

Abstract
Cell motility is regulated by a balance between forward protrusion and tail retraction. These phenomena are controlled by a spatial asymmetry in signals at the front and the back of the cell. We show here that the protein-tyrosine phosphatase, PTP-PEST, is required for the coupling of protrusion and retraction during cell migration. PTP-PEST null fibroblasts, which are blocked in migration, exhibit exaggerated protrusions at the leading edge and long, unretracted tails in the rear. This altered morphology is accompanied by changes in the activity of Rho GTPases, Rac1 and RhoA, which mediate protrusion and retraction, respectively. PTP-PEST null cells exhibit enhanced Rac1 activity and decreased RhoA activity. We further show that PTP-PEST directly targets the upstream regulators of Rac1 and RhoA, VAV2 and p190RhoGAP. Moreover, we demonstrate that the activities of VAV2 and p190RhoGAP are regulated by PTP-PEST. Finally, we present evidence indicating the VAV2 can be regulated by integrin-mediated adhesion. These data suggest that PTP-PEST couples protrusion and retraction by acting on VAV2 and p190RhoGAP to reciprocally modulate the activity of Rac1 and RhoA.
Cell motility is a concerted process involving protrusion of the leading edge, formation and turnover of cell-substrate adhesions, contraction of the cell body, and retraction of the trailing tail (1). These phenomena are controlled by Rho family GTPases which regulate the actin cytoskeleton (2, 3). Directed migration relies upon spatially restricted intracellular activities of Rho GTPases. Rac1 stimulates actin polymerization and the formation of lamellipodia to drive leading edge protrusion (4). RhoA promotes focal adhesion assembly, cell body contraction, and tail retraction. Fluorescence resonance energy transfer analysis in migrating cells reveals Rac1 is active at the leading edge (5, 6), where it recruits activated integrins (7). Using biochemical measurement of GTPase activity in isolated pseudopodia, Klemke and co-workers (8, 9) have suggested that both Rac1 and RhoA are active at the leading edge where they promote extension and retraction, respectively. Although low levels of RhoA are required to maintain adhesion to the substratum (10), perturbation of its activity results in impaired motility. Modest overexpression of activated RhoA or inactivation of p190RhoGAP leading to increased RhoA activity suppresses membrane protrusion at the leading edge (11, 12). During leukocyte transendothelial migration, RhoA activity is required to promote tail retraction (13) and to limit inappropriate protrusion at the back of the cell (14). Consistent with these observations, Ballestrem et al. (15) and others have proposed a model where active Rac1 functions at the leading edge, whereas active RhoA primarily functions at the trailing end (1). The intracellular mechanisms maintaining this asymmetry of Rac1 and RhoA activity are poorly understood. It is likely that integrins transmit distinct signals at the front and back of migrating cells to control local activity of Rac1 and RhoA.
Although integrins have been shown to modulate the activity of Rho GTPases (16–19), elucidation of the relevant signaling pathways downstream of integrins is only beginning to be addressed. Rho GTPases are regulated by their association with intracellular proteins that facilitate either nucleotide exchange (GEFs)2 or GTP hydrolysis (GAPs) (4, 20). Although numerous GEFs for Rho GTPases have been identified, their upstream regulation is not well understood. One mechanism for regulation is phosphorylation, which could be mediated through an integrin-dependent pathway. VAV2 is a GEF for multiple Rho GTPases (21–23) that is tyrosine-phosphorylated in response to soluble factors (21, 24), but its regulation by integrins is complex (23, 25). VAV2 plays a role in integrin-mediated cell spreading (26); however, its tyrosine phosphorylation is not enhanced by adhesive signals (23, 25). One possibility is that VAV2 contains both positive and negative phosphorylation sites; thus, a net change in phosphorylation may not reflect its activity state (27). Another possibility is that VAV2 associates with a tyrosine phosphatase. Overexpression of activated VAV2 in fibroblasts results in a highly protrusive phenotype with increased motility (23), suggesting that its activity is spatially regulated to restrict protrusions to the leading edge.
Like GEFs, numerous GAPs have been identified, many of which are tissue-specific, but few have been characterized (20). p190RhoGAP is ubiquitously expressed and one of the best-studied GAPs. Tyrosine phosphorylation of p190RhoGAP is associated with its activation (28). It is regulated by integrins (29), and its activity is required to down-regulate RhoA in response to cell-matrix adhesion (16). Overexpression of p190RhoGAP in fibroblasts leads to excessive protrusion of the leading edge in a wound assay (11), suggesting that its activity at the leading edge is regulated.
PTP-PEST is a cytoplasmic tyrosine phosphatase implicated in controlling membrane protrusion and focal adhesion turnover. Both over-expression of PTP-PEST (30) or its targeted deletion suppress cell motility (31). We recently showed that overexpression of PTP-PEST impairs membrane protrusion through inhibition of Rac1 (32). In this study we utilize PTP-PEST −/− fibroblasts (31) to further investigate mechanisms by which this protein-tyrosine phosphatase regulates motility. We find that PTP-PEST controls the balance of forward protrusion and tail retraction by differentially regulating Rac1 and RhoA activity. The PTP-PEST −/− fibroblasts plated on FN exhibit enhanced Rac1 activity and decreased RhoA activity compared with PTP-PEST −/− cells re-expressing PTP-PEST at wild type levels. We further show that PTP-PEST directly acts on upstream regulators of Rho family GTPases. Using substrate trapping, we demonstrate that VAV2 and p190RhoGAP are direct targets of PTP-PEST. Finally, we present evidence that the activity of VAV2 and p190RhoGAP are enhanced in PTP-PEST null fibroblasts. Our data show that PTP-PEST acts at multiple steps of the migration cycle and that PTP-PEST can modulate Rho GTPase activity by both direct and indirect mechanisms.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
PTP-PEST −/− and +/− and re-expressing (RE) fibroblasts were previously described (31) and maintained in DMEM (Sigma) containing 10% fetal bovine serum and antibiotics (penicillin, streptomycin, fungizone; Invitrogen). In some experiments wild type mouse embryo fibroblasts (a gift of Dr. J. Ceci, University of Texas Medical Branch) were used and maintained in DMEM containing 15% fetal bovine serum. CHOK1 cells were maintained in DMEM supplemented with 10% fetal bovine serum, antibiotics, and nonessential amino acids. Cells were transiently transfected using Lipofectamine plus (Invitrogen) as described (32).
DNA and Antibodies
Plasmids encoding green fluorescent protein-tagged Rho GTPases, N17Rac1 and L63RhoA, as well as pGEX vectors encoding GST fusion proteins for 15ARac1 and L63RhoA were gifts of Dr. Krister Wennerberg (University of North Carolina). PTP-PEST substrate trapping mutants were a gift of Dr. Michael D. Schaller (University of North Carolina). Myc-tagged wild type VAV2 in pcDNA was a gift of Dr. Michelle Booden (University of North Carolina). The T7-tagged dominant negative VAV2 mutant, VAV2 L342R/L343S, was a gift of Dr. Chris Carpenter (26). The p130cas mutant, casSD, was a gift of Dr. Kristiina Vuori (Burnham Institute). Dominant negative p190RhoGAP, p190RA, was a gift of Dr. Jeff Settleman (Massachusetts Institute of Technology) and was subcloned in-frame with green fluorescent protein as described (11). Expression plasmids encoding wild type and DDH2 DOCK180 were as described (65). The VAV2 antiserum was raised against a C-terminal GST fusion protein as described (23). Anti-serum recognizing mouse PTP-PEST was previously described (32). The anti-Myc 9E10 mAb was purchased from Covance. Antibodies against Rac1, p190RhoGAP, p120Ras GAP, and SOS-1 were purchased from BD Biosciences. Anti-DOCK 180 and RhoA antibodies were purchased from Santa Cruz.
