

Abstract
To unveil the still-elusive nature of metazoan replication origins, we identified them genome-wide and at unprecedented high-resolution in mouse ES cells. This allowed initiation sites (IS) and initiation zones (IZ) to be differentiated. We then characterized their genetic signatures and organization and integrated these data with 43 chromatin marks and factors. Our results reveal that replication origins can be grouped into three main classes with distinct organization, chromatin environment, and sequence motifs. Class 1 contains relatively isolated, low-efficiency origins that are poor in epigenetic marks and are enriched in an asymmetric AC repeat at the initiation site. Late origins are mainly found in this class. Class 2 origins are particularly rich in enhancer elements. Class 3 origins are the most efficient and are associated with open chromatin and polycomb protein-enriched regions. The presence of Origin G-rich Repeated elements (OGRE) potentially forming G-quadruplexes (G4) was confirmed at most origins. These coincide with nucleosome-depleted regions located upstream of the initiation sites, which are associated with a labile nucleosome containing H3K64ac. These data demonstrate that specific chromatin landscapes and combinations of specific signatures regulate origin localization. They explain the frequently observed links between DNA replication and transcription. They also emphasize the plasticity of metazoan replication origins and suggest that in multicellular eukaryotes, the combination of distinct genetic features and chromatin configurations act in synergy to define and adapt the origin profile.
In metazoans, DNA replication initiates at thousands of specific sites called DNA replication origins that are activated in a defined temporal order during each cell cycle (for review, see DePamphilis 1993; Aladjem 2007; Fragkos et al. 2015). No clear sequence specificity for origins has been found in multicellular organisms, but in Saccharomyces cerevisiae initiation sites (IS) are defined by an AT-rich consensus sequence (Marahrens and Stillman 1992). Conversely, recent analyses revealed the presence of GC-rich elements in eukaryotic origins, such as CpG islands (CGI) (Delgado et al. 1998; Prioleau 2009; Cayrou et al. 2011; Costas et al. 2011) and an Origin G-rich Repeated Element (OGRE) that can form G4 (Cayrou et al. 2011, 2012a). G4 were further detected in human origins (Besnard et al. 2012), where they might have a role in replication initiation (Valton et al. 2014).
Many potential origins are set before the S phase, but few are activated in a given cell cycle. Furthermore, some origins are activated only in specific developmental or environmental conditions, suggesting flexibility in their recognition or usage (DePamphilis 1993; Aladjem 2007; Nordman and Orr-Weaver 2012; Fragkos et al. 2015). Also, the timing of their activation during the cell cycle is flexible (Rhind and Gilbert 2013). More recently, the importance of the replication timing program variability in the genetic variations between individuals was underlined (Koren et al. 2014). However, these studies did not investigate replication origin positioning and organization along chromosomes. The chromatin environment may contribute to the flexibility in the choice of the origins to be activated as suggested by the presence of histone modifications at origins. However, a comprehensive “identity code” that could explain this flexibility and that takes into account the genetic and epigenetic signatures of origins genome-wide is still missing in vertebrates. This lack of basic knowledge in a crucial cellular mechanism hinders our understanding of the chromosome organization and probably of many diseases caused by unfaithful or illegitimate replication.
Here, we coupled purification of RNA-primed nascent DNA at origins with next-generation sequencing to identify consensus or cooperative genetic and chromatin features of origins in mouse embryonic stem cells (mESCs). Our results allow building a comprehensive landscape of the prereplication complex (pre-RC) and IS at origins and might help understanding how origin selection may adapt to different chromosome organizations linked to different cell behaviors.
Results
Origin maps reveal three different classes of origins defined by structural and functional genomic features
For genome-wide mapping of origins in mESCs, we coupled Nascent Strand (NS) purification with high-throughput sequencing (Supplemental Fig. S1A; Methods). In this approach, RNA-primed nascent DNA is first purified from replication origins, and then contaminated DNA is removed by lambda exonuclease (λ-Exo) digestion. We already described this method in detail (Cayrou et al. 2011, 2012b) and showed that it gives accurate and very reliable results, providing that the crucial λ-Exo digestion is performed in optimal conditions (Supplemental Figs. S1, S2; Cayrou et al. 2011; Li et al. 2014; Picard et al. 2014). For high-resolution analysis, we sequenced three independent NS samples (Supplemental Fig. S1C) and also an RNase A-treated NS sample for peak calling analyses (Supplemental Fig. S1B,C), which is an appropriate background control used in these experiments. Supplemental Figure S1C shows the data reproducibility and the low background obtained with sheared genomic DNA and the RNase A-digested NS sample before incubation with λ-Exo. Supplemental Figure S2 demonstrates that the presence of G4 in the sequence did not affect λ-Exo digestion in our conditions.
We combined two complementary software tools to detect origins. SICER identifies wide genomic zones enriched in reads, whereas SWEMBL highlights the precise position of peaks (Supplemental Fig. S1D; Methods). This joint analysis returned 65,019 high-confidence peaks that identified the major IS of DNA replication and 35,512 initiation zones (IZ) that encompassed one or several closely associated IS. Most origins contained one (60%) or two IS (22%) (Supplemental Fig. S1D). Therefore, an origin can correspond to a single IS or to an IZ, as previously described for the Lamin B2 (Abdurashidova et al. 2000) and DHFR origins (Kobayashi et al. 1998; Wang et al. 1998). Our new high resolution data are recapitulated by 80.2% of our previous data obtained on microarrays on mouse Chromosome 11 (Supplemental Fig. S1E; Cayrou et al. 2011).
We first investigated how origins are arranged in the genome. Our previous findings, based on microarray and DNA combing analysis of Chromosome 11 in mESCs, suggest that, on average, replication units cover 53 kb and contain four potential origins with an inter-origin spacing of 12.2 kb (Cayrou et al. 2011). Therefore, we analyzed the read density profiles in a 14-kb window, 7 kb each side of the 65,019 IS. Alternative window sizes did not give more substantial information (Methods). Based on k-means clustering of read densities in the 14-kb regions, we defined 12 IS subgroups. By sorting these subgroups according to the IS distance to the neighboring peak, we obtained a striking sigmoid-shaped profile (heatmap in Fig. 1A, left). We next annotated each IS relative to the presence of different genomic features (Fig. 1A, right): genes, promoters (1 kb upstream), and CGI. Based on the combination of read density profiles and genomic features, we could assemble the 12 IS subgroups in three qualitatively distinct IS classes (Fig. 1B) that correlated with specific genomic features (Fig. 1C).
Figure 1.

Three classes of replication origins. (A) Clustering of origins based on read densities around peaks. The left panel displays a heatmap of read densities in 7-kb regions on each side of the peak summit. It shows how an IS is positioned relative to its neighbors and indicates the signal strength (number of reads) and density at each IS. The brown intensity is proportional to the read counts per 100-bp bins. The numbers on the right of the heatmap indicate groups obtained by k-means clustering, and the left/right symmetry between cluster pairs is denoted by identical numbers followed by the L and R suffixes. The three classes of origins defined in the text are highlighted in boxes. The right panel indicates the overlap of IS (± 500 bp from the summit) with CpG islands (blue), promoters (red), and genes (black). (B) Read density mean profiles per class of origins. Each class is defined by assembling groups of IS characterized by a specific distance (dotted vertical lines) between two major IS, except for Class 1 origins, which have a single IS within a 14-kb region. The y-axis represents the average number of reads per peak. Class 1 profile is represented by subgroup 1; Class 2 by subgroups 5LR, 6LR; Class 3a by subgroups 8LR, 9LR; and Class 3b by 10LR, 11. (C) Genomic localization of origins. For each class, the bar plot indicates the proportion of IS associated with intergenic, CGi, promoters, and exons. (D) Distribution of late origins per class. The percentage of IS overlapping with the late-replicating regions defined in Hiratani et al. (2008) (Supplemental Table S1) is indicated for each class.
Class 1 was characterized by narrow and isolated peaks and included 44% of all IS. The three Class 1 subgroups (1, 2, and 3) (Fig. 1A, left) only differed by the IS strength. The two other origin classes, which formed the sigmoid-shaped profile, contained at least two major IS in the 14-kb window. We detected clear boundaries between subgroups 7 and 8 (Class 2/3a boundary) and between subgroups 9 and 10 (Class 3a/3b boundary). Class 2 and Class 3 were defined both by the drastic changes observed in the diagonal slope of read densities and by the associated genomic features. Class 2, 3a, and 3b profiles were arranged symmetrically with pairs of similarly spaced neighboring peaks appearing twice in the graph: once centered on the leftmost peak and once on the rightmost peak (for instance, 4L and 4R subgroups) (Fig. 1A, left).
