

Abstract
G protein-coupled receptor (GPCR) family C group 6 member A (GPRC6A) is a multiligand GPCR that is activated by cations, L-amino acids, and osteocalcin. GPRC6A plays an important role in the regulation of testosterone (T) production and energy metabolism in mice. T has rapid, transcription-independent (nongenomic) effects that are mediated by a putative GPCR. We previously found that T can activate GPRC6A in vitro, but the possibility that T is a ligand for GPRC6A remains controversial. Here, we demonstrate direct T binding to GPRC6A and construct computational structural models of GPRC6A that are used to identify potential binding poses of T. Mutations of the predicted binding site residues were experimentally found to block T activation of GPRC6A, in agreement with the modeling. Using Gpr6ca−/− mice, we confirmed that loss of GPRC6A resulted in loss of T rapid signaling responses and elucidated several biological functions regulated by GPRC6A-dependent T rapid signaling, including T stimulation of insulin secretion in pancreatic islets and enzyme expression involved in the biosynthesis of T in Leydig cells. Finally, we identified a stereo-specific effect of an R-isomer of a selective androgen receptor modulator that is predicted to bind to and shown to activate GPRC6A but not androgen receptor. Together, our data show that GPRC6A directly mediates the rapid signaling response to T and uncovers previously unrecognized endocrine networks.
Classically, testosterone (T) stimulates target gene expression through the nuclear androgen receptor (AR). T is converted by 5α-reductase to dihydrotestosterone, which binds with high affinity to AR to form a transcription factor complex that translocates to the nucleus to activate androgen-responsive genes. Emerging clinical observations indicate that the conversion of T to dihydrotestosterone is not obligatory and that T has clinically important functions distinct from activation of AR-mediated gene transcription in some tissues (1, 2).
One such alternative pathway is T activation of rapid, transcription-independent signaling responses through a putative membrane G protein-coupled receptor (GPCR). The identity of this T-sensing GPCR is not known and its physiological significance remains uncertain (3).
GPCR family C group 6 member A (GPRC6A) is a member of the class-C GPCR family (4–6), which includes the calcium-sensing receptor (CasR). Like CasR, GPRC6A is activated by cations and L-amino acids; however, the physiological ligand for GPRC6A is thought to be the bone-derived hormone osteocalcin (Ocn) (5, 7, 8). Both GPRC6A and Ocn are involved in the regulation of energy metabolism and T production in mice (9–11), and Gprc6a−/− and Ocn−/− mice are phenocopies (5, 12). Several studies have shown that Ocn activates GPRC6A in peripheral tissues, such as pancreatic β-cells and Leydig cells in the testes (11, 13, 14), but whether Ocn binds to and directly activates GPRC6A remains controversial.
GPRC6A is also a candidate for this T-sensing GPCR (15, 16). GPRC6A is widely expressed, including in β-cells, bone marrow stromal cells, monocytes, prostate cancer cells, skeletal muscle cells, keratinocytes, and Leydig cells, as well as in other tissues known to be targeted by T (12, 13, 15, 17–19). In this regard, overexpression of GPRC6A imparts the ability of extracellular T to illicit a rapid signaling response in HEK-293 cells, which lack the AR (14, 15). Moreover, GPRC6A loss-of-function attenuates T-stimulated rapid signaling in multiple tissues. Indeed, T rapid signaling is inhibited in bone marrow stromal cells derived from Gprc6a−/− mice and in 22Rv1 prostate cancer cells after siRNA-mediated knockdown of GPRC6A. There is also indirect evidence that T regulates LH secretion (15), stimulates insulin secretion in β-cells, and induces dihydronicotinamide-adenine dinucleotide phosphate oxidase isozymes in keratenocytes through GPRC6A (14, 16). The role of GPRC6A in mediating the effects of T also is controversial, because the binding sites for T in GPRC6A have not been identified, and some studies have failed to confirm T activation of GPRC6A in vitro (20). Moreover, the physiological roles of T activation of GPRC6A have not been defined. Indeed, the global Gprc6a−/− mouse phenotype is complex and has multiple hormonal abnormalities that might indirectly account for the effects attributed to T activation of GPRC6A (12).
To explore whether GPRC6A is a T-sensing GPCR, we have performed competitive binding assays demonstrating direct T binding to GPRC6A and have constructed computational structural models of GPRC6A to identify potential binding site and poses of T. Functional studies and mutagenesis experiments confirmed T activation of GPRC6A in cell models lacking AR. In addition, we have identified selective AR modulators (SARMs) that are predicted to bind to GPRC6A and that activate GPRC6A signaling in vitro. Finally, we directly demonstrated a physiological role of GPRC6A in mediating T effects in regulating β-cell and Leydig functions in cells derived from Gprc6a−/− mice.
Materials and Methods
Animals
Generation of global Gprc6a−/− mice has been previously reported (12). Mice were maintained and used in accordance with recommendations as described (National Research Council 1985; Guide for the Care and Use of Laboratory Animals DHHS Publication NIH 86–23, Institute on Laboratory Animal Resources, Rockville, MD) and following guidelines established by the University of Tennessee Health Science Center Institutional Animal Care and Use Committee. The animal study protocol was approved by the Institutional Review Board at University of Tennessee Health Science Center Institutional Animal Care and Use Committee.
Reagents and antibodies
Insulin (Mouse) Ultrasensitive ELISA kit and mouse C-peptide ELISA kit were obtained from ALPCO Diagnostics. T, glucose and insulin were purchased from Sigma.
Cell culture
All culture reagents were from Invitrogen. Human embryonic kidney HEK-293 cells were obtained from American Type Culture Collection. HEK-293 cells stably transfected with pcDNA3.mGPRC6A were created as previously described (15, 21).
Mouse islets isolation and ligand stimulation
Primary islets were isolated using modified method as described (22, 23). Briefly, after dissection and mincing, pancreata were digested with 3 mL/pancreas of a collagenase P (1 mg/mL; Roche) solution in complete Hank's Balanced Salt Solution (HBSS) (HBSS 1× supplemented with 20mM HEPES [pH 7.4] and 2mM CaCl2) for 15 minutes in a 37°C shaking water bath. Islets were subsequently purified through a Histopaque 1083 density centrifugation (Sigma). After centrifugation, the islet layer was transferred into petri dishes with wash buffer (HBSS with 10mM HEPES and 1% fetal bovine serum (FBS); Invitrogen), then handpicked and cultured in low-glucose medium (RPMI 1640 with 5.6mM glucose; Invitrogen) for 1 hour before being treated for 1 hour with T (80nM). The insulin stimulation index (SI) was calculated as the ratio of media insulin concentrations in T or Ocn divided by the insulin concentration in low glucose conditions.
Real-time RT-PCR
For quantitative real-time RT-PCR assessment of Cyp11a and Cyp17a gene expression, we isolated total RNA from the isolated Leydig cells of control and Gprc6aβ-cells cko mice by standard TRIzol method (Invitrogen) and reverse transcribed 2.0 μg of total RNAs using cDNA synthesis kit (Bio-Rad). PCRs contained 100 ng of template (cDNA or RNA), 300nM each of forward and reverse primer, and 1× iQ SYBR Green Supermix (Bio-Rad) in 50 μL. Samples were amplified for 40 cycles in an iCycler iQ Real-Time PCR Detection System (Bio-Rad) with an initial melt at 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. PCR product accumulation was monitored at multiple points during each cycle by measuring the increase in fluorescence caused by the binding of SybrGreen I to dsDNA. The threshold cycle of tested-gene product from the indicated genotype was normalized to the threshold cycle for cyclophilin A. The primers for mouse cytochrome P450, family 17, subfamily a, polypeptide 1 (Cyp17a1) consisted of mCyp17a.F56, agtcaaagacacctaatgccaag and mCyp17.R138, acgtctggggagaaacggt; for cytochrome P450, family 11, subfamily a, polypeptide 1 (Cyp11a1) consisted of mCyp11a.F127, aggtccttcaatgagatccctt and mCyp11a.R263, tccctgtaaatggggccatac; and for the cyclophilin A consisted of cyclophilin A.For, ctgcactgccaagactgaat and cyclophilin A.Rev, ccacaatgttcatgccttct. Dissociation analysis was used to confirm the presence of a single transcript and the lack of primer-dimer amplification in all PCRs.
Saturation analysis of [3H]T binding in HEK-293 cells expressing GPRC6A
Cells were dislodged from tissue culture plates by using a rubber police officer and crude membranes were prepared by Dounce homogenization in 50mM Tris-HCl (pH 7.4) at 25°C with 120mM NaCl, 1mM EDTA, 10mM MgCl2 and a protease inhibitor cocktail for mammalian tissue culture containing 104mM 4-(-2-Aminoethyl)benzenesulfonyl fluoride hydrochloride, 80μM aprotinin, 2mM leupeptin, 4mM bestatin, 1.5mM pepstatin A, and 1.4mM trans-epoxysuccinyl-l-leucylamido-(4-guanidino)butane. The homogenate was subjected to centrifugation at 14 000g for 10 minutes, and the resulting crude total cellular pellet was resuspended in the buffer above. The membranes were either used immediately for binding or flash frozen at −70°C. The membrane protein concentration was quantified by the method of Bradford, using BSA as the protein standard (24).
