

Summary
A unique 373 bp region (igr66) between grpE and dnaK of Streptococcus mutans lacks a promoter but is required for optimal production of DnaK. Northern blotting using probes specific to hrcA, igr66 or dnaK revealed multiple transcripts produced from the dnaK operon and 5′-RACE mapped 5′ termini of multiple dnaK transcripts within igr66. One product mapped to a predicted 5′-SL (stem-loop) and two others mapped just 5′ to Shine-Dalgarno (SD)-like sequences located immediately upstream to dnaK and to a predicted SL 120 bp upstream of the dnaK start codon (3′-SL). A collection of cat reporter-gene strains containing mutant derivatives of igr66 were engineered. Chloramphenicol acetyltransferase (CAT) activity varied greatly between strains, but there were no correlative changes in cat mRNA levels. Interestingly, mutations introduced into the SD-like sequences 5′ to the 3′-SL resulted in an 83–98% decrease in CAT activity. Markerless point mutations introduced upstream of dnaK in the SD-like sequences impaired growth at elevated temperatures and resulted in up to a 40% decrease in DnaK protein after heat shock. Collectively, these results indicate processing within igr66 enhances translation in a temperature dependent manner via non-canonical ribosome binding sites positioned > 120 bp upstream of dnaK.
Introduction
DnaK is a universally conserved protein of the Heat Shock Protein 70 family that, in conjunction with the co-chaperone DnaJ and nucleotide exchange factor GrpE, constitute the DnaK chaperone system. DnaK participates in folding of nascent proteins, refolding of damaged or misfolded proteins, protein secretion and presentation of damaged proteins to degradative pathways (Winter and Jakob, 2004; Konovalova et al., 2013; Castanié-Cornet et al., 2014). For bacteria living in multi-species biofilms within the oral cavity, chaperone proteins are integral to responses to common environmental stressors that cause mis-folding of proteins. For example, the damage induced by H2O2 that is generated by antagonistic commensal bacteria or the low pH conditions created by fermentation of dietary carbohydrates is, in part, dealt with by the DnaK chaperone machine (Lemos et al., 2005). The dental caries pathogen Streptococcus mutans is well adapted to life in oral biofilms and is able to rapidly adjust gene expression patterns and its physiology to the constant changes in nutrient availability and other environmental influences using a variety of adaptive strategies (Lemos and Burne, 2008; Lemos et al., 2013). DnaK plays essential roles in these adaptations. Of note, the dnaK gene of S. mutans is essential for viability and is induced in response to heat-shock and during the acid tolerance response; the latter contributing in major ways to the pathogenic potential of this organism (Jayaraman et al., 1997; Lemos et al., 2007).
In Gram-positive bacteria, the genes for the DnaK chaperone complex are usually encoded on a polycistronic transcript, with the HrcA repressor protein and the DnaK co-chaperones (hrcA-grpE-dnaK-dnaJ). HrcA regulates expression of the dnaK operon and the genes for the GroELS chaperonin by binding to a highly conserved target sequence known as a CIRCE (controlling inverted repeat of chaperone expression) in the promoter regions of these operons (Lemos et al., 2001; Schumann, 2003; Woodbury and Haldenwang, 2003; Kim et al., 2008). Regulation of the dnaK operon has been intensively studied in Bacillus subtilis and, in addition to transcriptional regulation by the HrcA repressor, the operon is regulated by processing of the mRNA to smaller transcripts with different stabilities (Homuth et al., 1997; 1999). Also, the presence of a CIRCE at the 5′ end of the dnaK operon transcript has a destabilizing effect on the primary transcript; cleavage of the mRNA between hrcA and grpE yields a more stable grpE-dnaK transcript (Homuth et al., 1999).
Similar to B. subtilis, the genes in the dnaK operon of S. mutans are arranged in the same order, the operon is transcribed from a σA-type promoter, there is a CIRCE 3′ to the promoter and inactivation of hrcA results in overexpression of the operon (Jayaraman et al., 1997). There is, however, a major difference between the genetic organization of the dnaK operon of S. mutans compared with the dnaK operons of B. subtilis and most other eubacteria. Specifically, the dnaK operon of S. mutans contains two large, highly conserved intergenic regions located between grpE and dnaK, and dnaK and dnaJ, designated as igr66 (373 bp) and igr67 (531 bp) (based on Oralgen designation), respectively. These regions, particularly the grpE-dnaK intergenic region, are substantially smaller in B. subtilis (Homuth et al., 1997) and many other bacteria. A previous study by our group showed that igr66 does not contain a promoter but is critical for optimal production of DnaK (Jayaraman et al., 1997; Lemos et al., 2007). In particular (Lemos et al., 2007), when 267 bp from the central region of igr66 was replaced by a polar kanamycin cassette (ΩKm) that was flanked by strong transcriptional terminators and followed by an outward reading promoter from the S. salivarius urease operon (PureI), DnaK protein levels were decreased to less than 5% of that found in the wild-type strain. However, when the same ΩKm-PureI construct was placed 5′ to igr66, leaving igr66 intact, DnaK protein levels were 50% of those found in the wild-type strain.
The purpose of the current study was to begin to understand the molecular basis for modulation of DnaK levels by igr66. Here we present evidence that, in addition to the HrcA repressor, DnaK production in S. mutans is regulated by processing of the primary transcript and by translational control involving a stable RNA structure and non-canonical ribosome binding sites (NC-RBS) located over 120 bp 5′ to the dnaK start codon.
Results
The dnaK gene of S. mutans is part of a four-gene operon regulated by the repressor protein HrcA, which binds to a CIRCE located three bases downstream of the dnaK transcriptional initiation site (Jayaraman et al., 1997; Lemos et al., 2001). Attempts to identify other promoters within this operon were unsuccessful, so the entire operon appears to be transcribed from the single promoter 5′ to the hrcA gene (Jayaraman et al., 1997). However, transcription from a single promoter does not always mean that the mRNA for genes in bacterial operons are present in equivalent amounts. In fact, when the frequency of reads for the dnaK operon was examined in RNAseq data from cells growing in non-stressed conditions [Fig. 1, http://strep-genome.cshl.edu (Zeng et al., 2013)], dnaK mRNA was present in significantly greater proportions than mRNAs from other genes in the operon: a ratio of 2:1 for dnaK : hrcA, 1.9:1 for dnaK : grpE, 1.5:1 for dnaK : dnaJ [based on RPKM (reads per kilobase per million) standardized read counts]. The greater number of dnaK reads compared with other genes in the operon, and the inability to demonstrate the existence of internal promoters, support that mechanisms in addition to HcrA-dependent repression of the operon control DnaK production in S. mutans.
Fig. 1.
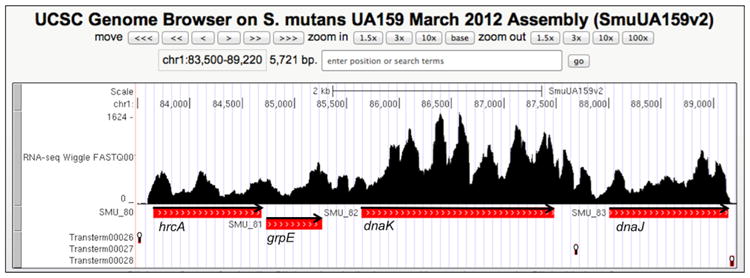
Read-mapping of RNA-Seq of the dnaK operon. The figure is a screen shot from the UCSC Genome Browser (http://strep-genome.cshl.edu) showing read frequencies on the y-axis mapped against the hrcA-grpE-dnaK-dnaJ genes from S. mutans UA159. Cells used for RNA isolation were grown to mid-exponential phase in TV broth (Burne et al., 1999) supplemented with 0.5% glucose (w/v). The locations of predicted rho-independent terminators (RIT) are indicated by stem-loop symbols below the genes, and the orientation of genes is indicated by arrows. RITs were predicted using TransTermHP (http://transterm.cbcb.umd.edu).
Northern blot analysis of the dnaK operon
Northern blot experiments were performed to identify the mRNA transcripts produced from the dnaK operon and to assess the stability of the transcripts in response to heat shock (Fig. 2). For mRNA stability experiments, cells were heat shocked at 42 °C for 3 min, rifampicin was added to stop transcription, incubation was continued at 42 °C and aliquots were removed at predetermined time points. A parallel experiment was performed with cells incubated at 37 °C instead of 42 °C (Figure S1). RNA was isolated as detailed in the methods section, and Northern blots were performed with probes specific for hrcA, igr66 or dnaK. The arrangement and the size of genes in the dnaK operon, as well as the predicted size and abundance of mRNA transcripts based on Northern blot results are depicted in Fig. 2A. All three probes recognized transcripts of 5.8 kb and 4.4 kb at both 37 °C and 42 °C, consistent with mRNA encoding hrcA-grpE-dnaK-dnaJ and hrcA-grpE-dnaK respectively (Fig. 2 and S1). The 5.8- and 4.4-kb transcripts appeared to be relatively stable at 42 °C, with only minor degradation noted after addition of rifampicin (Fig. 2C–E). While also present and fairly stable in experiments performed at 37 °C, the 5.8-and 4.4-kb transcripts were present in lower abundance than in cells incubated at 42 °C. Of note, the 217-bp igr66 probe was made using a biotin labeled oligonucleotide to allow for strand specific detection, whereas the dnaK (536-bp) and hrcA (350-bp) probes were labeled on both strands using a biotin labeling kit, and therefore the igr66 probe produced a weaker signal than the dnaK- or hrcA- specific probes. Also of relevance, anti-sense transcripts of igr66 could not be detected using Northern blotting (data not shown).