To evaluate the significance of this arrangement, we used the same in silico procedure to analyze 65,019 randomly selected 14-kb regions in the mouse genome (negative control) (Supplemental Fig. S3A). Most random fragments (n = 47,520) fell within IS-free regions or had no clear peak, and the high-density bins were restricted to a single peak per region (no secondary peak like for Classes 2, 3a, and 3b). These results validated the relevance of the topological features observed across Classes 2, 3a, and 3b.
This structural repartition correlated with specific genomic features. Class 1 IS were uniformly distributed along chromosomes (Supplemental Fig. S3B). As with all IS, Class 1 IS were more frequent in CGI-, promoter-, or gene-rich regions (P-value = 0) (Fig. 1C; Supplemental Table S1) than in randomly selected regions of the same size. However, they were poorly enriched at CpGi-, promoter-, or gene-rich regions relative to the other classes (Fig. 1C; Supplemental Table S1). Moreover, Class 1 included 75% of all late origins (Fig. 1D; Supplemental Fig. S3B), suggesting that late-replicated regions are essentially composed of thin and isolated origins. In contrast, the other two classes, which consisted of grouped IS, included mainly early origins and were more concentrated in regions rich in genes, promoters, and CGI. Indeed, Class 2 origins were more frequently positioned within genes, preferentially inside exons and introns (P-value = 7.9 × 10−141 and 8.65 × 10−53, respectively) (Fig. 1C). Like Class 1, Class 2 origins contained relatively inefficient IS (based on the density of reads/peak) (Fig. 1B), suggesting more flexibility in the choice of IS within these regions. Class 3 was characterized by two strong IS (mean density above 106 reads/peak versus 74 in Class 1), separated by <1.5 kb (Fig. 1B). It included 60% of the strongest IS, and 68% of origins associated with a promoter belonged to Class 3 (data not shown). This class was more likely to be associated with orphan CGI-type (Fenouil et al. 2012) and/or cryptic promoters than only with known promoters. Indeed, although it was significantly associated with promoters (25% of these IS; P-value <5.5 × 10−47) (Supplemental Table S1), more than 45% of Class 3 IS overlapped with a CGI. Moreover, Class 3 represented the vast majority of CGI-positive origins (58%). Reciprocally, CGI are likely good origin predictors as 75% of them overlapped with an IZ and 60% were specifically located on the IS.
These results reveal the existence of distinct classes of origins based on their different structural patterns and their association with specific genomic features.
Efficient replication initiation sites are prominent in open chromatin domains and facultative heterochromatin
To determine whether IS and IZ have a specific chromatin environment, we integrated 43 known marks/factors from published data on mESCs (Supplemental Table S1; Methods). Open chromatin-2 marks (i.e., marks linked to decondensed chromatin) were detected similarly in IS and IZ, indicating strict positioning on the IS inside the IZ (Fig. 2A, upper; Supplemental Fig. S4A–C; Supplemental Table S1). These marks included not only promoter-specific marks, such as H3K4me3, but also marks that are not strictly promoter-specific, such as H3K9ac and DNase I hypersensitive sites (DHS). Open chromatin marks were present at nearly half of the promoters and 5′ UTRs, but they were two- to threefold enriched when origins were also present in these regions (Supplemental Fig. S4D). A similar overrepresentation was observed in intronic and exonic regions that contained origins. In contrast to the association of origins with open chromatin marks, constitutive closed heterochromatin marks, such as H3K9me3, were poorly represented at origins (only 0.8% of IS). The high and sharp profile observed for H3K4me3 and H3K9ac at the IS center suggested a role in origin activation (Fig. 2C, left).
Figure 2.

Epigenetic marks and chromatin environment at IS. (A,B) Specific chromatin marks/factors associated with open chromatin-2 (blue boxes) and Polycomb complexes (brown boxes) at initiation sites. (A) Distribution of chromatin marks/factors relative to IS (dark green) and IZ (light green). (B) ChIP-seq signals for H3K4me3, H3K9ac, H3K27me3, and SUZ12 around ±7 kb from IS. (C) Hierarchical clustering of Pearson correlations between pairs of marks and/or chromatin factors for all IS. Marks and factors in the heatmap are organized according to the clustering described in Methods. Positive correlations are symbolized by a gradation of red that results from the localization of pairs of marks/factors at individual IS. Negative correlations are symbolized by a gradation of blue. Four significant groups of chromatin mark/factor are highlighted on the right: Open 1 chromatin marks/factors group (mostly associated with transcription initiation); Open 2 chromatin marks group (globally linked to decondensed chromatin); Enhancer marks (Enh.); and Polycomb group mark/factors (PcG). (D) More than 80% of H3K27me3 marks at IZ are associated with PRC1 or PRC2 proteins or are inside bivalent domains. (E) Overlap between marks/factors and origins.
We then compared the correlation between the position of these marks and that of IS (Fig. 2C) and of 1000 randomized sets of 65,019 regions. Open chromatin marks correlated more positively within IS than in randomly chosen genomic regions (Z-score for the correlation of each pair of marks in IS) (Supplemental Fig. S5A versus Supplemental Fig. S5C), with a strong confidence (Z-score >10). Notably, DHS (the most abundant open chromatin marks at origins: 49% of IS and 65% of IZ) (Supplemental Table S1) showed the highest correlations with IS (Z-score >14) (Supplemental Fig. S5E), suggesting the presence of a nucleosome-depleted region (NDR) as one of the main features of origins. DNase I sensitive sites were also the main determinants of DNA replication timing (Gindin et al. 2014). Moreover, 66% of the regions containing both H3K4me3 and H3K9ac could detect 32% of origins (Fig. 2E).
Less expected was the strong association of IS and IZ with marks/factors related to Polycomb (PcG) complexes (P-value = 0) (Fig. 2A, bottom; Supplemental Fig. S4A, B, left; Supplemental Table S1) that showed a distinct peak centered on the IS (Fig. 2B, right). PcG proteins are involved in gene silencing and form large complexes (PRC1 and PRC2) that can modify chromatin, notably by H3K27me3 deposition. Remarkably, PcG subunits were much more associated with origins than H3K27me3, the major PcG mark (Fig. 2D). Indeed, origins contained >80% of RNF2 binding sites (PRC1 subunit), 75% of SUZ12 binding sites (PRC2 subunit), and 90% of EZH2 sites (the PRC2 catalytic subunit) (Supplemental Fig. S4B, left). Finally, H3K27me3 distribution overlapped with that of H3K4me3 (Fig. 2D), shaping bivalent domains, which regulate transcriptional poising of developmentally regulated genes. As well as PRC1 and PRC2 sites, bivalent domains are strongly associated with origins in pluripotent mESCs (Bernstein et al. 2006). Therefore, PcG factors and open chromatin marks could play a synergic role in the choice of some origins in pluripotent cells (Fig. 2E).
Replication initiation zones are frequent in transcriptionally active domains
Conversely to IS, IZ were associated with marks/factors linked to transcription activity regulation that we define as open chromatin-1. Sites in which RNA Pol II and the active mark H3K27ac overlapped were more frequently found at IZ (Fig. 3A, upper; Supplemental Fig. S4B, right; Supplemental Fig. S4C), as indicated by the broad peaks centered on the IS (Fig. 3B, left). Interestingly, the IS strength positively correlated with the presence of RNA Pol II and DHS (148 and 132 reads/peak respectively, compared to 97 reads/peak for all IS), suggesting that replication initiation is favored in proximity of the transcription machinery. The superposition of the “paused” form of RNA Pol II (inhibition of RNA Pol II initiation by flavopiridol) with global RNA Pol II at origins (Supplemental Fig. S6) suggests that transcriptional pausing (Adelman and Lis 2012) favors a chromatin structure more suitable to origin positioning or activation.
Figure 3.

Epigenetic marks and chromatin factors at IZ. (A,B) Specific chromatin marks/factors associated with open chromatin-1 (light blue boxes) and enhancers (gray boxes) at initiation zones. (A) Distribution of chromatin marks/factors relative to IS (dark green) and IZ (light green). (B) ChIP-seq signals for H3K27ac, RNA Pol II, H3K4me1, and 5hmC around ±7 kb from the IS. (C) Hierarchical clustering of Pearson correlations between pairs of marks and/or chromatin factors at IZ. Marks and factors in the heatmap are ordered according to the clustering performed for the IS (see Fig. 2C). Positive correlations between pairs of marks/factors in individual IZ regions are symbolized by a gradation of red. Negative correlations are symbolized by a gradation of blue. (D) Percent overlap between poised (H3K4me1/H3K27me3) and active (H3K4me1/H3K27ac) enhancers within IZ. (E) Overlap between marks/factors and origins.