For saturation analysis of [3H]T, a range from 0nM–25nM [3H]T was used. Routinely, 50–75 μg of membrane protein were used per tube, and the analysis was performed in triplicate. The buffer consisting of 120mM NaCl, 1mM EDTA, and 10mM MgCl2 was used as the binding and wash buffer and varying concentrations of [3H]T were added and allowed to incubate for 2 hours at ambient room temperature with agitation. Nonspecific ligand binding was determined by the inclusion of 200nM cold T to a parallel set of tubes. The reactions were terminated by dilution with ice cold buffer and filtration through GF/C glass fiber membranes to retain bound ligand. The retained radioactivity was quantified by liquid scintillation counting.
For saturation analysis of [3H]T binding in the presence of Ca2+ (3mM CaCl2) was added to the binding and wash buffer and the assays performed as described.
The saturation data was analyzed using GraphPad Prism 6.0. For analysis of best fit, an extra sum of squares F test was performed to determine whether a one site or 2 site fit was superior for the saturation data.
GPRC6A homology modeling
Multiple sequence alignment (MSA) of the GPRC6A sequence with 8 family-C GPRCR sequences was performed using multiple alignment using Fast Fourier Transform and E-INS-i methods (25). The next family-C sequences were used: 1) human CasR, 2) human probable GPCR-158, 3) human retinoic acid-induced protein-3, 4) human taste receptor type-1 member-1, 5) human metabotropic glutamate receptor (mGluR)-1 (PDB code 4OR2), 6) human mGluR-5 (PDB code 4OO9), 7) mouse mGluR-3 (PDB code 2E4U), and 8) human γ-aminobutyric acid B receptor-1 (PDB code 4MQE). Sequences 1 through 4 have no available crystal structure, whereas sequences 5 and 6 have crystal structures for their transmembrane domains, and sequences 7 and 8 for their extracellular domains. In addition, in the multiple alignment using Fast Fourier Transform MSA, 7 family-A sequences were also included, with each of them possessing crystal structures of the short extracellular domain, transmembrane domain, and short cytoplasmic domain. These sequences were: bovine rhodopsin receptor (PDB code 3CAP), turkey β-1 adrenergic receptor (PDB code 2VT4), human β-2 adrenergic receptor (PDB code 2RH1), human adenosine A2A receptor (PDB code 2YDV), human 5-hydroxytryptamine receptor-2B (PDB code 4IB4), human 5-hydroxytryptamine receptor-1B (PDB code 4IAR), and rat neurotensin receptor type-1 (PDB code 4GRV).
From the MSA results, the structures of the mGlu-1 and mGlu-5 receptors (with crystal structures of transmembrane domains) were selected as templates for the transmembrane domain modeling. Missing regions in the cytoplasmic loop-2 (sequence 688–691) and C terminus (sequence 844–845) of the mGluR-1 template structure were modeled using mGluR-5, and missing regions in the cytoplasmic loop-2 (sequence 683–688) and the extracellular loop-2 (sequence 721–728) of the mGluR-5 template structure were modeled using mGluR-1. In this way, 10 main chain models with 10 side chains conformers per main chain model were generated for each template using the MOE-2012 (Molecular Operating Environment, 2013.08; Chemical Computing Group, Inc) homology modeling facility with the CHARMM27 force-field (26). The GPRC6A homology models were validated using PolyPhobius (27). The best-scoring homology models: 1 from using mGluR-1 as a template and 1 using mGluR-5, were selected for docking studies based on their predicted GB/VI scores (28), that rank the models based on Coulomb and Generalized Born interaction energies.
Exploration of potential ligand binding sites in GPRC6A models
The recently published crystal structures of mGluR-1 and mGluR-5 contain bound negative allosteric modulators (29, 30). The allosteric binding-site residues for these 2 receptors and the respective CasR and GPRC6A residues in the MSA are listed in Tables 1 and 2. In this site, Gly-667, Ser-669, Trp-795, Phe-798, and Tyr-802 in GPRC6A are found to be conserved in all 4 receptors, with other conserved or chemically similar residues present for residues Phe-650, Phe-666, Cys-673, Phe-752, Met-755, Leu-756, Ala-763, Ala-794, Ile-818, and Ile-825.
Table 1.
mGluR-1 Allosteric Binding Site Residues for Negative Allosteric Modulator (Antagonist) and Alignment With Other Receptors (35, 53)
| mGluR-1 Numbering | mGluR-1 | mGluR-5 | CasR | GPRC6A | Consensus (% Similarity) | Conservation (% Identical) |
|---|---|---|---|---|---|---|
| 648 | L | L-635 | F-668a | F-650 | 100 (nonpolar) | 50 (L, F) |
| 660 | Q | Q-647 | R-680a | R-662 | 50 (polar/charged) | 50 (Q, R) |
| 661 | R | R-648 | Q-681 | Q-663 | 50 (charged/polar) | 50 (R, Q) |
| 664 | V | I-651 | F-684a | F-666a | 100 (nonpolar) | 50 (F) |
| 668 | Sa | P-655 | F-688a | F-670a | 75 (nonpolar) | 50 (F) |
| 748 | T | T-735 | E-767 | E-746 | 50 (polar/charged) | 50 (T, E) |
| 753 | V | V-740 | L-773a | F-752 | 100 (nonpolar) | 50 (V) |
| 756 | Pa | P-743 | L-776a | M-755 | 100 (nonpolar) | 50 (P) |
| 757 | La | L-744 | I-777 | L-756 | 100 (nonpolar) | 75 (L) |
| 760 | Na | N-747 | T-780 | I-759a | 75 (polar) | 50 (N) |
| 761 | G | G-748 | C-781 | A-760 | 75 (nonpolar) | 50 (G) |
| 794 | T | T-781 | F-814 | Y-791 | 75 (polar) | 50 (T) |
| 797 | I | I-784 | V-817 | A-794 | 100 (nonpolar) | 50 (I) |
| 798 | Wa | W-785 | W-818a | W-795a | 100 (nonpolar) | 100 (W) |
| 801 | Fa | F-788 | F-821a | F-798 | 100 (nonpolar) | 100 (F) |
| 805 | Ya | Y-792 | Y-825 | Y-802 | 100 (polar) | 100 (Y) |
| 811 | K | K-798 | V-833 | V-810 | 50 (charged/nonpolar) | 50 (K, V) |
| 812 | I | I-799 | S-834 | P-811 | 75 (nonpolar) | 50 (I) |
| 815 | Ta | M-802 | E-837a | E-814a | 50 (charged) | 50 (E) |
| 818 | Aa | S-805 | A-840 | V-817 | 75 (nonpolar) | 50 (A) |
| 822 | S | S-809 | A-844 | S-821 | 75 (polar) | 75 (S) |
mGluR-1, CasR, and GPRC6A residues shown to be important for binding allosteric modulators by mutagenesis experiments.
Table 2.
mGluR-5 Allosteric Binding Site Residues for Negative Allosteric Modulator (Antagonist) and Alignment With Other Receptors (35, 53)
| mGluR-5 Numbering | mGluR-5 | mGluR-1 | CasR | GPRC6A | Consensus (% Similarity) | Conservation (% Identical) |
|---|---|---|---|---|---|---|
| 624 | G | G-637 | S-657a | C-639 | 50 (nonpolar/polar) | 50 (G) |
| 625 | I | I-638 | L-658 | H-640 | 75 (nonpolar) | 50 (I) |
| 628 | G | G-641 | C-661 | N-643 | 50 (nonpolar/polar) | 50 (G) |
| 651 | Ia | V-664 | F-684a | F-666a | 100 (nonpolar) | 50 (F) |
| 652 | G | G-665 | G-685 | G-667 | 100 (nonpolar) | 100 (G) |
| 654 | S | S-667 | S-687 | S-669 | 100 (polar) | 100 (S) |
| 655 | Pa | S-668 | F-688a | F-670a | 75 (nonpolar) | 50 (F) |
| 658 | Sa | C-671 | C-691 | C-673 | 100 (polar) | 75 (C) |
| 659 | Ya | Y-672 | I-692 | I-674 | 50 (polar/nonpolar) | 50 (Y, I) |
| 740 | V | V-753 | L-773a | F-752 | 100 (nonpolar) | 50 (V) |
| 743 | P | P-756 | L-776a | M-755 | 100 (nonpolar) | 50 (P) |
| 744 | L | L-757 | I-777 | L-756 | 100 (nonpolar) | 75 (L) |
| 747 | Na | N-760 | T-780 | I-759a | 75 (polar) | 50 (N) |
| 751 | I | I-764 | A-784 | A-763 | 100 (nonpolar) | 50 (I, A) |
| 781 | T | T-794 | F-814 | Y-791 | 75 (polar) | 50 (T) |
| 784 | I | I-797 | V-817 | A-794 | 100 (nonpolar) | 50 (I) |
| 785 | Wa | W-798 | W-818a | W-795a | 100 (nonpolar) | 100 (W) |
| 788 | Fa | F-801 | F-821a | F-798 | 100 (nonpolar) | 100 (F) |
| 802 | Ma | T-815 | E-837a | E-814a | 50 (charged) | 50 (E) |
| 805 | Sa | A-818 | A-840 | V-817 | 75 (nonpolar) | 50 (A) |
| 806 | V | V-819 | I-841a | I-818 | 100 (nonpolar) | 50 (V, I) |
| 809 | Sa | S-822 | A-844 | S-821 | 75 (polar) | 75 (S) |
| 810 | Aa | V-823 | S-845 | N-822 | 50 (nonpolar/polar) | 25 (A) |
| 813 | A | A-826 | L-848 | I-825 | 100 (nonpolar) | 50 (A) |
mGluR-5, CasR, and GPRC6A residues shown to be important for binding allosteric modulators by mutagenesis experiments.
