

Significance
Many enzymes perform desirable biochemical transformations, but are not suitable to use as biocatalysts outside of the cell. In particular, enzymes from heteromeric complexes typically have decreased activity when removed from their protein partners. We used directed evolution to restore the catalytic efficiency of the tryptophan synthase β-subunit (TrpB), which synthesizes l-tryptophan from l-serine and indole, surpassing the activity of the native complex. Experiments show that activating mutations promote catalysis through the same mechanism as partner protein binding, establishing that isolated subunits may be readily reactivated through directed evolution. Engineering TrpB for stand-alone function restored high activity with indole analogs, providing a simplified enzyme platform for the biocatalytic production of noncanonical amino acids.
Keywords: protein engineering, allostery, noncanonical amino acid, PLP
Abstract
Enzymes in heteromeric, allosterically regulated complexes catalyze a rich array of chemical reactions. Separating the subunits of such complexes, however, often severely attenuates their catalytic activities, because they can no longer be activated by their protein partners. We used directed evolution to explore allosteric regulation as a source of latent catalytic potential using the β-subunit of tryptophan synthase from Pyrococcus furiosus (PfTrpB). As part of its native αββα complex, TrpB efficiently produces tryptophan and tryptophan analogs; activity drops considerably when it is used as a stand-alone catalyst without the α-subunit. Kinetic, spectroscopic, and X-ray crystallographic data show that this lost activity can be recovered by mutations that reproduce the effects of complexation with the α-subunit. The engineered PfTrpB is a powerful platform for production of Trp analogs and for further directed evolution to expand substrate and reaction scope.
Heteromeric enzyme complexes catalyzing a rich array of useful reactions are often allosterically regulated by their protein partners, such that the catalytic subunits are much less active when isolated (1–3). Utilization of isolated enzyme subunits, however, is desirable for biosynthetic applications, where expressing large complexes increases the metabolic load on the host cell and complicates efforts to engineer activity, substrate specificity, stability, and other properties. A solution to these problems is to engineer the isolated subunit to function independently. However, it is unknown to what extent mutations in the amino acid sequence can reproduce the complex structural and functional changes that are induced by binding of a partner protein.
Tryptophan synthase (TrpS; EC 4.2.1.20) is a heterodimeric complex that catalyzes the formation of l-tryptophan (Trp, 1) from l-serine (Ser, 2) and indole glycerol phosphate (IGP, 3; Fig. 1A) (2). The mechanism of this transformation has been extensively studied for TrpS from Escherichia coli and Salmonella typhimurium, where it has been shown the enzyme consists of two subunits, TrpA (α-subunit) and TrpB (β-subunit), both of which have low catalytic efficiencies in isolation (4). The activities of both subunits increase upon complex formation and are further regulated by an intricate and well-studied allosteric mechanism (2). IGP binding to the α-subunit stimulates pyridoxal phosphate (PLP)-dependent aminoacrylate formation in the β-subunit [E(A-A); Fig. 1B], which in turn promotes retro-aldol cleavage of IGP in the α-subunit, releasing indole (4). This tightly choreographed mechanism serves to prevent the free diffusion of indole, which is only released from the α-subunit when the complex is in a closed conformation that forms a 25-Å tunnel through which indole diffuses into the β-subunit (5). Here, indole reacts with E(A-A) in a C–C bond-forming reaction, yielding l-tryptophan as product (Fig. 1B) (2, 5). These allosteric effects are mediated through the rigid-body motion of the communication (COMM) domain and a monovalent cation (MVC) binding site within the β-subunit (Fig. 1A), which undergo complex conformational transitions associated with open, partially closed, and fully closed states during the catalytic cycle (2, 4, 6).
Fig. 1.

Catalytic cycle of the native TrpS complex. (A) Indole (4) is released through a retro-aldol reaction in TrpA (red) and then diffuses through a 25-Å tunnel into TrpB (black), where a PLP-mediated β-substitution reaction occurs with l-serine (2), yielding l-tryptophan (1). The COMM domain is indicated in blue. Scheme is superimposed over PfTrpS with Lys-PLP internal aldimine, E(Ain), shown in green sticks. The native complex is an αββα heterotetramer; a single αβ pair is shown for clarity. (B) Mechanism of l-tryptophan formation. Transimination of l-serine to form an external aldimine, E(Aex1), followed by dehydration across Cα–Cβ through a quinonoid intermediate, E(Q1), is designated stage I of the overall reaction. A mechanistically similar process occurs in reverse for the addition of indole into E(A-A) and subsequent release of l-tryptophan, designated stage II of the reaction.
TrpS is a naturally promiscuous enzyme complex: the model system from S. typhimurium catalyzes its β-substitution reaction with most haloindoles, methylindoles, and aminoindoles, along with an assortment of nonindole nucleophiles for C–S, C–N, and C–C bond formation (7). Such noncanonical amino acids (NCAAs) have diverse applications in chemical biology (8), serve as intermediates in the synthesis of natural products (9, 10), and are privileged scaffolds for the development of pharmaceuticals (11). Despite its natural ability to produce these desirable compounds, TrpS has enjoyed only limited application (7). Optimized methods are restricted by low substrate concentrations and yields typically below 50% (7, 12, 13). To produce NCAAs, researchers have used the S. typhimurium TrpS complex (StTrpS), which suffers from poor thermostability and low tolerance to organic solvents (14). Although only the reactivity of the β-subunit, coupling of l-serine and indole, is necessary and desirable for synthetic applications, using TrpB as an isolated enzyme has not been feasible. Outside of its native complex, TrpB loses up to 95% of its native activity and is subject to inactivation (15, 16). These facts combine to present a compelling challenge: can we use directed evolution to recover the activity lost when TrpA is removed and create a highly active stand-alone TrpB enzyme? If so, what are the structural and kinetics effects of the mutations that harness this latent catalytic potential?
Results
Selection of the Parent Enzyme, TrpB, from Pyrococcus furiosus.
We focused the search for an engineering starting point on known thermophilic TrpS enzymes for three reasons: (i) higher operating temperatures afford increased solubility of the hydrophobic substrates, which is useful for preparative reactions; (ii) thermostable enzymes are more tolerant to the introduction of activating but potentially destabilizing mutations (17); and (iii) thermostable enzyme variants can be screened efficiently (described below). We compared published kinetic properties of TrpS from Thermotoga maritima (18), Thermococcus kodakaraensis (19), and Pyrococcus furiosus (20–22) and selected the last for its superior kinetic parameters and thermostability (Table S1). In our hands, PfTrpB heterologously expressed and purified from E. coli has a kcat of 0.31 s−1 and experiences a 12-fold increase in catalytic efficiency upon addition of purified P. furiosus TrpA (PfTrpA) to make the PfTrpS complex (Table 1), similar to values reported previously for E. coli TrpB (EcTrpB) (15). Notably, PfTrpB does not show the mechanism-based inactivation that inhibits use of StTrpB for preparation of NCAAs (23).
Table S1.
Comparison of TrpB thermophilic enzymes
| Host | kcat, s−1 | KM, mM l-Ser | KM, µM indole | kcat change with TrpA | Thermal stability indicators | Ref(s). |
| Thermotoga maritima | 4.2 | 110 | 40 | 2.4 | Kinetics measured at 80 °C | 18 |
| Thermococcus kodakarensis | 1.04 ± 0.03 | n/a | 63 ± 5 | 3.3 | 5% activity after 1 h at 80 °C | 19 |
| Pyrococcus furiosus | 0.31 ± 0.02 | 1.2 ± 0.1 | 77 ± 12 | 3.2 | T50 = 94.7 ± 1.2 | 20–22 |
Values for PfTrpB are those reported here; the references supply similar data measured by a different group. n/a, not available.
Table 1.
Biochemical characterization of tryptophan synthases
| Enzyme | Mutations | kcat,s−1 | KM, mM l-serine | KM, μM indole | kcat/KM, mM-1s⋅−1 indole | kcat change with TrpA* | T50 °C† |
| PfTrpS | — | 1.0 | 0.6 | 20 | 50 | — | > 95 |
| PfTrpB | — | 0.31 | 1.2 | 77 | 4 | 3.2 | 95 |
| PfTrpB2G9 | T292S | 1.1 | 0.84 | 14 | 78 | 0.34 | 95 |
| PfTrpB4D11 | E17G, I68V, T292S, F274S, T321A | 2.2 | 1.2 | 11 | 200 | 0.3 | 84 |
| PfTrpB0B2 | P12L, E17G, I68V, T292S, F274S, T321A | 2.9 | 0.7 | 8.7 | 330 | 0.04 | 87 |
All kinetic data are measured at 75 °C using a previously described (15) continuous assay for Trp production. Additional mutations relative to their parent enzyme are highlighted in bold.
The effect of PfTrpA was measured by addition of a fivefold stoichiometric excess of PfTrpA under conditions saturating in each substrate.
T50 is the temperature of half-maximal activity after incubation for 1 h. Assay parameters are described in Materials and Methods. SE for each measurement is given in Table S2.
Table S2.
