

Abstract
Replication timing is a crucial aspect of genome regulation that is strongly correlated with chromatin structure, gene expression, DNA repair, and genome evolution. Replication timing is determined by the timing of replication origin firing, which involves activation of MCM helicase complexes loaded at replication origins. Nonetheless, how the timing of such origin firing is regulated remains mysterious. Here, we show that the number of MCMs loaded at origins regulates replication timing. We show for the first time in vivo that multiple MCMs are loaded at origins. Because early origins have more MCMs loaded, they are, on average, more likely to fire early in S phase. Our results provide a mechanistic explanation for the observed heterogeneity in origin firing and help to explain how defined replication timing profiles emerge from stochastic origin firing. These results establish a framework in which further mechanistic studies on replication timing, such as the strong effect of heterochromatin, can be pursued.
The timing of DNA replication is a fundamental aspect of genome metabolism that correlates with, and has been proposed to regulate, chromatin structure, gene expression, DNA repair, and cellular differentiation (Goren and Cedar 2003; Gilbert et al. 2010). Replication timing is determined by the timing of replication origin firing (Rhind and Gilbert 2013). During the G1 phase of the cell cycle, the Origin Recognition Complex (ORC) binds origins and loads the ring-shaped MCM replicative helicase around DNA (Bell and Kaguni 2013). Activation of the MCM complex initiates replication and thus determines replication timing. Nonetheless, how the timing of such origin firing is regulated is unclear.
Although the average replication times of origins, as measured in population assays, is reproducible, the firing times of origins in individual cells are heterogeneous (Rhind and Gilbert 2013). In fact, single-molecule studies in both budding and fission yeast have shown that origin firing is stochastic (Patel et al. 2006; Czajkowsky et al. 2008). Nonetheless, if individual origins fire stochastically with a characteristic efficiency, they will exhibit reproducible average firing times, with more efficient origins firing earlier, on average (Bechhoefer and Rhind 2012). Therefore, understanding the timing of origin firing requires understanding what regulates the efficiency of origin firing.
One strong influence on replication timing is heterochromatin (Rhind and Gilbert 2013). In budding yeast, proximity to telomeric heterochromatin is both necessary and sufficient to delay origin firing (Ferguson and Fangman 1992). This effect is regulated by Rif1-dependent recruitment of protein phosphatase I, which may antagonize origin activation by the cyclin- and DBF4-dependent kinases (Lian et al. 2011; Davé et al. 2014; Hiraga et al. 2014; Mattarocci et al. 2014). Chromatin structure—in particular, histone deacetylation by Sir2 and Rpd3—also affects the timing of nontelomeric origins (Knott et al. 2009, 2012; Peace et al. 2014; Yoshida et al. 2014). However, the effect of chromatin structure on euchromatic origins is much weaker, and its mechanism is unclear.
Based on a mathematical analysis of replication kinetics of the budding yeast Saccharomyces cerevisiae, we previously proposed the multiple-MCM model, in which replication timing is influenced by the number of MCM complexes loaded at origins (Yang et al. 2010). In this model, early origins have more MCMs loaded and therefore are more likely to fire early in S phase. The prediction of the model that multiple MCMs be associated with origins is consistent with in vitro results from frog egg and yeast nuclear extracts that show multiple MCMs can be loaded at origins (Edwards et al. 2002; Bowers et al. 2004) and in vivo studies that show that there are many more MCMs loaded than the predicted number of origins (Donovan et al. 1997; Woodward et al. 2006). This model provides a mechanistic explanation for the observed heterogeneity in origin firing (Patel et al. 2006; Czajkowsky et al. 2008; Cayrou et al. 2011; Retkute et al. 2011) and helps to explain how defined replication timing profiles emerge from stochastic origin firing (Rhind et al. 2010).
The multiple-MCM model posits that MCM complexes are stochastically activated with similar probabilities across the genome, so that origins that have more MCM complexes loaded are more likely, on average, to fire early. The model makes three strong, testable predictions. First, early origins should have more MCMs loaded than late origins. Second, early origins should have more than one MCM complex loaded, and in particular, more MCMs loaded than ORC bound. Third, reducing the number of MCMs loaded at an origin should delay replication timing of that locus. We confirm all three predictions, supporting the model that replication timing is regulated by the number of MCMs loaded at each origin.
Results
To test the first prediction of the multiple-MCM model—that early origins have more MCMs loaded than late origins—we examined the genome-wide distribution of MCM by ChIP-seq in G1-arrested cells. As shown on Chromosome 10, the MCM ChIP-seq signal is concentrated at known origins, with more signal at the early origins ARS1012, ARS1014, ARS1018, and ARS1019, than at the late origins ARS1008, ARS1009, ARS1010, and ARS1016 (Fig. 1A). Surprisingly, there is a strong MCM ChIP-seq signal at the late-replicating telomere-proximal origins. The late replication of these origins depends on telomeric heterochromatin (Lian et al. 2011), suggesting that activation of MCM loaded at these origins may be repressed by local chromatin structure (Ferguson and Fangman 1992).
Figure 1.