A number of agonists/antagonists have been reported that bind to the transmembrane domain of the closest family C member, CasR (35). Residues involved in this binding in CasR identified by mutagenesis experiments (Ser-657, Phe-668, Arg-680, Phe-684, Phe-688, Leu-773, Leu-776, Trp-818, Phe-821, Glu-837, and Ile-841) are also conserved in GPCR6A except Ser-657, Leu-773, and Leu-776 which are replaced by Cys-639, Phe-752, and Met-755, respectively. CasR calcimimetic calindol and the calcilytic NPS2143 have been found to antagonize mouse GPRC6A (mGPRC6A) (35). The binding site residues for these antagonists in mGPRC6A found by mutagenesis experiments (Phe-666, Phe-670, Trp-797, and Glu-816) are conserved in both human GPRC6A and CasR. Several 2-phenyl-indole-derived allosteric antagonists have also been reported for mGPRC6A that are selective only for this receptor among those of family-C (53). A possible binding site residue reported for these antagonists in mGPRC6A (Ile-759) is conserved in human GPRC6A but not in CasR (Thr-780), mGluR-1 (Asn-760), or mGluR-5 (Asn-747).The above information was used for identifying putative binding sites in GPRC6A used in the present docking studies.
Docking of T
Docking of T to the transmembrane domain of the selected GPRC6A homology models was carried out using MOE-2012's Docking facility with the CHARMM27 force-field. CHARMM parameters for the ligands were generated by MOE from a fragment-based approach. The binding site was defined in MOE from the binding-site residues described above using MOE's Site Finder facility. Binding site residue side chains were allowed to be flexible during the docking using a tethering weight of 0.1 kcal/mol·A2. London dG-free energy scores, as implemented in MOE-2012, were used to rank poses of the docked ligand.
Measurement of cAMP accumulation
HEK-293 and HEK-293 transfected with mGPRC6A cDNA cells (105 cells/well) (15) were cultured in triplicate in 12-well plates in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (100 U/mL of penicillin and 100 μg/mL of streptomycin) for 48 hours followed by overnight incubation in DMEM/F12 containing 0.1% BSA to achieve quiescence. Quiescent cells were treated with vehicle control, 20mM L-Arg and T at concentration as indicated for 30 minutes at 37°C. Then, the reaction was stopped and the cells lysed with 0.5-mL 0.1N HCl. cAMP levels were measured by using Cyclic AMP EIA kit (Cayman Chemical) following the manufacture's protocol.
Leydig cells isolation
To isolate Leydig cells from wild-type and Gprc6a−/− mice, the testes from 10- to 12-week-old mice were obtained and enzymatically dispersed with the method as previously described (31). Briefly, the testes from wild-type and Gprc6a−/− mice were decapsulated and dispersed with 0.25-mg/mL collagenase (Invitrogen) in medium 199 for 10 minutes at 4°C with 70–90 rpm shaking. The separated cells were filtered through a 100-nm filter, centrifuged at 250g, and resuspended in 55% isotonic Percoll (Sigma). After density gradient centrifugation at 25 000g for 45 minutes at 4°C, the progenitor Leydig cells fraction was collected between densities of 1.064 and 1.070 g/mL. The cells were washed with HBSS (5 times of volume; Invitrogen), centrifuged at 250g, and the cells were washed with HBSS (5 times of volume), centrifuged at 250g, and resuspended in phenol red-free medium (DMEM/Ham's F-12, D-2906; Sigma) supplemented with 1-mg/mL BSA.
AR transactivation
HEK-293 cells were plated at 125 000 cells/well of a 24-well plate in DME+5% charcoal stripped FBS without phenol red. Cells were transfected with 0.25-μg glucocorticoid response element-luciferase, 10-ng cytomegalovirus promoter-Renilla luciferase, and 50-ng cytomegalovirus promoter-human androgen receptor using lipofectamine transfection reagent in optiMEM medium. Medium was changed 24 hours after transfection to DME+5% charcoal stripped FBS without phenol red and treated with a dose response of various drugs (1pM–10μM). Luciferase assay was performed 24 hours after treatment on a BioTek Synergy 4 plate reader. Firefly luciferase values were normalized to Renilla luciferase values.
Mouse islets isolation and ligand stimulation
Primary islets were isolated using a modified method as described (30, 31). Briefly, after dissection and mincing, pancreata were digested with 3 mL/pancreas of a collagenase P (1 mg/mL; Roche) solution in complete Hanks' balanced salt solution (1× supplemented with 20mM HEPES [pH 7.4] and 2mM CaCl2) for 15 minutes in a 37°C shaking water bath. Islets were subsequently purified through a Histopaque 1083 density centrifugation (Sigma). After centrifugation, the islet layer was transferred into petri dishes with wash buffer (Hanks' balanced salt solution with 10mM HEPES and 1% fetal bovine serum; Invitrogen), then handpicked and cultured in low-glucose medium (RPMI 1640 with 5.6mM glucose; Invitrogen) for 1 hour before being treated for 1 hour with 0.5μM DJ-I-267S as indicated. The insulin SI was calculated as the ratio of media insulin concentrations in high glucose divided by the insulin concentration in low-glucose conditions.
Statistics
We evaluated differences between groups by one-way ANOVA, followed by a post hoc Tukey's test. Significance was set at P < .05. All values are expressed as means ± SEM. All computations were performed using the Statgraphic statistical graphics system (STSC, Inc).
Results
Binding of T to GPRC6A
To examine whether T binds to GPRC6A, we performed saturation analysis of [3H]T binding in HEK293 cells overexpressing GPRC6A in the presence of calcium (Figure 1). Shown in Figure 1 is the total (solid circles), nonspecific (solid squares), and specific binding (open circles). We found that [3H]T binds to GPRC6A with an estimated Kd for [3H]T of 8.82 ± 1.02nM (n = 3) (Figure 1). We show that [3H]T binding to the transfected HEK-293 cells is saturable. Specific binding of [3HT] was only seen in HEK-293 cells overexpressing GPRC6A. No specific binding of [3H]T was observed in the parent nontransfected HEK-293 cells that do not express GPRC6A (data not shown). Specific binding of [3H]T to GPRC6A overexpressing HEK-293 was not affected by other ligands for GPRC6A, L-Arg (8, 32) and Ocn (5, 7, 13). Neither addition of 10mM–50mM L-Arg or 1- to 50-ng/mL decarboxylated Ocn (33) interfered with [3H]T binding in HEK293 cells overexpressing GPRC6A (data not shown).
Figure 1.
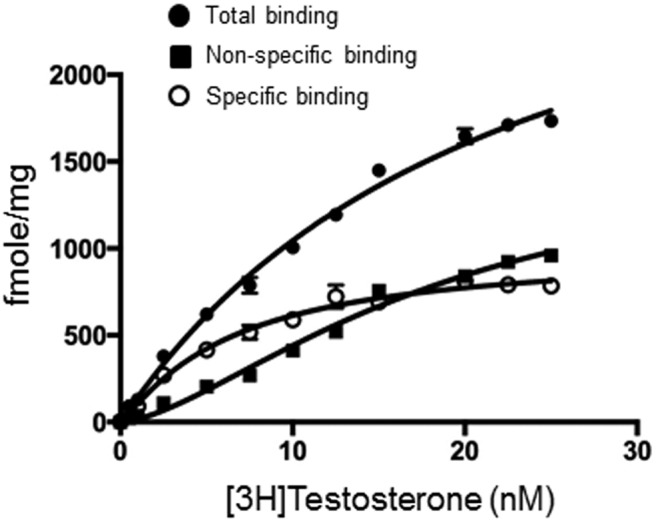
T binds to GPCR6A. Saturation analysis of [3H]T binding to GPCR6A in HEK-293 cells transfected with GPRC6A. Total binding (solid circle), nonspecific binding (solid square), and specific binding (open circles) components of [3H]T saturation binding. The total and NSB were used to do a global fit using GraphPad Prism to generate the specific binding. The binding was performed in the presence of 3mM Ca2+. Shown are the mean and SE from 3 separate binding experiments performed in triplicate.