Fig. 2.
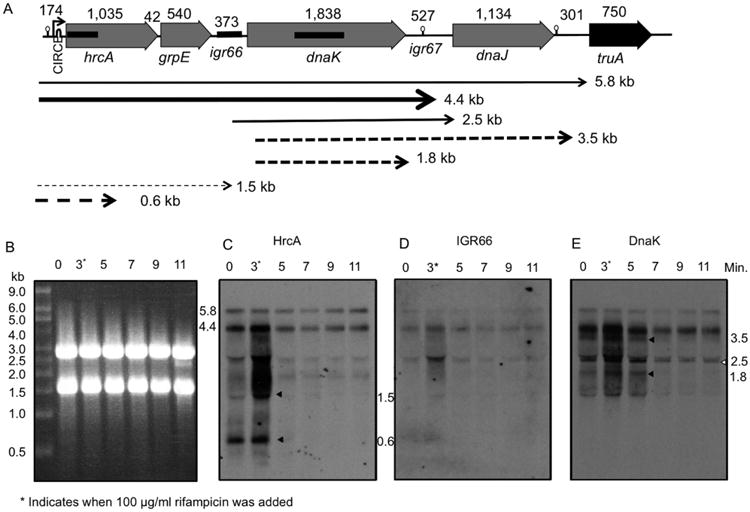
Northern blot analysis of dnaK operon transcripts.
A. The size in base pairs (bp) of genes and intergenic regions are indicated above the respective elements. The promoter, located 5′ to the hrcA gene, is denoted by an arrow with the approximate location of the CIRCE site labeled. The locations of rho independent terminators (RIT) are indicated by stem-loop symbols (see Fig. 1). The approximate location of probes for hrcA, igr66 and dnaK that were utilized in Northern blots are indicated by thicker black bars. The weight and size of arrows correspond to stability and relative abundance of transcripts based on Northern blot experiments in panels B–E. Dashed arrows represent less stable transcripts that are rapidly degraded, and solid arrows represent stable transcripts.
B. Ethidium bromide stained agarose gel, with 5 μg of total RNA loaded per lane. Northern analysis using a probe against (C) hrcA, (D) igr66 or (E) dnaK. Time in minutes displayed over each well specifies the time after heat-shock (42 °C) began. An asterisk next to the 3 min time point denotes the point at which rifampicin was added to cultures following heat shock. The size (kb) and locations of unstable transcripts are indicated by arrows (see text for additional detail).
Two apparent transcripts of approximately 2.7 kb and 1.4 kb were also recognized by all three probes. However, these apparent transcripts migrated just below the 23S rRNA and 16S rRNA, respectively, so they are probably artifacts arising from degradation of a larger transcript(s) coupled with ‘shadowing’ from the rRNAs. Importantly, the dnaK probe also hybridized to a 2.5 kb band present directly below the 2.7 kb 23S rRNA shadow (open arrow, Fig. 2E), likely corresponding to a transcript containing the dnaK gene and some of the surrounding intergenic regions. However, the lack of hybridization of the 217 bp igr66 probe with the 2.5 kb transcript may indicate that only a small portion of igr66 is present in this transcript, and therefore is not recognized by the igr66 probe. Notably, the 2.5 kb band was also present at 37 °C (Figure S1) and was still detected 11 min after heat-shock, i.e. 8 min after rifampicin treatment. Two additional transcripts of 3.5 and 1.8 kb were also recognized by the dnaK probe, but only at 42 °C, and diminished in abundance after the 5 min time point (Fig. 2E). The 3.5 kb transcript likely corresponds to a transcript encoding dnaK and dnaJ, whereas the 1.8 kb transcript corresponds to the size of dnaK (Fig. 2A). Additionally, the HrcA probe also recognized several smaller transcripts (Fig. 2C, black arrows), which disappeared rapidly after transcription was arrested with rifampicin. The 0.6 kb transcript was a curious finding, as the probe for hrcA recognizes the first 350 bp of the 1.0 kb hrcA gene, perhaps indicative of a ribonuclease cleavage site(s) within the hrcA coding sequence. This 0.6 kb transcript appeared to be more stable at 37 °C and was still evident at the 5 min time point in the parallel experiment performed at 37 °C (Figure S1). Collectively, these results indicate that processing of the primary transcript in S. mutans appears to be an important control point for the production of gene products encoded by the dnaK operon.
Mapping of the 5′ termini of dnaK transcripts
In order to determine whether any of the transcripts detected above could have arisen from cleavage of mRNA within igr66, 5′-RACE was performed as detailed in the methods section. Briefly, gene specific primers that annealed 0.45 and 0.42 kb into the dnaK mRNA were used for cDNA synthesis and subsequent 5′-RACE reactions respectively. Total RNA isolated from cells grown at 37 °C or heat-shocked at 42 °C for 3 min was used as the template. As can be seen in Figure S2, 5′-RACE yielded three products (0.8, 0.55, 0.4 kbp) from cells cultured at 37 °C or transiently exposed to 42 °C. The bands were gel purified and sequenced using a third gene specific primer complementary to sequences located 0.37 kbp into the dnaK structural gene. Figure 3 shows the location of the 5′ termini of the dnaK transcripts based on DNA sequence results of the 5′-RACE PCR products. The 0.8 kbp product mapped to the 5′ end of igr66, 7 nt from the grpE stop codon. The 0.55 kbp product yielded two different sequences, one that mapped 215 bp into igr66 within the 5′-SL (0.55 kbp product-1) and another that mapped to the unstructured region between the 5′-SL and 3′-SL, 245 bp into igr66 (0.55 kbp product-2). The 0.4 kbp product mapped just downstream of the 3′-SL, only 12 bp from the dnaK start codon. The results of the 5′ RACE experiments are consistent with what was observed in the Northern blot experiments, where smaller, less-stable transcripts were recognized by both the hrcA (1.8 kb, Fig. 2C) and dnaK probes (3.5 kb, 2.5 kb, 1.8 kb, Fig. 2E) but were not recognized by the igr66 probe. These findings provide additional evidence that transcripts containing dnaK are generated from processing of the primary transcript within igr66.
Fig. 3.

5′-RACE of igr66-dnaK transcripts. 5′ RACE to determine the 5′ termini of the dnaK transcript resulted in multiple products (Figure S2). Shown here is the DNA sequence of igr66 with bold underlining of nucleotides corresponding to the location of 5′ termini based on sequence results of 5′-RACE reactions. Two-headed arrows indicate the position of the 5 stem-loop structures predicted by mfold (Fig. 4) and described in the text.
Igr66 regulates dnaK production post-transcriptionally
Based on results from the mfold web server (version 3.6) (Zuker, 2003), which predicts secondary structure in single stranded RNA, igr66 RNA has the potential to form two very stable structures, designated here as the 5′-SL (stem:loop) and 3′-SL respectively (Fig. 4B). In addition, there are three much smaller SLs predicted for the 5′ portion of the igr66 RNA. Using the BLAT search feature on the Cornell University Streptococcus Genome Browser (http://strep-genome.cshl.edu) to compare the regions between grpE and dnaK in 57 isolates of S. mutans (Palmer et al., 2013) revealed that the sequence of igr66 is very highly conserved, with identities ranging from 96% to 100% with igr66 of S. mutans UA159. Figure S3 shows a ClustalW alignment of the grpE-dnaK intergenic region from the 10 most genomically diverse S. mutans clinical isolates. Based on the BLAT analysis, the region corresponding to the 3′-SL structure (nt 265 to 360 of igr66) appears to be particularly well conserved across isolates of S. mutans, with a maximum of only eight single nucleotide differences in the strain showing the highest sequence divergence from strain UA159 (Figure S3).
Fig. 4.
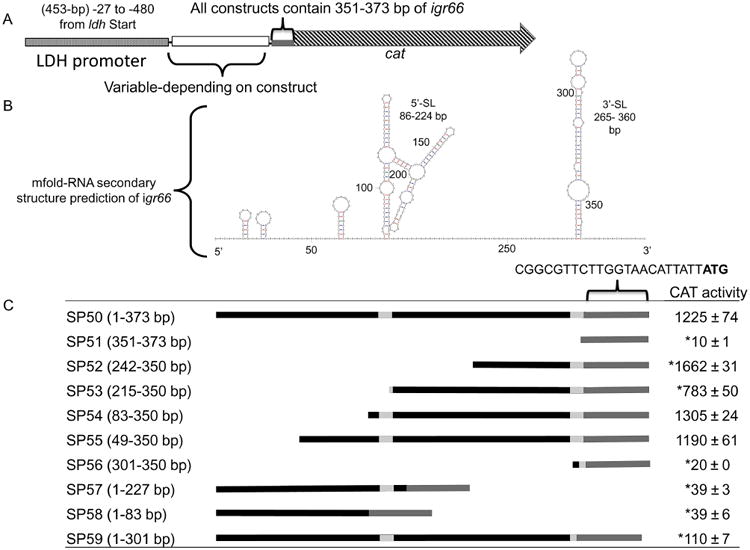
Predicted secondary structure of igr66 mRNA, and construction of reporter-gene igr66-deletion constructs, with respective CAT activities.