Biochemical characterization of native and engineered enzymes
| Enzyme | Mutations | kcat, s−1 | KM, mM l-Ser | KM, µM indole | kcat/KM, mM−1⋅s−1 (Ind) | kcat change with TrpA | T50 °C |
| PfTrpS | – | 1.0 ± 0.1 | 0.6 ± 0.1 | 20 ± 2 | 50 | – | >95 |
| PfTrpB | – | 0.31 ± 0.02 | 1.2 ± 0.1 | 77 ± 12 | 4 | 3.2 | 95.1 ± 1.3 |
| PfTrpB2G9 | T292S | 1.1 ± 0.2 | 0.84 ± 0.04 | 14 ± 3 | 78 | 0.34 | 94.7 ± 1.2 |
| PfTrpB4D11 | E17G, I28V, F274S, T292S, T321A | 2.2 ± 0.2 | 1.2 ± 0.2 | 11 ± 2 | 200 | 0.3 | 83.6 ± 2.4 |
| PfTrpB0B2 | P12L, E17G, I28V, F274S, T292S, T321A | 2.9 ± 0.2 | 0.7 ± 0.1 | 8.7 ± 1.8 | 330 | 0.04 | 87.3 ± 1.3 |
Assay parameters are described in SI Materials and Methods.
Directed Evolution of PfTrpB for Stand-Alone Function.
We performed random mutagenesis by error-prone PCR and screened a small PfTrpB mutant library (528 clones) for increased Vmax under saturating concentrations of substrate (l-serine and indole). The extreme thermostability of the parent enzyme permitted a 1-h heat treatment of the lysates at high temperature (75 °C) that precipitated the majority of E. coli proteins and ensured that any activated variants retained significant stability. Formation of l-tryptophan took place at 75 °C for up to 1 h (generation 1, 1 h; generations 2 and 3, 6 min). The reactions were then quenched in an ice-water bath, and the production of l-tryptophan was quantified at 290 nm with a plate reader. This procedure identified many activating mutations, with 3.8% (20/528) of the screened variants of generation 1 showing at least 40% greater product formation than the parent. A single mutation, T292S, gave rise to a 3.5-fold increase in kcat compared with PfTrpB, which completely recovered kcat and even exceeded the catalytic efficiency (kcat/KM) of the PfTrpS complex (Table 1). Twenty-six other mutations in 19 different variants also contributed activating effects (Fig. S1).
Fig. S1.

Distribution of activating mutations identified through random mutagenesis and screening. (A) Positions on PfTrpB where activating mutations were discovered are displayed as spheres. The mutations are W2R, G4C, V11A, E17G, K20E, E23V, F35S, N35S, Y41C, I68V, M123V, I127S, M144T, N150T, N166D, Y178C, H180R, Y181C, L182P, M233V, M233I, F274S, F274L, D284G, T292S, T321A, and T323A. Mutations located in the COMM domain are colored blue, those within 5 Å of PfTrpA (red) are cyan, those present in PfTrpB4D11 are yellow, and the remainder are gray. (B) Positions on PfTrpB where activating mutations were discovered using PfTrp4D11 as the parent for random mutagenesis. The mutations are G4D, E5K, Y10H, P12L, E13G, E21V, L59Q, K67I, L146V, D220E, N267S, G272D, and D284E. Color scheme is the same as A; yellow spheres are mutations present in the final variant PfTrpB0B2.
We recombined 12 of the most activating mutations identified in the first generation (SI Materials and Methods). Screening a total of 1,408 clones, we identified PfTrpB4D11 containing mutations E17G, I68V, F274S, T292S, and T321A. The kcat of PfTrpB4D11 was 2.2 s−1, a sevenfold improvement over wild type (Table 1). PfTrpB4D11 served as template for a final round of random mutagenesis, for which we screened 1,144 clones. PfTrpB0B2 carried one additional mutation, P12L, and had a kcat that was 9.4-fold higher than PfTrpB and threefold higher than PfTrpS. With PfTrpB0B2 we had surpassed our goal of engineering an isolated enzyme domain to be as active as in its native complex.
Biochemical Comparison of Evolved PfTrpB Enzymes with PfTrpS.
Kinetic analysis of PfTrpB and PfTrpS established that the 12-fold increase in the catalytic efficiency for indole upon complexation is driven by both an increase in kcat and decrease in KM (Table 1). Despite screening the mutant libraries under saturating conditions for both substrates, we observed a steady decrease in the KM for indole in the enzymes with greater activity, which mimics the behavior of the native complex. The KM for l-serine fluctuated during evolution, and in the final round the values for PfTrpB0B2 and PfTrpS were similar.
Though Michaelis–Menten kinetic analysis provides a simple readout of overall catalytic performance, activity changes upon complex formation and during directed evolution are associated with a shift in the populations of the intermediates in the TrpB catalytic cycle. The PLP cofactor absorbs in the UV-vis region, and different chemical intermediates have characteristic peaks that can be measured readily (Fig. 1B). Under turnover conditions, the recorded spectrum reflects the Boltzmann-weighted average of each of the intermediates in the catalytic cycle. Incubation of PfTrpB with l-serine results in a large increase in absorption at 428 nm, consistent with accumulation of E(Aex1) (Fig. 2A). A peak at 320 nm developed over several minutes, whereas the absorption at 428 nm remained constant (Fig. S2), which we attribute to the previously characterized serine deaminase activity that is an artifact of not having a competing nucleophile present (24). In contrast, l-serine-bound PfTrpS has a λmax near 350 nm, consistent with a shift in the equilibrium to populate the E(A-A) (Fig. 2B) (2). With the engineered proteins, the external aldimine absorbance at 428 nm decreases, and the absorbance bands at 350 nm grow in intensity over the course of directed evolution of Trp synthase activity (Fig. 2C and Fig. S2).
Fig. 2.

UV-vis absorption spectra of native and engineered PfTrpB. (A) The PLP absorption spectrum of PfTrpB (black) has a λmax = 412 nm, characteristic of E(Ain). Addition of 20 mM l-serine causes a red-shift to 428 nm (gray), consistent with E(Aex1) formation. (B) Addition of l-serine to PfTrpS causes a shift in λmax to 350 nm, which is attributed to the E(A-A). A residual peak at 428 nm indicates population of E(Aex1). (C) Like PfTrpS, addition of 20 mM l-serine to PfTrpB0B2 shows λmax = 350 nm as well as contributions from E(Aex1). All spectra were collected with 20 µM of enzyme. See Materials and Methods for details.
Fig. S2.

UV-vis absorption spectra for native and engineered TrpB. A, D, and E are the same as in Fig. 2. PLP absorption spectra for each enzyme are shown in black and share λmax = 412 nm, characteristic of E(Ain). Addition of 20 mM l-Ser causes a red-shift to 420 nm (red), consistent with E(Aex1) formation for PfTrpB and PfTrpB2G9 (B), which also has an absorbance band at ∼320 nm that we attribute to formation of pyruvate through serine deaminase activity. l-Ser addition to PfTrpS, PfTrpB4D11 (C), and PfTrpB0B2 causes a shift in λmax to 350 nm and a broad shoulder extending to 550 nm. The 350-nm absorption is attributed to the E(A-A), and the shoulder indicates a mixed population of E(Aex1) and E(A-A). All spectra collected with 20 µM enzyme. To limit the amount of pyruvate production, spectra were taken as quickly as possible (<10 s) after addition of l-Ser.
One question motivating this study was how mutational reactivation alters the allosteric regulation of PfTrpB by its TrpA protein partner. Adding purified PfTrpA to each TrpB variant gave a surprising result: instead of enhancing activity (or doing nothing), PfTrpA inhibited each improved TrpB, an effect that was stronger with each evolutionary step, to the point where PfTrpB0B2 had just 4% of its activity when PfTrpA was added (Table 1).
Structural Analysis of PfTrpB.
Whereas the residues that perform the TrpB chemistry are conserved throughout evolution, the conformational motions associated with allosteric signaling have not been characterized outside of StTrpS or in the absence of the α-subunit. We obtained high-resolution structures of wild-type PfTrpB in its ligand-free state, bound to its l-serine substrate, and bound to its l-tryptophan product (Table S3). Comparison of the ligand-free and Ser-bound structures (at 1.69 Å and 2.0 Å resolution, respectively) reveals motion of the COMM domain into a partially closed state upon serine binding, as well as a hydrogen bond between Asp300 and the Ser-hydroxyl of E(Aex1) (Fig. 3 A and B). This large conformational rearrangement is structurally equivalent to that characterized for StTrpS (25), with 0.5 Å rmsd between the E(Aex1) forms of TrpB (Fig. S3), despite the modest (59%) sequence identity and lack of an α-subunit. Combined with their similar biochemical properties, this structural conservation constitutes firm experimental support for applying insights gleaned from StTrpS and EcTrpS to the function of PfTrpB. Labeling studies have shown that β-substitution occurs with retention of stereochemistry, indicating the hydroxyl of E(Aex1) must rotate 180° from its crystallographically observed state to eliminate from the same face to which indole is added (26). The H-bond between E(Aex1) and Asp300 is, therefore, transient during the catalytic cycle and suggests an important role for the H-bond between Thr292 and Asp300 that we observed in the ligand-free structure. Notably, T292S was one of the most activating mutations identified in the first round of directed evolution (Fig. 4 A and B and Table 1). When l-Ser is incubated with PfTrpB2G9, the UV-vis spectrum clearly shows that the equilibrium is shifted toward E(A-A) (Fig. S2B). We hypothesize that this mutation alters the energetics of the Asp300-E(Aex1) interaction to favor the fully closed conformational state of the enzyme and thereby accelerate the reaction.