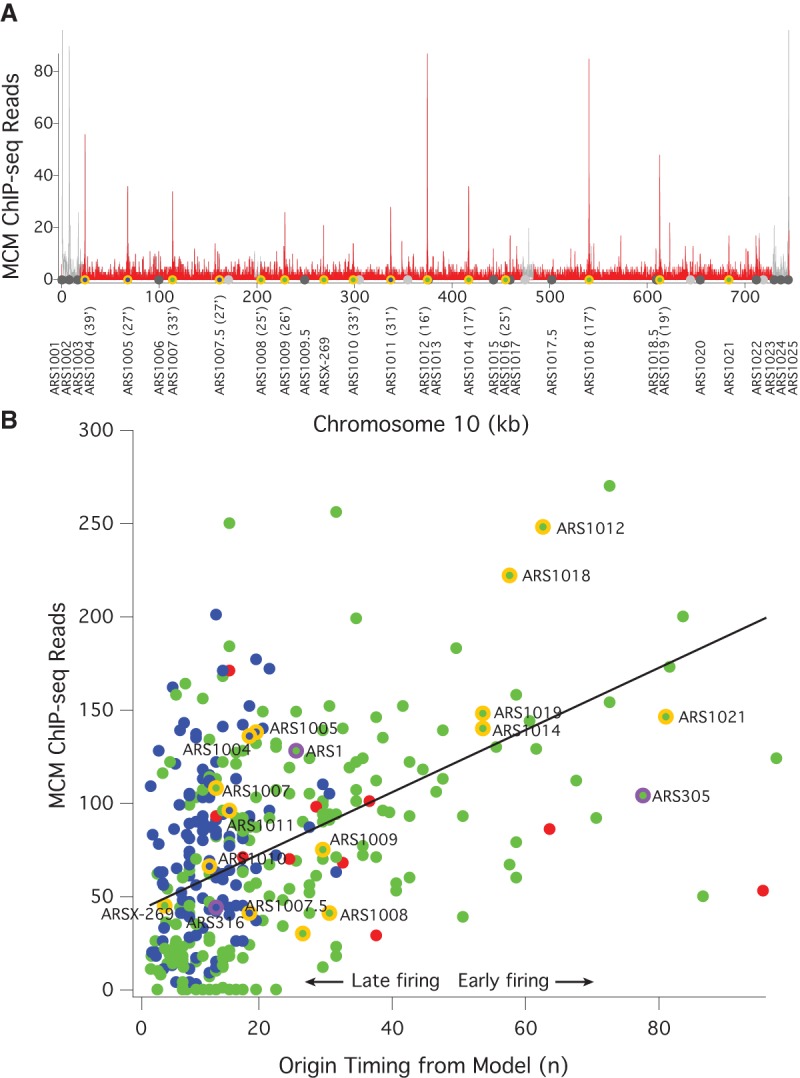
MCM ChIP-seq signal correlates with origin firing time. (A) Frequency of MCM ChIP-seq reads in 100-bp bins on Chromosome 10 from α-factor-arrested wild-type cells (yFS661). The red histogram represents uniquely mapped reads; the gray histogram represents multiply mapped reads that cannot be specifically placed at any one locus, but which demonstrate a high level of telomeric MCM binding. The locations of origins shown in B are shown with the color code used there. The firing time of these origins is given after their names. The locations of other confirmed or suspected origins (Siow et al. 2012) are indicated in dark gray and light gray, respectively. These origins are not included in B because they were not captured by the model used to determine n (Yang et al. 2010). (B) The correlation between the firing time of origins, as determined by the firing-time parameter n from a quantitative analysis of replication kinetics (Yang et al. 2010) and the number of MCM ChIP-seq reads in a 1-kb window around those origins. Blue dots represent origins repressed by telomere proximity (Lian et al. 2011), Rpd3 (Knott et al. 2009) activity, or Fkh1 (Knott et al. 2012); red dots represent centromeric origins and other origins activated by Ctf19 (Natsume et al. 2013); green dots represent all other origins. Chromosome 10 origins are indicated in orange; the origins used in Figure 2 and Supplemental Figure S2 are indicated in purple. ARS1004 is repressed by telomere proximity (Lian et al. 2011); ARS1005, ARS1007.5, and ARS1010 are repressed by Rpd3 (Knott et al. 2009); ARS1011 is repressed by Fkh1 (Knott et al. 2012). The line represents the best linear fit to the green dots (r = 0.54).
Across the genome, there is a significant correlation (r = 0.42, P < 10−5) between our MCM ChIP-seq signal and the previously inferred origin timing parameter, n (Fig. 1B; Yang et al. 2010). In our mathematical model of replication timing, n is the parameter that describes the firing-time distribution of origins. The model is based directly on the timing of origin firing, not on timing of origin replication (which is influenced both by origin firing and passive replication from the firing of neighboring origins), making n a more direct estimate of origin timing than metrics based on the timing of origin replication. One plausible interpretation of n is that it reflects the number of MCMs loaded at origins (Yang et al. 2010), leading directly to our prediction that MCM ChIP-seq signal should correlate with n.
Many origins fall along the MCM/timing diagonal, as predicted. However, many fall above the diagonal. These are origins that have more MCM loaded than would be expected, given their late firing times. These origins include telomeric origins (Fig. 1A), which are known to fire late in a heterochromatin-dependent manner (Ferguson and Fangman 1992; Lian et al. 2011). In addition, the Rpd3 HDAC is known to delay origin firing, presumably through modification of chromatin structure (Knott et al. 2009; Yoshida et al. 2014). Origins controlled by these factors also generally fall above the diagonal (Fig. 1B). Conversely, the Ctf19 kinetochore protein advances pericentromeric origin firing by facilitating the modification of centromeric heterochromatin (Natsume et al. 2013). The Fkh1 transcription factor also effects origin firing, delaying some origins and advancing others (Knott et al. 2012). We find that Fkh1-delayed origins tend to fall above the diagonal; Fhk1-advanced origins show no systematic effect. If these chromatin-influenced origins are excluded from the data set, the correlation between MCM ChIP-seq signal and origin timing increases significantly (r = 0.54, P < 10−5) (Fig. 1B). Comparing our origin timing data to other published MCM ChIP-seq data sets produces similar correlations (r = 0.44 to r = 0.61, P < 10−5) (De Piccoli et al. 2012; Belsky et al. 2015), as does comparing our MCM ChIP-seq data to other published estimates of origin timing and/or efficiency (r = 0.47 to r = 0.50, P < 10−5) (Supplemental Fig. S1; Hawkins et al. 2013; McGuffee et al. 2013). From these results, we conclude that the relative number of MCM complexes loaded at origins during G1 regulate their firing timing during S phase. However, these results only elucidate the relative number of MCMs at origins; they do not distinguish between models in which early origins have multiple MCMs (Yang et al. 2010) and those in which early origins have a single MCM complex loaded and late origins have substoichiometric MCM loading (de Moura et al. 2010; Retkute et al. 2011; Hawkins et al. 2013).