Immunofluorescence
Cells were plated on 12-mm round glass coverslips coated with FN (Invitrogen; 10 μg/ml) in serum-free medium (DMEM containing 2% bovine serum albumin) for the indicated times. Coverslips were fixed in 3.7% formaldehyde in PBS for 10 min and permeabilized in 0.5% Triton X-100 in PBS for 5 min at room temperature. F-actin was stained with Texas Red phalloidin or Alexa 594-phalloidin (Molecular Probes). Cells expressing the VAV2 R/S mutant were stained with an anti-T7 epitope tag antibody (Novagen) and Alexa 488-anti-mouse secondary antibody. Cells expressing p130cas mutant, CAS SD, or crkII mutants were immunostained with anti GST or anti-Myc antibodies, respectively. DOCK 180-transfected cells were detected with anti-DOCK 180 mAb, H-4 (Santa Cruz). Immunofluorescence was viewed on a Zeiss Axiophot epifluorescence microscope equipped with a MicroMax CCD digital camera (Southern Micro Systems, Marietta, GA), and images were obtained using Metamorph software.
Video Microscopy
PTP-PEST −/− cells or re-expressors were plated in 35-mm culture dishes containing a glass coverslip insert (MatTek Corp., Ashland, MA) coated with FN (10 μg/ml) in serum-containing medium. Video microscopy was performed on an Olympus IX-81 microscope with differential interference contrast (Nomarski) optics (60× objective, 1.45 N.A.). Images were acquired with a Hamamatsu C4880 cooled down CCD camera driven by Metamorph image analysis software (Universal Imaging Corp., Downingtown, PA). Cells were plated for 1 h and then filmed for 3 h with frames captured every 2 min. Cells were maintained in an environmental chamber at 37 °C and 5% CO2. Movies were converted to quick time format using Adobe Image ready software.
Immunoprecipitation and Western Blotting
For immunoprecipitations, cells were lysed in modified RIPA buffer (1% Triton X-100, 0.25% deoxycholate, 50 mM Tris, pH 7.6, 150 mM NaCl, 2 mM EGTA, 1 mM NaVO4, and protease inhibitors (1 mM PMSF, 10 μg/ml leupeptin and aprotinin)) on ice. Lysates were rotated at 4 °C for 15 min and then clarified by centrifugation at 14,000 RPM in a microcentrifuge for 10 min. Protein concentrations were determined using BCA method (Pierce). For VAV2 immunoprecipitations, cells were transfected with myc-wild type VAV2. Lysates were incubated with anti-myc monoclonal antibody, 9E10, for one h at 4 °C on a rotator. Anti-mouse agarose beads (20 μg, Sigma) were added for an additional hour. p190RhoGAP immunoprecipitations were identical except that lysates were incubated with anti-p190RhoGAP monoclonal antibody (Transduction Labs, BD). Immunoprecipitations were separated on 7.5% SDS-PAGE (33), transferred to nitrocellulose (34) and membranes were blocked in 5% cold fish gelatin in TBST. Filters were first immunoblotted for phosphotyrosine using PY20H diluted in 5% cold fish gelatin and then stripped and reprobed for VAV2 or p190RhoGAP with anti myc or anti p190RhoGAP monoclonal antibodies. To detect p120RasGAP co-precipitation, membranes from p190RhoGAP immunoprecipitations were reprobed with anti RasGAP mAb. Endogenous VAV2 was detected with a polyclonal anti-serum diluted 1:20,000 in 5% bovine serum albumin in TBST (23). Anti-mouse- or anti-rabbit secondary antibodies conjugated to horse radish peroxidase and chemiluminescence detection (Pierce super signal) on Kodak X-Omat AR x-ray film were used to visualize bands.
Substrate Trapping Assay
Phosphatase substrate trapping experiments were performed essentially as described (35, 36). Briefly, either 293T cells or HeLa cells were grown to subconfluence in growth medium and then pretreated with 50 mM pervanadate (100 mM NaVO4 was mixed with 12 ml of % H202 for each 100 ml of NaVO4 in PBS to a final stock concentration of 50 mM pervanadate) for 30 min in growth medium. For trapping of endogenous VAV2, lysates were prepared from re-expressing cells or PTP-PEST −/− cells in the absence of pervanadate pre-treatment. Cells were lysed in modified radioimmune precipitation assay buffer (see above) on ice, and lysates were clarified by centrifugation. Approximately 2 mg of lysate were incubated with 5–10 μg of GST fusion protein (GST alone) or GST fused to the catalytic domain of PTP-PEST (GST-wild type PTP-PEST, GST PTP-PESTC231S, or GSTPTP-PEST D199A) for 1 h at 4 °C with rotation. Beads were washed several times in lysis buffer and resuspended in SDS-PAGE sample buffer. Proteins bound to the beads were analyzed by SDS-PAGE followed by Western blotting as indicated above.
Rho GTPase, GEF, and GAP Activity Assays
RhoA activity assays were performed according to the method described by Ren et al. (19). PTP-PEST null or RE cells were serum-starved for 4 h, kept in suspension in serum-free DMEM containing 2% delipidated bovine serum albumin for 30 min, and then plated on FN-coated dishes in the same medium for various time points. Cells were lysed in buffer A, and lysates were clarified by centrifugation in a microcentrifuge at maximal speed at 4 °C for 10 min. GST Rhotekin beads were added (10 μg) and incubated with lysates for 30 min at 4 °C with gentle shaking. Pull-downs were washed in buffer B and resuspended in SDS sample buffer. Total lysate and pull-downs were analyzed by SDS-PAGE followed by Western blotting with anti-RhoA mAb (Santa Cruz). Rac1 activity was measured as described previously (37) using a p21-activated kinase binding domain pull-down assay. PTP-PEST null cells or RE cells were plated identically as for RhoA assays and were lysed in buffer B. Rac1 was detected using anti-Rac1 mAb (BD Transduction Laboratories).
VAV2 activity was assessed by affinity precipitation with a GST fusion protein encoding Rac1 with a point mutation at amino acid 15 (Gly-15 → Ala) to mimic a nucleotide-free state. PTP-PEST −/− cells, re-expressors, or CHOK1 cells transiently transfected with Myc wild-type VAV2 were plated on FN-coated tissue culture dishes or kept in suspension in serum-free medium for 1 h. Cells were lysed in 20 mM Hepes, pH 7.3, 1% Triton X-100, 150 mM NaCl, 5 mM MgCl2, 2 mM dithiothreitol, 1 mM NaVO4, and protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 10 μg/ml each leupeptin and aprotinin) on ice. Cleared lysates were incubated with 20 μg of fusion protein for 1 h at 4 °C on a rotator. After washes with lysis buffer, beads were resuspended in SDS sample buffer. The amount of VAV2 bound to the beads was determined by SDS-PAGE followed by Western blotting with anti-Myc antibody. Sos-1 activity was assayed under identical conditions. As a positive control, SOS-1 activity was measured in 293T cells that were serum-starved overnight and then stimulated with PDGF-BB (25 ng/ml) for 15 min. An analogous method was used to assay p190RhoGAP activity (38). A GST fusion protein encoding activated RhoA (L63 RhoA) was used to precipitate active p190RhoGAP from cell lysates. Plating and lysis conditions were identical to those used for VAV2.