In addition to promoters, RNA Pol II and H3K27ac were also present at enhancers that are currently defined by H3K4me1 occupancy (Creyghton et al. 2010). In contrast to open chromatin marks, enhancer marks were specifically enriched at IZ compared with IS, as illustrated by the wide and bimodal profile of 5-hydroxymethylcytosine (5hmC) and especially of H3K4me1 around the IS (Fig. 3B,D), a characteristic profile found also at promoters and enhancers (Koch et al. 2011). More than 51% of IZ contained at least one enhancer mark, and H3K4me1 alone determined 46% of all IZ (Supplemental Table S1; Fig. 3A, lower).
Enhancers can be characterized according to the levels of colocalization of H3K4me1 with H3K27ac/H3K9ac (active enhancers) or H3K27me3 (poised enhancers) (Creyghton et al. 2010; Rada-Iglesias et al. 2011). Surprisingly, we observed a significant correlation of IZ with poised enhancers, but not with active enhancers (Z-score >4.3 versus ≤1, respectively) (Fig. 3D; Supplemental Fig. S5B,D). However, this did not necessarily involve the presence of PcG proteins. These results suggest a cooperative activity of H3K4me1 and H3K27me3 to allow a chromatin environment suitable to initiate replication in some enhancer regions. Finally, the highest correlations in IZ included G4, in agreement with our previous work (Cayrou et al. 2012a). G4 clearly were the most frequent marks at IZ (79% of IZ) (Supplemental Table S1), and 47% of the regions containing enhancer marks and G4 predicted one-third of all origins (Fig. 3E).
Altogether, these findings suggest that epigenetic signatures and DNA structure/sequences specifically associated with active regulatory regions favor the IS choice within transcriptionally active domains.
The three main origin classes are defined by specific chromatin environments
Considering the different chromatin signatures found at origins, we then asked whether the three classes of origins were associated with specific sets of chromatin signatures. Class 1 is clearly very different from the two other classes; it is particularly poor in epigenetic marks, with only two positive correlations with the silenced chromatin marks H3K9me3 and fully methylated regions (ES FMR) (Fig. 4A; Stadler et al. 2011). This is consistent with the weaker link of this class with gene-rich regions and the finding that Class 1 includes mainly late replication origins.
Figure 4.
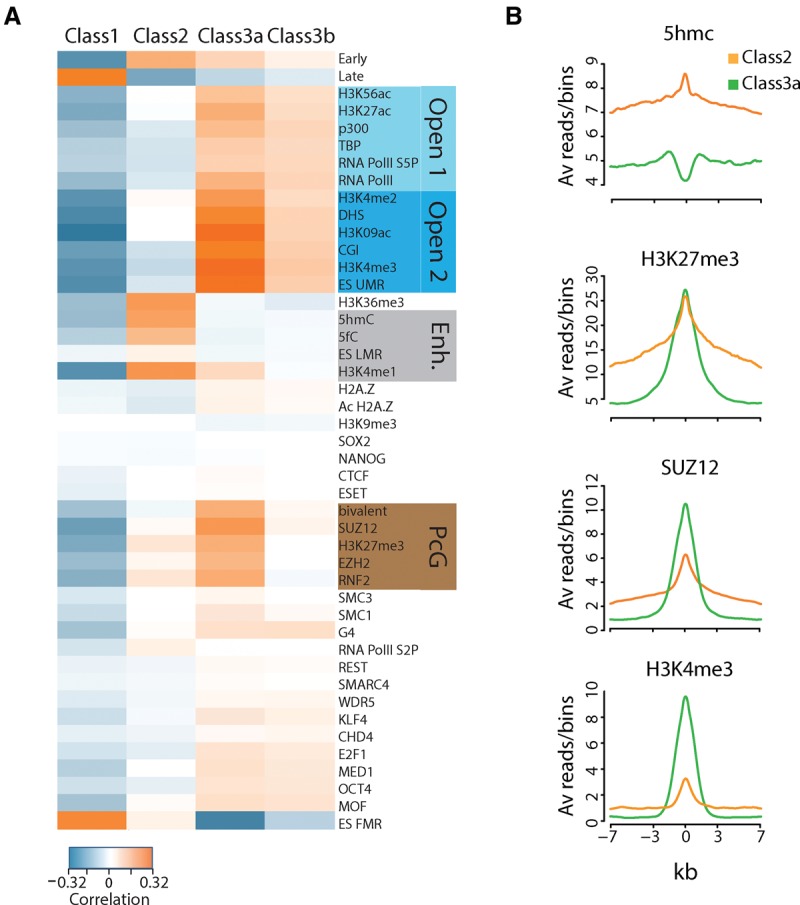
Origin classes are linked to specific chromatin signatures. (A) Association of chromatin marks/factors within each class. Pearson correlation between pairs of chromatin marks/factors for all IS in each class. Class 1 IS (n = 28,443) are correlated with closed chromatin marks and anticorrelated with all other marks/factors. Class 2 IS (n = 14,873) are the only IS positively correlated with enhancer features (Enh.), whereas Class 3a and 3b IS (n = 10,547) are associated with Open 2 and Polycomb group (PcG) chromatin marks/factors. (B) ChIP-seq signals for 5hmC, H3K27me3, SUZ12, and H3K4me3 around ±7 kb from the center of Class 2 and Class 3a IS.
Conversely, the other two classes are strongly associated with specific sets of chromatin marks. Class 2 exhibited weak correlations with open chromatin marks, but is the only class positively correlated with enhancer marks. Its enrichment in H3K4me1 (60% of IS) and 5hmC (45% of IS) (Supplemental Table S1) is also correlated with H3K36me3, suggesting a selection for enhancers inside active genes. We also found a broad and strong H3K27me3 enrichment in this class (Fig. 4B), in agreement with the detected connection with poised enhancers.
Class 3a origins have the strongest associations with open chromatin-2 marks, such as DHS, H3K9ac, and H3K4me3 (79%, 75%, and 66% of IS, respectively), and with open chromatin-1 marks/factors (Fig. 5A). Class 3a also includes many PcG-positive origins (37% of Class 3a IS were associated with SUZ12 binding sites), particularly those correlated with bivalent domains (correlation of 0.18 for Class 3a versus −0.02 for Class 2), in agreement with the H3K27me3 and SUZ12 enrichment at the peak center (Fig. 5B). Class 3b origins have a similar epigenetic profile, but less pronounced than in Class 3a origins.
Figure 5.

Sequence motifs at origins. (A,B) Distribution of OGRE sites around IS in mouse (A) and human (B) ES cells. PWM-based scanning of 1-kb regions on both sides of origin summits to detect OGRE motif instances. The x-axis indicates the position relative to the origin summit, the y-axis the number of predicted OGRE sites per 50-bp window, for the 65,019 mouse origins (A), and 149,791 human origins (B), respectively. (C) Clustering of oligonucleotide occurrence profiles. The left panel shows the hierarchical clustering of positional profiles of 8-mer occurrences in 50-bp non-overlapping windows over 1 kb on each side of peak summits. Each row corresponds to one specific k-mer. The color scale indicates the per k-mer normalized frequency. Local over- or underrepresentation is denoted by red or green hues, respectively. The right panels show detailed examples of individual position profiles for two clusters of k-mers. Colored bold lines represent the median profile of normalized frequencies for the entire cluster. The inset sequence logos were built by scanning sequences with matrices resulting from k-mer assemblies. (D) Profiles of origin-associated motifs. Distribution profiles of origin-associated motifs, obtained by scanning origin regions with the matrices identified in the previous step (C). The y-axis indicates the number of motif occurrences within each bin (300 bp). We combined all profiles in four different groups (G-rich, C-rich, GT/AC, and T/A). Sequence logos of the origin-associated motifs are displayed beside the corresponding profiles. (E) Proportion of origins with the representative motifs. Each origin containing at least one occurrence of the motif representative of each group is reported. The occurrence must be within the most enriched part within the origin (e.g., between +150 and +450 bp from the IS for cluster 2). (F) Proportion of origins positive for a specific motif per class. Origins containing at least one of the representative 8-mers are assigned according to their class. G/C-rich motif-positive origins are in Class 1, 2, and 3a/b (left), whereas origins enriched in AC or TG repeats are mainly in Class 1 (right).