To directly assay the number of MCM complexes loaded at origins and thereby test the second prediction—that early origins have multiple MCM complexes loaded—we used a single-origin purification strategy. We engineered different origins into the TALO8 plasmid affinity purification system (Unnikrishnan et al. 2010) and introduced a single binding site for the zinc-finger DNA binding protein Zif268, which binds to its 10-bp recognition site with sub-nanomolar affinity (Elrod-Erickson and Pabo 1999), as an internal control. We tagged MCM2 and ORC2 with the HA epitope and expressed an HA-tagged Zif268 (Supplemental Fig. S2A,B). We then purified TALO8 plasmids containing different origins from G1-arrested cells and determined by Western blotting how many MCM complexes were loaded in vivo, relative to the Zif268 control (Fig. 2A,B). On average, about three MCM complexes (with two molecules of Mcm2 in each MCM complex) are loaded on each plasmid containing the early origin ARS1 (Fig. 2B). Assuming that the loading of MCM complexes is not cooperative, ∼90% of the plasmids in the population should have between one and five MCM complexes loaded, and ∼5% should have no MCMs loaded, consistent with an ARS1-plasmid loss rate of ∼3% (Strich et al. 1986). Another early origin, ARS305, also has an average of more than two MCM complexes loaded (Supplemental Fig. S2C,D). Loading of MCMs is specific to G1, with only trace MCM signal seen in metaphase-arrested cells (Fig. 2A,B). Since ORC occupancy at ARS1 has been reported to be high in vivo (Diffley et al. 1994), we attribute the substoichiometric recovery of ORC to loss of ORC binding during purification. We compared the level of MCMs loaded between the early firing ARS1 and a late-firing origin, ARS316. ARS316 has, on average, less than one MCM complex loaded per plasmid (Fig. 2C,D), consistent with its later firing time, its low efficiency, and its high rate of plasmid loss (Poloumienko et al. 2001). These results demonstrate that multiple MCMs can be loaded at a single origin and provide additional evidence for the model that MCM loading regulates origin timing.
Figure 2.
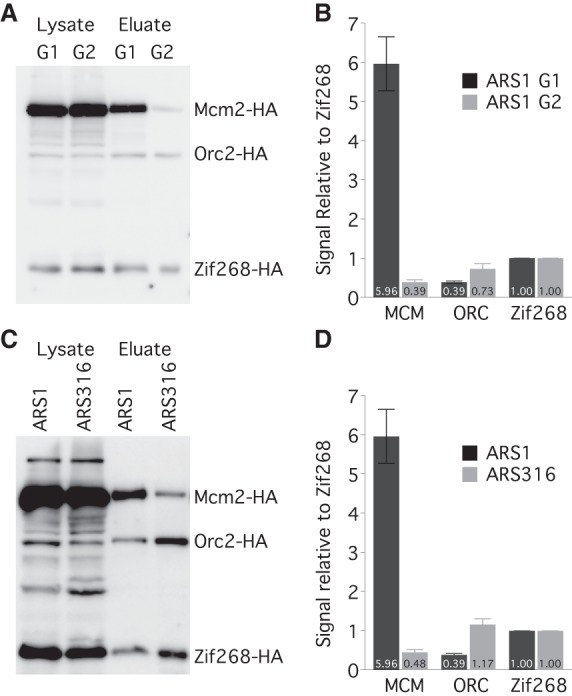
ARS1 has multiple MCM complexes loaded in vivo. (A) HA Western blot of protein from Mcm2-HA, Orc2-HA, Zif268-HA cells (yFS853) before and after affinity purification of an ARS1-containing TALO8 plasmid. G1 cells were arrested in α-factor; G2 cells were arrested in nocodazole. (B) Quantitation of eluate data in A: n = 7 for G1; n = 3 for G2; error bars represent SEM. (C) Western blot as in A from G1-arrested cells carrying an ARS1-containing (yFS853) or ARS316-containing (yFS855) TALO8 plasmid. (D) Quantitation of data in C: n = 7 for ARS1, n = 3 for ARS316, error bars represent SEM.
To test the third prediction—that reducing the number of MCMs loaded at an origin will delay its average replication time—we compared replication timing between ARS1 and an ARS1 mutation, ARS1-ΔB2, which reduces MCM loading without affecting ORC binding (Zou and Stillman 2000). We verified the MCM-loading defect of ARS1-ΔB2 using the TALO8 system (Fig. 3A,B) and tested replication timing of wild-type ARS1 and ARS1-ΔB2 using a copy number variation assay (Yabuki et al. 2002). Cells containing chromosomal alleles of ARS1 or ARS1-ΔB2 were released from a G1 arrest and sampled throughout S phase (Supplemental Fig. S3). Genomic DNA was prepared, and copy number at selected loci was determined using the NanoString nCounter approach (Geiss et al. 2008). The ΔB2 mutation causes a 13-min delay in the average replication timing (trep) of ARS1 (Fig. 3C), transforming ARS1 from an early origin to a late origin. These results suggest that the number of MCMs loaded at an origin directly affect its average firing time. Nonetheless, this conclusion rests on the manipulation of MCM levels at only one origin; the ability to manipulate MCM at other origins would increase the generality of this approach.