Computational modeling of T binding to GPRC6A
To investigate the molecular basis for T binding to GPRC6A, structural models of GPRC6A were constructed and used to identify potential T binding poses. The GPRC6A homology models developed are shown in Supplemental Figure 1. Helix regions in MSAs are highly conserved among GPCRs, whereas the loop regions exhibit low sequence similarity, as shown in Supplemental Figure 1A. The GPRC6A sequence exhibits similarities of 43.2% and 44.7% to the mGluR-1 and mGluR-5 receptors, respectively, the only sequences for which crystal structures of the transmembrane domain exist (sequences 2 and 3 in Supplemental Figure 1B). These receptor structures were selected as templates for the homology modeling calculations and the corresponding structural models selected for docking studies.
The highest scoring structural models are shown in Supplemental Figure 1, C and D. These models exhibit very similar structures, with a root mean square deviation of 1.92 Å2 between the backbone atoms of the transmembrane helices. These models were validated against Hidden Markov secondary structure prediction generated by PolyPhobius, and found to be consistent (Tables 3 and 4) with only 1 loop region and 1 transmembrane helix in the homology models exhibiting deviation from the PolyPhobius-predicted structure for 5 or more residues.
Table 3.
PolyPhobius Secondary Structure Prediction for GPRC6A Sequence
| Residue Number | Structure Prediction |
|---|---|
| 1–18 | Signal peptide |
| 19–592 | Noncytoplasmic |
| 593–616 | Transmembrane |
| 617–630 | Cytoplasmic |
| 631–651 | Transmembrane |
| 652–663 | Noncytoplasmic |
| 664–683 | Transmembrane |
| 684–702 | Cytoplasmic |
| 703–727 | Transmembrane |
| 728–750 | Noncytoplasmic |
| 751–773 | Transmembrane |
| 774–782 | Cytoplasmic |
| 783–804 | Transmembrane |
| 805–811 | Noncytoplasmic |
| 812–833 | Transmembrane |
| 834–926 | Cytoplasmic |
Table 4.
Secondary Structure Features of mGluR-1/mGluR-5 Models Generated in MOE-2012
| Residue Number | Structure |
|---|---|
| 1–18 | Signal peptide |
| 19–591 | Noncytoplasmic |
| 592–617 | Transmembrane |
| 618–633 | Cytoplasmic |
| 634–651 | Transmembrane |
| 652–657a | Noncytoplasmic |
| 658–686a | Transmembrane |
| 687–703 | Cytoplasmic |
| 704–725 | Transmembrane |
| 726–750 | Noncytoplasmic |
| 751–772 | Transmembrane |
| 773–778 | Cytoplasmic |
| 779–804 | Transmembrane |
| 805–811 | Noncytoplasmic |
| 812–837 | Transmembrane |
| 838–926 | Cytoplasmic |
Regions that show deviation from predicted structure for 5 or more residues.
Docking of T
Next, we docked T to the above homology models as described in Materials and Methods. The binding sites found in the mGluR-1- and mGluR-5-based models of GPRC6A are shown in Figure 2, A and B, respectively. The docking scores and the binding pocket residues in these sites are listed in Table 5 for the top-ranking binding poses of T. Thirteen residues in the mGluR-1-based model, and 12 in mGluR-5, were identified as possibly interacting with T. In both models, the hydrophobic core of T is surrounded by aliphatic side chains and/or aromatic Phe (F), Trp (W), or Tyr (Y) residues. The hydroxyl and carbonyl moieties of T are located in hydrogen bond acceptor-rich regions, either on side chains or on the backbones of residues in the binding pocket. Six of the predicted binding residues are found in both models, F-666, F-670, F-752, M-755, L-756, and L-759 (Figure 2 and Table 5).
Figure 2.

Docking of T to GPRC6A. A, top, Allosteric site in the mGluR-1-based template model, T is shown in yellow stick representation. Bottom, Residues surrounding T in the binding pocket. B, top, Allosteric site in the mGlur-5-based model, T is shown in yellow stick representation. Bottom, Residues surrounding T in the binding pocket.
Table 5.
Docking Scores (in kcal/mol) and Binding Pocket Residues for T binding in GPRC6A Models Generated in MOE-2012
| Model | Docking Score | Binding Site Residues |
|---|---|---|
| mGluR-1 | −11.58 | C-659, R-662, Q-663, F-666, G-667, F-670, E-746, A-751, F-752, M-755, L-756, I-759, E-814 |
| mGluR-5 | −12.31 | F-666, F-670, F-752, M-755, L-756, L-759, Y-791, A-794, W-795, F-798, V-817, S-821 |
Test of T binding and computational model by mutagenesis
To test the binding model, we compared the T activation of wild-type and selectively mutated GPRC6A transfected into HEK-293 cells.
First, we confirmed that T activates GPRC6A in a functional assay using HEK-293 cells. HEK-293 cells lack endogenous expression of both GPRC6A and the AR (14, 15). We observed a dose-dependent effect of T to stimulate cAMP accumulation (Figure 3A) and ERK phosphorylation (Figure 3B) in HEK-293 cells transfected with wild-type GPRC6A. A significant response was observed at 5nM and a maximal response at a T concentration of 60nM, using cAMP accumulation as the read out (Figure 3A). No response to T is observed in nontransfected HEK-293 cells in ERK phosphorylation (Figure 3B, low panel). Consistent with previous reports (13, 17), L-Arg (20mM) stimulated cAMP to a similar magnitude as T in (Figure 3C). We also found that the protein kinase A (PKA) inhibitor, H89, blocked GPRC6A-mediated T-stimulated ERK phosphorylation (Figure 3D), indicating that PKA is involved in the GPRC6A signaling pathway.
Figure 3.

Evidence for T activation of GPRC6A. Dose-dependent effects of T on cAMP response (A) and ERK phosphorylation (B) in HEK-293 cells expressing GPRC6A. C, Comparison of cAMP accumulation stimulated by T with other GPRC6A ligands, L-Arg (67) in HEK-293 cells transfected with and without GPRC6A. * and **, significant difference from control group and stimulated group at P < .05 and P < .01 (n ≥ 4). D, PKA inhibitor H89 blocked GPRC6A-mediated ERK phosphorylation stimulation. E, Functional effect of mutations affecting the F666A, E746A, W797A, and I820A mGPRC6A residues. HEK-293 cells were transfected with the mock plasmid or with a plasmid encoding the WT or F666A or E746A or W797A or I820A mGPRC6A mutants. F666A, E746A, W797A, and I820A mutants blocked GPRC6A-mediated T-stimulated ERK phosphorylation. F, Analysis of mGPRC6A expression by Western blotting was shown in right panel. Immunoblot analysis of whole-cell lysates (5-μg proteins) from HEK293 cells transiently transfected with an empty vector (MOCK), or a vector containing the WT or the indicated mGPRC6A mutant, was performed after 3–8% SDS polyacrylamide gel electrophoresis. mGPRC6A proteins were detected using the anti-Myc antibody (Cell Signaling). The position of the molecular mass markers is shown on the left (kDa). Arrowheads on the right indicate the molecular weight of 2 major bands corresponding to mGPRC6A. Black arrows on the right indicate major bands corresponding to the WT and mutant receptors, and white arrowhead indicates the dimer of the receptors, respectively. G, Loss T activation after GPRC6A knock down by CRISPR/Cas9 in PC-3 cells.
Next, we tested the effects of 4 distinct GPRC6A mutations predicted by the computational modeling. The computational modeling of T binding to GPRC6A suggested a number residues that might be particularly important in binding T, namely Phe-666, Glu-746, Trp-797, and Ile-820 (Figure 2 and Table 5). We therefore mutated these residues into alanine using site-directed mutagenesis (35). The individual mutants and the wild-type (WT) mGPRC6A were transiently transfected into HEK-293 cells. The F666A, E746A, W797A, and I820A mutants all showed a significant and reproducible decrease in T-stimulated ERK phosphorylation compared with that of the WT receptor in the transfected HEK-293 cells (Figure 3E). Interestingly, L-Arg, which is believed to bind to the venus fly trap (VFT) motif of GPRC6A, activated the F666A, E746A, W797A, and I820A mutants similarly to the wild-type receptor (Figure 3F). To confirm that the loss of response of mutant receptors was not due to differences in expression, we compared the expression of the wild-type and F666A mutant GPRC6A receptors by Western blotting using a Myc antibody, which recognized the Myc epitope located at the amino-terminal tail of the WT and mutant receptors (35). Expression of the F666A mutant receptor was comparable with that of the WT (Figure 3F). Thus, the mutational results are in agreement with the T binding hypothesis from the computational modeling.