Table S3.
Crystallographic data collection and refinement statistics
| Protein | PfTrpB | PfTrpB | PfTrpB | PfTrpS |
| PDB ID code | 5DVZ | 5DW0 | 5DW3 | 5E0K |
| Ligand | None | l-Serine | l-Tryptophan | None |
| Space group | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions, Å | a,b,c = 87.1, 111.9, 160.8 | a,b,c = 84.2, 109.1, 160.8 | a,b,c = 83.7, 108.9, 160.1 | a,b,c = 87.7, 225.0, 296.2 |
| Cell angles | α = β = γ = 90° | α = β = γ = 90° | α = β = γ = 90° | α = β = γ = 90° |
| Data collection | ||||
| Wavelength, Å | 0.9795 | 0.9795 | 0.9795 | 0.9795 |
| Beamline | SSRL 12.2 | SSRL 12.2 | SSRL 12.2 | SSRL 12.2 |
| Resolution, Å | 40–1.69 | 40–2.01 | 40–1.74 | 40–2.76 |
| Last bin (Å) | (1.72–1.69) | (2.05–2.01) | (1.77–1.74) | (2.81–2.76) |
| No. observations | 865,034 | 942,945 | 1,455,851 | 1,006,477 |
| Completeness (%) | 99.6 (99.9) | 98.0 (67.2) | 98.7 (75.0) | 99.3 (93.7) |
| Rpim (%) | 0.050 (0.899) | 0.034 (0.754) | 0.020 (0.752) | 0.082 (1.21) |
| CC(1/2) | 0.995 (0.439) | 0.999 (0.299) | 1.000 (0.425) | 0.990 (0.154) |
| I/σI | 7.3 (0.6) | 15.1 (0.9) | 18.0 (1.0) | 8.0 (0.6) |
| Redundancy | 4.9 (4.9) | 9.8 (5.6) | 9.8 (2.8) | 6.7 (5.3) |
| Refinement | ||||
| Total no. of reflections | 161,910 | 91,540 | 140,124 | 142,144 |
| Total no. of atoms | 12,278 | 11,976 | 12,282 | 28,872 |
| Final bin (Å) | (1.73–1.69) | (2.07–2.01) | (1.79–1.74) | (2.83–2.76) |
| Rwork (%) | 20.2 (39.1) | 17.6 (36.7) | 18.9 (36.1) | 20.4 (37.7) |
| Rfree (%) | 22.8 (39.0) | 22.1 (36.8) | 22.7 (38.4) | 23.8 (39.2) |
| Average B factor, Å2 | 31.8 | 42.7 | 40.5 | |
| Ramachandran plot favored, % | 96.8 | 96.2 | 96.6 | 97.3 |
| Allowed, % | 100 | 100 | 99.9 | 99.8 |
| Outliers, % | 0 | 0 | 0.1 | 0.2 |
Values in parentheses are for the highest-resolution shell. Rmerge is Σ|Io – I|/ΣIo, where Io is the intensity of an individual reflection, and I is the mean intensity for multiply recorded reflections; Rwork is Σ||Fo – Fc||/Fo, where Fo is an observed amplitude and Fc a calculated amplitude; Rfree is the same statistic calculated over a 5% subset of the data that has not been included.
Fig. 3.

Structural transitions upon ligand binding in PfTrpB. (A) Superimposition of PfTrpB-E(Ain) and PfTrpB-E(Aex1) in gray and cyan, respectively. Overlay shows the 2.1-Å displacement of the COMM domain upon E(Aex1) formation. This closure moves the side chain of Glu104 by 3.7 Å, toward its catalytic orientation (orange dashes). (B) Structure of Ser-bound PfTrpB with a Fo–Fc map of E(Aex1) contoured at 3.0 σ (green). (C) Structure of l-tryptophan–bound PfTrpB with Fo–Fc map of Trp ligand contoured at 3.0 σ.
Fig. S3.

Structural comparison between StTrpS and PfTrpB in E(Ain) and E(Aex1) forms. (A) Alignment of StTrpB (green; PDB ID code 1BKS) and PfTrpB (light blue) in their substrate-free [E(Ain)] forms yields an rmsd of 0.5 Å. Small differences exist in the structures near the α-subunit (red) binding site, and the COMM domain of PfTrpB (dark blue) is in a slightly more open conformation than for StTrpB. (B) Alignment of StTrpB (green; PDB ID code 2CLL) and PfTrpB (light blue) with l-serine covalently bound as the external aldimine, E(Aex1), yields an rmsd of 0.5 Å.
Fig. 4.

Distribution of PfTrpB0B2 mutations and interaction networks altered by mutational reactivation. (A) PfTrpB residues within 5 Å of PfTrpA (red) are colored cyan and the COMM domain is colored blue. (B) A hydrogen bond between D300 and E(Aex1) in the Ser-bound structure (orange) is formed transiently during the catalytic cycle. When this H-bond is severed, D300 may interact with T292 (no ligands, red, or Trp-bound, blue). This complex network is centered on a monovalent cation cofactor, shown here as Na+, which is known to mediate allosteric interactions between the α- and β-subunits (6). (C) Residues F274 and H275 undergo a rotameric shift upon substrate or product binding into an open state (blue). (D) In the closed state (red, no ligands bound), H275 blocks access to the active site.
The structure of PfTrpB with l-tryptophan bound in the active site at 1.74 Å resolution shows the product is not covalently linked to the PLP cofactor, but is in a novel ligand-binding pose (Fig. 3C). Residues Thr105 to His110 comprise a carboxylate binding motif (4) that we report also forms H-bonds to the primary amine of l-tryptophan through the backbone N–H of Ala106. This same residue also H-bonds with the Ser-hydroxyl of E(Aex1). Asp300, however, is not observed to interact with l-tryptophan. A hydrogen bond between Glu104 and the N-1 of l-tryptophan likely also occurs when indole binds. This interaction may serve to increase the nucleophilicity of indole by positioning C-3 close to the acrylate of E(A-A) and by increasing electron density of the arene (Fig. 1B) (2, 4). Glu104 is located in the COMM domain and shifts closer to the active site upon closure (Fig. 3A). In each of the substrate- and product-bound structures, we observed a rotameric shift of Phe274 and His275 (Fig. 4C) that may function as a gate within the indole tunnel (Fig. 4D). In the structure of PfTrpS, we observe a hydrogen bond between His275 and Asp43 from the α-subunit, indicating that PfTrpA binding stabilizes this open conformation (Fig. S4A). In StTrpS, NMR analysis has shown this motion is concerted, and our observation that it is conserved across species is consistent with it having an important role for regulating catalytic function (27). The location of the F274S mutation is therefore quite striking, because it may alter the energetics of this transition to favor higher activity.
Fig. S4.

Interactions between PfTrpA and PfTrpB that are disrupted in PfTrpB0B2. (A) Hydrogen bond between H275β (gray) and D43α (red) in the gate-open conformation is adjacent F274β (yellow), which is mutated to Ser in the PfTrpB4D11 and PfTrpB0B2 enzymes. (B) A salt bridge (dashes) between R148α (red) and E17β (yellow) is removed with the E17G mutation present in PfTrpB4D11 and PfTrpB0B2. P12β is located along the indole tunnel (C) between the subunits. Steric constraints indicate that the P12L mutation of PfTrpB0B2 would disrupt this tunnel, which might contribute to the large drop in Trp synthase activity of PfTrpS0B2.
Two mutations, E17G and P12L, map onto the α/β interface of a previously determined 3.0-Å structure of PfTrpS (22), but this low resolution precluded confident assessment of the side-chain interactions for these residues. We solved a higher-resolution structure at 2.76 Å that revealed a salt bridge between Glu17 of PfTrpB and Arg148 of PfTrpA (Fig. S4B). Pro12 of PfTrpB is an evolutionarily conserved residue that lies along the indole tunnel between the two subunits, where previous studies have found that mutation to bulkier residues inhibits substrate channeling (28). Such interactions are clearly disrupted at the α/β interface (Fig. S4C), but the strong inhibition of PfTrpB0B2 upon PfTrpA addition demonstrates that these proteins still associate. Finally, two mutations present in PfTrpB0B2, I68V and T321A, are distal to sites that undergo an observable structural change upon substrate binding or complex formation, and their contribution to rate enhancement is difficult to rationalize. Overall, 60% of the activating mutations identified through random mutagenesis were located within 5 Å of the α/β interface, the COMM domain, or regions that undergo observable motion upon transition to the closed conformation (Fig. S1). These positions comprise just 31% of the protein sequence, indicating modest enrichment within residues in the immediate spatial route between the α/β interface and the β-subunit active site. In contrast to the many mutations that have been identified as deleterious to the allosteric communication in TrpS (29, 30), the mutations identified here using directed evolution are the first reported to affect allosteric communication and increase the activity of TrpB in isolation.
PfTrpB0B2 Is a Stand-Alone Catalyst for Production of NCAAs.