A. All constructs were engineered to have the last 22 nt (351-373 bp) of the 3′ end of igr66 containing the RBS of the dnaK gene with expression driven by the lactate dehydrogenase promoter (Pldh).
B. The predicted secondary structure of igr66 with the lowest free energy (ΔG =−92.50) was obtained using the mfold Web Server (http://mfold.rna.albany.edu). Location of the 5′- and 3′-stem loop (SL) structures are indicated. See text for more detail.
C. Schematic diagrams of the various derivatives of igr66 and resulting Cat activity analyzed in this study. In the linear diagrams, the black bars represent the portion of igr66 included in each construct corresponding to the mfold prediction (above), with the light gray bars indicating inclusion of sequences located within secondary structures. CAT activity is expressed as nmoles of chloramphenicol acetylated (min × mgprotein)−1. Asterisk indicates a statistically significant difference compared with SP50 by one-way ANOVA (P-value < 0.05).
In order to determine the role that processing within igr66 plays in the regulation of DnaK levels, a collection of gene fusions were generated that contained various portions of igr66 fused to a chloramphenicol acetyltransferase (CAT) reporter gene lacking its own promoter and ribosome binding site (RBS) (Fig. 4). All constructs included a 22 bp sequence composed of DNA found immediately 5′ to the dnaK structural gene (nt 351–373 of igr66), such that translation of the cat gene should be driven by the cognate dnaK RBS (Kozak, 2005). The lactate dehydrogenase promoter (Pldh) from S. mutans was cloned upstream of the various igr66 sequences to drive expression of the igr66-cat translational fusions (Fig. 4A). These constructs were integrated into the chromosome of S. mutans strain UA159 in single copy, replacing the mannitol PTS locus by double cross-over recombination with the flanking phnA and glmS genes (Figure S4). Strain SP51 was used as a control and contained only the ldh promoter and the 22 bp with the predicted dnaK RBS (Fig. 4).
CAT assays were performed on lysates from cells grown to mid-exponential phase. Strains that contained the 3′-SL of igr66 displayed the highest CAT activity. Strain SP52, which had a 5′ terminus corresponding to the 0.5 kb 5′-RACE product-2 and lacked the first 241 bp of the 5′ end of igr66 (no 5′-SL, but including the entire 3′-SL), had significantly higher CAT activity than any other strain, including the strain that contained all of igr66, SP50 (Fig. 4C). Surprisingly, strain SP53, which had a 5′ terminus corresponding to the 0.5 kb 5′-RACE product-1 and was similar to SP52 in that it lacked the 5′-SL but had an additional 27 bp upstream of the 3′-SL, produced significantly less CAT activity than strains SP52 and SP50. Strains SP54 and SP55 lacked 81 and 48 bp from the 5′ end of igr66, respectively, and produced CAT activities similar to SP50. Thus, the 5′ end of igr66 had relatively little impact on production of CAT, perhaps consistent with the majority of nucleotide polymorphisms in igr66 among all isolates being restricted to this region (Figure S3). In addition, strains SP57 and SP58, which contained only the entire 5′-SL or 83 bp from the 5′ end of igr66, respectively, produced low levels of CAT activity, providing further evidence that the 3′-SL is essential for the function of igr66. Interestingly, strain SP59, which is only missing the region that encodes the 3′ half of the 3′-SL (nt 301–351) of igr66, produced more CAT activity (110 ± 7) than the strains missing the entire 3′ SL and portions of the unstructured region between the 5′ and 3′SL (SP56, SP57, SP58), so the region located immediately upstream of the 3′-SL is important for the activity of igr66.
In order to determine whether the differences seen in CAT activity were correlated with changes in the abundance of cat gene mRNA, quantitative real-time PCR (qRT-PCR) was performed on selected strains (Figure S5). No major differences in cat mRNA levels were seen between any of the strains tested, and the substantial differences in CAT activity seen in Fig. 4C were not correlated with the abundance of cat transcripts. CAT assays were also performed on selected strains following exposure to stress conditions for 15 min, including heat shock at 45 °C, acid shock at pH 5.0 and aeration at 200 r.p.m. In the case of acid shock and aeration, the CAT activity measured in strains subjected to stress did not differ from the same strains growing exponentially in rich medium (data not shown), whereas heat shock at 45 °C resulted in a decrease in CAT activity. However, the same decrease was also seen in a construct that drove cat expression from the ldh promoter and ldh RBS, suggesting that the decrease in CAT activity was due to an inherent sensitivity of the enzyme to elevated temperature, rather than differential expression associated with igr66. Therefore, the Cat protein is not a suitable reporter to assess whether igr66 functioned in a temperature dependent manner.
Efficient translation of the dnaK mRNA requires a non-canonical ribosome-binding site
For nearly all eubacterial mRNAs to be efficiently translated, the ribosome must first identify a translation start site through an interaction between the 3′ end of the 16S rRNA and Shine-Dalgarno (SD) sequence in the mRNA, coupled with recognition of a start codon (AUG, GUG or UUG) by the P-site of the 30S ribosomal subunit (Kozak, 2005). The SD sequence is usually located 5 to 8 nt upstream of the start codon and consists of 4 or 5 nt that are complementary to the 3′ end of the 16S rRNA (3′-UUCCUCCAC-5′). The sequence for the 3′ end of the 16S rRNA from S. mutans UA159 compared with E. coli K12 is shown in Figure S6. The 22 nt preceding the start codon of the dnaK gene are 5′-CGGCGTTCTTGGTAACATTATT ATG-3′, with the underlined GGT possibly serving as a weak SD located 10 nt from the start of the dnaK coding sequence. Interestingly, the 5′ termini identified in the 0.4-kbp 5′-RACE product coincides with the weak SD sequence (GGT) (Figs 3 and 5). However, mutating the GGT of this putative RBS to AAT in the full-length igr66-CAT construct resulted in only a 37% decrease in CAT activity (Fig. 5), indicating other nucleotides must contribute to optimal translational efficiency. Additionally, mutations introduced into this region that created what should have been a stronger RBS (GGAG) did not result in an increase in CAT activity compared with the construct containing wild-type igr66 (data not shown). In addition, we were surprised to find that the 5′ terminus of product-2 of the 0.5-kbp 5′-RACE product strongly resembled a canonical SD sequence (GUGGU), with 5 nt that were complimentary to the 3′ end of the S. mutans 16S rRNA (Figs 5 and S6). Also of note, there is another potential SD-like sequence (GAAAGGA, NC-RBS #2) located between this GAGGA (NC-RBS #1) and the beginning of the 3′-SL (Fig. 5) that has the potential to interact with the 3′ end of the 16S rRNA. In order to determine if these sequences contributed to modulation of translational efficiency by igr66, separate mutations were made in NC-RBS #1 and NC-RBS #2 in the full-length igr66 reporter cat-fusion construct by mutating the guanines to adenines. Mutations in NC-RBS #2 had the greater effect, with a 69% reduction in CAT activity compared to very little change in CAT activity when NC-RBS #1 was mutated (Fig. 5). Next, in order to determine if these NC-RBSs acted synergistically, simultaneous mutations of both NC-RBS #1 and #2 were created, which resulted in an 83% reduction in CAT activity; an additional 17% reduction in activity over mutation of NC-RBS #2 alone, suggesting that both NC-RBS #1 and #2 influence the efficiency of translation. Moreover, mutations where the guanines in NC-RBS #1 and #2 were mutated to cytosines resulted in a further decrease (98%) in CAT activity (963 ± 15 versus 23 ± 1). However, mutations of guanines to adenines were not predicted to affect the secondary structure of the region (Figs 5 and S7), but mutations of guanines to cytosines were predicted to modestly alter the secondary structure of the 3′-SL in a way that would allow some base-pairing between the weak contiguous RBS (C-RBS, GGT) and the introduced cytosines (Figure S7); perhaps explaining the further decrease in CAT activity seen with these particular mutations.
Fig. 5.
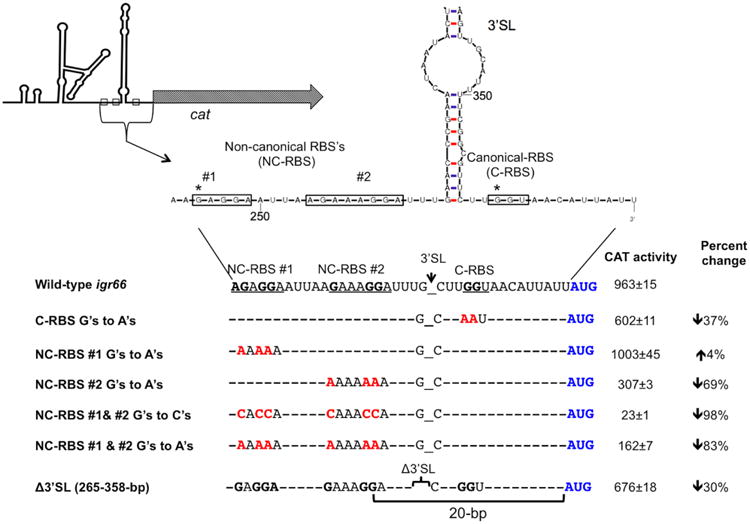
CAT activity of constructs with mutations in predicted RBS sequences or a deletion of the 3′-SL, compared with wild-type igr66. The top panel shows the location of non-canonical and contiguous-RBS sequences (boxes) in the context of the 3′-SL. An asterisk denotes the location of relevant 5′ termini identified in 5′RACE experiments. The bottom panel shows where mutations in guanines predicted to be required for efficient translation of dnaK were mutated to either cytosines or adenines (red text). The resulting CAT activity from these mutant strains and for a strain where the entire 3′-SL was deleted is shown (right column). The percent change in CAT activity compared with full-length wild-type igr66 is also indicated.