Figure 3.
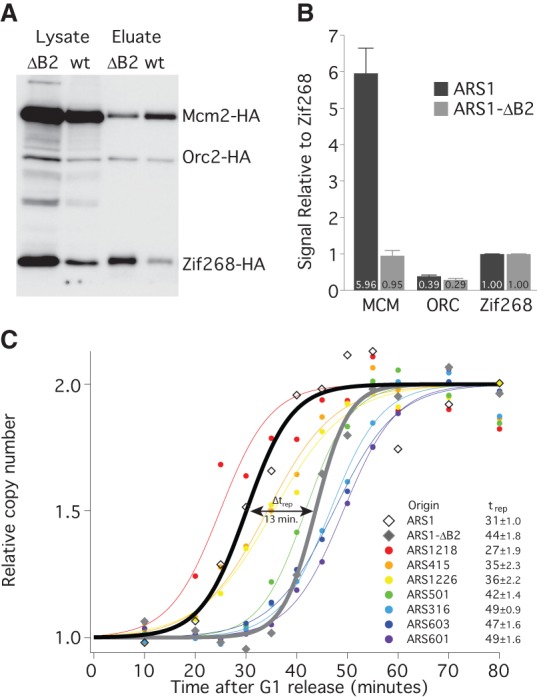
Reducing MCM loading delays the timing of origin firing. (A) Western blot as in Figure 2A from G1-arrested cells carrying an ARS1- (yFS853) or ARS1-ΔB2- (yFS854) containing TALO8 plasmid. (B) Quantitation of data in A: n = 7 for ARS1, n = 5 for ARS1-ΔB2, error bars represent SEM. (C) Replication timing of eight origins in cells with chromosomal alleles of ARS1 (yFS833) or ARS1-ΔB2 (yFS842). Cells were released from α-factor and sampled at various times during S phase. The copy number of each locus was assayed using a NanoString nCounter: n = 2 for ARS1 and ARS1-ΔB2, n = 4 for other origins. The inset table lists the average replication time (trep ± SEM) for each origin analyzed.
Discussion
The work presented here provides mechanistic insight into how replication timing is regulated in budding yeast. It supports a model (Rhind et al. 2010; Yang et al. 2010) in which replication timing is determined by the stochastic firing of origins that compete for rate-limiting activators (Fig. 4; Patel et al. 2008; Mantiero et al. 2011; Tanaka et al. 2011). In this model, origins that compete more efficiently for limiting activators have a higher probability of firing and thus, on average, replicate earlier. Our results suggest that the number of MCMs loaded at an origin contribute significantly to this competition and influence the origins efficiency and timing. Although our model posits that individual MCMs at an early origin are no more likely to fire than MCMs at a late origin, an origin with more MCMs is more likely to fire early, because the greater number of MCMs increases the chances of one of the MCM firing. The loading of multiple MCMs provides simple and biochemically plausible mechanisms for regulating origin efficiency and timing.
Figure 4.

Model for regulation of replication timing by MCM loading and heterochromatin. Origins at which many MCMs are loaded are more likely to fire in early S and therefore have an early average firing time; origins at which fewer MCMs are loaded are less likely to fire in early S and therefore have a later average firing time. High-affinity origins are bound by ORC for more of G1 and therefore may have more MCMs loaded. Heterochromatin provides a second layer of regulation, on top of our proposed MCM-based mechanism. Heterochromatin could delay origin firing at any step in the origin licensing/firing cycle—ORC binding, MCM loading, or MCM activation—but the observation that many MCMs are loaded at heterochromatically silenced telomeric origins suggests that one way heterochromatin delays replication time is by inhibiting activation of loaded MCMs.
An advantage of the multiple-MCM model is that it does not require an explicit timing mechanism during S phase. Once programed by MCM loading, origins will naturally fire at characteristic times in S phase without any mechanism that distinguishes early S from late S (Rhind et al. 2010). This mechanism can also naturally explain the observation that slowing the rate of fork progression proportionally slows the rate of origin firing (Alvino et al. 2007; Rhind 2008; Koren et al. 2010). If a rate limiting factor required for MCM activation travels with the replication fork, such as Cdc45 (Wu and Nurse 2009; Mantiero et al. 2011; Tanaka et al. 2011; Gindin et al. 2014), new origins would be unable to fire until previously initiated forks terminate, coupling origin firing timing to fork progression (Rhind 2008).
Another advantage of the multiple-MCM model is that it explains how events that impact origin licensing during G1 can affect the timing of origin firing during S phase. The timing and affinity of ORC binding during G1 affects the timing of origin activation (Wu and Nurse 2009; Hoggard et al. 2013). However, since ORC is only active during G1, it has been unclear how G1 ORC activity could affect origin activation during S phase after ORC is inactivated by CDK activity. The loading of multiple MCMs onto early origins provides a memory of G1 ORC activity and stably establishes the replication timing program.
Although our results suggest that the number of MCMs loaded at origins affect origin efficiency, other factors contribute as well, in particular chromatin structure (Rhind and Gilbert 2013). The correlation we see between MCM ChIP-seq signal and replication timing is <0.5 (Fig. 1; Supplemental Fig. S1), consistent with MCM number having a significant but not exclusive role in regulating origin timing. Furthermore, we see a similar level of correlation between other published MCM ChIP-seq and replication timing data sets (Supplemental Fig. S1; De Piccoli et al. 2012; Hawkins et al. 2013; McGuffee et al. 2013; Belsky et al. 2015), suggesting the result is robust to experimental details. The lack of correlation seen with older MCM ChIP-chip data sets (Wyrick et al. 2001; Xu et al. 2006) is presumably due to the lower dynamic range of those data sets.