These data highlight that the different origin classes, first defined in this study based on their structural patterns (Fig. 1A), are also characterized by specific chromatin features.
The G-rich motif is similarly distributed at origins of mouse and human ES cells
Genetic signatures at origins have remained elusive in multicellular organisms. We previously identified an Origin G-rich Repeated Element (OGRE) that is overrepresented upstream of the IS in mouse Chromosome 11 origins (Cayrou et al. 2011). We thus asked whether this motif was similarly distributed in this higher-resolution map of mESC origins. To account for the motif strand orientation, we scanned separately the Watson and Crick DNA strands and obtained symmetrical and consistent results. Analysis of the mESC sequencing data highlighted two close peaks of OGRE enrichment, located ∼225 and 75 bp upstream of the IS (Fig. 5A). Then, to determine whether OGRE occur also at IS of human ESCs (hESCs), we reanalyzed the raw data from Besnard et al. (2012) and for consistency applied the bioinformatics workflow used for mESC origin mapping. Because Besnard et al. (2012) did not include an experimental negative control, we sequenced fragmented genomic DNA from the same hESC line and subtracted the obtained values from the Besnard et al. (2012) data. The original article reported 250,000 origins in hESCs, but with our more stringent procedure, we found 149,791 potential IS that were contained in 30,056 IZ. This is similar to the result of a recent independent reanalysis of these data (Picard et al. 2014). We detected an OGRE motif upstream of human origins, and its occurrence distribution abruptly dropped at the precise location of the IS (Fig. 5B). The G-rich zone was wider in human (∼1 kb) than in mouse samples (<500 bp), consistent with the broader (6 kb in human versus 2.3 kb on average for mouse origins) and multi-IS composition (50% of origins have three or more IS) of hESC origins. We conclude that hESC origins also contain an oriented OGRE motif, like mESC origins.
Initiation sites are characterized by specific oligonucleotide profiles
Our previous analyses (Cayrou et al. 2011) relied on a generic motif discovery tool (MEME) (Bailey et al. 2015), based on the detection of overrepresented motifs. We extended this analysis by characterizing the positional distribution profile of each oligonucleotide (“k-mer”) around the IS. We used the RSAT position-analysis program (van Helden et al. 2000) to count the occurrences of all k-mers (from 1 to 8 b) in non-overlapping windows (50 bp) from −1 to +1 kb relative to the IS summits, according to the Watson and Crick strand. Observed occurrences were compared with the random expectation using a χ2 test. Significant k-mers were clustered according to their position profiles (Fig. 5C). k-mer clusters were further assembled and used as seeds to extract motifs (sequence logos) (Fig. 5D). The following analysis focused on 8-mer oligonucleotides, but the analysis of smaller k-mers gave comparable results (see supporting website).
The density heatmap (Fig. 5C) revealed groups of 8-mer oligonucleotides with highly significant positional biases around the IS. The first group was composed of G-rich 8-mer sequences (Supplemental Table S2) that showed a strong enrichment 225 bp on the left side of the summits on the Watson strand. This group could be further subdivided in two clusters (1 and 2 in Fig. 5C) composed of related, but distinct oligonucleotides. The positional profiles of these G-rich oligonucleotides (Fig. 5C, upper right) were in line with those described for the OGRE motif. As Watson and Crick strands were considered separately, the left-side enrichment in G repeats and the right-side enrichment in C repeats showed two symmetrical views of the same phenomenon: the enrichment for G-rich sequences on the 5′ side of the IS. We also detected highly significant oligonucleotide motifs centered on the IS, all related to the poorly complex 8-mer ACACACAC sequence (Fig. 5D, cluster 3) or its reverse complement TGTGTGTG (cluster 4). Finally, the analysis of positional biases also revealed a strong avoidance of poly-T and poly-A oligonucleotides at IS (clusters 7 and 8, respectively).
Based on the heatmap, we identified two well-separated regions around the IS. The region between 450 and 150 bp from the IS was enriched in G-rich repeats on the left of the IS (Fig. 5C,D, clusters 1 and 2) and in C-rich repeats on the right of the IS (clusters 5 and 6). Conversely, the central region (between −150 and +150 bp) contained more TG and AC repeats (clusters 3 and 4). We then counted the occurrences of each oligonucleotide in 300-bp-wide slices corresponding to these regions and computed the sum of occurrences per slice for each oligonucleotide cluster. We analyzed the combinations of these signals in individual peaks and found that 71% of origins displayed one or more TG and/or AC occurrence profiles and that 80% of them featured G- or C-rich oligonucleotides. Among these oligonucleotide-positive origins, a large part (66%) contained only one of the four oligonucleotide groups. These origins had either G-rich occurrences upstream or C-rich occurrences downstream, and only 12% had both (Fig. 5E). This subdivision was also true for the TG and AC repeats because only 4% of positive origins had a combined profile. The presence of G- or C-rich occurrences upstream of the IS did not exclude the occurrence of TG and AC repeats at the IS. However, only 25% of positive origins had both (P-value <0.0001), suggesting a specialization of G/C-rich oligonucleotides and of AC/TG repeats in the determination of different origin types.
We then asked whether specific sequence profiles were associated with the different classes of origins (Fig. 5F). G- or C-rich occurrences were not restricted to a particular class. Conversely, AC and TG repeats were clearly overrepresented in Class 1 origins. This result suggests that Class 1 origins, which are poor in epigenetic marks, are more associated with specific sequences than the other classes. In contrast, Class 3a origins, which are rich in chromatin marks, showed the lowest abundance of each oligonucleotide group (Fig. 5F). Consistently, the number of occurrences per origin decreased rapidly with the gain in chromatin marks (Supplemental Fig. S7). This observation was true for all chromatin marks associated with origins described in Figure 3. The overrepresentation of AC and TG repeats in Class 1 origins could contribute to specify replication initiation inside genomic regions devoid of chromatin marks.
A labile nucleosome is positioned at replication initiation sites
To assess nucleosome density at IS, we used two different data sets obtained by micrococcal nuclease digestion followed by sequencing (MNase-seq): MNase 1 (Fenouil et al. 2012) and MNase 2 (Fig. 6A, solid and dotted black lines, respectively; Teif et al. 2012). We observed a strong NDR on each side of the IS in both data sets, confirmed by the histone H3 position precisely overlapping the IS center (Fig. 6A, red line). Conversely, a nucleosome density peak was localized precisely at the IS, and its intensity varied according to the data set. This suggests that a nucleosome is positioned at the replication start and is surrounded by a NDR of ∼200 bp. This is in agreement with results based on the computational prediction of nucleosome occupancy at origins (Cayrou et al. 2012a) and with the MNase-seq results at six mammalian origins (Lombraña et al. 2013). The positioned nucleosome on the IS was present in all classes of origins (Supplemental Fig. S8B). Class 3a origins presented the stronger NDR profile, perfectly in line with the nucleosome mapping data in the highly efficient S. pombe origins (Lantermann et al. 2010). Because analysis of the raw data indicated that the MNase 2 set was more digested than the MNase 1 set (Supplemental Fig. S8A), we hypothesized that the nucleosome positioned at the IS is labile. Indeed, when looking at individual origins, we noticed that the nucleosome density in the two sets differed, particularly when the origin size and the number of IS increased (Supplemental Fig. S8B). This finding confirms the relationship between origin strength and chromatin structure.
Figure 6.

A labile nucleosome is present at the replication initiation site. (A) Nucleosome occupancy at origins. ChIP-seq signals for two different MNase digestion sets at ±1.5 kb around the IS (solid and dashed black lines). The global profile of origins centered on IS is in green. The two MNase profiles are matching, except on the IS, indicating a labile nucleosome. The histone H3 ChIP-seq signal is in red. (B) Distribution of histone H3 variants at origins. ChIP-seq signals for H3.1 (large dashed black line), H3.2 (medium dashed black line), H3.3 (fine dashed black line), and H2AZ (red line) at ±1.5 kb of IS (global profile in green). (C) Distribution of H3K64ac around IS. ChIP-chip signals for H3K64ac (50% top probes distribution) at ±1.5 kb of IS (global profile in green). (D,E) Nucleosome distribution around IS according to the G-rich sequence asymmetry. (D) A group of origins with G-rich asymmetry with initiation sites that show a highly significant enrichment (P-value 1 × 10−2) in either G-rich k-mers on the left side of the initiation sites (MNase-strand+, blue line, n = 9518) or in C-rich k-mers on the right side (MNase-strand−, red line, n = 9682). We thus oriented the right-side C-rich plot to systematically represent the features in the 5′ to 3′ direction. Note the striking consistency between MNase profiles obtained from left-side G-rich and right-side C-rich occurrences (MNase-merge, black line). The orange curve indicates the profile of G-rich occurrences around the IS considering the DNA strand on origins oriented according to the asymmetry of G-rich k-mer occurrences. (E) A group of nonasymmetric origins (n = 45,320) was defined by initiation sites with no significant enrichment for either of these two signals. Note the acute peak of MNase at the precise position of the IS and the absence of upstream depletion, which contrasts with G-rich asymmetric origins. (F) Schematic representation of the nucleosome distribution around IS. (G) Genome browser representations of Nascent Strands, MNase1, and MNase2 enrichment profiles are shown for a representative G-rich asymmetric origin.