In addition to the evidence that mutations in the SD sequences just upstream of the 3′-SL reduced CAT levels, deletion of the 3′-SL, in a way that kept intact the NC-RBS, only caused a 30% decrease in CAT activity (Fig. 5), providing evidence for the hypothesis that a primary function of the 3′-SL may be to bring the NC-RBS into close proximity to the dnaK start codon. Furthermore, despite the large differences in CAT activity, no correlative changes in cat mRNA expression were seen by qRT-PCR between constructs with point mutations or a deletion in the 3′-SL, and full length wild-type igr66 (Figure S8); providing further evidence that igr66 regulates dnaK expression at the level of translation.
The non-canonical RBS #2 is required for optimal DnaK protein production during heat stress
In order to determine how these putative NC-RBS sequences function to regulate DnaK, markerless point mutations were introduced into the chromosome of S. mutans upstream of the dnaK gene. Several attempts were made to introduce mutations in both NC-RBS #1 and #2, such that the guanines were mutated to adenines, but ultimately only mutations in NC-RBS#2 could be recovered (Fig. 6A). However, DNA sequence results revealed two additional mutations besides the mutations to NC-RBS#2. One was located in the 3′-SL and did not affect the predicted secondary structure (Figure S9), whereas the other mutation was a silent mutation located 57 bp downstream of the dnaK start codon. Attempts to identify other NC-RBS#2 mutants without additional mutations were unsuccessful and may indicate compensatory mutations are required in order to mutate NC-RBS#2. Notably, uncharacterized compensatory mutations were evident in the dnaK knockdown strain described previously (Lemos et al., 2007), further emphasizing the importance of this region to the production of DnaK and to S. mutans physiology.
Fig. 6.
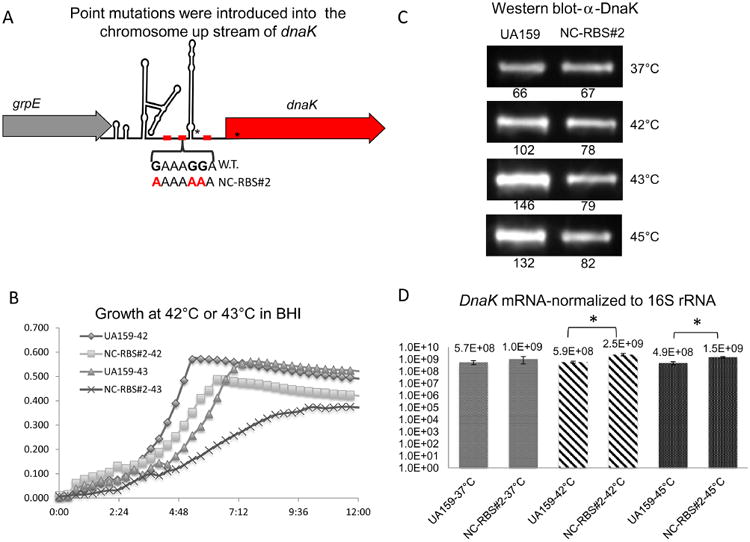
Effects of point mutations within NC-RBS#2 on growth and DnaK expression at elevated temperatures.
A. Schematic diagram of point mutations introduced on the chromosome upstream of the dnaK gene. Asterisks indicate the location of additional mutations (see text or Figure S9 for details).
B. Comparison of growth at 42 °C or 43 °C between wild-type UA159 and the NC-RBS#2 point mutant.
C. Western blot results with an anti-DnaK polyclonal antibody reacted against whole cell lysates from NC-RBS#2 point mutant compared with wild-type UA159 after exposure to indicated temperatures for 15 min. Numbers indicate the integrated density value for the band directly above for one representative blot. Experiments were performed in triplicate with similar results.
D. dnaK mRNA expression in UA159 compared with the NC-RBS#2 point mutant after exposure to the indicated conditions for 15 min.
Growth curves were performed to compare the growth characteristics of the NC-RBS#2 point mutant with those of wild-type S. mutans. While no differences in growth were observed at 37 °C in the NC-RBS#2 point mutant (data not shown), the growth rate at 42 °C was significantly decreased compared with S. mutans UA159, and even further impaired at 43 °C (Fig. 6B). Also, a Western immunoblot with an antibody against DnaK revealed reduced DnaK protein levels in response to heat shock in the NC-RBS#2 mutant compared with UA159 (Fig. 6C), with over 40% less DnaK protein produced at temperatures above 42 °C (Fig. 6C). Conversely, dnaK mRNA levels were at least threefold higher in the NC-RBS#2 point mutant, compared with the wild type at 42 °C and 45 °C (P-value ≤ 0.01) (Fig. 6D). Therefore, mutating only four nucleotides in igr66 (including the unintended mutation in the 3′-SL) resulted in a significant defect in DnaK protein production and growth impairment at elevated temperatures, indicating that NC-RBS#2 is critical to the proper function of igr66 during temperature stress in S. mutans.
Some insights into the basis for an influence of igr66 on DnaK production during heat shock were gained using an earlier version of the mfold program (version 2.6) that allowed for factoring temperature into the prediction of secondary structures of RNA (Fig. 7). In particular, it was discovered that at temperatures below 36 °C there is an additional predicted stem-loop involving the NC-RBS#1 and #2 (Fig. 7A). Conversely, at temperatures between 36 and 39 °C, this stem loop melts such that NC-RBS #1 is partially exposed (Fig. 7B). Interestingly, increasing the temperature in the algorithm above 40 °C resulted in the formation of a stem loop within NC-RBS #1, which leaves NC-RBS#2 completely exposed. It is important to note that in all predictions using this version of mfold, the C-RBS located 10 bp from the dnaK start codon forms a small stem loop, regardless of the temperature conditions used to predict secondary structures. Furthermore, using the sequence for the NC-RBS#2 point mutant as input and the same parameters to predict secondary structures at different temperatures, we found the same secondary structure prediction regardless of changes in temperature (Fig. 7D). Collectively, these observations indicate that, in addition to processing of mRNA within igr66, access to an apparent non-canonical ribosome-binding site(s) located within temperature dependent stem-loop structures play an important role in the production of DnaK during temperature stress.
Fig. 7.
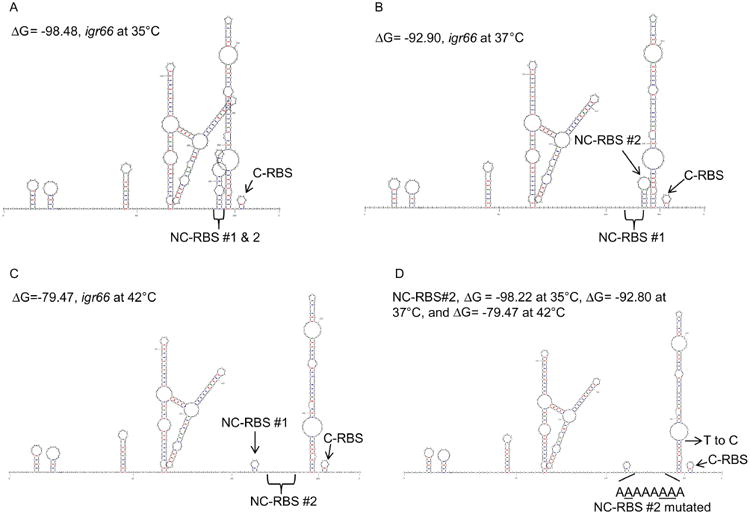
Temperature dependent secondary structure predictions of igr66. Resulting predicted secondary structure with the lowest ΔG values at (A) 35 °C, (B) 37 °C, or (C) 42 °C (see methods for detail). (D) Predicted secondary structures of igr66 with NC-RBS#2 point mutations at various temperatures (35–42 °C) with the indicated ΔG values. The location of the contiguous RBS (C-RBS), and the two non-canonical RBS sequences (NC-RBS #1 or #2) are indicated.