If we exclude origins that have been reported to be affected by the Rpd3 histone deacetylase, the KU telomere binding protein, the Fkh1 transcription factor, or the Ctf19 kinetochore protein, we find that the correlation improves to between 0.5 and 0.6. This effect suggests that these factors, all of which affect chromatin structure, modify origin firing independent of MCM number, but also suggest that other as yet unidentified factors also play an important role in regulating origin timing. Chromatin structure could affect origin efficiency at any point in the origin licensing and firing cycle: ORC binding, MCM loading, or MCM activation. However, the fact that we observe large numbers of MCMs loaded at late-replicating telomeres (Fig. 1A) suggests that heterochromatin can delay the firing of loaded MCMs, perhaps by counteracting the activity of rate-limiting activators (Hiraga et al. 2014; Mattarocci et al. 2014).
The multiple-MCM model raises the question of where multiple MCMs may be located after loading. Presumably, MCM is initially loaded in the nucleosome-free region next to ORC (Bell and Kaguni 2013). For multiple MCMs to be loaded, MCMs would need to diffuse away from this loading site into surrounding chromatin. Such diffusion has been observed on chromatinized templates in frog egg extracts (Edwards et al. 2002). Furthermore, recent high-resolution ChIP mapping of MCM at budding yeast origins suggests that MCM preferentially associates with origin-flanking nucleosomes (Belsky et al. 2015). Although, on average, the strongest MCM ChIP signal tends to be either at the +1 or −1 nucleosome, individual origins show signal across multiple flanking nucleosomes, allowing the possibility that multiple MCMs could associate with multiple nucleosomes in a heterogeneous manner.
The multiple-MCM model is silent as to why more MCMs are loaded at one origin than another. A simple hypothesis is that early origins have higher-affinity ORC binding sites, so that ORC spends more of G1 phase bound at those sites and can load more MCM (Fig. 4). This possibility is supported by a modest, but significant, correlation between ORC ChIP-seq and MCM ChIP-seq signal (r = 0.43) (Supplemental Fig. S1C; Eaton et al. 2010). However, the affinity of ORC for origins is likely determined by more than simply the local origin sequence, because for a significant number of origins, in vivo occupancy is affected by local chromatin structure, as well as the direct affinity of ORC for the origin sequence (Hoggard et al. 2013). Furthermore, other factors, such as trans-acting regulators of origin efficiency and chromatin structure, may also affect the number of MCMs loaded at origins.
Recent results suggest a conceptually similar mechanism may regulate origin timing in human cells (Gindin et al. 2014; Rhind 2014). In human cells, the density of DNase I hypersensitive sites is an excellent predictor of replication timing, with regions dense in DNase I hypersensitive sites replicating early (Gindin et al. 2014). Moreover, a model that uses DNase I hypersensitive sites as a proxy for licensed origins, which are fired by a hypothetical rate limiting activator that moves with replication forks, accurately predicts developmentally regulated replication timing. Importantly, the model does not require that early firing origins have a higher probability of firing than late-firing origins; the higher density of origins in early firing regions is sufficient to increase the chance of such regions replicating early. Thus, in this model, the effect of the high density of origins in early firing regions of the human genome is conceptually similar to the effect of the high density of MCMs at early origins in the budding yeast genome in the multiple-MCM model, albeit at a much longer length scale (Rhind 2014).
Methods
Yeast strains, derivatives, growth conditions, and synchronizations
Saccharomyces cerevisiae yeast strains used in this study—listed in Supplemental Table S2—were derived from W303, grown in rich (YPAD) or selective (CM−Leu−Trp) media at 30°C and manipulated using standard methods (Guthrie and Fink 2002). G1 synchronization with α-factor (Sigma) was carried out for 3 h. For bar1Δ strains, 0.025 µg/mL α-factor was used. For BAR1 strains, 5 µg/mL α-factor was used. For release from G1 arrest, Pronase (Sigma) was added to a final concentration of 0.2 µg/µL. G2 synchronization was carried out using 15 µg/mL Nocodazole (Sigma). Flow cytometry was carried out as described (Haase 2004). yFS661 was used for ChIP-seq experiments. TALO8 strain constructions were done as follows: yFS806 contains the original TALO8 plasmid bearing eight copies of lacO repeats with ARS1 (pFS408). Mcm2 was C-terminally tagged in yFS806 with HA using a KanMX6-HA plasmid (pFS152) and primers spd58 and spd59, resulting in yFS844. Orc2 was C-terminally tagged in yFS806 with HA using an HphMX6-HA plasmid (pFS268) and primers spd60 and spd61, forming yFS845. A double-tagged Mcm2-HA Orc2-HA strain was created (yFS846) using similar strategy by sequential tagging. BAR1 was deleted in all three strains using a NatMX6 plasmid (pFS274) and primers spd114 and spd115 creating yFS847, yFS848, and yFS849, respectively. yFS850 was created by curing yFS849 of pFS408. yFS833 was used for the NanoString-based replication timing assays. An ARS1-ΔB2 derivative of yFS833 (yFS842) was created using the plasmid pFS415 as described (Marahrens and Stillman 1994).
Plasmid construction
Plasmids and oligonucleotides used in this study are listed in Supplemental Tables S3 and S4, respectively. Oligonucleotide-mediated site-directed mutagenesis using primers spd69 and spd70 was used to introduce EagI and MluI restriction sites into pFS408 to create pFS414. Two complimentary oligonucleotides, spd130 and spd133, were annealed to introduce a single Zif268 binding site in pFS414 to form pFS410. TALO8 derivatives were generated as follows: ARS1 was amplified from yFS661 using primers spd134 and spd135 and cloned into EagI-, MluI-cut pFS410 to create pFS411. ARS316 was amplified from yFS661 using primers spd126 and spd127 and cloned into EagI-, MluI-cut pFS410 to create pFS412. ARS305 was amplified from yFS661 using primers spd138 and spd139 and cloned into EagI-, MluI-cut pFS410 to create pFS416. ARS1-ΔB2 was amplified from yFS842 using primers spd134 and spd135 and cloned into EagI-, MluI-cut pFS410 to create pFS413.