Moreover, we detected one oriented NDR relative to the positioned nucleosome (Supplemental Fig. S8C, red dot), suggesting a specific orientation of this NDR relative to individual origins. Several recent studies have shown that the nucleotide sequence could play a role in the nucleosome positioning. Particularly in human, some GC-rich regions seem to be nucleosome-depleted, differently from yeast, where it is the AT-rich regions that are most often associated with a NDR. Therefore, we analyzed the nucleosome distribution around the IS relative to the strand of the G-rich occurrences found in Figure 5. We observed a strict NDR only on the origin part that is enriched in G-rich occurrences, upstream of the IS. (Fig. 6D). The same result was obtained with the H3 profile around the IS, considering the DNA strand on which the G-rich occurrences are located (Supplemental Fig. S9). In contrast, we did not detect any NDR on origins without significant enrichment in G-rich k-mers on their 5′ side (Fig. 6E). This analysis also confirmed the presence of a nucleosome at the IS, both present in G-rich asymmetric regions or G-rich nonasymmetric regions (Fig. 6F,G).
In transcriptionally active chromatin, unstable nucleosomes contain both H2AZ and H3.3 variants (Jin and Felsenfeld 2007). Therefore, to further investigate the nucleosome lability at IS, we analyzed ChIP-seq data on histone variants (Jia et al. 2012). However, the presence of histone variants at IS did not convincingly explain the observed nucleosome lability, as H3.1 was the main H3 variant at IS. Moreover, we only observed a small H3.3 peak and, as expected, no H3.2 (Fig. 2B). H2AZ was broadly enriched at replication origins (Fig. 6B, red line), but 10% of the H2AZ sites only covered 8.6% of IS (Supplemental Table S1). We then investigated whether specific epigenetic marks could explain nucleosome lability. A recent work suggested a role of the previously uncharacterized H3K64ac mark in nucleosome stability and dynamics (Di Cerbo et al. 2014). We observed that 86% of origins covered by the H3K64ac ChIP assay were associated with at least one positive probe (Supplemental Fig. S8D). Moreover, H3K64ac (top 50% stronger probes) (Supplemental Fig. S8E) displayed a high and sharp profile centered on the labile nucleosome at IS and also a broad profile around the IS (Fig. 6C) in all origin classes (Supplemental Fig. S8F). These profiles were detected inside genes as well as in intergenic regions (Supplemental Fig. S8G).
In summary, each IS contains a positioned labile nucleosome flanked by a NDR 200 bp upstream of the IS. This nucleosome is particularly enriched in H3K64ac, suggesting that this mark has a potential new role in initiation of DNA replication.
Discussion
Altogether our data bring new insights into the nature and flexibility of metazoan origins. Origins appear to be organized in a bipartite fashion, with a nucleosome-free, G-rich region upstream of a nucleosome positioned at the IS. They can be classified in three classes with different structural organization, nucleotide composition, epigenetic properties, and genomic distribution.
Coexistence of three different origin classes
The structural organization of origins within each of the three classes differs from that of neighboring origins. This organization is remarkably paralleled by specific features within each class. We emphasize that these three classes could be further subdivided without altering the main conclusions reached in this study.
Class 1 is the main origin class (44% of all IS). They are located in gene-poor regions and contain limited epigenetic signatures, consistent with being mainly late replicated origins. Nevertheless, they are enriched in strand-asymmetric sequences, such as ACACACAC or TGTGTGTG repeats, at the IS. Conversely, the two other classes include early origins located in gene-rich regions with specific chromatin marks. This is reminiscent of a recent analysis that classified S. cerevisiae origins in two main groups (“DNA-dependent” and “chromatin-dependent”) based on their in vivo and in vitro DNA-ORC affinity (Hoggard et al. 2013). Accordingly, Class 1 origins, poor in chromatin marks and 77% of which are associated with at least one preferential sequence, might represent “sequence-dependent” origins, whereas the other two classes, associated with specific chromatin features, might define “chromatin-dependent” origins. Class 1 “sequence-dependent” origins, like the “DNA-dependent” origins in yeast, account for 75% of late origins, in contrast to “chromatin- dependent” origins that are mainly early firing origins. This observation suggests the preservation in eukaryotes of a sequence-dependent mechanism for origin selection and positioning in late-replicated regions, possibly to compensate the deficit in chromatin marks.
G-rich or G4 signatures at replication origins and nucleosome organization
Most origins contain a G-rich or the complementary C-rich octamer. The distribution of these motifs, relative to the IS, is comparable to that of the OGRE motif (Cayrou et al. 2011). Moreover, we also found the G-rich motif in hESCs. The G-rich sequence is not at the IS, but 250 bp upstream of it, reflecting the fact that this motif is oriented relative to the IS. We previously proposed that these G-rich elements could form G4 (Cayrou et al. 2012a). It was subsequently suggested that G4 occur in human origins as well (Besnard et al. 2012), and they influence replication initiation in chicken cells (Valton et al. 2014). We confirm here that 78% of mESC origins contain at least one potential G4 structure on the leading strand template.
How can these OGRE/G4 elements favor replication initiation? Their position strongly suggests a role in origin recognition at the pre-RC site rather than in origin opening at the IS. In support of this hypothesis, G-rich elements were identified as a binding site for ORC in mouse rDNA (Zellner et al. 2007), and a recent biochemical analysis showed that purified ORC could bind to G4 RNA or DNA (Hoshina et al. 2013). In the Drosophila genome, ORC binding sites contain a central GC-rich region surrounded by AT-rich sequences (Vorobyeva et al. 2013). G-rich motifs/G4 may help in destabilizing the proximal double-stranded DNA structure (König et al. 2013), facilitating the MCM2-7 helicase access to the leading single-stranded DNA template.
In addition, G-rich regions, particularly G4, correlate with nucleosome depletion in many eukaryotes (Rhodes 1979; Iyer and Struhl 1995; Huppert and Balasubramanian 2007; Halder et al. 2009; Wong and Huppert 2009; Fenouil et al. 2012). Mouse origins contain a NDR, like other eukaryote origins (Mavrich et al. 2008; Berbenetz et al. 2010; Eaton et al. 2010; MacAlpine et al. 2010; Lubelsky et al. 2011; Givens et al. 2012), and here we show that this region overlaps with the G-rich/G4 element upstream of the IS and not with the IS itself. This is in agreement with the hypothesis that this G-rich nucleosome-free region may be the pre-RC assembly site. Interestingly, Saccharomyces japonicus are characterized by high GC content, while AT content is a negative predictor of origin function (Xu et al. 2012), similar to our observation in mouse ES cells.
As G4 can inhibit replication fork progression (Sarkies et al. 2010; Paeschke et al. 2011), it could be asked how a replication inhibitory element might also stimulate replication. DNA replication initiation occurs in two temporal steps. Pre-RCs are formed in G1, and only 20%–30% of these pre-RCs are activated in each cell during S phase. In their folded form, G4 could negatively regulate initiation of DNA replication at pre-RCs; however, when unfolded, they could trigger DNA replication at the subpopulation of pre-RCs that are activated during S phase. G4 formation may be favored by formation of an R-loop on the C strand (Fragkos et al. 2015), and G4/OGRE elements could function as a structural switch: folded or unfolded. Alternatively, replication origins could be activated at pre-RCs and then transiently stall at the proximal G4 on the leading strand, a phenomenon similar to transcriptional pausing (Liu et al. 2015). This mechanism could be used to start initiation on the opposite strand, similarly to the model proposed for the DBF4 replication origin (Romero and Lee 2008). Alternatively, it might control the timing of origin activation, as Rif1 has recently been found to bind G4 (Kanoh et al. 2015).