Regulatory elements similar to those in igr66 are found in certain other streptococci
The intergenic regions between grpE and dnaK in other streptococcal species vary in size from 180 bp in S. pyogenes to 479 bp in S. pneumonia, but all are substantially larger than in most other eubacteria. When igr66 from S. mutans was used in a BLASTn search against the database of Oral Microbial Genomes annotated by the Human Oral Microbiome Database (HOMD), S. anginosus, S. constellatus, S. intermedius, S. mitis and S. oralis shared the greatest degree of homology. Sequence identity among species was localized primarily to the regions that correspond to nt 5–30 and 85–220 of igr66, which correspond to one of the minor 5′-SLs and the large 5′-SL respectively (Figs 4 and S10). A ClustalW (version 2.1) sequence alignment between the streptococcal species with completed genomes that are most closely related to S. mutans is shown in Figure S11. The location of a proposed processing site (Figure S11), as identified from the 0.55 kbp 5′-RACE product-1, appears to be conserved in the primary sequence and even, in some cases, in secondary structure (Figure S12). In S. anginosus, the grpE-dnaK intergenic region shares 87% sequence identity to that of S. mutans igr66 and includes the non-canonical RBS sequences and the GGT sequence, as well as two predicted stem-loop structures similar to the 5′-SL and 3′-SL of igr66 of S. mutans (Figure S12). In contrast, S. mitis and S. oralis share 88% and 87% identity, respectively, to a portion of igr66 that corresponds to the 5′-SL (nt 86–224 of igr66) and have a similar predicted secondary structure (Figures S11 and S12). Interestingly, S. mitis and S. oralis both have strong RBS sequences (AGGAG) located 5 to 7 nt from the start codon of dnaK but lack the region that corresponds to the 3′-SL of igr66 and the apparent non-canonical RBSs. Therefore, streptococci have fairly large grpE-dnaK intergenic regions, with most containing sequence and structural similarity to the 5′-SL structures of igr66, perhaps related to conservation in the processing of this RNA. However, the species that lack a strong consensus SD-sequence contiguous to dnaK all harbor potential non-canonical RBS sequences in approximately the same location with respect to a structure that is similar to the 3′-SL in igr66 of S. mutans.
Discussion
There are numerous examples of post-transcriptional regulation of gene expression in bacteria, governed by a variety of mechanisms that include differential mRNA stability and the efficiency of ribosome binding to control translational initiation (Boehringer and Ban, 2007; Marzi et al., 2007; Condon and Bechhofer, 2011; Storz et al., 2011; Sesto et al., 2012). Similarly, it is clear that bacteria can regulate the efficiency of translation of genes in polycistronic transcripts to control the production of proteins that are required in different stoichiometries (Quax et al., 2013). While much work has gone into dissecting transcriptional control of gene expression in S. mutans (Smith and Spatafora, 2012), explorations of the posttranscriptional regulation of gene expression in this organism are in their infancy (Merritt et al., 2014). The results presented here describe a novel mechanism of posttranscriptional regulation of DnaK production that appears to be highly conserved in all sequenced strains of S. mutans, but that also appears to be conserved in a subset of other streptococci.
Northern blot results revealed that the primary transcript of the dnaK operon of S. mutans is processed to yield multiple smaller transcripts that display different stabilities in heat shocked cells, similar to what has been reported for B. subtilis (Homuth et al., 1999). Additionally, our results showed that dnaK transcripts that include igr66 were generally more stable than those without the intergenic region (Fig. 2). Specifically, both the hrcA and dnaK probes recognized a number of less-stable transcripts that were not recognized by the igr66 probe; therefore, igr66 may be required for processing and stabilization of transcripts that contain the dnaK coding sequence. In support of this idea, mapping of the 5′ terminus of dnaK transcripts by 5′ RACE revealed multiple discrete products originating within igr66, presumably as a result of cleavage by ribonuclease(s) with specificity for this region (Fig. 3). It was postulated that RNase III, which has specificity for dsRNA (Arraiano et al., 2010), is responsible for the processing of a stem-loop structure located within the hrcA-grpE intergenic region in B. subtilis (Homuth et al., 1999). Intriguingly, one of the 5′ RACE products containing dnaK mRNA mapped within the 5′-SL, possibly from RNase III cleavage. However, a number of other ribonucleases have been identified in S. mutans that could be involved in processing of igr66 and other transcripts (Jester et al., 2012; Merritt et al., 2014). Future experiments will be targeted at identification of the pathways that generate the processed mRNAs for the DnaK chaperone complex.
Our results support that the 5′-SL and 3′-SL contribute to the capacity of igr66 to influence the amount of DnaK protein that is produced. A wide range of CAT activity was observed in the various reporter-fusion constructs containing igr66 derivatives in spite of the fact that qRT-PCR showed no corresponding changes in cat mRNA levels between strains (Figs 4 and S5). Although, there may be additional mechanisms regulating the stability or translation of the dnaK transcript, igr66 had a profound influence on translation of the cat reporter gene (Fig. 4) and dnaK (Lemos et al., 2007). CAT activity was highest in strain SP52, which contained the 3′-SL and a portion of the unstructured region 5′ to the 3′-SL that contains SD-like sequences (GAGG and GAAAGGA); both are present in product-2 of the 0.55 kbp 5′ RACE experiment (Fig. 3). Notably, CAT activity was significantly lower in strain SP53, which only differs from SP52 in that it contains an additional 27 bp from the 5′ end of igr66, with the 5′ terminus corresponding to that of product-1 of the 0.55-kbp 5′ RACE. Moreover, strain SP54, which contained both the 5′- and 3′-SLs, displayed CAT activity equal to SP50, which contained all of igr66. Taken together, these results suggest that the rate at which mRNAs within igr66 are processed may affect DnaK production. Consistent with this idea, CAT activity in strain SP52 was higher, presumably because processing of the mRNA was not required for efficient translation. Conversely, CAT activity in strain SP53 was lower because the sequence recognized by the ribonuclease is missing, slowing or preventing the processing that is required for optimal gene expression. It cannot be discerned from these data, however, what the relative impacts of processing versus modulation of translational efficiency of these transcripts have on DnaK levels and, ultimately, on the physiology of S. mutans.
While the discovery that processing of the dnaK transcript yields mRNAs with different stabilities is not entirely surprising, the findings that mutations that alter the sequence, or the position with respect to the dnaK start codon, of the putative non-canonical RBS greatly affect translation, are indeed novel and unusual. In particular, strain SP51, which contains the putative cognate RBS of the dnaK gene, produced very little CAT activity, whereas introduction of mutations into both non-canonical RBS sequences that reside just 5′ to the 3′-SL (Fig. 5) caused an 83%–98% reduction in CAT activity, depending on the mutations. It was somewhat surprising that mutations in NC-RBS #1 had no effect on CAT activity, but mutations in NC-RBS #2 alone reduced CAT activity by 68%. Moreover, when the same mutations to NC-RBS#2 were recapitulated on the chromosome upstream of dnaK, a substantial growth defect at elevated temperatures was evident (Fig. 6B). In addition, despite a threefold increase in dnaK mRNA in the NC-RBS#2 point mutant, 40% less DnaK protein was produced after heat shock in the NC-RBS#2 mutant, compared with wild-type strain (Fig. 6). The higher levels of dnaK transcription associated with lower DnaK protein levels are likely attributable to regulation of HrcA stability by the DnaK chaperone system.
Using an older version of mfold, which allowed for temperature variation in secondary structure predictions, an additional stem-loop structure in igr66 at temperatures of 35 °C or below involving both NC-RBS #1 and #2 was predicted (Fig. 7A). Notably, this stem-loop structure partially melted at 37 °C exposing NC-RBS#1, whereas in predications conducted at temperatures above 40 °C a stem-loop formed in NC-RBS#1, leaving NC-RBS#2 completely exposed (Fig. 7B and C). Further still, these temperature-dependent structural changes were not present in models using the sequence from the NC-RBS#2 point mutant (Fig. 7D). In fact, the secondary structures of the NC-RBS#2 point mutant transcript resembled those of wild-type igr66 at 42 °C, which contained a stem-loop obscuring NC-RBS#1. Remarkably, neither the 5′-SL nor 3′-SL structures were predicted to change in response to increased temperature (Fig. 7). In addition, complete deletion of the 3′-SL only reduced CAT activity by 30%, suggesting the primary function of the 3′-SL maybe to hold the NC-RBS in close proximity to the start codon. Collectively, these results indicate that in S. mutans, igr66 regulates DnaK levels by multiple mechanisms, likely in a temperature or stress-dependent manner.
The presence of stem-loop structures located near or overlapping with RBSs are not uncommon (Kozak, 2005; Malys and McCarthy, 2010), and there are a number of examples in bacteriophage T4 genes where the SD sequence is separated from the ATG start codon by a stable mRNASL (Nivinskas et al., 1999). In bacteriophage T4 genes, the hairpin loops are thought to position the SD sequence closer to the ATG start codon: from 22 nt to 5 nt in Gene 38, and from 27 nt to 11 nt in Gene 25. However, based on the mfold predictions for the igr66 3′-SL, the NC-RBS sequences are between 32 and 22 nt from the ATG start codon, presumably too far to function as canonical SD sequences. It has been proposed (de Smit and van Duin, 2003) that ribosome-standby sites can be located in unstructured regions upstream of secondary structures in mRNAs. More specifically, it was posited that these sites allow the 30S ribosomal subunits to bind, possibly through ribosomal-protein S1 binding of single-stranded RNA, which causes the mRNAto unfold and translation initiation to proceed. In support of the hypothesis that such a mechanism may be relevant to igr66, there is an A/U-rich region directly upstream of the putative non-canonical-RBS (NC-RBS) sequences (Fig. 4), a characteristic of known S1 binding sites. However, the S1 proteins of firmicutes do not contain the D1 domain responsible for binding to ribosomal-protein S2 (Salah et al., 2009), and therefore it is unclear if or how S1 interacts with the ribosome to initiate translation in low G + C containing Gram-positive bacteria.