RESULTS
PTP-PEST Is Required for Tail Retraction
PTP-PEST is a cytoplasmic tyrosine phosphatase implicated in controlling membrane protrusion and focal adhesion turnover. Both the overexpression of PTP-PEST (30) or its targeted deletion (31) suppress cell motility. We recently showed that overexpression of PTP-PEST impairs membrane protrusion through inhibition of Rac1 (32). Here, we utilize PTP-PEST −/− fibroblasts to further investigate mechanisms by which this protein-tyrosine phosphatase regulates motility. As previously reported, PTP-PEST −/− fibroblasts are unable to migrate in a wound assay (31). We confirmed these findings by comparing the ability of wild type mouse embryo fibroblasts, PTP-PEST +/− fibroblasts, and PTP-PEST −/− fibroblasts that stably re-express PTP-PEST and PTP-PEST −/− cells to migrate in a wound assay (supplemental Fig. 1A). Only PTP-PEST −/− cells were unable to close a wound after 24 h. To determine the relative levels of PTP-PEST expression in these various cells, we immunoblotted cell lysates with an antiserum that recognizes mouse PTP-PEST. The cDNA used to re-express PTP-PEST is a mouse cDNA (31); therefore, this antiserum is useful to compare total levels of PTP-PEST. Both wild type mouse embryo fibroblasts and re-expressors show equivalent expression, +/− cells have reduced expression, and PTP-PEST null cells are negative for protein expression (supplemental Fig. 1B). These data establish that wild type mouse embryo fibroblasts, re-expressors, and +/− cells migrate to a similar extent. When we examined the morphology of the re-expressors and +/− cells, the re-expressors most closely resembled the wild type mouse embryo fibroblasts. Furthermore, the +/− cells showed a heterogeneous morphology (not shown). Therefore, we chose to further examine the differences between PTP-PEST −/− cells and those that re-express PTP-PEST at wild type levels.
We first examined their morphology on FN-coated coverslips over a time course of 3–4 h. PTP-PEST −/− cells show enhanced spreading within 30 min to 1 h after plating (Fig. 1, right upper panels) when compared with heterozygous controls (+/−) (not shown) or null cells that re-express PTP-PEST (RE) (Fig. 1, left upper panels). However, when PTP-PEST −/− fibroblasts are plated on FN-coated coverslips for greater than 90 min they undergo a dramatic change in morphology (Fig. 1, right lower panels). They form exaggerated membrane ruffles and lamellipodia at the leading edge but also develop long, thin tails in the rear. Some cells also show a bipolar morphology with at least two prominent lamellae.
FIGURE 1. Absence of PTP-PEST uncouples protrusion and retraction.

PTP-PEST null (−/−) or control fibroblasts that re-express PTP-PEST were plated on FN coated cover-slips in serum-free medium for the times indicated. Cells were fixed and stained with Texas Red phalloidin to visualize F-actin. The panels on the left show that the control cells do not significantly alter their morphology over the time course. In contrast, the −/− cells (right-hand panels) undergo substantial changes in shape. Initially, PTP-PEST null cells have enhanced protrusions (30 and 60 min) but at later times begin to develop tails, with a “fishtail” appearance (2 h) and long, thin tails at one end and large, highly exaggerated lamellipodia at the opposite end (3 h).
To distinguish between a loss of polarity or a defect in tail retraction, we utilized time-lapse video microscopy to monitor the cells over a longer time course of migration (Fig. 2, see video images). Fig. 2A shows the time-lapse frames of the development of tails in PTP-PEST −/− cells. Cells were plated on FN-coated dishes for 1 h and then filmed for 3 h. Compared with RE controls, which exhibit minimal membrane activity with modest movement, PTP-PEST null cells initially show enhanced membrane activity along with a flatter, pancake-like morphology. By 1–2 h, the cells begin to form tails in the rear that become highly elongated after 3–4 h. Once a tail develops, the cell body continues to protrude and move forward, but migration is impeded due to an inability of the tail to detach from the substrate (Fig. 2B, see video images). The fraction of cells that formed tails was quantitated (Fig. 2D). Nearly 70% of null cells were observed to form tails after 3 h of plating, whereas only 10% of control cells formed tails.
FIGURE 2. PTP-PEST is required for tail retraction.

A, to visualize the development of tails, control or PTP-PEST −/− cells were videotaped using differential interference contrast microscopy. Cells were plated for 1 h on FN-coated dishes in serum-containing medium, then filmed for 3 h. Frames were captured every 90 s. Control fibroblasts (top panel, supplemental video 1) show little change in shape or movement; however, PTP-PEST −/− fibroblasts (bottom panel, supplemental video 2) exhibit increased membrane activity. After 2 h of plating, tails develop and continue to elongate as the cell body moves forward (supplemental video 2). B, by 4 h tails have formed in the PTP-PEST null cells, but they are unable to retract (see arrows and video 3). C, PTP-PEST −/− cells are partially able to migrate in a haptotaxis migration assay but are unable to move through a transwell filter coated on the top and bottom side, indicating an ability to form protrusions but an inability to release adhesions formed at the cell rear. D, control or PTP-PEST −/− cells were scored over a time course of plating on FN for the percentage of cells that developed tails. Although the re-expressers do not develop tails, 60–70% of the −/− cells form tails within 90 min after plating.
We also performed migration assays using transwell filters to compare directional versus random migration. As shown in Fig. 2C, in a random transwell filter assay (where the top and bottom of the filter are coated with FN), PTP-PEST −/− cells are completely blocked in migration. However, a greater fraction of the null cells are able to undergo haptotaxis, where only the bottom of the filter is coated. This observation suggests that PTP-PEST −/− cells are able to protrude toward a directional cue but are unable to detach the cell body from the upper surface when both are coated. Together with the video data, these results demonstrate that PTP-PEST is required to mediate tail retraction.
PTP-PEST Reciprocally Regulates Rac1 and RhoA Activity
The phenotype described above suggested that PTP-PEST may differentially regulate Rho GTPase activity. We recently reported that overexpression of PTP-PEST blocks membrane protrusion through down-regulation of Rac1 (32). We also showed that Rac1 activity is enhanced in PTP-PEST −/− fibroblasts plated on a FN substrate (Ref. 32, Fig. 3A). This observation is consistent with the increased protrusive behavior observed at the leading edge of PTP-PEST −/− cells (Fig. 2). Because RhoA activity is required for tail retraction (13, 14, 39, 40) and to limit membrane protrusion (11, 12), we next measured RhoA activity in PTP-PEST −/− cells. In control cells (RE), RhoA activity shows a typical biphasic response to adhesion on a FN substrate (Fig. 3B, left panel). RhoA activity is decreased after cell attachment and then is increased above basal levels after 1 h. In PTP-PEST −/− cells, RhoA activity is initially suppressed and remains suppressed throughout the time course (Fig. 3B, right panel). Thus, PTP-PEST is required to activate RhoA after cell attachment. To functionally correlate a change in Rac1 and RhoA activity with the PTP-PEST −/− phenotype, we expressed dominant negative Rac1 (N17 Rac1) or constitutively active RhoA (L63RhoA) in PTP-PEST −/− cells. As shown in Fig. 3C, dominant negative Rac1 and activated RhoA were able to rescue the null cell phenotype. These cells resemble RE controls and do not form excessive protrusions or tails after 3 h of plating on FN. Nearly 80–90% of null cells expressing N17Rac1 (% tails = 19%) or L63RhoA (% tails = 12.5%) showed a normal morphology compared with null cells expressing green fluorescent protein only (% tails = 61%). Activated and dominant negative forms of Cdc42 had little effect on the PTP-PEST null phenotype nor did we observe changes in Cdc42 activity (data not shown). These results indicate that PTP-PEST regulates membrane protrusion and tail retraction by differentially modulating the activity of Rac1 and RhoA.
FIGURE 3. PTP-PEST reciprocally regulates Rac1 and RhoA activity.

PTP-PEST −/− cells or those re-expressing PTP-PEST were kept in suspension or plated on FN for the times indicated and assayed for RhoA activity (A) using a GST-Rhotekin pull-down assay or for Rac1 activity (B) using a GST-PAK binding domain pull-down assay. A, PTP-PEST is required for the activation of RhoA. In control cells, re-expressers, RhoA activity is initially suppressed and then activated by 60 min after adhesion to FN. RhoA activity remained elevated. However, in PTP-PEST −/− cells, whereas RhoA activity was initially suppressed, activation was not observed. B, Rac1 activity is enhanced in PTP-PEST −/− cells. In control re-expressers Rac1 was transiently activated within 30 min after plating on FN and then returned to base-line levels. In PTP-PEST −/− cells, Rac1 activity is elevated and sustained at all time points tested. C, expression of dominant negative Rac1 (N17Rac) or activated RhoA (L63 RhoA) restored a normal morphology to PTP-PEST −/− cells after 3 h of plating on FN-coated coverslips.