To test the function of GPRC6A in another cell culture model that expresses endogenous GPRC6A, we investigated the response of human prostate cancer cell line, PC-3. PC-3 cells highly express GPRC6A (36, 37) but lack AR expression (38). To confirm the importance of GPRC6A-mediated T signaling in this prostate cancer cell line, PC-3, we assessed the response to GPRC6A ligands after knock down of GPRC6A using CRISPR/Cas9 system (Supplemental Figure 2). We assessed ERK activity in PC-3 cells transfected with GPRC6A single guide RNA-3 (sgRNA-3) and compared these responses with control groups consisting of mock-transfected cells with a random negative control sgRNA and GPRC6A sgRNA-3 knock down PC-3 cells. We observed decreased levels of mRNA expression of GPRC6A and protein levels in GPRC6A sgRNA-3 knock down PC-3 cells, compared with controls (Supplemental Figure 2, D and E). T stimulated phospho-ERK activity in mock-transfected PC-3 cells, and this response was significantly decreased in PC-3 human prostate cancer cell knock down by GPRC6A sgRNA-3 (Figure 3G).
Loss of T responses in Gprc6a−/− mice
To evaluate the role of GPRC6A in sensing T in vivo, we examined early growth response protein 1 (Egr-1) expression in the pancreas and testis of Gprc6a−/− mice and wild-type littermates after the administration of 200-mg/kg T by ip injection. We found that T administration significantly increased Egr-1 expression in the pancreas and testis of wild-type mice, whereas Gprc6a−/− mice showed no response to T (Figure 4A).
Figure 4.

β-Cell and Leydig cells dysfunction in Gprc6a−/− mice. A, Effects of systemic administration of T on Egr-1 expression in pancreas and testis. T (200 mg/kg) or vehicle was injected into mice ip, Egr-1 mRNA abundance in various mouse tissue 1 hour after injection was determined by real-time PCR. Values represent the mean ± SEM. *, significant difference from control group and stimulated group at P < .05 (n ≥ 3). B, Effects of T on insulin SI in islets isolated from wild-type and Gprc6a−/− mice. Values represent the mean ± SEM. *, significant difference from control group and stimulated group at P < .05 (n ≥ 3); #, significant different between T-treated control group and Gprc6a−/− mice (P < .05; n ≥ 3). C, RT-PCR analysis showing that Gprc6a message is expressed in INS-1 cells. A similar band was observed in rat kidney and pancreas, which are known to express GPRC6A. The primers sequence are rGPRC6A.For535, aaaatccgctttccttcgttr; and GPRC6A.Rev1400, tgggcatcaaaatgaaatgar. D, Effects of T to stimulate insulin secretion in rat β-cells INS-1. Values represent the mean ± SEM. * and **, significant difference from control group and stimulated group at P < .05 and P < .01 (n = 4). E, Effects of T on message expression of enzymes regulating T biosynthesis in isolated Leydig cells. Values represent the mean ± SEM. *, significant difference from control group and stimulated group at P < .05 (n ≥ 3).
To test this biological response ex vivo, we examined the effect of GPRC6A activation in isolated islets and Leydig cells from wild-type and Gprc6a−/− mice. We found that T increased the insulin SI in islets isolated from wild-type but not Gprc6a−/− mice (Figure 4B). Next, we confirmed that GPRC6A is expressed in β-cells. We found that Gprc6a transcripts were present in rat kidney, rat pancreas, and INS-1 rat β-cell line (Figure 4C) (39). In addition, we observed a dose-dependent effect of T to stimulate insulin secretion in INS-1 β-cells (Figure 4D). Finally, we found that 100nM T significantly stimulated T biosynthesis enzymes, cholesterol side-chain cleavage enzyme (Cyp11a) and steroid 17-α-monooxygenase (Cyp17a) expression in Leydig cells from wild-type mice but not in Gprc6a−/− mice (Figure 4E).
Compounds that activate GPRC6A but not the AR
In spite of the above evidence of T-binding to and activation of GPRC6A, because T can bind to AR and because of the possibility that some of membrane effects of T are mediated by membrane associated AR, we sought additional small compounds that are specific ligands for GPRC6A and do not activate AR.
We have previously reported on a series of S-type derivatives of 3-phenoxy-2-hydroxy-N-(4-nitro or cyano-3-(trifluoromethyl) phenyl)propanamide (40–50) that function as SARMs that have a range of binding affinities for AR and are stereospecific for AR. We screened 9 SARM-like compounds (Supplemental Table 1) for their ability to activate GPRC6A. Seven of these SAMRs also activated GPRC6A.
Because SARMs activation of AR is stereospecific, we next examined the effects of SARM enantiomers on GPRC6A and AR activation. Initially, we measured ERK activation in HEK-293 cells expressing GPRC6A compared with HEK-293 cells lacking GPRC6A. We found that both DJ-I-267S and DJ-I-267R activated GPRC6A in nanomolar amounts. DJ-I-267 is not stereospecific for GPRC6A, because DJ-I-267S or DJ-I-267R or a racemic mixture of DJ-I-267S+R activate GPRC6A (Figure 5, A–C). In contrast, DJ-I-267S activates AR, but DJ-I-267R does not (Figure 5, D and E). Thus, we have identified a compound that activates GPRC6A but not AR (51, 52).
Figure 5.

Lead compounds that activates GPRC6A but not the AR. Lead compounds stimulated GPRC6A (A–C). Lead compound DJ-I-267S activated AR, but DJ-I-267R not (D and E) in vitro. F, Effects of DJ-I-267R on serum of insulin levels in wild-type mice. Serum insulin level were measured 1 hour after ip injection of 10-mg/kg DJ-I-27R in vehicle (5% DMSO + 95% PEG). G, Effects of DJ-I-267S on insulin SI in islets isolated for wild-type and Gprec6a−/− mice. Primary islets were isolated as described in Materials and Methods.
To test the biological activity of the GPRC6A selective ligand, we found that DJ-I-267R at the dose of 10 mg/kg stimulated insulin secretion in wild-type mice after 1 hour ip administration (Figure 5F). Thus, the DJ-I-267R compound that does not activate AR but activates GPRC6A-stimulated insulin secretion. Next, we examined the response of pancreatic islets isolated from wild-type and Gprc6a−/− mice to DJ-I-267S. The DJ-I-267S increased insulin SI in wild-type isolated pancreatic islets was attenuated in islets from Gprc6a −/− mice (Figure 5G). The result shows that DJ-I-267S signaling in islets requires GPRC6A.
Finally, the potential binding models of DJ-I-267R and S to the transmembrane domain of the GPRC6A homology models were investigated using the same docking protocol as described for T in Materials and Methods. The binding site found for the DJ-I-267R and S isomers in the mGluR-1- and mGluR-5-based models is shown in Figure 6, A and B, respectively. The docking scores and the binding pocket residues in these sites are listed in Table 6 and shown in Supplemental Figures 3 and 4. Both the stereoisomers bind with similar binding modes and predicted binding affinities in both protein models. The major differences in the binding modes lie in the orientation of the chiral center and flipping of the aromatic rings. In both models, aromatic rings in DJ-I-267R and S are surrounded by Phe or Tyr residues, forming π-π and π-H bonds. The substituted groups on the aromatic rings are located in hydrogen bond acceptor and donor-rich regions, either on side chains or on the backbone of residues in the binding pocket.
Figure 6.
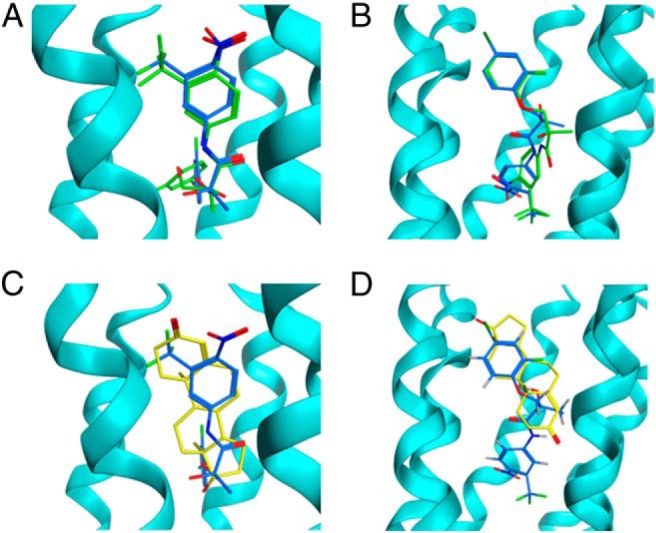
Docking of DJ-I-267 to GPRC6A and comparison of binding modes. A, Top ranked binding poses of DJ-I-267R and DJ-I-267S in the mGluR-1-based model and (B) mGluR-5-based model allosteric binding site. DJ-I-267R is shown in blue and DJ-I-267S in green stick representation. C, Comparison of top ranked binding poses of T and DJ-I-267R in the mGluR-1-based model and (D) mGluR-5-based model. T is shown in yellow and DJ-I-267R shown in blue.