Our selection of TrpB to evolve for high activity outside of its native complex was motivated by practical considerations. Previous studies have shown that StTrpS is a promiscuous enzyme, capable of synthesizing diverse analogs of l-tryptophan (7, 13). These analogs require subtly different transition state stabilization for catalysis, but the role of allostery in promoting this desirable chemistry has not been studied. We selected a representative panel of substrate analogs to study promiscuous activity in the native and engineered proteins (Fig. 5A). The StTrpS complex reacts with most halogenated and methylated indoles, which only modestly change the steric and electronic properties of the indole ring and afford reactivity at C3. Alternatively, indazole (5) and indoline (6), which have substantially altered electronic properties, react at N1 for C–N bond formation. We measured the relative rate of PfTrpB compared with PfTrpS and observed much larger rate enhancements for TrpB on the indole derivatives than on the native substrate (Fig. 5B), up to a 100-fold increase in the rate of NCAA synthesis with 5-bromoindole (7). These results raised an important question about our objective to engineer PfTrpB for function as an independent subunit by directed evolution: will screening for higher activity with indole translate into increased activity on alternative substrates, or will evolution generate an enzyme that is highly active on indole but less active toward other substrates, as has been observed in directed evolution of other enzymes (31, 32)?
Fig. 5.

Substrate profile of native and engineered TrpB enzymes. (A) Indole analogs that have been reported to react with StTrpS were tested for reactivity with the PfTrpB, PfTrpS, and PfTrpB0B2 enzymes. The nucleophilic atom is indicated with a gray circle. (B) Relative activities of enzyme complex PfTrpS (black) and PfTrpB0B2 (gray) compared with PfTrpB. Reactions performed in duplicate with 20 mM of each substrate and varying enzyme concentrations to ensure incomplete conversion after 1 h. Products were later confirmed in scaled-up reactions using PfTrpB0B2. See Materials and Methods for details.
We measured the activity of PfTrpB0B2 on the substrate panel (Fig. 5B) and were pleased to observe a substantial increase in activity relative to PfTrpB for all nucleophiles tested. The activity profile is broadly similar to that of PfTrpS, with a few exceptions. The PfTrpS complex reacts approximately eightfold faster than PfTrpB0B2 with bromoindole 7, whereas PfTrpB0B2 reacts approximately sixfold faster than PfTrpS with indoline 6 and threefold faster with indazole 5. The identity of each product was confirmed by mass spectrometry and NMR in reactions using PfTrpB0B2. Because we screened for increases in Vmax during laboratory evolution, the variants we selected also have improved expression levels relative to PfTrpB. E. coli cultures produce threefold more soluble and active PfTrpB0B2 than PfTrpB, exceeding 230 mg enzyme per liter culture, facilitating preparative reactions and future use of PfTrpB0B2 as a biocatalyst.
SI Materials and Methods
Cloning, Expression, and Purification of PfTrpA and PfTrpB.
The gene encoding PfTrpB (UNIPROT ID Q8U093) was obtained as a single gBlock, codon-optimized for E. coli, and cloned using Gibson Assembly (40) into pET22(b)+ between restriction sites NdeI and XhoI in frame with the C-terminal his6-tag for expression in E. coli BL21 E. cloni EXPRESS cells (Lucigen). The gene coding for PfTrpA (UNIPROT ID Q8U094) was treated in the same manner as described above, but the his6-tag was omitted. For heterologous protein expression of PfTrpA and PfTrpB, 5 mL TBamp was inoculated with single BL21 E. cloni colonies and incubated overnight at 37 °C and 250 rpm. The overnight cultures were used to inoculate 500-mL TBamp expression cultures to an initial OD600 of 0.1. After shaking at 250 rpm and 37 °C for ∼3 h or until an OD600 of 0.8 was reached, the cultures were chilled on ice for 20 min. The expression was induced by the addition of 500 mM IPTG to a final concentration of 1 mM in the chilled cultures that then continued to grow at 250 rpm and 20 °C for another 20 h. Cells were harvested at 4 °C and 5,000 × g for 10 min; the pellets were frozen at –20 °C until further use. For purification, frozen cell pellets of PfTrpB were thawed at room temperature and resuspended in 50 mM phosphate buffer, pH 8, with 20 mM imidazole and 100 mM NaCl (buffer A) with 100 µM PLP. After lysis with BugBuster (Novagen) according to the manufacturer’s recommendations, the cells were centrifuged at 20,000 × g and 4 °C for 10 min. The lysate was then incubated at 75 °C for 10 min, centrifuged again as described above, and applied to a 1-mL HisTrap HP column. The purification was performed with an AKTA purifier FPLC system (GE Healthcare). PfTrpB eluted during a linear gradient from buffer A to buffer B (50 mM phosphate buffer with 500 mM imidazole and 100 mM NaCl, pH 8) at 140 mM imidazole. Purified PfTrpB was desalted into 50 mM phosphate buffer, pH 8, frozen in liquid N2, and stored at −80 °C until further use.
Frozen cell pellets containing heterologously expressed PfTrpA were thawed at room temperature and resuspended in 50 mM Tris (pH 8.0). After lysis with BugBuster, heat treatment, and centrifugation as described for PfTrpB, the cleared lysate was passed manually through a 1-mL Q HP HiTrap column, and the flow-through containing PfTrpA was collected. Solid ammonium sulfate was slowly added to the PfTrpA-containing flow through while stirring on ice. After a final concentration of 35% was reached, the lysate was allowed to stir for another 2 h on ice and was then centrifuged at 20,000 × g and 4 °C for 30 min. The cleared ammonium sulfate-treated solution was loaded on a 5-mL pre-equilibrated [buffer B: 35% ammonium sulfate, 50 mM phosphate buffer (pH 8)] phenyl Sepharose HP HiTrap column using an AKTA purifier FPLC. TrpA eluted in a linear gradient with buffer A [50 mM phosphate buffer (pH 8)] at 2.5% ammonium sulfate and was then buffer exchanged into 50 mM phosphate buffer, pH 8, and frozen at –80 °C until further use. To obtain highly pure wild-type complex (TrpA and B), we combined purified TrpB with purified TrpA in a 1:2 ratio, applied this mixture to a 1-mL HisTrap HP column, and purified the formed complex via TrpB’s his6-tag (Fig. S6).
Fig. S6.

SDS/PAGE of PfTrpA, PfTrpB, and PfTrpS. 1, PfTrpA; 2, PfTrpB; 3–5, aliquots from 1.5-mL fractions from PfTrpA pull-down using His-tagged PfTrpB; 4–20% gradient, SDS/PAGE. Molecular weights from ColorPlus Prestained Protein Ladder (New England Biosystems) listed on the right.
Protein concentrations were determined via the Bradford assay (Bio-Rad).
Library Construction.
The error-prone PCR library for generation 1 was constructed with 300 µM MnCl2 and had an error rate of 1.7 missense mutations per gene among sequenced hits. Initial error-prone PCR libraries for generation 3 were constructed with the Mutazyme II kit (Stratagene) using 100–300 ng DNA. A larger library using 200 ng DNA was selected based on its retention of function rate: 40% of clones had activity not less than 40% less than wild type. This library had an error rate of 1.8 missense mutations per gene among sequenced hits. DNA shuffling was performed to recombine twelve activating mutations, which were distributed over two gBlocks (block 1: W2R, F35S, M123V, N166D, M233V, and T292S; block 2: E17G, I68V, M144T, L182P, F274S, and T312A). These two gBlocks and the wild-type gBlock served as template for DNA shuffling following a modified protocol of Stemmer (41). The resulting library was cloned into pET22(b)+ between restriction sites NdeI and XhoI in frame with the C-terminal his-tag for expression in E. coli BL21 E. cloni EXPRESS cells.
Due to low recombination frequency of the DNA shuffling library, a recombination library of mutations found at residues M123, M144, N166, L182, M233, F274, and T292 (generation 2) was constructed using SOE PCR (42). The mutagenesis primers encoded the desired mutations and also the corresponding wild-type sequences. Because the distances between some of the target sites would have resulted in very short fragments, we built the library in two steps. During the first step, we generated five fragments encompassing residues M123, N166, M233, and T292 using Phusion polymerase according to the manufacturer’s recommendations. The fragments were then DpnI-digested, gel-purified, and used as template for the subsequent assembly PCR using the flanking primers only. The assembly PCR product served as template for a second round of PCRs generating the fragments carrying the remaining mutations, followed by the same procedure as described above. The final assembly product was cloned into pET22(b)+ between restriction sites NdeI and XhoI in frame with the C-terminal his-tag for expression in E. coli BL21 E. cloni EXPRESS cells.
High-Throughput Screening.