VAV2 Is a Substrate of PTP-PEST
PTP-PEST could modulate Rho GTPase activity through either direct or indirect mechanisms. We next determined whether PTP-PEST directly acts on upstream regulators of Rho family GTPases. VAV2 is a guanine nucleotide exchange factor for Rac1 and RhoA that is both positively and negatively regulated by tyrosine phosphorylation (27). When overexpressed in fibroblasts, activated VAV2 produces a protrusive, highly motile phenotype (23) similar to the leading edge of PTP-PEST −/− cells. We also observed that VAV2-associated tyrosine-phosphorylated proteins could only be immunoprecipitated in the presence of a tyrosine phosphatase inhibitor.3 This finding suggested that VAV2 might associate with a tyrosine phosphatase. To determine whether VAV2 is a direct target of PTP-PEST, we utilized a substrate trapping technique (35, 36). A GST fusion protein containing the catalytic domain of PTP-PEST, mutated in its active site, was used as an affinity matrix to isolate potential substrates from 293 cells pretreated with the tyrosine phosphatase inhibitor, pervanadate. Immunoblot analysis shows that VAV2 binds to the trapping mutant but not the wild type phosphatase or GST alone (Fig. 4A). The trapping assay was confirmed by showing that p130cas, a known substrate, is precipitated by the trapping mutant, whereas focal adhesion kinase, which is not a substrate, did not bind the trapping mutant. Under the conditions of the assay, focal adhesion kinase should be heavily tyrosine-phosphorylated, showing that the trapping assay is specific. To demonstrate that VAV2 is a bona fide substrate of PTP-PEST, we determined if endogenous VAV2 could be trapped from PTP-PEST −/− fibroblasts. Fig. 4B shows that endogenous VAV2 binds to the trapping mutant but not to GST or the wild type fusion protein. Finally, we determined if the tyrosine phosphorylation state of VAV2 was altered in PTP-PEST −/− cells. VAV2 was immunoprecipitated from RE or −/− cells and then immunoblotted with anti-phosphotyrosine antibodies. Fig. 4C shows that the level of VAV2 tyrosine phosphorylation is elevated in −/− cells compared with RE controls. These data demonstrate that VAV2 is a substrate of PTP-PEST.
FIGURE 4. VAV2 is a substrate of PTP-PEST.

A, to determine whetherVAV2 was a direct target of PTP-PEST, a substrate trapping technique was employed. A GST fusion protein containing the catalytic domain of PTP-PEST mutated in its active site was used as an affinity matrix to “trap” tyrosine-phosphorylated substrates. 293T cells were treated with pervanadate and then lysed. Trapping mutants or wild type (WT) PTP-PEST was used to pull down VAV2. The pull-downs were blotted (WB) with an anti-serum raised against human VAV2. Two different trapping mutants were used, PTP-PEST-CS (Cys-231 → Ser (CS)) and PTP-PEST-DA (Asp-199 → Ala (DA)), to trap VAV2. LYS, lysate. As shown in the upper blot, VAV2 is present in pull-downs using the trapping mutant but not GST alone or wild type PTP-PEST. Below is a positive control for p130CAS, a known PTP-PEST substrate, and a negative control for focal adhesion kinase, which is not a substrate. FAK, focal adhesion kinase. B, substrate trapping of endogenous VAV2 from PTP-PEST −/− fibroblasts. C, tyrosine phosphorylation of VAV2 is enhanced in PTP-PEST −/− cells. IP, immunoprecipitation.
PTP-PEST Regulates VAV2
The above observations led us to pursue whether PTP-PEST regulates the exchange factor activity of VAV2. Previous studies showed that an increase in VAV2 tyrosine phosphorylation corresponds to an increase in exchange activity (21, 23–25, 41). However, both positive and negative regulatory tyrosine sites have been identified on VAV2; thus, a change in total phosphotyrosine content is difficult to interpret (27). Direct measurement of GEF activity by immunoprecipitation has not been feasible due to low activity. Therefore, we turned to an alternate approach to assess the exchange activity of VAV2 in PTP-PEST −/− cells. A useful property of GEFs is that once activated, they bind to nucleotide-free Rho proteins with high affinity (42–45). To indirectly assay VAV2 activity, we utilized a point mutant of Rac1 (glycine15 mutated to alanine) that mimics the nucleotide-free state to affinity precipitate VAV2 from cell lysates (42, 46, 47). Glutathione-Sepharose beads bound to a GST fusion protein containing G15A Rac1 (15ARac1) are incubated with lysates, and active Rac1 exchange factors selectively bind. A change in the amount of Rac1 exchange factor precipitated by the beads reflects a change in activity.
To validate this approach, we tested if we could detect a change in the amount of VAV2 precipitated in response to cell adhesion. For this experiment we utilized CHOK1 cells transiently transfected with Myc-tagged wild type VAV2. Compared with control CHOK1 cells held in suspension, more VAV2 was precipitated from CHOK1 cells plated on FN (Fig. 5A), suggesting that VAV2 can be activated by cell adhesion. Using this assay, we next determined whether VAV2 was regulated in PTP-PEST −/− cells. We compared the amount of VAV2 precipitated by GST-15ARac1 from RE controls or PTP-PEST −/− cells expressing wild type Myc-tagged VAV2 that were kept in suspension or plated on FN. Fig. 5B shows increased VAV2 precipitated in the PTP-PEST −/− cells plated on FN compared with those in suspension. In addition, the amount of VAV2 precipitated from PTP-PEST −/− cells was substantially increased relative to RE controls. To demonstrate that VAV2 activity is specific, we measured the activity of SOS-1, an exchange factor for Ras and Rac1. Sos-1 activity was not detected in PTP-PEST −/− or RE cells. To verify that active SOS-1 could be precipitated by the 15A mutant, we measured SOS-1 activity in 293T cells treated with platelet-derived growth factor, since SOS-1 should be activated by growth factor stimulation. Fig. 5C shows that increased SOS-1 can be precipitated by 15A Rac1 in response to platelet-derived growth factor. These results together indicate that VAV2 activity is regulated by cell adhesion and is further enhanced in cells lacking PTP-PEST.
FIGURE 5. Increased VAV2 precipitation in PTP-PEST null cells.

A, affinity precipitation with a G15A mutant of Rac1 that mimics nucleotide-free Rac1 (NF Rac1) was used to indirectly assess GEF activity. CHOK1 cells were transfected with Myc-tagged wild type VAV2, then held in suspension or plated on FN for 60 min. GST-15ARac1 was used to pull down VAV2. The amount of VAV2 precipitated by NF-Rac1 when cells were plated on FN was increased relative to cells in suspension. WB, Western blot. S, suspension. B, NF-Rac1 precipitates an increased amount of VAV2 from PTP-PEST −/− cells compared with re-expressers in response to adhesion to FN. C, SOS-1 activity is not affected by PTP-PEST and serves as a negative control. However, SOS-1 can be activated by platelet-derived growth factor (PDGF) stimulation in 293 cells. D, expression of a VAV2 mutant that is altered in its DH domain, VAV2 Arg → Ser, restores a normal phenotype to PTP-PEST −/− cells. Note the arrows show protrusions in untransfected null cells, whereas arrowheads point to cells that expressing the VAV2 mutant with a morphology resembling control re-expressers. All cells were plated for 3 h on FN-coated coverslips in serum-free medium.
To functionally link VAV2 with the PTP-PEST −/− phenotype, we expressed a dominant negative form of VAV2 that is in the DH domain (VAV2 L342R/L343S). This mutant has been shown to block cell spreading and Rac activity in response to adhesion on FN (26). Expression of this mutant in PTP-PEST −/− cells reverted the phenotype. Whereas 65–70% of null cells or those expressing wild type VAV2 (not shown) formed tails, only 10% of the cells expressing the VAV2 mutant formed tails, resembling RE controls (Fig. 5D). Thus, blocking the GEF activity of VAV2 alone is sufficient to restore a normal morphology to PTP-PEST −/− cells.