Table 6.
Docking Scores (in kcal/mol) and Binding Pocket Residues for DJ-I-267R and S Binding in GPRC6A Models
| Model | Ligand | Docking Score | Binding Site Residues |
|---|---|---|---|
| mGluR-1 | DJ-I-267R | −15.1 | F-650, R-662, Q-663, F-666, G-667, F-670, Q-715, E-746, A-751, F-752, M-755, L-756, I-759 A-794, W-795, F-798, E-814, V-817 |
| mGluR-1 | DJ-I-267S | −14.88 | R-662, Q-663, F-666, G-667, F-670, Q-715, E-746, A-751, F-752, M-755, L-756, I-759 A-794, W-795, F-798, E-814, I-818, V-817 |
| mGluR-5 | DJ-I-267R | −17.98 | C-639, H-640, N-643, F-666, S-669, F-670, C-673, I-674, F-752, M-755, L-756, I-759, Y-791, W-795, F-798, V-817, I-818, S-821, N-822, I-825 |
| mGluR-5 | DJ-I-267S | −18.01 | C-639, H-640, N-643, F-666, S-669, F-670, C-673, I-674, F-752, M-755, L-756, I-759, Y-791, A-794, W-795, F-798, V-817, I-818, S-821, N-822, I-825 |
Comparison of binding modes
The superimposed binding poses of T and DJ-I-267R with the best docking scores in the homology models are shown in Figure 6, C and D, respectively. In both homology models, T binds in a similar orientation as DJ-I-267R with the cyclohexene group of T superimposing well with the aromatic group in DJ-I-267R. The major differences in the binding modes arise due to the differences in the sizes of the 2 ligands, with DJ-I-267R having 2 aromatic rings connected by a flexible linker region, whereas T being a much more rigid ligand. This results in DJ-I-267R extending deeper into the binding pocket in the transmembrane domain of GPRC6A compared with T.
Discussion
The diverse ligand specificity of GPRC6A and the lack of understanding of the structural basis for ligand/receptor interactions have resulted in controversy regarding the possibility that GPRC6A mediates the rapid, nongenomic response to T (15, 34). In the current study, we developed a structural model exhibiting sites in the heptahelical domain of GPRC6A that are predicted to bind T. Also, we showed that T specifically binds to GPRC6A using [3H]T radioligand binding assays and that mutagenesis of the residues in the T binding site identified by the modeling results in the inhibition of T activation of GPRC6A in vitro, in agreement with the model. We also found that DJ-I-267, a SARM that stereo-selectively activates AR, exhibits nonstereo-specificity for GPRC6A. Indeed, DJ-I-267R fails to activate AR-signaling, but is a potent agonist of GPRC6A. The predicted binding poses for DJ-I-267R overlapped the site predicted for T in our structural model. Finally, we used Gprc6a−/− mice to define specific roles of GPRC6A in mediating the rapid T responses in vitro and in vivo. Specifically, we identified a role of T activation of GPRC6A in the regulation of insulin secretion by β-cells and enzymes controlling T production by Leydig cells. We found that DJ-I-267 regulated insulin secretion in pancreatic islets isolated from wild-type but not Gprc6a−/− mice. Moreover, injection of DJ-I-267 increased circulating insulin levels in wild-type mice. Collectively, these data indicate that GPRC6A is physiologically relevant T-sensing receptor that mediates the nonclassical, rapid membrane signaling responses to T as wells chemical compounds belonging to the SARM family.
Our model of GPRC6A suggests that the potential binding pocket for T, and DJ-I-267, corresponds to the common allosteric site for class C GPCRs located in the transmembrane domain. Previous mutagenesis-based studies of class C GPCR allosteric sites showed that positions of the potential T binding-pocket residues have also been implicated in the binding of mGluR-1, mGluR-5, and CasR allosteric modulators (Tables 1 and 2). Phe-666 and Glu-746, which are found to be important in T activation of GPRC6A by mutagenesis studies, also overlaps the site for the calcimimetic and calcilytic binding to the closely related CasR (35). High concentrations of calcimimetics and calcilytic also, respectively, stimulate and antagonize mGPRC6A (35). Antagonists may also interact with this binding site in GPRC6A (53). Of note, the 2-phenyl-indole-derived allosteric antagonists, which are specific for only GPRC6A among the class-C GPCRs, are found to interact with Ile-759. This residue is not conserved in other class-C GPCRs and may be important in conferring specificity to this receptor.
Our current model focuses on the heptahelical domain of GPRC6A and does not include the VFT extracellular domain. Calcium activates GPRC6A through binding to this VFT motif (54). Our findings that calcium augments T activation of GPRC6A functions but does not enhance T binding to this receptor is consistent with distinct binding sites for ligands and allosteric modulators in the VFT and heptahelical domains. Future modeling that includes the VFT may be able to identify the most stable binding modes of ligands and allow a detailed structure-based description of binding poses (55), as well as identification of novel small molecules that selectively modulate GPRC6A.
The identification of a GPCR mediating the rapid response to T also permits us for the first time, to define specific physiological processes regulated by this T-sensing GPCR. First, we showed that GPRC6A is an important GPCR for T signaling in β-cells. In this regard, T dose-dependently stimulated insulin secretion in INS-1 cells and activated cAMP-dependent pathways in HEK-293 cells expressing GPRC6A. More importantly, T administration stimulated rapid Egr-1 signaling in the pancreas of wild-type mice, but this response was lost in the pancreas of Gprc6a−/− mice. Pancreatic islets from Gprc6a−/− mice had a diminished insulin secretion index. Although no relationship between T and β-cell mass has been described (56), T has been shown to protect against glucotoxicity-induced apoptosis of pancreatic β-cells (57) and act through nongenomic pathways to affect adiponectin levels in humans (58). T deficiency can also contribute to the development of metabolic syndrome (59). Thus, T may have effects on pancreatic β-cell function through activation of GPRC6A (59).
Our novel result implicating GPRC6A as a receptor for T is controversial for several reasons. First, our studies differ from a recent report that neither T nor Ocn, another proposed GPRC6A ligand, activates GPRC6A (20). This negative studies differ from ours in several respects. The studies use different cells models for functional assays, ie, Flp-in-CHO in the negative studies vs HEK-293 cells overexpressing GPRC6A, PC-3 cells with GPRC6A knock down, and isolated pancreatic islets from wild-type and Gprc6a null mice in studies showing that T activates GPRC6A. In addition, different GPRC6A cDNA constructs where used, ie, a chimeric GPRC6A receptor in which the signal peptide of mGluR5 sequence was added to the N-terminus in pcDNA5/FRT/V5-His-Topo vector in the negative studies, vs wild-type GPRC6A with an N-terminal c-Myc tag in pcDNA3.1 vector in our studies (20, 21). Thus, differences in cell models and functions of transfected GPRC6 cDNA constructs might account for the negative findings. Although we do not have a precise explanation for the difference in the results, our findings that GPRC6A regulates insulin secretion and is activated by Ocn have been independently verified by another laboratory (7, 10, 11, 13). Second, our findings challenge the prevailing hypotheses that the rapid signaling response to T are mediated by “membrane-associated ARs” (60), or T conversion to estradiol and activation of rapid ERK-dependent membrane signaling (61), or nonreceptor mechanism (62). Indeed, we observed T activation of GPRC6A in 2 different cell models lacking AR and identified a novel compound that activates GPRC6A but not AR and cannot be converted to estradiol, suggesting that membrane associated nuclear receptors are not relevant to T rapid signaling responses we observed. Although additional efforts to reconcile the disparate data and establishing the relevance of additional rapid signaling mechanisms are needed, the preponderance of evidence suggest that GPRC6A is a receptor for T, as well as Ocn.
We also show that T, like Ocn, (13, 15), activates GPRC6A in Leydig cells. Isolated Leydig cells from Gprc6a−/− mice exhibited diminished expression of the T-stimulated Cyp11a and Cyp17a, which regulate T biosynthesis. In addition, T stimulated rapid Egr-1 signaling in the testes of wild-type mice but not in the testes of Gprc6a−/− mice. These findings suggest the presence of a positive feedback loop, whereby T secreted by Leydig cells regulates its own production. This autocrine loop would provide a mechanism for the heretofore unexplained finding that testicular interstitial fluid has effects on Leydig cell T secretion that are independent of LH and human chorionic gonadotropin (63), as well as the finding that Leydig cell production of T and Cyp17a1 expression are unaffected by the loss of AR (64).