For high-throughput expression, BL21 E. cloni EXPRESS cells carrying PfTrpB wild-type and variant plasmids were grown in 96-well deep-well plates in 300 μL TBamp at 37 °C and 80% humidity with shaking at 250 rpm overnight. TBamp expression cultures (630 μL) were inoculated with 20 μL of the overnight cultures and continued to grow at 37 °C and 80% humidity with shaking at 250 rpm for 3 h. Expression was induced with the addition of ITPG to a final concentration of 1 mM to cold-shocked (20 min on ice-water bath) cultures. The expression continued for another 20 h at 20 °C with shaking at 250 rpm. Cells were then centrifuged at 4,000 × g for 10 min and frozen at −20 °C overnight. For screening, cells were allowed to thaw at room temperature and then lysed in lysis buffer (400 μL per well of 200 mM phosphate buffer, pH 8, with 1 mg/mL lysozyme, 40 μM PLP, and ∼0.05 mg/mL DNase I) for 1 h at 37 °C. After centrifugation at 5,000 × g for 20 min, a 160-μL aliquot of the lysate was transferred into PCR plates (USA Scientific), heat-treated for 1 h at 75 °C, and then spun again at 1,000 × g and 4 °C for 30 min. After the addition of 160-μL assay buffer (200 mM phosphate buffer, 20 mM indole, 0.75 mM l-serine, pH 8) to 40 μL of cleared, heat-treated lysates in UV-transparent assay plates (Evergreen Scientific), formation of l-tryptophan took place at 75 °C for up to 1 h (generation 1:1 h; generations 2 and 3:6 min). The reactions were then quenched in an ice-water bath, and the amount of l-tryptophan formed was recorded at 290 nm with a plate reader (Tecan Infinite M200).
Kinetics.
PfTrpB activity (kcat) was measured by monitoring tryptophan formation in a UV1800 Shimadzu spectrophotometer (Shimadzu) at 75 °C over 1 min at 290 nm using Δε290 = 1.89 mM−1⋅cm−1 (15). The assay buffer contained 200 mM potassium phosphate, pH 8, and 5 μM PLP. Michaelis–Menten constants (KM) for indole were determined using a concentration range from 0.4 to 0.015 mM indole with the concentration of l-serine fixed at 25 mM. KM values for l-serine were measured with a concentration range from 50 to 0.5 mM l-serine with the concentration of indole fixed at 250 µM. Data were fit using an in-house nonlinear least-squares algorithm to the Michaelis–Menten equation implemented in MATLAB.
UV-Vis Spectroscopy.
Spectra were collected between 550 and 250 nm on a UV1800 Shimadzu spectrophotometer using 20 µM of enzyme in 200 mM potassium phosphate (pH 8.0) in a quartz cuvette. Samples were incubated at 75 °C for >3 min to ensure a stable temperature was reached. Stage 1 of the reaction was initiated by addition of 20 mM l-serine, and the spectra were measured in <10 s to limit production of pyruvate from deamination of l-serine, which absorbs at 320 nm.
Substrate Selectivity.
Purified enzyme was used to measure the relative rate of PfTrpS and PfTrpB0B2 NCAA production compared with PfTrpB. The 225-µL reactions were set up in duplicate with 20 mM l-serine and 20 mM indole analog (Fig. 5A) in 200 mM potassium phosphate (pH 8.0) with 5% DMSO. Assays were set up in glass HPLC vials (to prevent absorption of indole into the reaction vessel) and incubated at 75 °C for 1 h and quenched with addition of 775 µL 50% acetonitrile. Quenched reactions were transferred to an Eppendorf tube, spun for 10 min at 14,000 × g and 4 °C to remove precipitated protein, and the supernatant was transferred to fresh HPLC vials. The concentration of enzyme was adjusted to prevent reactions from running to completion. Relative rates were computed by measuring the ratio of the product peaks measured via UHPLC-MS (Agilent 1290 with 6140 MS detector) at 280 nm and then normalizing for the enzyme concentration.
Thermostability Measurements.
To determine the temperature where 50% of enzymes are rendered inactive (T50s), 5 μM of PfTrpB wild type and 1 μM of its variants were incubated at varying temperature ranges (wild type: 80–99 °C; variants 75–95 °C) for 1 h. After quenching on ice, the residual activity was measured as described above.
Crystallography.
Freshly purified PfTrpB (described above) was buffer exchanged against 20 mM Hepes (pH 7.85) with 5% glycerol and frozen at −80 °C until further use. Sparse matrix screening was performed using an Art Robbins Gryphon Nano crystallization robot using 0.2 µL of 11 mg/mL PfTrpB with 40 µM PLP and 0.2 µL of well solution. Crystals were found in a solution of 25% PEG3350 and 0.1 M Na Hepes (pH 7.5) after 3 d. Crystals were then routinely grown as sitting drops against a 1-mL reservoir of 15–25% PEG3350 and 0.1 M Na Hepes (pH 7.85) with mother liquor comprised of 1.5 µL of 8.0 mg/mL PfTrpB and 1.5 µL of well solution. Ligand-bound structures were determined by soaking crystals of PfTrpB with 15 mM l-tryptophan or 100 mM l-serine for 2 min. Crystals were cryoprotected through oil immersion in Fomblin Y (Sigma) and flash-frozen in liquid N2 until diffraction. Diffraction data were collected remotely at the Stanford Synchrotron Radiation Laboratories on beamline 12-2. Crystals routinely diffracted at or below 2.0 Å, and the data were integrated and scaled using XDS (43) and AIMLESS (44). A resolution cutoff of CC1/2 > 0.3 was applied along the strongest axis of diffraction (44, 45). These data contributed to model quality as judged by Rfree in the final bin <0.4. Structures were solved using molecular replacement with PHASER, as implemented in CCP4 (46, 47). The search model comprised a single monomer of PfTrpB (PDB ID code 1V8Z) (21) subjected to 10 cycles of geometric idealization in Refmac5 and removal of all ligands. Model-building was performed in Coot (48) beginning with data processed at 2.4 Å, followed by subsequent inclusion of increasingly higher-resolution shells of data with relaxed geometric constraints. This procedure was particularly important for the structures of l-tryptophan- and l-serine-bound PfTrpB, which contained a large rigid body motion of the COMM domain. Refinement was performed using REFMAC5 (49). The MolProbity server was used to identify rotamer flips and to identify clashes (50). After the protein, ligand, and solvent atoms were built, TLS operators were added to refinement, which resulted in substantial improvements in Rfree for the models. Crystallographic and refinement statistics are reported in Table S3.
A similar procedure was used for the crystal structure of PfTrpS, with a protein concentration of 4.0 mg/mL. Sparse matrix screening identified crystals grew in 20% PEG 3350 and 0.2 M Na citrate. Crystals were routinely grown in 24-well format with 16–20% PEG 3350 and 0.15–0.2 M Na citrate. Cryoprotection was achieved by the addition of 25% glycerol and flash-frozen in liquid N2. Crystals regularly diffracted to ∼3.0 Å, and a single crystal was found that diffracted to 2.76 Å. A molecular replacement search model was made using an αβ-dimer of PfTrpS (PDB ID code 1WDW) (22). Model-building proceeded as described above. Data in the final bin were weak, but contributed to model quality as measured by Rfree < 0.4 in the final bin. Coordinates are deposited in the Protein Data Bank with ID codes 5E0K (PfTrpS), 5DVZ (PfTrpB), 5DW0 (Ser-bound PfTrpB), and 5DW3 (Trp-bound PfTrpB).
Identification of Nonnatural Amino Acid Products.
The identities of the amino acid products were confirmed by 1H NMR and LRMS. Proton NMR spectra were recorded on a Varian 300-MHz or Bruker 400-MHz spectrometer. Proton chemical shifts are reported in parts per million (δ) relative to tetramethylsilane and calibrated using the residual solvent resonance (DMSO, δ 2.50 ppm; CD3OD, δ 3.31 ppm; D2O, δ 4.79 ppm). Data are reported as follows: chemical shift (multiplicity [singlet (s), doublet (d), doublet of doublets (dd), doublet of doublets of doublets (ddd), triplet (t), triplet of doublets (td)], coupling constants [Hz], integration). Fluorine NMR spectra were recorded on a 300-MHz (282 MHz) spectrometer without proton decoupling. Fluorine chemical shifts are reported in ppm relative to FCCl3 (δ 0.00 ppm) and were calibrated automatically by the spectrometer using the solvent deuterium lock signal. Low-resolution mass spectra were obtained using an Agilent 1290 UHPLC-LCMS. The optical purity of the products was estimated by derivatization with FDNP-alanamide as described below.
Reaction with heat-treated lysate.
All reactions were conducted using PfTrpB0B2, which was prepared by heat treatment as described in SI Materials and Methods, Cloning, Expression, and Purification of PfTrpA and PfTrpB. The protein was used as a solution in potassium phosphate buffer (50 mM, pH 8) and was found to have a concentration of 134 μM, as determined by specific activity (Table 1). Serine and PLP were used as aqueous solutions (500 mM and 15 mM, respectively).
A 6-mL crimp vial was charged sequentially with the indole analog (50 μmol), dimethyl sulfoxide (50 μL), serine (100 μL), and PLP (3.3 μL). The resulting suspension was diluted with 772 μL of potassium phosphate buffer (200 mM, pH 8). Finally, the enzyme solution was added, and the vial was sealed and immersed in an oil bath that had been equilibrated to 75 °C. After 12 h, the reaction mixture was allowed to cool to room temperature, and then purified directly on C-18 silica (20 mL column volume) with 0–50% methanol/H2O.
Determination of optical purity.