PTP-PEST Directly Targets p190RhoGAP to Modulate RhoA Activity
We next sought to identify a mechanism whereby PTP-PEST modulates RhoA activity. p190RhoGAP is a negative regulator of RhoA that is both tyrosine-phosphorylated (28, 29) and involved in controlling membrane protrusion (11). In addition, p190RhoGAP activity is required to suppress RhoA activity in response to integrin engagement (16). To determine whether p190RhoGAP was a target of PTP-PEST, we performed substrate trapping with catalytically inactive PTP-PEST. As shown in Fig. 6A, p190RhoGAP binds to the C231S trapping mutant. We next assayed the tyrosine phosphorylation state of p190RhoGAP in PTP-PEST −/− cells compared with RE controls. p190RhoGAP immunoprecipitated from PTP-PEST −/− cells shows increased tyrosine phosphorylation compared with control cells (Fig. 6B, top). These observations indicate that p190RhoGAP is a substrate of PTP-PEST.
FIGURE 6. p190RhoGAP is a substrate of PTP-PEST.

A, lysates of HeLa cells pretreated with pervanadate were incubated with fusion proteins corresponding to PTP-PEST trapping mutants, C231S (CS), or D199A (DA) or wild type (WT) PTP-PEST. LYS, lysate. Western blotting (WB) shows that p190RhoGAP is present in the pull down using the trapping mutant (CS) but not wild type fusion protein. B, tyrosine phosphorylation of p190RhoGAP is enhanced in PTP-PEST −/− cells. In addition, the association of p190RhoGAP and p120RasGAP is increased in PTP-PEST −/− cells. IP, immunoprecipitate.
We next ascertained if the GAP activity of p190RhoGAP was altered in PTP-PEST −/− cells. One indicator of enhanced GAP activity is elevated tyrosine phosphorylation (16, 28, 29). As shown above (Fig. 6B), we observe enhanced tyrosine phosphorylation of p190RhoGAP in the PTP-PEST −/− cells. An increase in p190RhoGAP activity is also coincident with its increased interaction with p120RasGAP (48–50). p190RhoGAP immunoprecipitated from PTP-PEST −/− cells shows an increased interaction with p120RasGAP compared with RE controls (Fig. 6B, bottom), whereas total levels of both proteins are equivalent in −/− and RE cells. As an additional method to assess p190RhoGAP activity, we utilized an affinity precipitation assay with activated RhoA (L63) (38). This approach is analogous to the method described above to measure GEF activity with nucleotide free Rac1 except that activated RhoA (L63RhoA) is used by virtue of its enhanced affinity for GAPs. Because we found that RhoA activity in PTP-PEST null cells was inhibited in response to adhesion, p190RhoGAP was precipitated from RE controls or PTP-PEST null cells that were either kept in suspension or plated on a FN substrate for 1 h. An increased amount of p190RhoGAP was precipitated from null cells plated on FN with reduced levels detected in RE or null cells in suspension (Fig. 7A). These data together indicate that p190RhoGAP activity is elevated in PTP-PEST null cells. To verify that the increased GAP activity was functionally responsible for the phenotype of PTP-PEST null cells, we expressed a GAP-dead mutant, p190RA in the −/− cells. This mutant was able to revert the −/− cells to a normal phenotype (Fig. 7B; % tails = 8.5%). PTP-PEST −/− cells expressing p190RA did not exhibit exaggerated membrane protrusions or tails.
FIGURE 7. p190RhoGAP activity is enhanced in PTP-PEST −/− cells.

A, as in Fig. 5, Rho proteins cycle between different nucleotide bound states. A Q63L mutant of RhoA mimics activated, GTP-RhoA and binds with high affinity to GAPs. To implicate p190RhoGAP in PTP-PEST regulation of RhoA, we utilized an affinity precipitation assay to measure in vivo activity of p190RhoGAP in response to adhesion to FN. A GST fusion protein of Q63LRhoA was incubated with cell lysates, and the pull-downs were blotted (WB) for p190RhoGAP. Q63LRhoA precipitates an increased amount of p190RhoGAP in PTP-PEST −/− cells plated on FN compared with suspension and also compared with re-expresser controls. S, suspension. B, a dominant negative mutant of p190RhoGAP that lacks GAP activity, p190RA, was expressed in PTP-PEST −/− cells. This mutant reverts the phenotype of PTP-PEST −/− cells to normal cells. Cells were plated on FN-coated coverslips for 3 h in the absence of serum. GFP, green fluorescent protein.
DISCUSSION
In this study we utilized PTP-PEST −/− fibroblasts to examine how targeted deletion of this protein-tyrosine phosphatase inhibits cell migration. Targeted deletion of PTP-PEST results in a morphology where the leading edge is highly protrusive but the trailing tail is unable to retract. Here we present evidence that PTP-PEST reciprocally modulates the activity of Rho GTPases, Rac1 and RhoA. Excessive Rac1 activity combined with inhibited RhoA activity perturbs the balance between protrusion and retraction needed for proper migration. PTP-PEST mediates these effects by directly targeting upstream regulators of Rho GTPases. PTP-PEST modulates Rac1 activity by targeting the exchange factor VAV2. PTP-PEST also acts on p190RhoGAP to control RhoA activity (Fig. 8A). Our results demonstrate that PTP-PEST is required to couple forward protrusion with tail retraction.
FIGURE 8. Model for proposed action of PTP-PEST in coupling membrane protrusion and tail retraction.
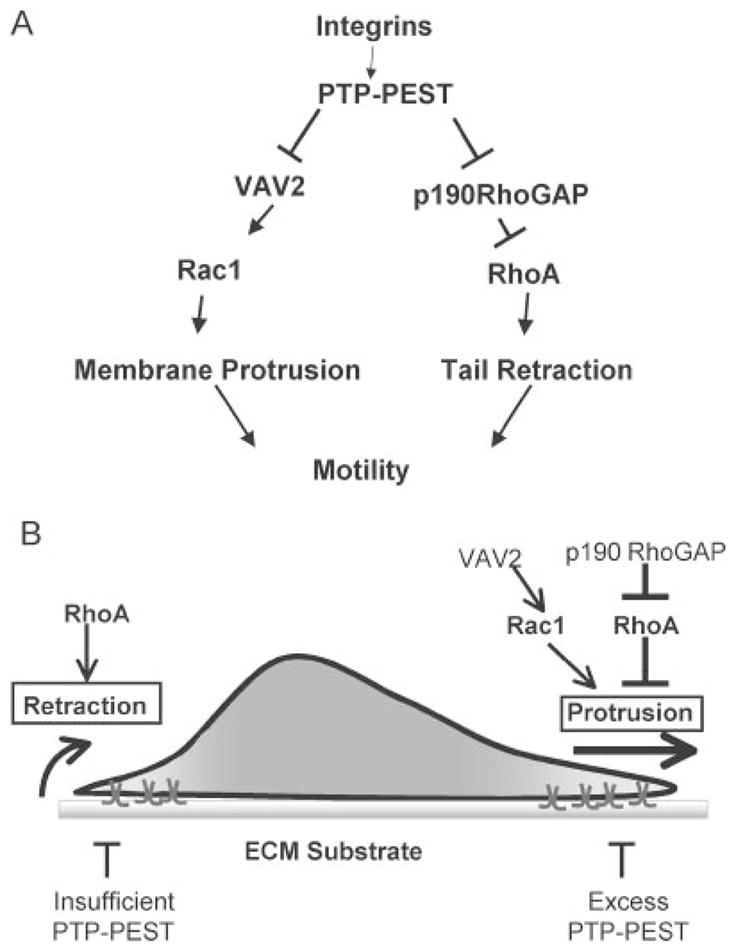
PTP-PEST functions at the front and back of a migrating cell. At the cell front PTP-PEST controls the extent of membrane protrusion. At the cell rear PTP-PEST is required for tail retraction. Excess PTP-PEST blocks membrane protrusion. The absence of PTP-PEST results in excessive protrusion, the formation of multiple leading edges, and a lack of tail retraction. PTP-PEST mediates its effect through reciprocally modulating the localized activity of Rac1 and RhoA through direct regulation of the activity and/or sub-cellular targeting of VAV2 and p190RhoGAP. ECM, extracellular cell matrix.