The wide tissue expression of GPRC6A and the fact that it is activated by T, as well as by multiple structurally distinct ligands, predicts additional metabolic functions and integration of endocrine networks involving multiple organs. Indeed, global Gprc6a−/− mice, in addition to abnormalities of β-cells, Leydig cells, skin, and prostate, also have abnormalities in liver, bone, muscle, and adipocyte function. Because GPRC6A and AR are also coexpressed in many of these tissues, there may be other organs where differences in rapid and classical T effects may be physiologically important. Recent clinical studies show that graded doses of T administration to patients treated with a 5α-reductase inhibitor has effects on lean body and muscle mass, sexual functions, hematocrit, cholesterol and other biological effects that are consistent with the predicted functions of GPRC6A derived from the phenotype of Gprc6a−/− mice (65). Previous studies have also shown that GPRC6A is increased in prostate cancer cells and is linked to prostate cancer progression in genome wide associative studies (17, 66). Ablation of GPRC6A attenuates prostate cancer progression in a mouse model (17), suggesting that the AR-independent effects of T in resistant prostate cancer might be mediated by GPRC6A. GPRC6A has also been shown to mediate the functional response to T in skin keratinocytes (16).
In conclusion, an understanding of the tissue specific functions of GPRC6A and the structural basis for its binding to T are revealing new connections between endocrine networks that heretofore were not thought to be related. Further, the knowledge that T activates GPRC6A has led to the discovery of a SARM isomer that selectively activates GPRC6A but not AR. Future studies that conditionally delete GPRC6A and AR in specific tissues are now possible and will be necessary to differentiate between GPRC6A and AR in mediating the physiologic effects of T. The structural modeling presented here also represents a first step towards developing agonists and antagonists for GPRC6A that might be useful for treating a wide variety of clinical disorders, ranging from metabolic syndrome (agonists) to prostate cancer (antagonist).
Additional material
Supplementary data supplied by authors.
Acknowledgments
We thank Dr C. Newgard, S. Stedman Center for Nutritional and Metabolic Studies, Duke University, Durham, NC, for the clonal cell line INS1 832/13. GPRC6A mutants from site mutagenesis were kindly provided from Dr Martial Rua, Institut de Neurobiologie Alfred Fessard-IFR 2118, Signal Transduction and Developmental Neuropharmacology Team, Gif-sur-Yvette, France.
Author contributions: M.P., K.K., S.E.S., S.K.N., J.B., J.C.S., and L.D.Q. contributed to the research design and writing of the manuscript; M.P., K.K., R.Y., R.N., S.E.S., and Y.W. performed the research; and S.K.N., D.-J.H., and D.D.M. provided experimental materials.
This work was supported by National Institutes of Health Grant R01-AR37308 and Americans Diabetes Association Grant 1-13-BS-149-BR.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AR
- androgen receptor
- CasR
- calcium-sensing receptor
- cyp11a1
- cytochrome P450, family 11, subfamily a, polypeptide 1
- cyp17a1
- cytochrome P450, family 17, subfamily a, polypeptide 1
- Egr-1
- early growth response protein 1
- FBS
- fetal bovine serum
- GPCR
- G protein-coupled receptor
- GPRC6A
- GPCR family C group 6 member A
- HBSS
- Hank's Balanced Salt Solution
- mGluR
- metabotropic glutamate receptor
- mGPRC6A
- mouse-GPRC6A
- MSA
- multiple sequence alignment
- Ocn
- osteocalcin
- PKA
- protein kinase A
- SARM
- selective AR modulator
- sgRNA-3
- single guide RNA-3
- SI
- stimulation index
- T
- testosterone
- VFT
- venus fly trap
- WT
- wild-type.
References
- 1. Bhasin S, Jasuja R. Selective androgen receptor modulators as function promoting therapies. Curr Opin Clin Nutr Metab Care. 2009;12(3):232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Asuthkar S, Demirkhanyan L, Sun X, et al. The TRPM8 protein is a testosterone receptor: II. Functional evidence for an ionotropic effect of testosterone on TRPM8. J Biol Chem. 2015;290(5):2670–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Benten WP, Lieberherr M, Stamm O, Wrehlke C, Guo Z, Wunderlich F. Testosterone signaling through internalizable surface receptors in androgen receptor-free macrophages. Mol Biol Cell. 1999;10(10):3113–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Karsenty G, Oury F. Regulation of male fertility by the bone-derived hormone osteocalcin. Mol Cell Endocrinol. 2014;382(1):521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oury F, Khrimian L, Denny CA, et al. Maternal and offspring pools of osteocalcin influence brain development and functions. Cell. 2013;155(1):228–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wellendorph P, Bräuner-Osborne H. Molecular cloning, expression, and sequence analysis of GPRC6A, a novel family C G-protein-coupled receptor. Gene. 2004;335:37–46. [DOI] [PubMed] [Google Scholar]
- 7. Wei J, Hanna T, Suda N, Karsenty G, Ducy P. Osteocalcin promotes β-cell proliferation during development and adulthood through Gprc6a. Diabetes. 2014;63(3):1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pi M, Faber P, Ekema G, et al. Identification of a novel extracellular cation-sensing G-protein-coupled receptor. J Biol Chem. 2005;280(48):40201–40209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fulzele K, Riddle RC, DiGirolamo DJ, et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell. 2010;142(2):309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ferron M, Wei J, Yoshizawa T, et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. 2010;142(2):296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oury F, Sumara G, Sumara O, et al. Endocrine regulation of male fertility by the skeleton. Cell. 2011;144(5):796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pi M, Chen L, Huang MZ, et al. GPRC6A null mice exhibit osteopenia, feminization and metabolic syndrome. PLoS One. 2008;3(12):e3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pi M, Wu Y, Quarles LD. GPRC6A mediates responses to osteocalcin in β-cells in vitro and pancreas in vivo. J Bone Miner Res. 2011;26(7):1680–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lanzino M, Sisci D, Morelli C, et al. Inhibition of cyclin D1 expression by androgen receptor in breast cancer cells–identification of a novel androgen response element. Nucleic Acids Res. 2010;38(16):5351–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pi M, Parrill AL, Quarles LD. GPRC6A mediates the non-genomic effects of steroids. J Biol Chem. 2010;285(51):39953–39964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ko E, Choi H, Kim B, et al. Testosterone stimulates Duox1 activity through GPRC6A in skin keratinocytes. J Biol Chem. 2014;289(42):28835–28845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pi M, Quarles LD. Multiligand specificity and wide tissue expression of GPRC6A reveals new endocrine networks. Endocrinology. 2012;153(5):2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pi M, Zhang L, Lei SF, et al. Impaired osteoblast function in GPRC6A null mice. J Bone Miner Res. 2010;25(5):1092–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsuka S, Aonuma F, Higashi S, et al. Promotion of insulin-induced glucose uptake in C2C12 myotubes by osteocalcin. Biochem Biophys Res Commun. 2015;459(3):437–442. [DOI] [PubMed] [Google Scholar]
- 20. Jacobsen SE, Nørskov-Lauritsen L, Thomsen AR, et al. Delineation of the GPRC6A receptor signaling pathways using a mammalian cell line stably expressing the receptor. J Pharmacol Exp Ther. 2013;347(2):298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuang D, Yao Y, Lam J, Tsushima RG, Hampson DR. Cloning and characterization of a family C orphan G-protein coupled receptor. J Neurochem. 2005;93(2):383–391. [DOI] [PubMed] [Google Scholar]
- 22. Liu M, Shapiro ME. A new method for isolation of murine islets with markedly improved yields. Transplant Proc. 1995;27(6):3208–3210. [PubMed] [Google Scholar]
- 23. Gerling IC, Serreze DV, Christianson SW, Leiter EH. Intrathymic islet cell transplantation reduces β-cell autoimmunity and prevents diabetes in NOD/Lt mice. Diabetes. 1992;41(12):1672–1676. [DOI] [PubMed] [Google Scholar]
- 24. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. [DOI] [PubMed] [Google Scholar]
- 25. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. MacKerell AD, Bashford D, Bellott M, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102(18):3586–3616. [DOI] [PubMed] [Google Scholar]
- 27. Kall L, Krogh A, Sonnhammer EL. An HMM posterior decoder for sequence feature prediction that includes homology information. Bioinformatics. 2005;21(suppl 1):i251–i257. [DOI] [PubMed] [Google Scholar]
- 28. Labute P. The generalized Born/volume integral implicit solvent model: estimation of the free energy of hydration using London dispersion instead of atomic surface area. J Comput Chem. 2008;29(10):1693–1698. [DOI] [PubMed] [Google Scholar]