Based on our results, we propose the following working model (Fig. 8) in which igr66-dependent regulation of translation occurs via modulation of mRNA processing that not only influences transcript stability, but also modulates the access of the ribosome to the non-canonical and canonical RBSs in a temperature-dependent manner. The model predicts that in circumstances where cells are growing exponentially in non-stressed conditions, lower levels of the primary transcript are present in the cell and DnaK is produced at a level sufficient to cope with folding of nascent proteins and baseline misfolding of proteins associated with growth, secretion processes and so forth. When cells are exposed to heat-shock or other stressors, increased expression of the primary transcript occurs, resulting in increased expression of all the genes within the dnaK operon. However, processing within the 5′-SL of igr66 allows for more efficient translation of the dnaK transcript, yielding higher levels of DnaK protein. Another layer of control is enacted through the temperature-dependent secondary structures present between the 5′-SL and 3′-SL, which affect access of the ribosome to NC-RBS #1 and #2. Whereas a weak stem-loop is formed when temperatures are below 36 °C, obscuring NC-RBS #1 and #2 and reducing translational initiation (Fig. 8B, top panel), exposure to temperatures between 37 °C and 39 °C may allow the stem-loop to partially melt, giving the ribosome access to NC-RBS#1 (Fig. 8B, middle panel). At temperatures around 40 °C or above, this stem-loop melts further, allowing access to NC-RBS#2 and enhancing translation initiation (Fig. 8B, bottom panel). In the model, both the 5′-SL and 3′-SL are involved in fine-tuning of DnaK protein production (Fig. 8B and C), with processing of the 5′-SL resulting in enhanced translation initiation and protein production (Fig. 8C), whereas processing of the 3′-SL results in decreased translation initiation and transcript stability (3.5 kb and 1.8 kb transcripts). Although the 5′-SL appears to contain a ribonuclease-processing site, the 3′-SL is required for optimal expression of the downstream gene. Based on our Northern blot results, all three dnaK transcripts are present in the cell during heat stress, but we propose that the 2.5 kb transcript is the most stable and translationally active form based on the fact that this transcript likely contains the NC-RBSs that were experimentally shown to be required for optimal production of CAT and DnaK. We also propose that the primary function of the 3′-SL is to allow for proper spacing between the NC-RBSs and the start codon. At this time, however, it cannot be excluded that the 3′-SL may interact with an RNA binding protein, or with the ribosome itself, to enhance mRNA stability, translation initiation or both.
Fig. 8.
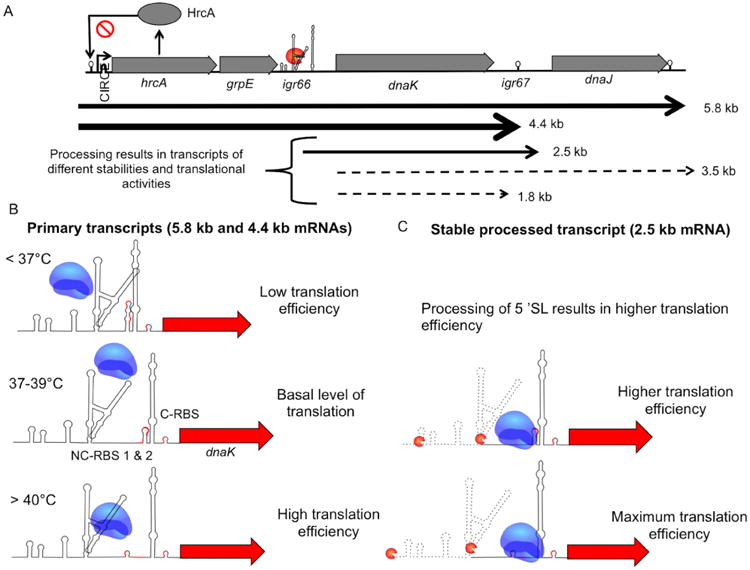
Proposed model of how igr66 regulates translation of dnaK transcript.
A. The primary dnaK mRNA is processed, producing mRNA transcripts with various stabilities and translational activities, with the most stable transcripts indicted by solid black arrows, and the less stable transcripts indicted by dotted arrows.
B. In our model the translational efficiency of the primary mRNAs (5.8 kb and 4.4 kb) depends on ribosome access to NC-RBS #1 and #2, which is controlled through temperature dependent stem-loops structures. Also, interference by the 5′-SL slows ribosomal recognition of the SD-sequences required for efficient initiation of dnaK translation, resulting in basal levels of DnaK protein.
C. Processing of the 5′-SL allows the ribosome better access to the non-canonical-RBS's (NC-RBS) increasing DnaK production. Processing of the 3′-SL leads to the less stable 3.5 and 1.8 kb transcripts, resulting in decreased transcript stability and less efficient translation through the canonical RBS (C-RBS).
The high level of conservation of igr66 within the species S. mutans is intriguing and implies that the presence of igr66 could confer to this organism a competitive advantage within the human oral cavity; possibly under conditions favorable to the development of dental caries when the proportions of S. mutans in oral biofilms increases. One way igr66 could impart this selective advantage is to allow cells to fine-tune DnaK protein production under conditions that do not necessarily trigger changes in transcription initiation via HrcA or other potential regulatory pathways. Such a mechanism could offer bioenergetic advantages, e.g. diminishing the need for de novo transcription, thereby enhancing growth and persistence under particular environmental conditions. Additional genetic and biochemical studies will be required to establish a comprehensive understanding of how this very unusual genetic arrangement influences regulation of DnaK production and the physiologic and ecologic basis for why igr66 was so highly conserved during evolution of S. mutans and certain other streptococci.
Experimental procedures
Bacterial strains and culture conditions
sStreptococcus mutans UA159 and its derivatives (Table S1) were grown in brain heart infusion (BHI) broth at 37 °C in a 5% CO2 aerobic atmosphere. When needed, kanamycin (1 mg ml −1) or erythromycin (10 μg ml −1) was added to broth or BHI agar. E. coli was grown in Luria-Broth supplemented with kanamycin (50 μg ml −1), when necessary. For CAT assays and quantitative reverse transcriptase real-time PCR (qRT-PCR) experiments, cultures were grown in 50 ml of BHI broth to mid-exponential phase (OD600 = 0.4), with a 12 ml aliquot removed and treated with Bacterial RNAprotect™ (Qiagen). Cells were then harvested by centrifugation (3400 × g) and cell pellets frozen at −80 °C until CAT assays or RNA extraction was performed. For mRNA stability experiments, cells were grown to OD600 = 0.4 at 37 °C, at which point an aliquot was removed and combined with an equal volume of Bacterial RNAprotect, serving as the zero time point. The remaining cultures was placed in a 42 °C or 37 °C water bath for 3 min before rifampicin was added to a final concentration of 100 μg−1. Immediately after the rifampicin was added, an aliquot was removed to a tube containing an equal volume of Bacterial RNAprotect. Additional aliquots were then removed at 5-, 7-, 9- and 11 min after treatment began and added to Bacterial RNAprotect. All aliquots were then harvested by centrifugation (3400 × g) and frozen at −80 °C until RNA extractions were performed.
RNA isolation and Northern blotting
Total RNA was isolated from S. mutans strains (Zeng and Burne, 2008) using the RNeasy Kit (Qiagen) with on-column digestion with DNase1 treatment repeated once. The resulting RNA was immediately frozen at −80 °C until use for Northern blotting or qRT-PCR. Northern blotting was done using the NorthernMax® Kit (Ambion®-Life Technologies) following the supplier's protocol. Briefly, 5 μg of total RNA was separated on a denaturing 1.0% agarose gel and transferred to a BrightStar®-Plus positively charged nylon membrane using capillary transfer. Following transfer, the membrane was UV cross-linked twice using the auto-cross-link setting in a UV StrataLinker™ 1800 (Stratagene, La Jolla, CA, USA). Membranes were pre-hybridized in pre-warmed (68 °C) ULT-RAhyb® Ultrasensitive Hybridization buffer (AmbionTM, Life Technologies, Grand Island, NY, USA) for 30 min at 42 °C DNA probes were diluted 1/5 into 10 mM EDTA and heat denatured at 90 °C for 10 min before they were added to the pre-hybridization buffer at a final concentration of 1–2 ng ml−1, depending on the probe, and incubated for approximately 15 h at 42 °C. Membranes were washed under high stringency conditions according to the NorthernMax protocol for DNA probes larger than 50 bp. Blots were then developed using the BrightStar® Kit (Ambion) following the protocol for the least amount of background and then exposed to film for visualization. After each experiment, membranes were stripped by placing the membrane in a boiling solution of 0.1% SDS and allowing the liquid to cool to room temperature before blots were pre-hybridized for 30 min, and re-reprobed.
The igr66 DNA probe (217 bp) was created by PCR amplification using primers IGR66-Fwd and IGR66-Rev, where the reverse primer contained a biotin molecule at the 5′ end (Integrated DNA Technologies). The igr66 probe was used at a concentration of 2 ng ml−1. Both the dnaK (547 bp) and hrcA (350 bp) probes were created using PCR amplification, with primers DnaK-Fwd and DnaK-Rev and HrcA-Fwd and HrcA-Rev, respectively, and biotin labeled using the BrightStar® Psorlalen-Biotin Nonisotopic Labeling Kit (Invitrogen™) according to the supplier's directions. The dnaK and hrcA probes were both used at a final concentration of 1 ng ml−1. Northern blots were repeated multiple times, and the results shown are of representative consistent results.