The amino acids were used as 0.2-M solutions in 1 N aqueous (aq.) HCl. FDNP-alanamide was used as a solution in acetone (33 mM). In a 2-mL vial, the amino acid (2.5 μL, 0.50 μmol) was diluted in 1 M aq. NaHCO3 (67 μL). FDNP-alanamide (30 μL, 1 μmol) was added, and then the vial was immersed in an oil bath that had been equilibrated to 40 °C. After 12 h, the reaction mixture was allowed to cool to room temperature, and then diluted with 1:1 water/acetonitrile (900 μL). The resulting solution was subjected to centrifugation (20,000 × g, 4 °C, 5 min). The supernatant was analyzed directly by LC-MS. Each amino acid was derivatized with both racemic and enantiopure FDNP-alanamide for comparison. All amino acid products were enantiopure. Absolute stereochemistry was inferred by analogy to l-tryptophan.
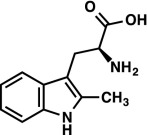 |
1H NMR (400 MHz, CD3OD) δ 7.60 (d, J = 7.1 Hz, 1H), 7.26 (d, J = 7.4 Hz, 1H), 7.06–6.97 (m, 2H), 3.83 (dd, J = 9.9, 4.2 Hz, 1H), 3.26 (ABX, JAX = 9.9 Hz, JBX = 4.2 Hz, JAB = 15.2 Hz, νAB = 164.0 Hz, 2H), 2.42 (s, 3H). LRMS (ESI) (m/z) for [M+H]+ C12H15N2O2 requires 219.1, observed 219.1. |
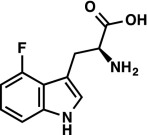 |
1H NMR (300 MHz, DMSO-d6) δ 11.40 (s, 1H), 7.23 (s, 1H), 7.18 (d, J = 8.1 Hz, 1H), 7.00 (td, J = 7.9, 5.2 Hz, 1H), 6.69 (ddd, J = 11.5, 7.8, 0.8 Hz, 1H), 3.52–3.36 (m, 2H), 2.90 (dd, J = 15.8, 11.1, 1H). 19F NMR (282 MHz, DMSO-d6) δ –124.26 (dd, JF–H = 11.3, 5.2 Hz). LRMS (ESI) (m/z) for [M+H]+ C11H12FN2O2 requires 223.1, observed 223.1. |
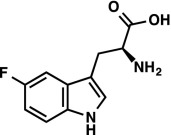 |
1H NMR (300 MHz, DMSO-d6) δ 11.19 (s, 1H), 7.36–7.25 (m, 3H), 6.87 (td, J = 9.2, 2.5 Hz, 1H), 3.37 (dd, J = 7.5, 4.1, 1H), 3.04 (ABX, JAX = 8.1 Hz, JBX = 4.2 Hz, JAB = 14.6 Hz, νAB = 85.8 Hz, 2H). 19F NMR (282 MHz, DMSO-d6) δ –125.84 (td, JF–H = 9.8, 4.6 Hz). LRMS (ESI) (m/z) for [M+H]+ C11H12FN2O2 requires 223.1, observed 223.1. |
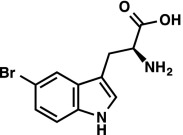 |
1H NMR (400 MHz, CD3OD) δ 7.91 (d, J = 1.8 Hz, 1H), 7.29 (d, J = 8.5 Hz, 1H), 7.23 (s, 1H), 7.21 (dd, J = 8.6, 1.9 Hz, 1H), 3.82 (dd, J = 9.1, 4.1 Hz, 1H), 3.29 (ABX, JAX = 9.1 Hz, JBX = 4.2 Hz, JAB = 15.2 Hz, νAB = 124.0 Hz, 2H). LRMS (ESI) (m/z) for [M+H]+ C11H12BrN2O2 requires 283.0 and 285.0, observed 283.0 and 285.0. |
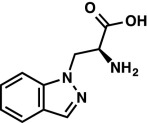 |
As the HCl salt: 1H NMR (400 MHz, D2O) δ 8.16 (s, 1H), 7.84 (d, J = 8.2 Hz, 1H), 7.62 (d, J = 8.6 Hz, 1H), 7.52 (t, J = 7.7 Hz, 1H), 7.25 (t, J = 7.5 Hz, 1H), 4.99 (ABX, JAX = 4.1 Hz, JBX = 6.0 Hz, JAB = 15.6 Hz, νAB = 44.0 Hz, 2H), 4.54 (dd, J = 6.0, 4.1 Hz, 1H). LRMS (ESI) (m/z) for [M+H]+ C10H12N3O2 requires 206.1, observed 206.1. |
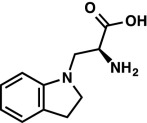 |
As the HCl salt: 1H NMR (400 MHz, D2O) δ 7.22 (d, J = 7.3 Hz, 1H), 7.16 (t, J = 7.7 Hz, 1H), 6.83 (t, J = 7.4 Hz, 1H), 6.70 (d, J = 7.9 Hz, 1H), 4.25 (q, J = 6.0 Hz, 1H), 3.70–3.56 (m, 2H), 3.46 (q, J = 8.3 Hz, 1H), 3.37 (q, J = 8.4 Hz, 1H), 3.00 (t, J = 8.2 Hz, 2H). LRMS (ESI) (m/z) for [M+H]+ C11H14N2O2 requires 207.1, observed 207.1. |
 |
1H NMR (400 MHz, DMSO-d6) δ 10.48 (d, J = 2.2 Hz, 1H), 7.30 (d, J = 8.5 Hz, 1H), 6.95 (d, J = 2.2 Hz, 1H), 6.69 (d, J = 2.1 Hz, 1H), 6.51 (dd, J = 8.5, 2.1 Hz, 1H), 3.38 (dd, J = 9.2, 3.6 1H), 3.02 (ABX, JAX = 9.1 Hz, JBX = 3.8 Hz, JAB = 15.0 Hz, νAB = 155.5 Hz, 2H). LRMS (ESI) (m/z) for [M+H]+ C11H13N2O3 requires 221.1, observed 221.1. |
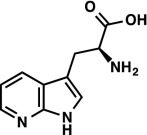 |
1H NMR (300 MHz, D2O) δ 8.16 (dd, J = 5.0, 1.4 Hz, 1H), 8.06 (dd, J = 7.9, 1.5 Hz, 1H), 7.31 (s, 1H), 7.13 (dd, J = 7.9, 4.9 Hz, 1H), 3.96 (dd, J = 7.3, 5.1 Hz, 1H), 3.30 (ABX, JAX = 7.3 Hz, JBX = 5.2 Hz, JAB = 15.4 Hz, νAB = 33.0 Hz, 2H). LRMS (ESI) (m/z) for [M+H]+ C10H12N3O2 requires 206.1, observed 206.1. |
Discussion
We evolved PfTrpB for independent function by selecting mutations that increased its Vmax in heat-treated E. coli lysates. We found that activating mutations were unusually common, which could in part be attributed to the high thermal stability of the parent enzyme that increases tolerance to the introduction of random mutations (17). However, it is clear that many mutations have the potential to recover activity lost when TrpB is removed from its native complex. The single T292S mutation raised the catalytic efficiency of PfTrpB on indole by almost 20-fold and completely restored activity to TrpS-like levels (Table 1). Further evolution resulted in the identification of PfTrpB0B2, which retained T292S as well as five additional mutations. This variant has an 83-fold increase in catalytic efficiency on indole compared with PfTrpB. The T292S and F274S mutations occur in dynamic regions of TrpB related to catalytic function, as discussed above. However, the majority of the mutations in PfTrpB0B2, and 40% of all activating mutations that we identified, are at positions that do not undergo any observable structural changes upon formation of the partially closed state, nor do the corresponding residues in StTrpS move upon formation of the fully closed state (2). The location of many activating mutations outside of any observed allosteric site raises the central question of this study: did directed evolution increase activity through the same mechanism(s) through which TrpA binding regulates function? If so, we posit that increases in kcat, which was under selective pressure during directed evolution, would be coupled to other kinetic and spectroscopic characteristics of the native complex for which there was no selective pressure.
To a first approximation, the open–close equilibrium of TrpB can be inferred from its KM for indole because TrpA binding stabilizes the closed conformation (2) and decreases the KM (Table 1). Each generation of evolution of PfTrpB produced a lower KM for indole, to values below those in PfTrpS, despite screening under saturating conditions (Table 1). UV-vis spectroscopy affords a more-sensitive probe of the active site environment because the steady-state distribution of intermediates in the catalytic cycle can be directly observed. Measurement of the steady-state population of the stage I intermediates in PfTrpB shows that the electrophilic E(A-A) state is stabilized relative to E(Aex1) when PfTrpA binds. This same stabilization of E(A-A) is observed in TrpB enzymes evolved for independent function (Fig. 2). These data reflect the properties of PfTrpB in its native reaction. Activity on indole analogs is also significantly increased by PfTrpA binding, with the rate enhancements differing by two orders of magnitude between diverse indole analogs (Fig. 5). Because each of these substrates requires slightly different transition state stabilization, the rate enhancement upon effector addition is a sensitive metric for changes in the conformational ensemble that are relevant to catalysis. Consistent with the data measured on the native reaction, we observed that increases in activity with indole analogs caused by mutation were similar to the effects of TrpA addition (Fig. 5). Taken together, these data support the hypothesis that increases in PfTrpB activity from directed evolution arose through the same mechanism by which effector binding activates catalysis.