Previous studies with PTP-PEST −/− cells demonstrated its requirement for migration (31); however, a mechanism for this inhibition was not addressed. We extended these findings by identifying both cellular and biochemical mechanisms that can account for the observed phenotype. The cellular mechanism consists of two striking morphological characteristics; that is, the highly protrusive nature at one end and the long, thin tails at the opposite end. The broad, flat lamellipodia at the leading edge are reminiscent of those seen in highly motile cells. The PTP-PEST −/− cells exhibit increased membrane activity as observed by video microscopy (Fig. 2A). In many cases, PTP-PEST −/− cells develop competing leading edges. The inability to polarize and undergo directional migration could entirely account for a block in migration. This observation led us to compare the ability of these cells to undergo random versus haptotactic migration. A greater fraction of −/− cells were able to migrate in a haptotaxis assay, suggesting that they can respond to a directional migration cue. The nearly complete inhibition of migration when both sides of the transwell are coated is consistent with an inability to release the cell body or tail from the upper membrane. Indeed, immunofluorescence staining of the membranes showed cells to be trapped in the pores.4 The elongated tails observed by immunofluorescence (Fig. 1) suggested a retraction defect. Video microscopy confirmed the development of the tails (Fig. 2A and supplemental videos 1–3) and the inability to release from the substrate once formed (Fig. 2B and supplemental videos 1–3). The videos also show that the development of the tails is driven by excessive protrusion. In normal cells forward protrusion is coupled to rear release such that the rate of protrusion is equal to the rate of retraction, resulting in a net forward locomotion (1). In PTP-PEST −/− cells the degree of protrusion exceeds the degree of retraction. Thus, the defect in tail retraction accounts for the inability of PTP-PEST −/− cells to migrate.
Our findings point to a biochemical mechanism for PTP-PEST that corroborates the morphological data. We found that PTP-PEST reciprocally modulates the activity of Rac1 and RhoA. To strike a balance between protrusion and retraction that is permissive for motility, Rac1 and RhoA activity must be regulated both spatially and temporally (1, 2, 15). It is likely that PTP-PEST modulates the localized activity of Rac1 and RhoA (see the model, Fig. 8B). Enhanced Rac1 activity is consistent with the increased protrusive behavior of PTP-PEST −/− cells. Rac1 activity at the leading edge is required for the formation of lamellipodia (3, 10), whereas high levels promote invasion (51). Thus, the probable function of PTP-PEST is to limit Rac1 activity to control the extent of protrusion (32). On the other hand, whereas a low level of RhoA is required to maintain adhesion to the substratum (10), blocking RhoA activity impairs tail retraction (13) and results in the formation of competing leading edges (14). High levels of RhoA promote membrane retraction and suppress membrane activity (12). Decreased activity of RhoA in PTP-PEST −/− cells is consistent with increased protrusion as well as a lack of tail retraction. Our results demonstrate that PTP-PEST is necessary to activate RhoA at the cell rear to promote retraction and to restrict the formation of protrusions to the leading edge.
Our findings are in agreement with a number of studies showing a functional antagonism between Rac1 and RhoA indicating that elevated Rac1 suppresses RhoA through biochemical cross-talk (12, 52–55). Our data support a model where PTP-PEST directly acts on upstream regulators of Rac1 and RhoA. We show that the guanine nucleotide exchange factor, VAV2, is a substrate of PTP-PEST. This conclusion is supported by substrate trapping of VAV2 by PTP-PEST (Fig. 4, A and B) and by elevated tyrosine phosphorylation of VAV2 in PTP-PEST −/− cells (Fig. 4C). This result suggests that PTP-PEST modulates VAV2 activity.
Because VAV2 can be positively or negatively regulated by tyrosine phosphorylation (21, 23, 25, 27) and immunoprecipitated GEFs often show low activity, we employed an alternate method to assess VAV2 activity. A useful property of activated GEFs is their high affinity binding to Rho proteins in the nucleotide-free state (42–44). Meller et al. (45) have used nucleotide-free Rho proteins to biochemically isolate novel exchange factors. Point mutants of Rho proteins that mimic the nucleotide-free state have been used to affinity precipitate GEFs from cell lysates (42, 46, 47). To indirectly ascertain VAV2 activity, we used a mutant Rac1 (Gly-15 → Ala) as a GST fusion protein (15ARac1) to affinity precipitate VAV2 from cell lysates. Using this pull-down assay, we observe increased precipitation of VAV2 from PTP-PEST −/− cells compared with controls (Fig. 5), suggesting enhanced activity. This finding implicates PTP-PEST in negatively regulating VAV2.
Our data further suggest that VAV2 activity can be regulated by integrin-mediated adhesion. The increased precipitation of VAV2 with the 15ARac1 pull-down in response to adhesion to FN (Fig. 5A) was both surprising and intriguing. Previous efforts to implicate VAV2 in integrin signaling were inconclusive since an increase in tyrosine phosphorylation was not observed (23, 25). Subsequently, integrin-dependent cell spreading and Rac1 activation were shown to depend on VAV2 exchange activity (Marignani and Carpenter (26)). One explanation to reconcile these results is that VAV2 associates with a tyrosine phosphatase in response to adhesion that masks changes in phosphotyrosine levels. Our present study indicates that PTP-PEST may be the relevant phosphatase. Ongoing efforts are aimed at identifying the sites on VAV2 targeted by PTP-PEST. It is conceivable that PTP-PEST affects both the activity and localization of VAV2 at the leading edge (see the model in Fig. 8B). Preliminary studies suggest that PTP-PEST may act on tyrosine both in the N and C terminus of VAV2.4
PTP-PEST could also mediate its effects on membrane protrusion indirectly via its substrate p130cas (56, 57) or through its interaction with paxillin (66). In PTP-PEST null cells, p130cas tyrosine phosphorylation is elevated along with its interaction with crk (31). This association correlates with increased motility, protrusion, and Rac1 activation through its interaction with the exchange factor, DOCK180 (58–62). We found that expression of dominant negative cas or crk mutants was unable to rescue the PTP-PEST null phenotype.4 We also found that expression of a dominant negative mutant of DOCK 180, DOCK 180DDH2, did not rescue the PTP-PEST null phenotype. This mutant is deficient in Rac activation (65). Instead, we found that increased expression of either wild type DOCK180 or the DH mutant in PTP-PEST −/− cells led to a multinucleated phenotype.5 This result made it difficult to assess the contribution of DOCK180 to the migration phenotype. Interestingly, we did not observe this effect when DOCK180 or the DH mutant were expressed in the re-expressors. In contrast, a DH domain mutant of VAV2 (L342R/L343S) (26) was able to fully rescue the PTP-PEST null phenotype. Our findings together with previous studies suggest that PTP-PEST contributes to regulation of Rac1 through both direct and indirect mechanisms. It is likely that the interaction of PTP-PEST with paxillin or p130cas targets PTP-PEST to the correct location in the cell. Interestingly, a recent study suggests that VAV2 is also targeted to the leading edge by a Rap1-dependent mechanism (67). Our study implicates PTP-PEST in direct activation of Rac1 by VAV2. It is likely that this activation may functionally differ from Rac1 activation downstream of p130cas or suppression of Rac1 downstream of paxillin. Sorting out the contribution of each pathway to the overall activation of Rac1 and their effects on motility will require detailed structure/function studies using mutants of PTP-PEST that specifically knock-out one or more binding interactions.