5′-RACE
5′-RACE was performed essentially as described in the Invitrogen 5′-RACE system for Rapid Amplification of cDNA ends version 2.0 manual, with only minor modifications. Briefly, cDNA was generated using gene-specific primers (dnaK_GSP1) and the iScript cDNA Synthesis kit from Bio-Rad according to the supplier's protocol. cDNA was purified using the Qiagen nucleotide removal kit and a poly-C tail was added using the TdT enzyme (Invitrogen™) and dCTP. 5′-RACE PCR was performed using nested primers located 5′ to dnaK_GSP1 (dnaK_GSP2 or dnaK_GSP3) and the 5′-RACE Abridged Anchor Primer or Abridged Universal Amplification Primer (AUAP) described in the Invitrogen manual. 5′-RACE products were gel purified using the Qiagen gel extraction kit and sequenced at the University of Florida ICBR core facilities.
RNA secondary structure predictions
The mfold web server (http://mfold.rna.albany.edu) (Zuker, 2003) was used to predict secondary structures within igr66 using the default settings except for the indicated changes to the following parameters: Maximum distance between paired bases limited to 150, structure draw mode set to natural angles, exterior loop type set to flat, regularization angle in degrees set to 90 and structure rotation angle set to 90 degrees. For temperature-dependent predictions, the previous version of mfold [RNA Folding Form (version 2.3 energies)] was used, which allows for changes in folding temperatures from 0 to 100 °C. For temperature-dependent folding, the same changes to folding parameters were made as indicated above, with the folding temperature set to either: 35 °C, 37 °C, 40 °C, 42 °C or 45 °C, as indicated in each reported structure. In all cases, reported structures are the result with the lowest (ΔG) free energy value.
Cloning and strain construction
Expression of cat in the CAT-reporter gene fusions was driven by the lactate dehydrogenase promoter (Pldh) from S. mutans UA159, with igr66 and its derivatives inserted between the promoter and cat reporter gene. The lactate dehydrogenase promoter (Pldh), −480 to −27 bp from the ldh start codon, was PCR amplified using primers SP27F and SP27R (Table S2). The 373 bp intergenic grpE-dnaK region (igr66) was amplified from S. mutans UA159 using primers SP28F and SP28R. The cat gene from Staphylococcus aureus was amplified from plasmid pJL105 (Lemos and Burne, unpublished) using primers SP29F and SP29R. All three PCR products were then gel purified (QIAquick Gel Extraction kit from Qiagen) and recombination PCR with 100 ng of each fragment was performed to combine the Pldh promoter, igr66 and the cat genes using SP27F and SP29R, as described elsewhere (Palmer et al., 2012). The resulting recombinant PCR product (Pldh-igr66-cat) was cloned into the pGEM-T Easy vector system (Promega) and digested with Sph1 and BamHI, resulting in a 1.5 kb fragment that was gel purified. The 1.5 kb product was then ligated to pJL105, digested with Sph1 and BamHI (removing the spectinomycin and cat genes from the original vector) and used to transform chemically competent E. coli JM109 cells (Promega), followed by selection on L agar supplemented with kanamycin. The resulting plasmid was designated pSP01-2 and is a derivative of the integration vector pJL84 (Santiago et al., 2012), which was designed to insert promoter-cat gene fusions into the mannitol PTS locus of S. mutans (Figure S4). Plasmid pSP01-2 was used as the template in Q5 Mutagenesis (New England Biolabs) reactions using primers described in Table S2, which were designed using the NEBaseChanger website http://nebasechanger.neb.com. The resulting plasmids were used to transform S. mutans UA159 with selection on BHI agar plates supplemented with kanamycin. Plasmid pSP01-2 also contains an erythromycin resistance gene on the opposite side of the vector (Figure S4) for counter selection against single crossover variants, so colonies that resulted from transformation were screened for erythromycin sensitivity. All constructs were confirmed by DNA sequence analysis.
Chloramphenicol acteyltransferase assays
CAT assays were performed in triplicate as described previously (Zeng and Burne, 2009) with triplicate biological samples. Assay results were standardized by protein concentration determined using the BCA Protein Assay Kit (Thermo Scientific) with BSA (bovine serum albumin) as a standard. Results are expressed as nmol chloramphenicol acetylated per (min × mg protein)−1. Statistical differences were determined using one-way ANOVA.
Generation of cDNA and quantitative real-time PCR (qRT-PCR)
cDNA was generated using the Superscript® III First-Strand Synthesis System (Invitrogen™) using random hexamers and 1 μg total RNA as template. Control reactions with no RT enzyme were included. qRT-PCR was performed essentially as described elsewhere (Zeng and Burne, 2009) with the following modifications. The Bio-Rad Sso-Advanced™ Universal SYBR® Green Supermix was used in 20 μ l reactions (with primers described in Table S2) according to the suppliers recommendations. Real-time PCR was carried out using the Bio-Rad CFX96™ System in a C1000 Touch Thermal Cycler. Subsequently, the concentration of each cDNA template was determined using a combination of absolute and relative quantification methods, such that a threshold cycle was determined based on a standard curve, and then normalized by 16s rRNA. To this end, a standard curve was created for each gene using eight 10-fold serial dilutions of a starting concentration of 108 copies μl−1, as described in Ahn et al. (2005). The concentration of purified PCR products were estimated at OD260, and the number of copies/ml for standard curves were calculated based on the following formula: copies/ml = (6.023 × 1023 × C × OD260)/MWt, where C is 5 × 10−5 g ml−1 for DNA and MWt is the molecular weight of PCR product (base pairs × 6.58 × 102 g). Results shown are the average of triplicate assays of three biological replicates, standardized by 16s rRNA levels, where indicated.
Markerless mutations
Point mutations were introduced into the chromosome using a markerless approach, as described in Kaspar et al. (2015). Briefly, two overlapping PCR fragments were created using primers SP80F and SP105R, and SP105F and SP80R (Table S2), where primers SP105F and SP105R were complimentary and contained the desired mutations in NC-RBS #2. The resulting PCR products were gel extracted and combined by splice overlap extension PCR using primers SP80F and SP80R. The resulting 2.0 kb product was isolated by gel extraction and mixed at a ratio of 100:1 with a suicide vector, which contained an internal fragment to the lacG gene from S. mutans and an ermR marker. The resulting mixture was used to transform S. mutans strain UA159 followed by selection on BHI plates containing erythromycin. The resulting colonies were sequenced to confirm the presence of the desired mutations, and once confirmed were cured of the suicide vector DNA by growth on minimal media containing lactose, and counter selected for sensitivity to erythromycin.
Growth experiments
Wild-type S. mutans UA159 and the NC-RBS#2 point mutant were inoculated in triplicate from single colonies and grown overnight before being diluted 1:100 into fresh BHI media. For growth experiments, 300 μl of each diluted culture was loaded in triplicate wells of a Bioscreen C 100-well plate (Helsinki, Finland) and overlaid with 100 μl of sterile mineral oil. The Bioscreen plate was placed in a Bioscreen C lab system (Helsinki, Finland) set to record the optical density at 600 nm (OD600) every 20 min, with shaking for 10 s before reading for 24 h. The temperature was initially set to 37 °C for 2 h to allow cells to begin to grow, before switching to either 42 °C or 43 °C for the remaining 22 h. Wells containing un-inoculated BHI with mineral oil were included as negative controls, and the resulting OD600 values were subtracted as background from wells containing inoculated media.
Western immunoblot
Whole cell lysates were prepared from triplicate 10 ml cultures grown to early-exponential phase (OD600 = 0.25) and then exposed to either: 37 °C, 42 °C, 43 °C, or 45 °C water baths for 15 min. After treatment, EDTA was added to a final concentration of 1 mM to inhibit protease activity and immediately chilled on ice. Cells were pelleted at 3000 × g for 10 min at 4 °C and washed once in 10 mM Tris-HCL, pH 7.4. Pellets were stored at −80 °C, until whole cell lysates could be prepared. Pellets were suspended in 400 μl 10 mM Tris-HCL, pH 7.4, combined with 250 μl glass beads (average diameter 0.1 mm) and lysed using a Mini Bead Beater (Biospec Products) for two 30 s intervals and placed on ice for 2 min between cycles. Lysates were then cleared by centrifugation at 7000 × g for 10 min at 4 °C. Protein concentration of each lysate was determined by BCA assay (Thermo Scientific), with BSA used as a standard. Cell lysates were combined with 4× Laemmli sample buffer (Bio-Rad), heated to 99 °C for 10 min and centrifuged at 7000 × g for 2 min. Ten micrograms of each sample was separated on a 10% TGX Bio-Rad gel and transferred to a PDVF membrane using the Bio-Rad Trans-Blot transfer system according to manufacturers directions. Western blots were performed according to the SuperSignal West Pico Chemiluminescent Substrate kit (Thermo Scientific) and visualized with a FluorChem 8900 imaging system (Alpha Innotech, USA). The primary polyclonal DnaK antibody (Jayaraman and Burne, 1995) was used at 1:10 000 dilution, and a secondary peroxidase-labeled goat anti-rabbit (IgG) antibody (Kirkegaard & Perry Laboratories, USA) at 1:10 000. The Western blot shown is a representative of triplicate assays. Densitometry values were calculated based on the integrated density value of the respective bands using the FluorChem 8900 software, and represents a single experiment.
Supplementary Material
Footnotes
Supporting information: Additional supporting information may be found in the online version of this article at the publisher's web-site.