A perplexing observation about mutational reactivation is that addition of PfTrpA to the engineered proteins did not result in allosteric activation, but instead inhibited catalysis. A simple open–closed model of the ensemble might imply that TrpA binding and mutations that stabilize the closed state would be additive in their effects on catalysis. Though the introduction of mutations may change the nature of the allosteric signaling that results from TrpA binding, no such assumption is required to explain the inhibition. It is well documented that stabilization of the closed state by effector binding alters the energetics of multiple transition states of TrpB (2). These changes in transition-state energy result in a lower overall barrier to the reaction. Doubling these energetic perturbations (e.g., TrpA and the mutations combined) would lower the energy of the original rate-limiting step, but could end up impeding catalysis by increasing the energy of a different step above the original reaction barrier (Fig. S5). With this explanation, the inhibitory effect of TrpA becomes consistent with the demonstration that the mutations and effector addition accelerate catalysis through the same mechanism.
Fig. S5.

Hypothetical reaction coordinate diagram to illustrate inversion of TrpA effector activation to inhibition. Native enzyme, such as TrpB (blue), with multiple reaction transition states. Effector addition or introduction of mutations (green) increases the rate of the reaction by lowering the free energy of the rate-limiting step (RLS), while simultaneously raising the energy of a different transition state thereby generating a new RLS. The same changes applied a second time (red), as might occur with PfTrpA addition to the engineered TrpB, can lead to a free-energy barrier higher than for the native enzyme and a reduction in overall reaction rate.
Allostery is common in enzymes and has been targeted in previous protein engineering endeavors (vide infra). To compare the role of allostery in these diverse efforts, we define the change in activity that arises through effector binding as an enzyme’s allosteric potential. In this view, it has been shown that allosteric potential can be decreased by using mutagenesis to disrupt specific interactions between fructose-1,6-bisphosphatase and its effector, adenosine monophosphate (33). Recent studies have also shown that engineering can increase allosteric potential, enhancing inhibition of phosphofructokinase by phosphoenolpyruvate (34). An allosteric potential can even be engineered into enzymes where previously one did not exist (35). Nature has found mutations that constitutively activate allosterically regulated enzymes, as is well known in phosphorylation cascades contributing to tumorigenesis (36). Finding such mutations for engineering purposes has proved challenging. The native substrate of the acyl-transferase LovD is covalently bound to an acyl carrier protein, LovF, which also functions as an allosteric activator. LovD was subjected to nine rounds of directed evolution for altered substrate specificity, thermal stability, and tolerance to organic solvent to generate a biocatalyst for production of the drug simvastatin (37). Insight into whether increases in activity were related to allosteric regulation was gleaned from microsecond molecular dynamics simulations, which suggested that LovF promotes the stability of a closed and catalytically active conformation of LovD and that directed evolution identified mutations that increased activity in a similar fashion (37). Our kinetic, structural, and spectroscopic data provide firm experimental support for a similar effect in TrpB, demonstrating that allosteric potential can be readily converted into catalytic activity through directed evolution.
We advocate that future efforts to recoup allosteric potential not be limited to making mutations within a hypothetical allosteric pathway (38). Residues whose interactions are not changed upon effector addition can nonetheless tune the cooperative network of interactions that transfer allosteric signals (39). More generally, allostery enables proteins to respond to environmental cues, and often only small energetic differences separate active conformations from inactive or less-active ones. The protein ensembles of such enzymes are likely to be sensitive to the effects of widely distributed mutations. Indeed, a single conservative mutation (T292S) was sufficient to raise the catalytic efficiency of PfTrpB to the level of the TrpS complex, and many other mutations contributed smaller activating effects. Therefore, we anticipate that recovering allosteric potential with directed evolution will be broadly achievable.
These insights into the modulation of protein allosteric regulation are of both fundamental and practical interest. Our observation that the activity of PfTrpB was enhanced on a range of substrates is consistent with the hypothesis that the mutations alter transition-state stabilization in a manner similar to PfTrpA binding; it also establishes a greatly simplified enzyme platform for production of NCAAs. This demonstration that isolated subunits may be readily reactivated through directed evolution provides a useful tool for the chemical biology community and expands the scope of enzymes accessible for biocatalysis.
Materials and Methods
Detailed experimental methods are presented in SI Materials and Methods.
Cloning, Expression, and Purification of PfTrpA and PfTrpB.
The genes encoding PfTrpB (UNIPROT ID Q8U093) and PfTrpA (UNIPROT ID Q8U094) were obtained as gBlocks and cloned into pET22(b)+ for expression in E. coli BL21 E. cloni EXPRESS cells (Lucigen). Heterologous protein expression of PfTrpA and PfTrpB was performed in Terrific Broth with 100 μg/mL ampicillin (TBamp) and induced with 500 mM IPTG (final concentration 1 mM). PfTrpB was purified via a HisTrap HP column. PfTrpA was purified via a Q HP HiTrap column (flow-through), followed by ammonium sulfate precipitation, and hydrophobic interaction chromatography on a phenyl Sepharose HP HiTrap column (Fig. S6).
Library Construction and High-Throughput Screening.
Error-prone PCR libraries were constructed using standard protocols with either MnCl2 or Mutazyme II (Stratagene). DNA shuffling and site-directed mutagenesis by overlap extension (SOE) PCR were performed to recombine activating mutations. The resulting libraries were cloned into pET22(b)+ with the C-terminal his-tag for expression in E. coli BL21 E. cloni EXPRESS cells. High-throughput expression and screening were performed on 96-well scale. Formation of l-tryptophan was recorded at 290 nm.
Kinetics and UV-Vis Spectroscopy.
Data were collected between 550 and 250 nm on a UV1800 Shimadzu spectrophotometer (Shimadzu) using 0.25–20 µM of enzyme in 200 mM potassium phosphate (pH 8.0) in a quartz cuvette. Samples were incubated at 75 °C for >3 min to ensure a stable temperature was reached. PfTrpB activity (kcat) was measured by monitoring tryptophan formation at 290 nm using Δε290 = 1.89 mM−1⋅cm−1 (15).
Substrate Selectivity.
The relative rate of NCAA production was measured using 20 mM l-serine and 20 mM indole analog (Fig. 5A) in 200 mM potassium phosphate (pH 8.0) with 5% (vol/vol) DMSO. Reactions were incubated at 75 °C for 1 h, quenched, and the relative rate of production formation was measured by comparing the ratio of the product peaks measured via ultra HPLC-MS (UHPLC-MS) Agilent 1290 with 6140 MS detector at 280 nm and then normalizing for the enzyme concentration.
Crystallography.
Crystals of PfTrpB and PfTrpS were grown using the sitting-drop vapor diffusion method, and cryoprotected before diffraction at the Stanford Synchrotron Radiation Laboratories on beamline 12-2. Ligand-bound crystals of PfTrpB were prepared by soaking preformed crystals with a concentrated solution of l-Ser or l-Trp. Structures were determined by molecular replacement and models were built using standard procedures.
Identification of Nonnatural Amino Acid Products.
Preparative-scale reactions were conducted using PfTrpB0B2, which was prepared as a heat-treated lysate. Products were purified directly on C-18 silica, and their identities confirmed by 1H NMR and low-resolution mass spectrometry (LRMS). The optical purity of the products was estimated by derivatization with N-(5-fluoro-2,4-dinitrophenyl)alanamide (FDNP-alanamide).
Acknowledgments
The authors thank Jackson Cahn, Dr. Robert Trachman, Dr. Mike Chen, and Belinda Wenke for helpful discussions and comments on the manuscript. We thank the staff of the Caltech Molecular Observatory, Dr. Jens Kaiser, Dr. Julie Hoy, and Pavle Nikolovski for crystallographic support. The Molecular Observatory is supported by the Gordon and Betty Moore Foundation, the Beckman Institute, and the Sanofi-Aventis Bioengineering Research Program at Caltech. This work was funded through the Jacobs Institute for Molecular Engineering for Medicine; Ruth Kirschstein NIH Postdoctoral Fellowship F32GM110851 (to A.R.B.); and the Alfonso Martín Escudero Foundation (J.M.C.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 5DVZ, 5DW0, 5DW3, and 5E0K).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1516401112/-/DCSupplemental.
