer as a possible aromatase inhibitor-resistance mechanism. Breast Cancer Res. Treat. 139, 731–740. [DOI] [PubMed] [Google Scholar]
- Haynes B.P., Straume A.H., Geisler J., et al. (2010). Intratumoral estrogen disposition in breast cancer. Clin. Cancer Res. 16, 1790–1801. [DOI] [PubMed] [Google Scholar]
- Hu R., Dawood S., Holmes M.D., et al. (2011). Androgen receptor expression and breast cancer survival in postmenopausal women. Clin. Cancer Res. 17, 1867–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur J., Chesnes J., Coser K.R., et al. (2004). The Bik BH3-only protein is induced in estrogen-starved and antiestrogen-exposed breast cancer cells and provokes apoptosis. Proc. Natl Acad. Sci. USA 101, 2351–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jänicke R.U. (2009). MCF-7 breast carcinoma cells do not express caspase-3. Breast Cancer Res. Treat. 117, 219–221. [DOI] [PubMed] [Google Scholar]
- Jemal A., Bray F., Center M.M., et al. (2011). Global cancer statistics. CA Cancer J. Clin. 61, 69–90. [DOI] [PubMed] [Google Scholar]
- Jonat W., Pritchard K.I., Sainsbury R., et al. (2006). Trends in endocrine therapy and chemotherapy for early breast cancer: a focus on the premenopausal patient. J. Cancer Res. Clin. Oncol. 132, 275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan V.C. (2003). Tamoxifen: a most unlikely pioneering medicine. Nat. Rev. Drug Discov. 2, 205–213. [DOI] [PubMed] [Google Scholar]
- Krazeisen A., Breitling R., Imai K., et al. (1999). Determination of cDNA, gene structure and chromosomal localization of the novel human 17β-hydroxysteroid dehydrogenase type 7. FEBS Lett. 460, 373–379. [DOI] [PubMed] [Google Scholar]
- Labrie F. (1991). Intracrinology. Mol. Cell. Endocrinol. 78, C113–C118. [DOI] [PubMed] [Google Scholar]
- Labrie F. (2003). Extragonadal synthesis of sex steroids: intracrinology. Ann. Endocrinol. 64, 95–107. [PubMed] [Google Scholar]
- Lanzino M., Sisci D., Morelli C., et al. (2010). Inhibition of cyclin D1 expression by androgen receptor in breast cancer cells—identification of a novel androgen response element. Nucleic Acids Res. 38, 5351–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante Y., Cadot C., Fournier M.A., et al. (2008). Estradiol and estrone C-16 derivatives as inhibitors of type 1 17β-hydroxysteroid dehydrogenase: blocking of ER+ breast cancer cell proliferation induced by estrone. Bioorg. Med. Chem. 16, 1849–1860. [DOI] [PubMed] [Google Scholar]
- Laplante Y., Rancourt C., Poirier D. (2009). Relative involvement of three 17β-hydroxysteroid dehydrogenases (types 1, 7 and 12) in the formation of estradiol in various breast cancer cell lines using selective inhibitors. Mol. Cell. Endocrinol. 301, 146–153. [DOI] [PubMed] [Google Scholar]
- Liehr J.G. (2000). Is estradiol a genotoxic mutagenic carcinogen? Endocr. Rev. 21, 40–54. [DOI] [PubMed] [Google Scholar]
- Lin S.X., Chen J., Mazumdar M., et al. (2010). Molecular therapy of breast cancer: progress and future directions. Nat. Rev. Endocrinol. 6, 485–493. [DOI] [PubMed] [Google Scholar]
- Lin S.X., Poirier D., Adamski J. (2013). A challenge for medicinal chemistry by the 17β-hydroxysteroid dehydrogenase superfamily: an integrated biological function and inhibition study. Curr. Top. Med. Chem. 13, 1164–1171. [DOI] [PubMed] [Google Scholar]
- Marijanovic Z., Laubner D., Moller G., et al. (2003). Closing the gap: identification of human3-ketosteroid reductase, the last unknown enzyme of mammalian cholesterol biosynthesis. Mol. Endocrinol. 17, 1715–1725. [DOI] [PubMed] [Google Scholar]
- Martin L.A., Dowsett M. (2013). BCL-2: a new therapeutic target in estrogen receptor-positive breast cancer? Cancer Cell 24, 7–9. [DOI] [PubMed] [Google Scholar]
- Miller W.R., Larionov A.A. (2012). Understanding the mechanisms of aromatase inhibitor resistance. Breast Cancer Res. 14, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve R.M., Chin K., Fridlyand J., et al. (2006). A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeier L.A., Dabbs D.J., Beriwal S., et al. (2010). Androgen receptor in breast cancer: expression in estrogen receptor-positive tumors and in estrogen receptor-negative tumors with apocrine differentiation. Mod. Pathol. 23, 205–212. [DOI] [PubMed] [Google Scholar]
- Nokelainen P., Peltoketo H., Vihko R., et al. (1998). Expression cloning of a novel estrogenic mouse 17β-hydroxysteroid dehydrogenase/17-ketosteroid reductase (m17HSD7), previously described as a prolactin receptor-associated protein (PRAP) in rat. Mol. Endocrinol. 12, 1048–1059. [DOI] [PubMed] [Google Scholar]