References
- Ahn SJ, Lemos JAC, Burne RA. Role of HtrA in growth and competence of Streptococcus mutans UA159. J Bacteriol. 2005;187:3028–3038. doi: 10.1128/JB.187.9.3028-3038.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arraiano CM, Andrade JM, Domingues S, Guinote IB, Malecki M, Matos RG, et al. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol Rev. 2010;34:883–923. doi: 10.1111/j.1574-6976.2010.00242.x. [DOI] [PubMed] [Google Scholar]
- Boehringer D, Ban N. Trapping the ribosome to control gene expression. Cell. 2007;130:983–985. doi: 10.1016/j.cell.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Burne RA, Wen ZT, Chen YY, Penders JE. Regulation of expression of the fructan hydrolase gene of Streptococcus mutans GS-5 by induction and carbon catabolite repression. J Bacteriol. 1999;181:2863–2871. doi: 10.1128/jb.181.9.2863-2871.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanié-Cornet MP, Bruel N, Genevaux P. Chaperone networking facilitates protein targeting to the bacterial cytoplasmic membrane. Biochim Biophys Acta. 2014;1843:1442–1456. doi: 10.1016/j.bbamcr.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Condon C, Bechhofer DH. Regulated RNA stability in the Gram positives. Curr Opin Microbiol. 2011;14:148–154. doi: 10.1016/j.mib.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homuth G, Masuda S, Mogk A, Kobayashi Y, Schumann W. The dnaK operon of Bacillus subtilis is heptacistronic. J Bacteriol. 1997;179:1153–1164. doi: 10.1128/jb.179.4.1153-1164.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homuth G, Mogk A, Schumann W. Posttranscriptional regulation of the Bacillus subtilis dnaK operon. Mol Microbiol. 1999;32:1183–1197. doi: 10.1046/j.1365-2958.1999.01428.x. [DOI] [PubMed] [Google Scholar]
- Jayaraman GC, Burne RA. DnaK expression in response to heat shock of Streptococcus mutans. FEMS Microbiol Lett. 1995;131:255–261. doi: 10.1111/j.1574-6968.1995.tb07785.x. [DOI] [PubMed] [Google Scholar]
- Jayaraman GC, Penders JE, Burne RA. Transcriptional analysis of the Streptococcus mutans hrcA, grpE and dnaK genes and regulation of expression in response to heat shock and environmental acidification. Mol Microbiol. 1997;25:329–341. doi: 10.1046/j.1365-2958.1997.4671835.x. [DOI] [PubMed] [Google Scholar]
- Jester BC, Romby P, Lioliou E. When ribonucleases come into play in pathogens: a survey of Gram-Positive bacteria. Intl J Microbiol. 2012;2012:1–18. doi: 10.1155/2012/592196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar J, Ahn SJ, Palmer SR, Choi SC, Stanhope MJ, Burne RA. A unique open reading frame within the comX gene of Streptococcus mutans regulates genetic competence and oxidative stress tolerance. Mol Microbiol. 2015;96:463–482. doi: 10.1111/mmi.12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SN, Bae YG, Rhee DK. Dual regulation of dnaK and groE operons by HrcA and Ca++ in Streptococcus pneumoniae. Arch Pharm Res. 2008;31:462–467. doi: 10.1007/s12272-001-1179-4. [DOI] [PubMed] [Google Scholar]
- Konovalova A, Søgaard-Andersen L, Kroos L. Regulated proteolysis in bacterial development. FEMS Microbiol Rev. 2013;38:493–522. doi: 10.1111/1574-6976.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene. 2005;361:13–37. doi: 10.1016/j.gene.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Lemos JA, Burne RA. A model of efficiency: stress tolerance by Streptococcus mutans. Microbiology. 2008;154:3247–3255. doi: 10.1099/mic.0.2008/023770-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JA, Luzardo Y, Burne RA. Physiologic effects of forced down-regulation of dnaK and groEL expression in Streptococcus mutans. J Bacteriol. 2007;189:1582–1588. doi: 10.1128/JB.01655-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JA, Quivey RG, Koo H, Abranches J. Streptococcus mutans: a new Gram-positive paradigm? Microbiology. 2013;159:436–445. doi: 10.1099/mic.0.066134-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JAC, Chen YYM, Burne RA. Genetic and physiologic analysis of the groE operon and role of the HrcA repressor in stress gene regulation and acid tolerance in Streptococcus mutans. J Bacteriol. 2001;183:6074–6084. doi: 10.1128/JB.183.20.6074-6084.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JAC, Abranches J, Burne RA. Responses of cariogenic streptococci to environmental stresses. Curr Issues Mol Biol. 2005;7:95–107. [PubMed] [Google Scholar]
- Malys N, McCarthy JEG. Translation initiation: variations in the mechanism can be anticipated. Cell Mol Life Sci. 2010;68:991–1003. doi: 10.1007/s00018-010-0588-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzi S, Myasnikov AG, Serganov A, Ehresmann C, Romby P, Yusupov M, Klaholz BP. Structured mRNAs regulate translation initiation by binding to the platform of the ribosome. Cell. 2007;130:1019–1031. doi: 10.1016/j.cell.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Merritt J, Chen Z, Liu N, Kreth J. Posttranscriptional regulation of oral bacterial adaptive responses. Curr Oral Health Rep. 2014;1:50–58. doi: 10.1007/s40496-013-0005-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivinskas R, Malys N, Klausa V, Vaiskunaite R, Gineikiene E. Post-transcriptional control of bacteriophage T4 gene 25 expression: mRNA secondary structure that enhances translational initiation. J Mol Biol. 1999;288:291–304. doi: 10.1006/jmbi.1999.2695. [DOI] [PubMed] [Google Scholar]
- Palmer SR, Crowley PJ, Oli MW, Ruelf MA, Michalek SM, Brady LJ. YidC1 and YidC2 are functionally distinct proteins involved in protein secretion, biofilm formation and cariogenicity of Streptococcus mutans. Microbiology. 2012;158:1702–1712. doi: 10.1099/mic.0.059139-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer SR, Miller JH, Abranches J, Zeng L, Lefebure T, Richards VP, et al. Phenotypic heterogeneity of genomically-diverse isolates of Streptococcus mutans. PLoS ONE. 2013;8:e61358. doi: 10.1371/journal.pone.0061358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quax TEF, Wolf YI, Koehorst JJ, Wurtzel O, van der Oost R, Ran W, et al. Differential translation tunes uneven production of operon-encoded proteins. Cell Rep. 2013;4:938–944. doi: 10.1016/j.celrep.2013.07.049. [DOI] [PubMed] [Google Scholar]
- Salah P, Bisaglia M, Aliprandi P, Uzan M, Sizun C, Bontems F. Probing the relationship between Gram-negative and Gram-positive S1 proteins by sequence analysis. Nucleic Acids Res. 2009;37:5578–5588. doi: 10.1093/nar/gkp547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago B, MacGilvray M, Faustoferri RC, Quivey RG. The branched-chain amino acid aminotransferase encoded by ilvE is involved in acid tolerance in Streptococcus mutans. J Bacteriol. 2012;194:2010–2019. doi: 10.1128/JB.06737-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann W. The Bacillus subtilis heat shock stimulon. Cell Stress Chaperones. 2003;8:207–217. doi: 10.1379/1466-1268(2003)008<0207:tbshss>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesto N, Wurtzel O, Archambaud C, Sorek R, Cossart P. The excludon: a new concept in bacterial antisense RNA-mediated gene regulation. Nat Rev Microbiol. 2012;11:75–82. doi: 10.1038/nrmicro2934. [DOI] [PubMed] [Google Scholar]
- de Smit MH, van Duin J. Translational standby sites: how ribosomes may deal with the rapid folding kinetics of mRNA. J Mol Biol. 2003;331:737–743. doi: 10.1016/s0022-2836(03)00809-x. [DOI] [PubMed] [Google Scholar]
- Smith EG, Spatafora GA. Gene regulation in S. mutans: complex control in a complex environment. J Dent Res. 2012;91:133–141. doi: 10.1177/0022034511415415. [DOI] [PubMed] [Google Scholar]
- Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell. 2011;43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter J, Jakob U. Beyond transcription – new mechanisms for the regulation of molecular chaperones. Cri Rev Biochem Mol Biol. 2004;39:297–317. doi: 10.1080/10409230490900658. [DOI] [PubMed] [Google Scholar]
- Woodbury R, Haldenwang WG. HrcA is a negative regulator of the dnaK and groESL operons of Streptococcus pyogenes. Biochem Biophys Res Commun. 2003;302:722–727. doi: 10.1016/s0006-291x(03)00254-7. [DOI] [PubMed] [Google Scholar]
- Zeng L, Burne RA. Multiple sugar: phosphotransferase system permeases participate in catabolite modification of gene expression in Streptococcus mutans. Mol Microbiol. 2008;70:197–208. doi: 10.1111/j.1365-2958.2008.06403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Burne RA. Transcriptional regulation of the cellobiose operon of Streptococcus mutans. J Bacteriol. 2009;191:2153–2162. doi: 10.1128/JB.01641-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Choi SC, Danko CG, Siepel A, Stanhope MJ, Burne RA. Gene regulation by CcpA and catabolite repression explored by RNA-Seq in Streptococcus mutans. PLoS ONE. 2013;8:e60465. doi: 10.1371/journal.pone.0060465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.