References
- 1.Du L, Lou L. PKS and NRPS release mechanisms. Nat Prod Rep. 2010;27(2):255–278. doi: 10.1039/b912037h. [DOI] [PubMed] [Google Scholar]
- 2.Dunn MF. Allosteric regulation of substrate channeling and catalysis in the tryptophan synthase bienzyme complex. Arch Biochem Biophys. 2012;519(2):154–166. doi: 10.1016/j.abb.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davidsen JM, Bartley DM, Townsend CA. Non-ribosomal propeptide precursor in nocardicin A biosynthesis predicted from adenylation domain specificity dependent on the MbtH family protein NocI. J Am Chem Soc. 2013;135(5):1749–1759. doi: 10.1021/ja307710d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niks D, et al. Allostery and substrate channeling in the tryptophan synthase bienzyme complex: Evidence for two subunit conformations and four quaternary states. Biochemistry. 2013;52(37):6396–6411. doi: 10.1021/bi400795e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hyde CC, Ahmed SA, Padlan EA, Miles EW, Davies DR. Three-dimensional structure of the tryptophan synthase alpha 2 beta 2 multienzyme complex from Salmonella typhimurium. J Biol Chem. 1988;263(33):17857–17871. [PubMed] [Google Scholar]
- 6.Dierkers AT, Niks D, Schlichting I, Dunn MF. Tryptophan synthase: Structure and function of the monovalent cation site. Biochemistry. 2009;48(46):10997–11010. doi: 10.1021/bi9008374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillips RS. Synthetic applications of tryptophan synthase. Tetrahedron Asymmetry. 2004;15(18):2787–2792. [Google Scholar]
- 8.Lang K, Chin JW. Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem Rev. 2014;114(9):4764–4806. doi: 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- 9.Barry SM, et al. Cytochrome P450–catalyzed L-tryptophan nitration in thaxtomin phytotoxin biosynthesis. Nat Chem Biol. 2012;8(10):814–816. doi: 10.1038/nchembio.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kieffer ME, Repka LM, Reisman SE. Enantioselective synthesis of tryptophan derivatives by a tandem Friedel-Crafts conjugate addition/asymmetric protonation reaction. J Am Chem Soc. 2012;134(11):5131–5137. doi: 10.1021/ja209390d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel RN. Biocatalytic synthesis of chiral alcohols and amino acids for development of pharmaceuticals. Biomolecules. 2013;3(4):741–777. doi: 10.3390/biom3040741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith DRM, et al. The first one-pot synthesis of L-7-iodotryptophan from 7-iodoindole and serine, and an improved synthesis of other L-7-halotryptophans. Org Lett. 2014;16(10):2622–2625. doi: 10.1021/ol5007746. [DOI] [PubMed] [Google Scholar]
- 13.Goss RJM, Newill PLA. A convenient enzymatic synthesis of L-halotryptophans. Chem Comm. 2006;47:4924–4925. doi: 10.1039/b611929h. [DOI] [PubMed] [Google Scholar]
- 14.Ahmed SA, Miles EW. Aliphatic alcohols stabilize an alternative conformation of the tryptophan synthase alpha 2 beta 2 complex from Salmonella typhimurium. J Biol Chem. 1994;269(23):16486–16492. [PubMed] [Google Scholar]
- 15.Lane AN, Kirschner K. The catalytic mechanism of tryptophan synthase from Escherichia coli. Kinetics of the reaction of indole with the enzyme–L-serine complexes. Eur J Biochem. 1983;129(3):571–582. doi: 10.1111/j.1432-1033.1983.tb07087.x. [DOI] [PubMed] [Google Scholar]
- 16.Lane AN, Kirschner K. The mechanism of binding of L-serine to tryptophan synthase from Escherichia coli. Eur J Biochem. 1983;129(3):561–570. doi: 10.1111/j.1432-1033.1983.tb07086.x. [DOI] [PubMed] [Google Scholar]
- 17.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Protein stability promotes evolvability. Proc Natl Acad Sci USA. 2006;103(15):5869–5874. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hettwer S, Sterner R. A novel tryptophan synthase beta-subunit from the hyperthermophile Thermotoga maritima. Quaternary structure, steady-state kinetics, and putative physiological role. J Biol Chem. 2002;277(10):8194–8201. doi: 10.1074/jbc.M111541200. [DOI] [PubMed] [Google Scholar]
- 19.Hiyama T, Sato T, Imanaka T, Atomi H. The tryptophan synthase β-subunit paralogs TrpB1 and TrpB2 in Thermococcus kodakarensis are both involved in tryptophan biosynthesis and indole salvage. FEBS J. 2014;281(14):3113–3125. doi: 10.1111/febs.12845. [DOI] [PubMed] [Google Scholar]
- 20.Yamagata Y, et al. Entropic stabilization of the tryptophan synthase alpha-subunit from a hyperthermophile, Pyrococcus furiosus. X-ray analysis and calorimetry. J Biol Chem. 2001;276(14):11062–11071. doi: 10.1074/jbc.M009987200. [DOI] [PubMed] [Google Scholar]
- 21.Hioki Y, et al. The crystal structure of the tryptophan synthase beta subunit from the hyperthermophile Pyrococcus furiosus. Investigation of stabilization factors. Eur J Biochem. 2004;271(13):2624–2635. doi: 10.1111/j.1432-1033.2004.04191.x. [DOI] [PubMed] [Google Scholar]
- 22.Lee SJ, et al. Conformational changes in the tryptophan synthase from a hyperthermophile upon alpha2beta2 complex formation: Crystal structure of the complex. Biochemistry. 2005;44(34):11417–11427. doi: 10.1021/bi050317h. [DOI] [PubMed] [Google Scholar]
- 23.Ahmed SA, Ruvinov SB, Kayastha AM, Miles EW. Mechanism of mutual activation of the tryptophan synthase alpha and beta subunits. Analysis of the reaction specificity and substrate-induced inactivation of active site and tunnel mutants of the beta subunit. J Biol Chem. 1991;266(32):21548–21557. [PubMed] [Google Scholar]
- 24.Miles EW, McPhie P. Evidence for a rate-determining proton abstraction in the serine deaminase reaction of the beta 2 subunit of tryptophan synthetase. J Biol Chem. 1974;249(9):2852–2857. [PubMed] [Google Scholar]
- 25.Ngo H, et al. Allosteric regulation of substrate channeling in tryptophan synthase: Modulation of the L-serine reaction in stage I of the beta-reaction by alpha-site ligands. Biochemistry. 2007;46(26):7740–7753. doi: 10.1021/bi7003872. [DOI] [PubMed] [Google Scholar]
- 26.Miles EW, Houck DR, Floss HG. Stereochemistry of sodium borohydride reduction of tryptophan synthase of Escherichia coli and its amino acid Schiff’s bases. J Biol Chem. 1982;257(23):14203–14210. [PubMed] [Google Scholar]
- 27.McDowell LM, Lee M, McKay RA, Anderson KS, Schaefer J. Intersubunit communication in tryptophan synthase by carbon-13 and fluorine-19 REDOR NMR. Biochemistry. 1996;35(10):3328–3334. doi: 10.1021/bi9518297. [DOI] [PubMed] [Google Scholar]
- 28.Schlichting I, Yang XJ, Miles EW, Kim AY, Anderson KS. Structural and kinetic analysis of a channel-impaired mutant of tryptophan synthase. J Biol Chem. 1994;269(43):26591–26593. [PubMed] [Google Scholar]
- 29.Rowlett R, et al. Mutations in the contact region between the alpha and beta subunits of tryptophan synthase alter subunit interaction and intersubunit communication. Biochemistry. 1998;37(9):2961–2968. doi: 10.1021/bi972286z. [DOI] [PubMed] [Google Scholar]
- 30.Raboni S, Bettati S, Mozzarelli A. Identification of the geometric requirements for allosteric communication between the alpha- and beta-subunits of tryptophan synthase. J Biol Chem. 2005;280(14):13450–13456. doi: 10.1074/jbc.M414521200. [DOI] [PubMed] [Google Scholar]
- 31.Aharoni A, et al. The ‘evolvability’ of promiscuous protein functions. Nat Genet. 2005;37(1):73–76. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- 32.Bloom JD, Romero PA, Lu Z, Arnold FH. Neutral genetic drift can alter promiscuous protein functions, potentially aiding functional evolution. Biol Direct. 2007;2:17. doi: 10.1186/1745-6150-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J-S, Seo SW, Jang S, Jung GY, Kim S. Rational engineering of enzyme allosteric regulation through sequence evolution analysis. PLOS Comput Biol. 2012;8(7):e1002612. doi: 10.1371/journal.pcbi.1002612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGresham MS, Reinhart GD. Enhancing allosteric inhibition in Thermus thermophilus Phosphofructokinase. Biochemistry. 2015;54(3):952–958. doi: 10.1021/bi501127a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee J, et al. Surface sites for engineering allosteric control in proteins. Science. 2008;322(5900):438–442. doi: 10.1126/science.1159052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torkamani A, Verkhivker G, Schork NJ. Cancer driver mutations in protein kinase genes. Cancer Lett. 2009;281(2):117–127. doi: 10.1016/j.canlet.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiménez-Osés G, et al. The role of distant mutations and allosteric regulation on LovD active site dynamics. Nat Chem Biol. 2014;10(6):431–436. doi: 10.1038/nchembio.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reynolds KA, McLaughlin RN, Ranganathan R. Hot spots for allosteric regulation on protein surfaces. Cell. 2011;147(7):1564–1575. doi: 10.1016/j.cell.2011.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hilser VJ, Dowdy D, Oas TG, Freire E. The structural distribution of cooperative interactions in proteins: Analysis of the native state ensemble. Proc Natl Acad Sci USA. 1998;95(17):9903–9908. doi: 10.1073/pnas.95.17.9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 41.Stemmer WPC. DNA shuffling by random fragmentation and reassembly: In vitro recombination for molecular evolution. Proc Natl Acad Sci USA. 1994;91(22):10747–10751. doi: 10.1073/pnas.91.22.10747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 43.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr. 2013;69(Pt 7):1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336(6084):1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 49.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- 50.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]