An AC/TG asymmetric repeated motif and a labile-positioned nucleosome at the initiation site
We also identified an AC (or complementary TG) repeat precisely at the IS center in 40% of origins, particularly in late-replicated origins (Class 1). CA/GT repeats are strongly enriched at nucleosome-positioning sequences in the mouse (Widlund et al. 1997), and they overlap with the nucleosome positioned at IS in our study. This is reminiscent of the nucleosome positioned downstream from the ORC binding site described in budding yeast origins (Eaton et al. 2010) and in a panel of six mouse origins (Lombraña et al. 2013). Moreover, TG-rich sequences can form Z-DNA structures (Wahls et al. 1990, 1991; Majewski and Ott 2000) that have been detected in positioned nucleosomes, where they facilitate their remodeling (Liu et al. 2006; Maruyama et al. 2013). Remarkably, MCM helicase activity is weak on a nucleosome template, and the chromatin remodeling complex FACT is necessary to promote DNA unwinding (Tan et al. 2006). In agreement, we found a strong enrichment of H3K64ac strictly centered on the nucleosome at the IS, a new chromatin mark associated with labile nucleosomes in mESCs (Di Cerbo et al. 2014). These results suggest a new role for this mark in positioning a labile nucleosome that prevents the accessibility to the IS outside the required time and that can be remodeled when the pre-RC is activated.
Plasticity and synergy of origin selection by specialized epigenetic signatures
Two of the three classes of origins are correlated with a specific chromatin environment, consistent with the identification of “chromatin-dependent” origins in S. cerevisiae (Hoggard et al. 2013). Each class is characterized by a specific origin structure and relationship with the surrounding chromatin. Class 2 origins correspond to large initiation zones with many inefficient initiation sites and are overrepresented in enhancer regions (identified by H3K4me1), possibly offering more flexibility to initiate DNA replication. Interestingly, poised enhancers are frequently associated with origins (75%), whereas active enhancers are poorly represented (34%). Indeed, association of H3K4me1 with H3K27 methylation, deposited on chromatin by EZH2 (PRC2 catalytic subunit) (Kuzmichev et al. 2002), may provide a chromatin environment suitable for replication initiation. Moreover, the low SUZ12 abundance in Class 2 origins (Fig. 5B) suggests that H3K27me3 alone ensures flexible initiation rather than strict initiation in poised enhancers.
Conversely, strong SUZ12 enrichment was found in the highly efficient Class 3a origins that replicate early in S phase. This SUZ12 enrichment centered on the IS (Fig. 3B) suggests that PcG proteins are more linked to origin activation than to pre-RC establishment. Moreover, loss of SUZ12 (Pasini et al. 2004) or of EZH2 impairs entry in S phase, and the number of origins is increased in EZH2-deficient cells (Piunti et al. 2014). The explanation given was that DNA replication fork stalling promotes the activation of dormant origins. However, the distribution profile of inter-origin distances in Ezh2−/− cells is strikingly similar to the profile predicted for “Random origin firing” in mESCs (Cayrou et al. 2011), in which all potential origins are randomly activated, leading to an increase in the number of very small, but also very large inter-origin distances. The partial effect on inter–origin distances observed by Piunti et al. (2014) is consistent with the small number of PcG sites in the genome, and thereby with the number of origins affected by EZH2 deficiency. These observations suggest that PcG proteins structurally restrict the position of origin activation inside Polycomb domains.
Our analysis also shows that open chromatin marks, such as H3K4me3, are major features of origins in mESCs, notably of Class 3a origins, the most representative “chromatin-dependent” origins. The low rate of origin localization near transcription start sites (31% of Class 3a origins) cannot explain the overrepresentation of this mark at origins (66%). On the other hand, the high level of histone acetylation (75% for the H3K9ac level) in these origins may favor their accessibility (Görisch et al. 2005). The high efficiency of Class 3a origins and the absence of any other strong origin in the vicinity imply a constraint to initiate replication at specific locations in these regulatory regions.
Class 3a origins also show high CGI content that can create bivalent domains in mESCs (Wachter et al. 2014). These domains (80.6% of bivalent domains are in mESC origins) are composed of an activating (H3K4me3) and a repressive mark (H3K27me3) (Bernstein et al. 2006) and contain promoters of developmentally important genes that are kept poised for activation until lineage specification. Consistently, we found poised RNA Pol II at mESC origins, suggesting that the regulation of transcription and origin firing are linked. For instance, G/C-rich sequences and open/bivalent chromatin marks could act as a platform to recruit chromatin remodeling or modification factors, both for the replication origin program and the transcription program.
Genetic and epigenetic guidance of replication initiation sites
Our analysis shows that combinations of specific genomic signatures regulate the localization of IS in complex eukaryotes. Importantly, specificity appears to be achieved by the synergic action of sequence motifs and chromatin modifications to allow the flexible selection of replication origins in different genomic contexts. In gene-poor regions that are replicated late in the cell cycle, origin selection seems to be more dictated by sequence-specific features. Conversely, epigenetic signatures appear more involved in origin selection in early replicating gene-rich regions, in agreement with their open chromatin configuration.
Our study also shows that IS are characterized by specific signatures, such as a well-positioned nucleosome with specific sequences and epigenetic marks. In contrast, the upstream sequence contains a G-rich element inside a nucleosome-depleted region, which is likely to be the pre-RC site.
The existence of several classes of origins, with distinct sets of features to control origin positioning, appears to be an important mechanism to regulate origin usage. This flexibility in elements that can drive origin positioning could be used by eukaryotic cells to adapt to the environment, to the constraints linked to the complex structure and variety of conformations of chromatin and chromosomes, and finally to different tissue-specific transcription programs.
Methods
Cell culture
CGR8 mESCs were cultured on gelatin-coated dishes (feeder-free, to avoid DNA contamination by MEF cells) in GMEM medium supplemented with 10% of FBS and 1000 units/mL of LIF.
Nascent strand isolation
Nascent strands (NS) were purified as previously described in detail (Cayrou et al. 2012b). Briefly, after extraction with DNAzol, NS were first separated from genomic DNA by sucrose gradient. Several fractions of interest containing NS DNA (0.5–2 kb in size) were phosphorylated by T4 polynucleotide kinase and digested twice with λ-Exo to eliminate contaminating DNA. The first round of digestion was with 100–150 units λ−Exo (i.e., 300–500 units/μg DNA) overnight at 37°C. The resulting DNA was precipitated with ethanol, phosphorylated again, and digested again in the same conditions. A third λ−Exo digestion (100 units) can also be performed for 2 h at 37°C. Three samples were purified from three independent cell cultures. The RNase A controls were obtained by treating NS-containing fractions with 50–100 μg/mL RNase A before λ−Exo digestion to remove RNA primers at the 5′ end of the NS. As a control, we also used genomic DNA sonicated to the same size as the NS DNA and treated by λ−Exo as described for the NS samples. This protocol differs in several ways from a recently described method (Foulk et al. 2015) in which short NS were not purified by sucrose gradient before λ−Exo digestion. Instead, a large amount of whole replicating DNA was used. Moreover, as a negative control in the above study, a large amount of total fragmented DNA (150 μg) was digested with λ−Exo in strong limiting conditions (0.7 units of λ-Exo/μg of DNA). This is 500- to 1000-fold less than in our previous works (Supplemental Fig. S2; Cayrou et al. 2012b).
Sequencing and read mapping
Single-stranded NS DNA was then converted into double-stranded DNA by random priming using DNA polymerase I (Klenow fragment). Heat-denatured sheared genomic DNA and RNase A-digested samples were also random primed before library preparation using the Illumina ChIP-seq DNA Sample Prep Kit, according to the manufacturer's instructions. Samples were sequenced using the Illumina HiSeq 2000 at the MGX GenomiX facility (Montpellier). For each sample, sequenced reads were mapped as in Supplemental Material.
Peak calling
After a thorough evaluation (Supplemental Material), we combined two peak calling programs with complementary properties (Fig. 1B). SWEMBL (https://www.ebi.ac.uk/~swilder/SWEMBL/) identifies well-delimited peaks, whereas SICER (Zang et al. 2009) returns wider regions enriched in reads. All SICER regions that overlapped with at least one SWEMBL peak were retained and defined as IZ. Reciprocally, all SWEMBL peaks falling within a SICER region were identified as IS.
Random peak selection
As a negative control for the subsequent analyses (peak clustering, coverage of chromatin marks, motif discovery), the RSAT random-genome-fragments program was used to select random genomic regions of the same number and sizes as the origin peaks.