As discussed above, elevated levels of Rac1 can lead to a suppression of RhoA activity (52, 54). Our data show that p190RhoGAP, a negative regulator of RhoA, is a direct target of PTP-PEST by virtue of its binding to a PTP-PEST trapping mutant (Fig. 6A) and its enhanced tyrosine phosphorylation (Fig. 6B). The tyrosine phosphorylation state of p190RhoGAP directly correlates with its GAP activity. The increased precipitation of p190RhoGAP by activated RhoA (Fig. 7A) further supports a role for PTP-PEST in regulation of p190RhoGAP. Together, these data show that PTP-PEST negatively regulates p190RhoGAP. It is unclear in which cellular compartment p190RhoGAP activity is localized. Evidence for p190-RhoGAP involvement in protrusion (11) suggests that GAP activity may reside at the cell front. p190RhoGAP has been implicated in regulating localized RhoA activity through its interaction with paxillin (63), a protein that also binds PTP-PEST. It is conceivable that p190RhoGAP activity is modulated in distinct regions of the cell depending on whether RhoA needs to be active, such as at the cell rear where PTP-PEST could suppress GAP activity, or at the cell front, where RhoA activity needs to be maintained at a low level. PTP-PEST could act as a switch to control local activity of p190RhoGAP.
Relative to other tyrosine phosphatases that regulate cell motility, PTP-PEST appears to be unique in its function. Fibroblasts lacking PTP1B, PTPα or SHP-2, which are also blocked in migration, are unable to spread or form protrusions. It is likely that individual tyrosine phosphatases act at different steps of the migration cycle (64); thus, the localized activity is key to its function during cell migration. An outstanding and important question is where does the activity of PTP-PEST or other protein-tyrosine phosphatases reside in a migrating cell? The development of methods to measure in vivo tyrosine phosphatase activity will greatly enhance our understanding of the function of these enzymes in cell motility.
Collectively the data presented here along with our previous studies (32) suggest a model for PTP-PEST in regulating cell migration (Fig. 8B). PTP-PEST contributes to cell migration both at the front and back of the cell through reciprocal regulation of Rac1 and RhoA. At the front of the cell Rac1 must be active, and RhoA must be low to facilitate forward protrusion. At the back of the cell RhoA must be active, and Rac1 activity must be low or excluded. By modulating the activity of VAV2 and p190RhoGAP in these two cellular compartments, PTP-PEST can maintain the appropriate asymmetry of Rac1 and RhoA activity to permit forward movement. When excess PTP-PEST is present, protrusion is blocked. In the absence of PTP-PEST expression, migration is deregulated at both the leading edge and the cell rear, with excess protrusion and a lack of tail retraction. In this way PTP-PEST couples forward protrusion with tail retraction. Future studies are aimed at determining how PTP-PEST is targeted to the front or back of the cell where it can regulate Rac1 or RhoA, respectively.
Supplementary Material
Acknowledgments
We thank Mike Schaller, Bill Arthur, Krister Wennerberg, Shawn Ellerbroek, and Becky Worthylake for valuable discussions and Lisa Sharek and Rosario Espejo for technical help. We are grateful to Dr. Kristiina Vuori, Dr. Michael Schaller, and Dr. Chris Carpenter for valuable reagents.
Footnotes
This work was supported by National Institutes of Health Grants GM29860 and HL45100 (to K. B.) and Canadian Institutes of Health Research MOP 12466 (to M. T.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1 and Videos 1–3.
The abbreviations used are: GEF, guanine nucleotide exchange factor; GAP, GTPase-activating protein; FN, fibronectin; RE, re-expressor; DMEM, Dulbecco’s modified Eagle’s medium; GST, glutathione S-transferase; mAb, monoclonal antibody.
B. Liu, unpublished observations.
S. Sastry, unpublished observations.
S. K. Sastry, unpublished observations.
References
- 1.Ridley A, Schwartz M, Burridge K, Firtel R, Ginsberg M, Borisy G, Parsons JT, Horwitz AF. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 2.Etienne-Manneville S, Hall A. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 3.Ridley A. J Cell Sci. 2001;114:2713–2722. doi: 10.1242/jcs.114.15.2713. [DOI] [PubMed] [Google Scholar]
- 4.Burridge K, Wennerberg K. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 5.Gardiner E, Pestonjamasp K, Bohl B, Chamberlain C, Hahn K, Bokoch G. Curr Biol. 2002;12:2029–2034. doi: 10.1016/s0960-9822(02)01334-9. [DOI] [PubMed] [Google Scholar]
- 6.Kraynov V, Chamberlain C, Bokoch G, Schwartz M, Slabaugh S, Hahn K. Science. 2000;290:333–337. doi: 10.1126/science.290.5490.333. [DOI] [PubMed] [Google Scholar]
- 7.Kiosses W, Shattil S, Pampori N, Schwartz M. Nat Cell Biol. 2001;3:316–320. doi: 10.1038/35060120. [DOI] [PubMed] [Google Scholar]
- 8.Brahmbhatt A, Klemke R. J Biol Chem. 2003;278:13016–13025. doi: 10.1074/jbc.M211873200. [DOI] [PubMed] [Google Scholar]
- 9.Cho S, Klemke R. J Cell Biol. 2002;156:725–736. doi: 10.1083/jcb.200111032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nobes C, Hall A. J Cell Biol. 1999;144:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arthur W, Burridge K. Mol Biol Cell. 2001;12:2711–2720. doi: 10.1091/mbc.12.9.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cox E, Sastry S, Huttenlocher A. Mol Biol Cell. 2001;12:265–277. doi: 10.1091/mbc.12.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Worthylake R, Lemoine S, Watson J, Burridge K. J Cell Biol. 2001;154:147–160. doi: 10.1083/jcb.200103048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Worthylake R, Burridge K. J Biol Chem. 2003;278:13578–13584. doi: 10.1074/jbc.M211584200. [DOI] [PubMed] [Google Scholar]
- 15.Ballestrem C, Hinz B, Imhof B, Wehrle-Haller B. J Cell Biol. 2001;155:1319–1332. doi: 10.1083/jcb.200107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arthur W, Petch L, Burridge K. Curr Biol. 2000;10:719–722. doi: 10.1016/s0960-9822(00)00537-6. [DOI] [PubMed] [Google Scholar]
- 17.O’Connor K, Nguyen B, Mercurio A. J Cell Biol. 2000;148:253–258. doi: 10.1083/jcb.148.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price L, Leng J, Schwartz M, Bokoch G. Mol Biol Cell. 1998;9:1863–1871. doi: 10.1091/mbc.9.7.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ren X, Kiosses W, Schwartz M. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt A, Hall A. Genes Dev. 2002;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- 21.Bustelo X. Mol Cell Biol. 2000;20:1461–1477. doi: 10.1128/mcb.20.5.1461-1477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abe K, Rossman K, Liu B, Ritola K, Chiang D, Campbell S, Burridge K, Der CJ. J Biol Chem. 2000;275:10141–10149. doi: 10.1074/jbc.275.14.10141. [DOI] [PubMed] [Google Scholar]