MCM chromatin immunoprecipitation
yFS661 was synchronized at G1, formaldehyde cross linked and subjected to MNase ChIP with rabbit anti-Mcm2-7 polyclonal antibody (UM185, gift from Steve Bell) (Bowers et al. 2004) and protein G agarose (Sigma) as described (Liu et al. 2005). Briefly, 5 × 109 G1-arrested cells were cross linked with 0.1% formaldehyde for 15 min and quenched with 125 mM glycine. MNase-digested chromatin was prepared by spheroplasting cells with Zymolyase 100T (Seikagaku), recovering chromatin by centrifugation, and treating with MNase (Worthington) to digest it to mononucleosomal and sub-mononucleosomal fragments.
Sequence data analysis
The MCM ChIP-seq library was prepared from MCM-ChIPed DNA and sequenced on the Illumina GAII platform according to the manufacturer's protocols using commercially available reagents (Epicentre). Reads were filtered for quality and aligned to the sacCer1 (R27, October 1, 2003) version of the S. cerevisiae genome using Bowtie 1.0.0 (Langmead et al. 2009), producing 1,376,249 uniquely mapped reads. Reads were output in SAM format and manipulated using SAMtools 0.1.19 (Li et al. 2009). Read density within 1 kb of every origin listed in OriDB (Siow et al. 2012) were calculated using custom Perl scripts. Origins previously identified (Yang et al. 2010) were mapped to OriDB origins by identifying reciprocal nearest neighbors using custom Perl scripts and manual curation. Pearson correlations were calculated in Igor 6.34 (Wavemetrics) and tested for significance using a two-tailed t-test.
TALO8 plasmid purification
Strains containing TALO8 derivatives were grown overnight in CM−Leu−Trp raffinose. At an OD610 of 0.2, cells were filtered and inoculated in YP raffinose. After 2 h of growth, α-factor was added; after another 1 h, 20% galactose was added to induce Zif268-HA for the last 2 h of synchronization. A similar strategy was used for cells undergoing metaphase arrest except that cells were subjected to nocodazole for 2.5 h. G1 and G2 arrests were confirmed by flow cytometry. For each sample, 400 ODs of cells were collected by centrifugation, washed once with water, and suspended in 5 mL buffer H150 (25 mM HEPES KOH 7.6, 2 mM MgCl2, 0.5 mM EGTA, 0.1 mL EDTA, 10% Glycerol, 150 mM KCl, 0.02% NP40, 1 mM PMSF, and 1× Complete Protease Inhibitor cocktail [Roche]). This slurry was frozen as small pellets in liquid nitrogen and lysed at −196°C using a Retsch MM301 ball mill. Cell powder was thawed in 5 mL H150 lysis buffer on ice and cleared by centrifugation at 27,000 rpm for 90 min. As the input for Western blots, 1/40th of the supernatant was used. Dynabead Protein G (Life Technologies) coupled to FLAG antibody (Sigma) was incubated with the supernatant for 2 h at 4°C. The antibody coupled magnetic beads were washed four times with potassium glutamate buffer (HEPES KOH 7.6, 5 mM magnesium acetate, 1 mM EDTA, 1 mM EGTA, 10% Glycerol, 200 mM potassium glutamate, 0.1% Triton X-100). The protein complexes were eluted with elution buffer (50 mM ammonium bicarbonate, 0.1% SDS), run on a 0.8% polyacrylamide gel, transferred to PVDF membrane (Millipore), and probed with peroxidase conjugated HA antibody (Roche). The membrane was developed using the SuperSignal West Dura extended Duration Substrate kit (Thermo Scientific) and imaged by chemiluminescence on an LAS300 (Fujifilm). Images were quantitated using ImageJ (NIH).
Replication timing assays
Wild-type (yFS833) and ARS1-ΔB2 mutant (yFS849) strains grown in rich medium (YPAD) at 30°C were synchronized at G1, filtered, and released into fresh medium in the presence of pronase. Samples were collected every 5 min between 10 and 80 min. Two ODs of cells were collected for genomic DNA isolation and 0.2 ODs for flow cytometry. Copy number was assayed on a NanoString nCounter using a custom CodeSet and the manufacturer's protocols (Geiss et al. 2008). The sigmoidal function f(t) = a + (d/(1 + exp[−b(t−c)])) was fit to the flow cytometry and copy number data in R (R Core Team 2014) using a nonlinear least squares fitting. The base (a), height (d), and midpoint (trep, c) of the curves were extracted and used to individually normalize the data for each curve between unreplicated (1N) and replicated (2N). In addition, the copy number data in each experiment was normalized in time to the average replication time of its experiment, as determined by flow cytometry, to control for the variation in replication timing between the experiments. The data points shown in Figure 3C are the average of all experiments, and the curves shown are fit to the average data. The trep shown in the inset table are the average and standard error of the treps extracted from the individual experiments, to show the variation between experiments.
Data access
The sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession number SRP040498.
Acknowledgments
We thank David MacAlpine and Jason Belsky for sharing unpublished data and analyses, Steve Bell for the MCM antibody, Toshi Tsukiyama for TALO8 constructs and advice, Scot Wolfe for Zif268 constructs and advice, and Conrad Nieduszynski for strains. This work was supported by National Institute of General Medical Sciences (NIH) grant GM098815 to N.R. and by a National Sciences and Engineering Research Council of Canada (NSERC) grant to J.B.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.195305.115.