Read density maps
Read occurrence profiles were obtained by extracting 7 kb on each side of the summit of each peak (10-kb data provided in Supplemental Material) and counting the reads per 100-bp bins. Data processing was done in R (R Core Team 2014) by applying k-means clustering on the read occurrence profiles with various numbers of clusters. For visual purposes, the resulting clusters were sorted according to their average density profile to highlight the relationships between the central peak and its first closest neighbor. On the density map, pairs of neighbor peaks appear twice: once centered on the leftmost peak, and once on the rightmost peak. Consistently, our clustering procedure revealed symmetrical clusters. We labeled clusters as “L” or “R,” respectively, to denote the left-side or right-side location of the secondary peak. For further analysis, we regrouped the symmetrical pairs of clusters (e.g., cluster 10L and 10R form cluster 10).
We developed a custom R library (ocpR; available in Supplemental Materials and http://tagc.univ-mrs.fr/origins/) to automate data processing and test the impact of different parameters (k, summit flanking size) that allow data clustering, ordering, and visualization. Based on the visual inspection of the resulting heatmaps, we retained k = 19 clusters. Clustering results with other k values are in Supplemental Material.
To assess the clusters’ relevance, we performed two negative controls: (1) k-means clustering on randomized bins (random permutation of read count values inside each row independently); and (2) random selection of genome fragments using the RSAT program random-genome-fragments.
Overlap with genomic features and data from other sources
Genomic annotations were downloaded from the UCSC Table Browser tool (Mouse genome version NCBI37/mm9 NCBI37). Overlapping of origins with the genomic annotations was done as in the Supplemental Material. Data on histone marks and transcription factors in mESCs were downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/). The accession numbers are detailed in Supplemental Table S1.
Heatmap, correlations, and clustering
Comparison between ChIP-seq data sets (histone marks and TFBS BED files) and peaks (IS) or zones (IZ) was computed using BEDTools annotateBed (Quinlan and Hall 2010) that allows the annotation of multiple BED files (histone marks, ChIP-seq marks) against a reference (IS and IZ) and returns a matrix of overlapping marks. A Pearson's correlation matrix was computed in R, from which a correlation distance (d = 1 − r) was derived. Mark clustering and heatmap visualization were computed with the R function gplots: heatmap.2. Pairwise comparisons of genomic coordinates were computed with BEDTools intersectBed.
Ab initio motif discovery
The RSAT program position-analysis (van Helden et al. 2000) was used to detect oligonucleotides (k-mers) with positional biases around peak summits. The occurrence of all k-mers from 1 to 8 nt were counted in 50-bp-wide non-overlapping windows from −1 to +1 kb relative to origin summits. The difference between observed and expected distribution (window-specific Bernoulli model) was measured with a χ2 test, and the top 100 ranking significant k-mers were clustered based on their positional profiles of occurrences. Clusters of co-occurring k-mers were used to define position-specific signals, which were used to orientate each peak for the polarity-oriented analysis of MNase occupancy.
Data access
The high-throughput sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE68347. A supporting website with the details of the analysis is available at http://tagc.univ-mrs.fr/origins/.
Acknowledgments
The research leading to these results received funding from the European Research Council (FP7/2007-2013 Grant Agreement no. 233339). This work was also supported by the Fondation pour la Recherche Médicale (FRM) and the European Commission Network of Excellence EpiGeneSys (HEALTH-F4-2010-257082).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.192799.115.
References
- Abdurashidova G, Deganuto M, Klima R, Riva S, Biamonti G, Giacca M, Falaschi A. 2000. Start sites of bidirectional DNA synthesis at the human lamin B2 origin. Science 287: 2023–2026. [DOI] [PubMed] [Google Scholar]
- Adelman K, Lis JT. 2012. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13: 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aladjem MI. 2007. Replication in context: dynamic regulation of DNA replication patterns in metazoans. Nat Rev Genet 8: 588–600. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Johnson J, Grant CE, Noble WS. 2015. The MEME Suite. Nucleic Acids Res 43: W39–W49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbenetz NM, Nislow C, Brown GW. 2010. Diversity of eukaryotic DNA replication origins revealed by genome-wide analysis of chromatin structure. PLoS Genet 6: e1001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326. [DOI] [PubMed] [Google Scholar]
- Besnard E, Babled A, Lapasset L, Milhavet O, Parrinello H, Dantec C, Marin JM, Lemaitre JM. 2012. Unraveling cell type–specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat Struct Mol Biol 19: 837–844. [DOI] [PubMed] [Google Scholar]
- Cayrou C, Coulombe P, Vigneron A, Stanojcic S, Ganier O, Peiffer I, Rivals E, Puy A, Laurent-Chabalier S, Desprat R, et al. 2011. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res 21: 1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Coulombe P, Puy A, Rialle S, Kaplan N, Segal E, Méchali M. 2012a. New insights into replication origin characteristics in metazoans. Cell Cycle 11: 658–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Grégoire D, Coulombe P, Danis E, Méchali M. 2012b. Genome-scale identification of active DNA replication origins. Methods 57: 158–164. [DOI] [PubMed] [Google Scholar]
- Costas C, de la Paz Sanchez M, Stroud H, Yu Y, Oliveros JC, Feng S, Benguria A, López-Vidriero I, Zhang X, Solano R, et al. 2011. Genome-wide mapping of Arabidopsis thaliana origins of DNA replication and their associated epigenetic marks. Nat Struct Mol Biol 18: 395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado S, Gómez M, Bird A, Antequera F. 1998. Initiation of DNA replication at CpG islands in mammalian chromosomes. EMBO J 17: 2426–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePamphilis ML. 1993. Origins of DNA replication that function in eukaryotic cells. Curr Opin Cell Biol 5: 434–441. [DOI] [PubMed] [Google Scholar]
- Di Cerbo V, Mohn F, Ryan DP, Montellier E, Kacem S, Tropberger P, Kallis E, Holzner M, Hoerner L, Feldmann A, et al. 2014. Acetylation of histone H3 at lysine 64 regulates nucleosome dynamics and facilitates transcription. eLife 3: e01632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton ML, Galani K, Kang S, Bell SP, MacAlpine DM. 2010. Conserved nucleosome positioning defines replication origins. Genes Dev 24: 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenouil R, Cauchy P, Koch F, Descostes N, Cabeza JZ, Innocenti C, Ferrier P, Spicuglia S, Gut M, Gut I, et al. 2012. CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters. Genome Res 22: 2399–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulk MS, Urban JM, Gerbi SA. 2015. Characterizing and controlling intrinsic biases of lambda exonuclease in nascent strand sequencing reveals phasing between nucleosomes and G-quadruplex motifs around a subset of human replication origins. Genome Res 25: 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkos M, Ganier O, Coulombe P, Méchali M. 2015. DNA replication origin activation in space and time. Nat Rev Mol Cell Biol 16: 360–374. [DOI] [PubMed] [Google Scholar]
- Gindin Y, Valenzuela MS, Aladjem MI, Meltzer PS, Bilke S. 2014. A chromatin structure-based model accurately predicts DNA replication timing in human cells. Mol Syst Biol 10: 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givens RM, Lai WK, Rizzo JM, Bard JE, Mieczkowski PA, Leatherwood J, Huberman JA, Buck MJ. 2012. Chromatin architectures at fission yeast transcriptional promoters and replication origins. Nucleic Acids Res 40: 7176–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görisch SM, Wachsmuth M, Tóth KF, Lichter P, Rippe K. 2005. Histone acetylation increases chromatin accessibility. J Cell Sci 118: 5825–5834. [DOI] [PubMed] [Google Scholar]