- 23.Liu B, Burridge K. Mol Cell Biol. 2000;20:7160–7169. doi: 10.1128/mcb.20.19.7160-7169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pandey A, Podtelejnikov A, Blagoev B, Bustelo X, Mann M, Lodish H. Proc Natl Acad Sci U S A. 2000;97:179–184. doi: 10.1073/pnas.97.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moores S, Selfors L, Fredericks J, Breit T, Fujikawa K, Alt F, Brugge J, Swat W. Mol Cell Biol. 2000;20:6364–6373. doi: 10.1128/mcb.20.17.6364-6373.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marignani P, Carpenter C. J Cell Biol. 2001;154:177–186. doi: 10.1083/jcb.200103134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez-Lago M, Lee H, Cruz C, Movilla N, Bustelo X. Mol Cell Biol. 2000;20:1678–1691. doi: 10.1128/mcb.20.5.1678-1691.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang JH, Gill S, Settleman J, Parsons S. J Cell Biol. 1995;130:355–368. doi: 10.1083/jcb.130.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakahara H, Mueller SC, Nomizu M, Yamada Y, Yeh Y, Chen WT. J Biol Chem. 1998;273:9–12. doi: 10.1074/jbc.273.1.9. [DOI] [PubMed] [Google Scholar]
- 30.Garton A, Tonks N. J Biol Chem. 1999;274:3811–3818. doi: 10.1074/jbc.274.6.3811. [DOI] [PubMed] [Google Scholar]
- 31.Angers-Lousteau A, Cote JF, Charest A, Dowbenko D, Spencer S, Lasky L, Tremblay M. J Cell Biol. 1999;144:1019–1031. doi: 10.1083/jcb.144.5.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sastry S, Lyons P, Schaller M, Burridge K. J Cell Sci. 2002;115:4305–4316. doi: 10.1242/jcs.00105. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli U. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Towbin H, Staehlin T, Gordon J. Proc Natl Acad Sci U S A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flint A, Tiganis T, Barford D, Tonks N. Proc Natl Acad Sci U S A. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyons P, Dunty J, Schaefer E, Schaller M. J Biol Chem. 2001;276:24422–24431. doi: 10.1074/jbc.M011080200. [DOI] [PubMed] [Google Scholar]
- 37.Glaven J, Whitehead I, Bagrodia S, Kay R, Cerione R. J Biol Chem. 1999;274:2279–2285. doi: 10.1074/jbc.274.4.2279. [DOI] [PubMed] [Google Scholar]
- 38.Noren N, Arthur W, Burridge K. J Biol Chem. 2003;278:13615–13618. doi: 10.1074/jbc.C200657200. [DOI] [PubMed] [Google Scholar]
- 39.Alblas J, Ulfman L, Hordijk P, Koenderman L. Mol Biol Cell. 2001;12:2137–2145. doi: 10.1091/mbc.12.7.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Somlyo A, Bradshaw D, Ramos S, Murphy C, Myers C, Somlyo A. Biochem Biophys Res Commun. 2000;269:652–659. doi: 10.1006/bbrc.2000.2343. [DOI] [PubMed] [Google Scholar]
- 41.Crespo P, Schuebel K, Ostrom A, Gutkind J, Bustelo X. Nature. 1997;385:169–172. doi: 10.1038/385169a0. [DOI] [PubMed] [Google Scholar]
- 42.Feig LA. Nat Cell Biol. 1999;1:25–27. doi: 10.1038/10018. [DOI] [PubMed] [Google Scholar]
- 43.Hart M, Powers S. Methods Enzymol. 1995;255:129–135. doi: 10.1016/s0076-6879(95)55016-x. [DOI] [PubMed] [Google Scholar]
- 44.Hart MJ, Sharma S, el Masry N, Qiu RG, McCabe P, Polakis P, Bollag G. J Biol Chem. 1996;271:25452–25458. doi: 10.1074/jbc.271.41.25452. [DOI] [PubMed] [Google Scholar]
- 45.Meller N, Irani-Tehrani M, Kiosses W, del Pozo M, Schwartz M. Nat Cell Biol. 2002;4:639–647. doi: 10.1038/ncb835. [DOI] [PubMed] [Google Scholar]
- 46.Arthur WT, Ellerbroek SM, Der CJ, Burridge K, Wennerberg K. J Biol Chem. 2002;277:42964–42972. doi: 10.1074/jbc.M207401200. [DOI] [PubMed] [Google Scholar]
- 47.Reuther GW, Lambert QT, Booden MA, Wennerberg K, Becknell B, Marcucci G, Sondek J, Caligiuri MA, Der CJ. J Biol Chem. 2001;276:27145–27151. doi: 10.1074/jbc.M103565200. [DOI] [PubMed] [Google Scholar]
- 48.Hu KQ, Settleman J. EMBO J. 1997;16:473–483. doi: 10.1093/emboj/16.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kulkarni SV, Gish G, van der Geer P, Henkemeyer M, Pawson T. J Cell Biol. 2000;149:457–470. doi: 10.1083/jcb.149.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roof RW, Haskell MD, Dukes BD, Sherman N, Kinter M, Parsons SJ. Mol Cell Biol. 1998;18:7052–7063. doi: 10.1128/mcb.18.12.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keely PJ, Westwick JK, Whitehead IP, Der CJ, Parise LV. Nature. 1997;390:632–636. doi: 10.1038/37656. [DOI] [PubMed] [Google Scholar]
- 52.Nimnual A, Taylor L, Bar-Sagi D. Nat Cell Biol. 2003;5:236–241. doi: 10.1038/ncb938. [DOI] [PubMed] [Google Scholar]
- 53.Rottner K, Hall A, Small JV. Curr Biol. 1999;9:640–648. doi: 10.1016/s0960-9822(99)80286-3. [DOI] [PubMed] [Google Scholar]
- 54.Sander EE, ten Klooster JP, van Delft S, van der Kammen RA, Collard JG. J Cell Biol. 1999;147:1009–1022. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsuji T, Ishizaki T, Okamoto M, Higashida C, Kimura K, Furuyashiki T, Arakawa Y, Birge R, Nakamoto T, Hirai H, Narumiya S. J Cell Biol. 2002;157:819–830. doi: 10.1083/jcb.200112107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garton A, Flint AJ, Tonks N. Mol Cell Biol. 1996;16:6408–6418. doi: 10.1128/mcb.16.11.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cote JF, Charest A, Wagner J, Tremblay ML. Biochemistry. 1998;37:13128–13137. doi: 10.1021/bi981259l. [DOI] [PubMed] [Google Scholar]
- 58.Klemke R, Leng J, Molander R, Brooks P, Vuori K, Cheresh D. J Cell Biol. 1998;140:961–972. doi: 10.1083/jcb.140.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brugnera E, Haney L, Grimsley C, Lu M, Walk S, Tosello-Trampont A, Macara I, Madhani H, Fink G, Ravichandran K. Nat Cell Biol. 2002;4:574–582. doi: 10.1038/ncb824. [DOI] [PubMed] [Google Scholar]
- 60.Grimsley CM, Kinchen JM, Tosello-Trampont AC, Brugnera E, Haney LB, Lu M, Chen Q, Schubert D, Klingele D, Hengartner MO, Ravichandran KS. J Biol Chem. 2003;279:6087–6097. doi: 10.1074/jbc.M307087200. [DOI] [PubMed] [Google Scholar]
- 61.Kiyokawa E, Hashimoto Y, Kobayashi S, Sugimura H, Kurata T, Matsuda M. Genes Dev. 1998;12:3331–3336. doi: 10.1101/gad.12.21.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kiyokawa E, Hashimoto Y, Kurata T, Sugimura H, Matsuda M. J Biol Chem. 1998;273:24479–24484. doi: 10.1074/jbc.273.38.24479. [DOI] [PubMed] [Google Scholar]
- 63.Tsubouchi A, Sakakura J, Yagi R, Mazaki Y, Schaefer E, Yano H, Sabe H. J Cell Biol. 2002;159:673–683. doi: 10.1083/jcb.200202117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Larsen M, Tremblay M, Yamada K. Nature. 2003;4:700–711. doi: 10.1038/nrm1199. [DOI] [PubMed] [Google Scholar]
- 65.Cote JF, Motoyama A, Bush J, Vuori K. Nat Cell Biol. 2005;7:797–807. doi: 10.1038/ncb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jamieson J, Tumbarello D, Halle M, Brown M, Tremblay M, Turner CE. J Cell Sci. 2005;118:5835–5847. doi: 10.1242/jcs.02693. [DOI] [PubMed] [Google Scholar]
- 67.Arthur WT, Quilliam LA, Cooper JA. J Cell Biol. 2004;167:111–122. doi: 10.1083/jcb.200404068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.