- 29. Wu H, Wang C, Gregory KJ, et al. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science. 2014;344(6179):58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Doré AS, Okrasa K, Patel JC, et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature. 2014;511(7511):557–562. [DOI] [PubMed] [Google Scholar]
- 31. Xiao YC, Hardy DO, Sottas CM, Li XK, Ge RS. Inhibition of LH-stimulated androgen production in rat immature Leydig cells: effects on nuclear receptor steroidogenic factor 1 by FGF2. Growth Factors. 2010;28(1):1–9. [DOI] [PubMed] [Google Scholar]
- 32. Christiansen B, Hansen KB, Wellendorph P, Bräuner-Osborne H. Pharmacological characterization of mouse GPRC6A, an L-α-amino-acid receptor modulated by divalent cations. Br J Pharmacol. 2007;150(6):798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sabek OM, Nishimoto SK, Fraga D, Tejpal N, Ricordi C, Gaber AO. Osteocalcin effect on human β-cells mass and function. Endocrinology. 2015;156(9):3137–3146. [DOI] [PubMed] [Google Scholar]
- 34. Clemmensen C, Smajilovic S, Wellendorph P, Brauner-Osborne H. The G protein-coupled receptor, class C, group 6, subtype A (GPRC6A) receptor: from cloning to physiological function. Br J Pharmacol. 2013;171(5):1129–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Faure H, Gorojankina T, Rice N, et al. Molecular determinants of non-competitive antagonist binding to the mouse GPRC6A receptor. Cell Calcium. 2009;46(5–6):323–332. [DOI] [PubMed] [Google Scholar]
- 36. Pi M, Quarles LD. GPRC6A regulates prostate cancer progression. Prostate. 2012;72(4):399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dreaden EC, Gryder BE, Austin LA, et al. Antiandrogen gold nanoparticles dual-target and overcome treatment resistance in hormone-insensitive prostate cancer cells. Bioconjug Chem. 2012;23(8):1507–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tilley WD, Wilson CM, Marcelli M, McPhaul MJ. Androgen receptor gene expression in human prostate carcinoma cell lines. Cancer Res. 1990;50(17):5382–5386. [PubMed] [Google Scholar]
- 39. Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes. 2000;49(3):424–430. [DOI] [PubMed] [Google Scholar]
- 40. Bohl CE, Wu Z, Chen J, et al. Effect of B-ring substitution pattern on binding mode of propionamide selective androgen receptor modulators. Bioorg Med Chem Lett. 2008;18(20):5567–5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen J, Hwang DJ, Bohl CE, Miller DD, Dalton JT. A selective androgen receptor modulator for hormonal male contraception. J Pharmacol Exp Ther. 2005;312(2):546–553. [DOI] [PubMed] [Google Scholar]
- 42. Chen J, Hwang DJ, Chung K, et al. In vitro and in vivo structure-activity relationships of novel androgen receptor ligands with multiple substituents in the B-ring. Endocrinology. 2005;146(12):5444–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hwang DJ, Yang J, Xu H, et al. Arylisothiocyanato selective androgen receptor modulators (SARMs) for prostate cancer. Bioorg Med Chem. 2006;14(19):6525–6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jones A, Chen J, Hwang DJ, Miller DD, Dalton JT. Preclinical characterization of a (S)-N-(4-cyano-3-trifluoromethyl-phenyl)-3-(3-fluoro, 4-chlorophenoxy)-2-hydroxy-2-methyl-propanamide: a selective androgen receptor modulator for hormonal male contraception. Endocrinology. 2009;150(1):385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jones A, Hwang DJ, Duke CB, 3rd, et al. Nonsteroidal selective androgen receptor modulators enhance female sexual motivation. J Pharmacol Exp Ther. 2010;334(2):439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jones A, Hwang DJ, Narayanan R, Miller DD, Dalton JT. Effects of a novel selective androgen receptor modulator on dexamethasone-induced and hypogonadism-induced muscle atrophy. Endocrinology. 2010;151(8):3706–3719. [DOI] [PubMed] [Google Scholar]
- 47. Kim J, Wu D, Hwang DJ, Miller DD, Dalton JT. The para substituent of S-3-(phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamides is a major structural determinant of in vivo disposition and activity of selective androgen receptor modulators. J Pharmacol Exp Ther. 2005;315(1):230–239. [DOI] [PubMed] [Google Scholar]
- 48. Li W, Hwang DJ, Cremer D, et al. Structure determination of chiral sulfoxide in diastereomeric bicalutamide derivatives. Chirality. 2009;21(6):578–583. [DOI] [PubMed] [Google Scholar]
- 49. Mohler ML, Bohl CE, Jones A, et al. Nonsteroidal selective androgen receptor modulators (SARMs): dissociating the anabolic and androgenic activities of the androgen receptor for therapeutic benefit. J Med Chem. 2009;52(12):3597–3617. [DOI] [PubMed] [Google Scholar]
- 50. Mohler MLN, Vipin A, Hwang DJ, et al. Nonsteroidal tissue selective androgen receptor modulators: a promising class of clinical candidates. Expert Opin Ther Pat. 2005;15(11):1565–1585. [Google Scholar]
- 51. Narayanan R, Coss CC, Yepuru M, Kearbey JD, Miller DD, Dalton JT. Steroidal androgens and nonsteroidal, tissue-selective androgen receptor modulator, S-22, regulate androgen receptor function through distinct genomic and nongenomic signaling pathways. Mol Endocrinol. 2008;22(11):2448–2465. [DOI] [PubMed] [Google Scholar]
- 52. Narayanan R, Ahn S, Cheney MD, et al. Selective androgen receptor modulators (SARMs) negatively regulate triple-negative breast cancer growth and epithelial:mesenchymal stem cell signaling. PLoS One. 2014;9(7):e103202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gloriam DE, Wellendorph P, Johansen LD, et al. Chemogenomic discovery of allosteric antagonists at the GPRC6A receptor. Chem Biol. 2011;18(11):1489–1498. [DOI] [PubMed] [Google Scholar]
- 54. Silve C, Petrel C, Leroy C, et al. Delineating a Ca2+ binding pocket within the venus flytrap module of the human calcium-sensing receptor. J Biol Chem. 2005;280(45):37917–37923. [DOI] [PubMed] [Google Scholar]
- 55. Ellingson SR, Miao Y, Baudry JY, Smith JC. Multi-conformer ensemble docking to difficult protein targets. J Phys Chem B. 2014;119(3):1026–1034. [DOI] [PubMed] [Google Scholar]
- 56. Meier JJ, Butler AE, Saisho Y, et al. β-Cell replication is the primary mechanism subserving the postnatal expansion of β-cell mass in humans. Diabetes. 2008;57(6):1584–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hanchang W, Semprasert N, Limjindaporn T, Yenchitsomanus PT, Kooptiwut S. Testosterone protects against glucotoxicity-induced apoptosis of pancreatic β-cells (INS-1) and male mouse pancreatic islets. Endocrinology. 2013;154(11):4058–4067. [DOI] [PubMed] [Google Scholar]
- 58. Høst C, Gormsen LC, Hougaard DM, Christiansen JS, Pedersen SB, Gravholt CH. Acute and short-term chronic testosterone fluctuation effects on glucose homeostasis, insulin sensitivity, and adiponectin: a randomized, double-blind, placebo-controlled, crossover study. J Clin Endocrinol Metab. 2014;99(6):E1088–E1096. [DOI] [PubMed] [Google Scholar]
- 59. Zitzmann M. Testosterone deficiency, insulin resistance and the metabolic syndrome. Nat Rev Endocrinol. 2009;5(12):673–681. [DOI] [PubMed] [Google Scholar]
- 60. Boonyaratanakornkit V, Edwards DP. Receptor mechanisms mediating non-genomic actions of sex steroids. Semin Reprod Med. 2007;25(3):139–153. [DOI] [PubMed] [Google Scholar]
- 61. Bukulmez O, Hardy DB, Carr BR, et al. Androstenedione up-regulation of endometrial aromatase expression via local conversion to estrogen: potential relevance to the pathogenesis of endometriosis. J Clin Endocrinol Metab. 2008;93(9):3471–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Loss ES, Jacobus AP, Wassermann GF. Rapid signaling responses in Sertoli cell membranes induced by follicle stimulating hormone and testosterone: calcium inflow and electrophysiological changes. Life Sci. 2011;89(15–16):577–583. [DOI] [PubMed] [Google Scholar]
- 63. Sharpe RM. Intratesticular regulation of testosterone secretion: comparison of the effects and interactions of hCG, an LHRH agonist and testicular interstitial fluid on Leydig cell testosterone secretion in vitro. Mol Cell Endocrinol. 1985;41(2–3):247–255. [DOI] [PubMed] [Google Scholar]
- 64. O'Hara L, McInnes K, Simitsidellis I, et al. Autocrine androgen action is essential for Leydig cell maturation and function, and protects against late-onset Leydig cell apoptosis in both mice and men. FASEB J. 2015;29(3):894–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bhasin S, Travison TG, Storer TW, et al. Effect of testosterone supplementation with and without a dual 5α-reductase inhibitor on fat-free mass in men with suppressed testosterone production: a randomized controlled trial. JAMA. 2012;307(9):931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Takata R, Akamatsu S, Kubo M, et al. Genome-wide association study identifies five new susceptibility loci for prostate cancer in the Japanese population. Nat Genet. 2010;42(9):751–754. [DOI] [PubMed] [Google Scholar]
- 67. Bhindi B, Locke J, Alibhai SM, et al. Dissecting the association between metabolic syndrome and prostate cancer risk: analysis of a large clinical cohort. Eur Urol. 2014;67(1):64–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.