- Halder K, Halder R, Chowdhury S. 2009. Genome-wide analysis predicts DNA structural motifs as nucleosome exclusion signals. Mol Biosyst 5: 1703–1712. [DOI] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang CW, Lyou Y, Townes TM, Schübeler D, Gilbert DM. 2008. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol 6: e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggard T, Shor E, Müller CA, Nieduszynski CA, Fox CA. 2013. A link between ORC-origin binding mechanisms and origin activation time revealed in budding yeast. PLoS Genet 9: e1003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshina S, Yura K, Teranishi H, Kiyasu N, Tominaga A, Kadoma H, Nakatsuka A, Kunichika T, Obuse C, Waga S. 2013. Human origin recognition complex binds preferentially to G-quadruplex-preferable RNA and single-stranded DNA. J Biol Chem 288: 30161–30171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert JL, Balasubramanian S. 2007. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res 35: 406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer V, Struhl K. 1995. Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J 14: 2570–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Zheng X, Hu G, Cui K, Zhang J, Zhang A, Jiang H, Lu B, Yates J III, Liu C, et al. 2012. Regulation of pluripotency and self-renewal of ESCs through epigenetic-threshold modulation and mRNA pruning. Cell 151: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Felsenfeld G. 2007. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev 21: 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanoh Y, Matsumoto S, Fukatsu R, Kakusho N, Kono N, Renard-Guillet C, Masuda K, Iida K, Nagasawa K, Shirahige K, et al. 2015. Rif1 binds to G quadruplexes and suppresses replication over long distances. Nat Struct Mol Biol 10.1038/nsmb.3102. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Rein T, DePamphilis ML. 1998. Identification of primary initiation sites for DNA replication in the hamster dihydrofolate reductase gene initiation zone. Mol Cell Biol 18: 3266–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch F, Fenouil R, Gut M, Cauchy P, Albert TK, Zacarias-Cabeza J, Spicuglia S, de la Chapelle AL, Heidemann M, Hintermair C, et al. 2011. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat Struct Mol Biol 18: 956–963. [DOI] [PubMed] [Google Scholar]
- König SL, Huppert JL, Sigel RK, Evans AC. 2013. Distance-dependent duplex DNA destabilization proximal to G-quadruplex/i-motif sequences. Nucleic Acids Res 41: 7453–7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Handsaker RE, Kamitaki N, Karlić R, Ghosh S, Polak P, Eggan K, McCarroll SA. 2014. Genetic variation in human DNA replication timing. Cell 159: 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. 2002. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 16: 2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantermann AB, Straub T, Strålfors A, Yuan GC, Ekwall K, Korber P. 2010. Schizosaccharomyces pombe genome-wide nucleosome mapping reveals positioning mechanisms distinct from those of Saccharomyces cerevisiae. Nat Struct Mol Biol 17: 251–257. [DOI] [PubMed] [Google Scholar]
- Li B, Su T, Ferrari R, Li JY, Kurdistani SK. 2014. A unique epigenetic signature is associated with active DNA replication loci in human embryonic stem cells. Epigenetics 9: 257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Mulholland N, Fu H, Zhao K. 2006. Cooperative activity of BRG1 and Z-DNA formation in chromatin remodeling. Mol Cell Biol 26: 2550–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kraus WL, Bai X. 2015. Ready, pause, go: regulation of RNA polymerase II pausing and release by cellular signaling pathways. Trends Biochem Sci 40: 516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombraña R, Almeida R, Revuelta I, Madeira S, Herranz G, Saiz N, Bastolla U, Gómez M. 2013. High-resolution analysis of DNA synthesis start sites and nucleosome architecture at efficient mammalian replication origins. EMBO J 32: 2631–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubelsky Y, Sasaki T, Kuipers MA, Lucas I, Le Beau MM, Carignon S, Debatisse M, Prinz JA, Dennis JH, Gilbert DM. 2011. Pre-replication complex proteins assemble at regions of low nucleosome occupancy within the Chinese hamster dihydrofolate reductase initiation zone. Nucleic Acids Res 39: 3141–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlpine HK, Gordân R, Powell SK, Hartemink AJ, MacAlpine DM. 2010. Drosophila ORC localizes to open chromatin and marks sites of cohesin complex loading. Genome Res 20: 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski J, Ott J. 2000. GT repeats are associated with recombination on human chromosome 22. Genome Res 10: 1108–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marahrens Y, Stillman B. 1992. A yeast chromosomal origin of DNA replication defined by multiple functional elements. Science 255: 817–823. [DOI] [PubMed] [Google Scholar]
- Maruyama A, Mimura J, Harada N, Itoh K. 2013. Nrf2 activation is associated with Z-DNA formation in the human HO-1 promoter. Nucleic Acids Res 41: 5223–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, Pugh BF. 2008. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res 18: 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordman J, Orr-Weaver TL. 2012. Regulation of DNA replication during development. Development 139: 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paeschke K, Capra JA, Zakian VA. 2011. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 145: 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. 2004. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 23: 4061–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Cadoret JC, Audit B, Arneodo A, Alberti A, Battail C, Duret L, Prioleau MN. 2014. The spatiotemporal program of DNA replication is associated with specific combinations of chromatin marks in human cells. PLoS Genet 10: e1004282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Rossi A, Cerutti A, Albert M, Jammula S, Scelfo A, Cedrone L, Fragola G, Olsson L, Koseki H, et al. 2014. Polycomb proteins control proliferation and transformation independently of cell cycle checkpoints by regulating DNA replication. Nat Commun 5: 3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prioleau MN. 2009. CpG islands: starting blocks for replication and transcription. PLoS Genet 5: e1000454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470: 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Gilbert DM. 2013. DNA replication timing. Cold Spring Harb Perspect Med 3: 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes D. 1979. Nucleosome cores reconstituted from poly (dA-dT) and the octamer of histones. Nucleic Acids Res 6: 1805–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero J, Lee H. 2008. Asymmetric bidirectional replication at the human DBF4 origin. Nat Struct Mol Biol 15: 722–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkies P, Reams C, Simpson LJ, Sale JE. 2010. Epigenetic instability due to defective replication of structured DNA. Mol Cell 40: 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. 2011. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480: 490–495. [DOI] [PubMed] [Google Scholar]
- Tan BC, Chien CT, Hirose S, Lee SC. 2006. Functional cooperation between FACT and MCM helicase facilitates initiation of chromatin DNA replication. EMBO J 25: 3975–3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teif VB, Vainshtein Y, Caudron-Herger M, Mallm JP, Marth C, Höfer T, Rippe K. 2012. Genome-wide nucleosome positioning during embryonic stem cell development. Nat Struct Mol Biol 19: 1185–1192. [DOI] [PubMed] [Google Scholar]
- Valton AL, Hassan-Zadeh V, Lema I, Boggetto N, Alberti P, Saintomé C, Riou JF, Prioleau MN. 2014. G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J 33: 732–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helden J, del Olmo M, Pérez-Ortín JE. 2000. Statistical analysis of yeast genomic downstream sequences reveals putative polyadenylation signals. Nucleic Acids Res 28: 1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorobyeva NE, Mazina MU, Golovnin AK, Kopytova DV, Gurskiy DY, Nabirochkina EN, Georgieva SG, Georgiev PG, Krasnov AN. 2013. Insulator protein Su(Hw) recruits SAGA and Brahma complexes and constitutes part of Origin Recognition Complex-binding sites in the Drosophila genome. Nucleic Acids Res 41: 5717–5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachter E, Quante T, Merusi C, Arczewska A, Stewart F, Webb S, Bird A. 2014. Synthetic CpG islands reveal DNA sequence determinants of chromatin structure. eLife 3: e03397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahls WP, Wallace LJ, Moore PD. 1990. The Z-DNA motif d(TG)30 promotes reception of information during gene conversion events while stimulating homologous recombination in human cells in culture. Mol Cell Biol 10: 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahls WP, Swenson G, Moore PD. 1991. Two hypervariable minisatellite DNA binding proteins. Nucleic Acids Res 19: 3269–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Dijkwel PA, Hamlin JL. 1998. Lagging-strand, early-labelling, and two-dimensional gel assays suggest multiple potential initiation sites in the Chinese hamster dihydrofolate reductase origin. Mol Cell Biol 18: 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlund HR, Cao H, Simonsson S, Magnusson E, Simonsson T, Nielsen PE, Kahn JD, Crothers DM, Kubista M. 1997. Identification and characterization of genomic nucleosome-positioning sequences. J Mol Biol 267: 807–817. [DOI] [PubMed] [Google Scholar]
- Wong HM, Huppert JL. 2009. Stable G-quadruplexes are found outside nucleosome-bound regions. Mol Biosyst 5: 1713–1719. [DOI] [PubMed] [Google Scholar]
- Xu J, Yanagisawa Y, Tsankov AM, Hart C, Aoki K, Kommajosyula N, Steinmann KE, Bochicchio J, Russ C, Regev A, et al. 2012. Genome-wide identification and characterization of replication origins by deep sequencing. Genome Biol 13: R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W. 2009. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25: 1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellner E, Herrmann T, Schulz C, Grummt F. 2007. Site-specific interaction of the murine pre-replicative complex with origin DNA: assembly and disassembly during cell cycle transit and differentiation. Nucleic Acids Res 35: 6701–6713. [DOI] [PMC free article] [PubMed] [Google Scholar]