References
- Alvino GM, Collingwood D, Murphy JM, Delrow J, Brewer BJ, Raghuraman MK. 2007. Replication in hydroxyurea: It's a matter of time. Mol Cell Biol 27: 6396–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechhoefer J, Rhind N. 2012. Replication timing and its emergence from stochastic processes. Trends Genet 28: 374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SP, Kaguni JM. 2013. Helicase loading at chromosomal origins of replication. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsky JA, MacAlpine HK, Lubelsky Y, Hartemink AJ, MacAlpine DM. 2015. Genome-wide chromatin footprinting reveals changes in replication origin architecture induced by pre-RC assembly. Genes Dev 29: 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers JL, Randell JC, Chen S, Bell SP. 2004. ATP hydrolysis by ORC catalyzes reiterative Mcm2-7 assembly at a defined origin of replication. Mol Cell 16: 967–978. [DOI] [PubMed] [Google Scholar]
- Cayrou C, Coulombe P, Vigneron A, Stanojcic S, Ganier O, Peiffer I, Rivals E, Puy A, Laurent-Chabalier S, Desprat R, et al. 2011. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res 21: 1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czajkowsky DM, Liu J, Hamlin JL, Shao Z. 2008. DNA combing reveals intrinsic temporal disorder in the replication of yeast chromosome VI. J Mol Biol 375: 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davé A, Cooley C, Garg M, Bianchi A. 2014. Protein phosphatase 1 recruitment by Rif1 regulates DNA replication origin firing by counteracting DDK activity. Cell Rep 7: 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Moura AP, Retkute R, Hawkins M, Nieduszynski CA. 2010. Mathematical modelling of whole chromosome replication. Nucleic Acids Res 38: 5623–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Piccoli G, Katou Y, Itoh T, Nakato R, Shirahige K, Labib K. 2012. Replisome stability at defective DNA replication forks is independent of S phase checkpoint kinases. Mol Cell 45: 696–704. [DOI] [PubMed] [Google Scholar]
- Diffley JF, Cocker JH, Dowell SJ, Rowley A. 1994. Two steps in the assembly of complexes at yeast replication origins in vivo. Cell 78: 303–316. [DOI] [PubMed] [Google Scholar]
- Donovan S, Harwood J, Drury LS, Diffley JF. 1997. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc Natl Acad Sci 94: 5611–5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton ML, Galani K, Kang S, Bell SP, Macalpine DM. 2010. Conserved nucleosome positioning defines replication origins. Genes Dev 24: 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards MC, Tutter AV, Cvetic C, Gilbert CH, Prokhorova TA, Walter JC. 2002. MCM2–7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. J Biol Chem 277: 33049–33057. [DOI] [PubMed] [Google Scholar]
- Elrod-Erickson M, Pabo CO. 1999. Binding studies with mutants of Zif268. Contribution of individual side chains to binding affinity and specificity in the Zif268 zinc finger-DNA complex. J Biol Chem 274: 19281–19285. [DOI] [PubMed] [Google Scholar]
- Ferguson BM, Fangman WL. 1992. A position effect on the time of replication origin activation in yeast. Cell 68: 333–339. [DOI] [PubMed] [Google Scholar]
- Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, et al. 2008. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 26: 317–325. [DOI] [PubMed] [Google Scholar]
- Gilbert DM, Takebayashi SI, Ryba T, Lu J, Pope BD, Wilson KA, Hiratani I. 2010. Space and time in the nucleus: developmental control of replication timing and chromosome architecture. Cold Spring Harb Symp Quant Biol 75: 143–153. [DOI] [PubMed] [Google Scholar]
- Gindin Y, Valenzuela MS, Aladjem MI, Meltzer PS, Bilke S. 2014. A chromatin structure-based model accurately predicts DNA replication timing in human cells. Mol Syst Biol 10: 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goren A, Cedar H. 2003. Replicating by the clock. Nat Rev Mol Cell Biol 4: 25–32. [DOI] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. 2002. Methods in enzymology Vol. 351. Guide to yeast genetics and molecular and cell biology: part C. Academic Press, New York. [Google Scholar]
- Haase SB. 2004. Cell cycle analysis of budding yeast using SYTOX Green. Curr Protoc Cytom Chapter 7: Unit 7.23 10.1002/0471142956.cy0723s26. [DOI] [PubMed] [Google Scholar]
- Hawkins M, Retkute R, Müller CA, Saner N, Tanaka TU, de Moura AP, Nieduszynski CA. 2013. High-resolution replication profiles define the stochastic nature of genome replication initiation and termination. Cell Rep 5: 1132–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraga S, Alvino GM, Chang F, Lian HY, Sridhar A, Kubota T, Brewer BJ, Weinreich M, Raghuraman MK, Donaldson AD. 2014. Rif1 controls DNA replication by directing Protein Phosphatase 1 to reverse Cdc7-mediated phosphorylation of the MCM complex. Genes Dev 28: 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggard T, Shor E, Müller CA, Nieduszynski CA, Fox CA. 2013. A link between ORC-origin binding mechanisms and origin activation time revealed in budding yeast. PLoS Genet 9: e1003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott SR, Viggiani CJ, Tavaré S, Aparicio OM. 2009. Genome-wide replication profiles indicate an expansive role for Rpd3L in regulating replication initiation timing or efficiency, and reveal genomic loci of Rpd3 function in Saccharomyces cerevisiae. Genes Dev 23: 1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott SR, Peace JM, Ostrow AZ, Gan Y, Rex AE, Viggiani CJ, Tavaré S, Aparicio OM. 2012. Forkhead transcription factors establish origin timing and long-range clustering in S. cerevisiae. Cell 148: 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Soifer I, Barkai N. 2010. MRC1-dependent scaling of the budding yeast DNA replication timing program. Genome Res 20: 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian HY, Robertson ED, Hiraga S, Alvino GM, Collingwood D, McCune HJ, Sridhar A, Brewer BJ, Raghuraman MK, Donaldson AD. 2011. The effect of Ku on telomere replication time is mediated by telomere length but is independent of histone tail acetylation. Mol Biol Cell 22: 1753–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CL, Kaplan T, Kim M, Buratowski S, Schreiber SL, Friedman N, Rando OJ. 2005. Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol 3: e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantiero D, Mackenzie A, Donaldson A, Zegerman P. 2011. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J 30: 4805–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marahrens Y, Stillman B. 1994. Replicator dominance in a eukaryotic chromosome. EMBO J 13: 3395–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattarocci S, Shyian M, Lemmens L, Damay P, Altintas DM, Shi T, Bartholomew CR, Thomä NH, Hardy CF, Shore D. 2014. Rif1 controls DNA replication timing in yeast through the PP1 phosphatase Glc7. Cell Rep 7: 62–69. [DOI] [PubMed] [Google Scholar]
- McGuffee SR, Smith DJ, Whitehouse I. 2013. Quantitative, genome-wide analysis of eukaryotic replication initiation and termination. Mol Cell 50: 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Müller CA, Katou Y, Retkute R, Gierliński M, Araki H, Blow JJ, Shirahige K, Nieduszynski CA, Tanaka TU. 2013. Kinetochores coordinate pericentromeric cohesion and early DNA replication by Cdc7-Dbf4 kinase recruitment. Mol Cell 50: 661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PK, Arcangioli B, Baker SP, Bensimon A, Rhind N. 2006. DNA replication origins fire stochastically in fission yeast. Mol Biol Cell 17: 308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PK, Kommajosyula N, Rosebrock A, Bensimon A, Leatherwood J, Bechhoefer J, Rhind N. 2008. The Hsk1(Cdc7) replication kinase regulates origin efficiency. Mol Biol Cell 19: 5550–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peace JM, Ter-Zakarian A, Aparicio OM. 2014. Rif1 regulates initiation timing of late replication origins throughout the S. cerevisiae genome. PLoS One 9: e98501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poloumienko A, Dershowitz A, De J, Newlon CS. 2001. Completion of replication map of Saccharomyces cerevisiae chromosome III. Mol Biol Cell 12: 3317–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Retkute R, Nieduszynski CA, de Moura A. 2011. Dynamics of DNA replication in yeast. Phys Rev Lett 107: 068103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N. 2008. An intrinsic checkpoint model for regulation of replication origins. Cell Cycle 7: 2619–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N. 2014. The three most important things about origins: location, location, location. Mol Syst Biol 10: 723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Gilbert DM. 2013. DNA replication timing. Cold Spring Harb Perspect Biol 5: a010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Yang SC, Bechhoefer J. 2010. Reconciling stochastic origin firing with defined replication timing. Chromosome Res 18: 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siow CC, Nieduszynska SR, Müller CA, Nieduszynski CA. 2012. OriDB, the DNA replication origin database updated and extended. Nucleic Acids Res 40: D682–D686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strich R, Woontner M, Scott JF. 1986. Mutations in ARS1 increase the rate of simple loss of plasmids in Saccharomyces cerevisiae. Yeast 2: 169–178. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Nakato R, Katou Y, Shirahige K, Araki H. 2011. Origin association of Sld3, Sld7, and Cdc45 proteins is a key step for determination of origin-firing timing. Curr Biol 21: 2055–2063. [DOI] [PubMed] [Google Scholar]
- Unnikrishnan A, Gafken PR, Tsukiyama T. 2010. Dynamic changes in histone acetylation regulate origins of DNA replication. Nat Struct Mol Biol 17: 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward AM, Göhler T, Luciani MG, Oehlmann M, Ge X, Gartner A, Jackson DA, Blow JJ. 2006. Excess Mcm2–7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol 173: 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu PY, Nurse P. 2009. Establishing the program of origin firing during S phase in fission yeast. Cell 136: 852–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyrick JJ, Aparicio JG, Chen T, Barnett JD, Jennings EG, Young RA, Bell SP, Aparicio OM. 2001. Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science 294: 2357–2360. [DOI] [PubMed] [Google Scholar]
- Xu W, Aparicio JG, Aparicio OM, Tavaré S. 2006. Genome-wide mapping of ORC and Mcm2p binding sites on tiling arrays and identification of essential ARS consensus sequences in S. cerevisiae. BMC Genomics 7: 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuki N, Terashima H, Kitada K. 2002. Mapping of early firing origins on a replication profile of budding yeast. Genes Cells 7: 781–789. [DOI] [PubMed] [Google Scholar]
- Yang SC, Rhind N, Bechhoefer J. 2010. Modeling genome-wide replication kinetics reveals a mechanism for regulation of replication timing. Mol Syst Biol 6: 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Bacal J, Desmarais D, Padioleau I, Tsaponina O, Chabes A, Pantesco V, Dubois E, Parrinello H, Skrzypczak M, et al. 2014. The histone deacetylases Sir2 and Rpd3 act on ribosomal DNA to control the replication program in budding yeast. Mol Cell 54: 691–697. [DOI] [PubMed] [Google Scholar]
- Zou L, Stillman B. 2000. Assembly of a complex containing Cdc45p, replication protein A, and Mcm2p at replication origins controlled by S-phase cyclin-dependent kinases and Cdc7p-Dbf4p kinase. Mol Cell Biol 20: 3086–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]