- Ohnesorg T., Keller B., HrabédeAngelis M., et al. (2006). Transcriptional regulation of human and murine 17β-hydroxysteroid dehydrogenase type-7 confers its participation in cholesterol biosynthesis. J. Mol. Endocrinol. 37, 185–197. [DOI] [PubMed] [Google Scholar]
- Pasqualini J.R. (2004). The selective estrogen enzyme modulators in breast cancer: a review. Biochim. Biophys. Acta 1654, 123–143. [DOI] [PubMed] [Google Scholar]
- Pasqualini J.R., Chetrite G., Blacker C., et al. (1996). Concentrations of estrone, estradiol, and estrone sulfate and evaluation of sulfatase and aromatase activities in pre-and post-menopausal breast cancer patients. J. Clin. Endocrinol. Metab. 81, 1460–1464. [DOI] [PubMed] [Google Scholar]
- Payne A.H., Hales D.B. (2004). Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 25, 947–970. [DOI] [PubMed] [Google Scholar]
- Pearson O.H., Manni A., Arafah B.M. (1982). Antiestrogen treatment of breast cancer: an overview. Cancer Res. 42, 3424–3429. [PubMed] [Google Scholar]
- Poirier D. (2010). 17β-Hydroxysteroid dehydrogenase inhibitors: a patent review. Expert Opin. Ther. Pat. 20, 1123–1145. [DOI] [PubMed] [Google Scholar]
- Quent V.M., Loessner D., Friis T., et al. (2010). Discrepancies between metabolic activity and DNA content as tool to assess cell proliferation in cancer research. J. Cell. Mol. Med. 14, 1003–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes D.R., Yu J., Shanker K., et al. (2004). ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 6, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring A., Dowsett M. (2004). Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer 11, 643–658. [DOI] [PubMed] [Google Scholar]
- Rixe O., Fojo T. (2007). Is cell death a critical end point for anticancer therapies or is cytostasis sufficient? Clin. Cancer Res. 13, 7280–7287. [DOI] [PubMed] [Google Scholar]
- Russo I.H., Russo J. (1998). Role of hormones in mammary cancer initiation and progression. J. Mammary Gland Biol. Neoplasia 3, 49–61. [DOI] [PubMed] [Google Scholar]
- Shehu A., Mao J., Gibori G.B., et al. (2008). Prolactin receptor-associated protein/17β-hydroxysteroid dehydrogenase type 7 gene (Hsd17b7) plays a crucial role in embryonic development and fetal survival. Mol. Endocrinol. 22, 2268–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehu A., Albarracin C., Devi Y.S., et al. (2011). The stimulation of HSD17B7 expression by estradiol provides a powerful feed-forward mechanism for estradiol biosynthesis in breast cancer cells. Mol. Endocrinol. 25, 754–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora M.J., Cordero K.E., Larios J.M., et al. (2009). The androgen metabolite 5α-androstane-3β,17β-diol (3βAdiol) induces breast cancer growth via estrogen receptor: implications for aromatase inhibitor resistance. Breast Cancer Res. Treat. 115, 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanway S.J., Purohit A., Woo L.W., et al. (2006). Phase I study of STX 64 (667 Coumate) in breast cancer patients: the first study of a steroid sulfatase inhibitor. Clin. Cancer Res. 12, 1585–1592. [DOI] [PubMed] [Google Scholar]
- Thiantanawat A., Long B.J., Brodie A.M. (2003). Signaling pathways of apoptosis activated by aromatase inhibitors and antiestrogens. Cancer Res. 63, 8037–8050. [PubMed] [Google Scholar]
- Törn S., Nokelainen P., Kurkela R., et al. (2003). Production, purification, and functional analysis of recombinant human and mouse 17β-hydroxysteroid dehydrogenase type 7. Biochem. Biophys. Res. Commun. 305, 37–45. [DOI] [PubMed] [Google Scholar]
- Vera-Badillo F.E., Templeton A.J., de Gouveia P., et al. (2014). Androgen receptor expression and outcomes in early breast cancer: a systematic review and meta-analysis. J. Natl Cancer Inst. 106, 319. [DOI] [PubMed] [Google Scholar]
- Williams S.J. (2013). Sulfatase inhibitors: a patent review. Expert Opin. Ther. Pat. 23, 79–98. [DOI] [PubMed] [Google Scholar]
- Woolcott C.G., Shvetsov Y.B., Stanczyk F.Z., et al. (2010). Plasma sex hormone concentrations and breast cancer risk in an ethnically diverse population of postmenopausal women: the Multiethnic Cohort Study. Endocr. Relat. Cancer 17, 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Chen Y., Liang J., et al. (2005). Hypomethylation-linked activation of PAX2 mediates tamoxifen-stimulated endometrial carcinogenesis. Nature 438, 981–987. [DOI] [PubMed] [Google Scholar]
- Yager J.D., Davidson N.E. (2006). Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 354, 270–282. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.