

Abstract
Sickle cell disease is a risk factor for invasive bacterial infections, and splenic dysfunction is believed to be the main underlying cause. We have previously shown that the liberation of heme in acute hemolysis can induce heme oxygenase-1 during granulopoiesis, impairing the ability of developing neutrophils to mount a bactericidal oxidative burst, and increasing susceptibility to bacterial infection. We hypothesized that this may also occur with the chronic hemolysis of sickle cell disease, potentially contributing to susceptibility to infections. We found that neutrophil oxidative burst activity was significantly lower in treatment-naïve children with sickle cell disease compared to age-, gender- and ethnicity-matched controls, whilst degranulation was similar. The defect in neutrophil oxidative burst was quantitatively related to both systemic heme oxygenase-1 activity (assessed by carboxyhemoglobin concentration) and neutrophil mobilization. A distinct population of heme oxygenase-1-expressing cells was present in the bone marrow of children with sickle cell disease, but not in healthy children, with a surface marker profile consistent with neutrophil progenitors (CD49dHi CD24Lo CD15Int CD16Int CD11b+/−). Incubation of promyelocytic HL-60 cells with the heme oxygenase-1 substrate and inducer, hemin, demonstrated that heme oxygenase-1 induction during neutrophilic differentiation could reduce oxidative burst capacity. These findings indicate that impairment of neutrophil oxidative burst activity in sickle cell disease is associated with hemolysis and heme oxygenase-1 expression. Neutrophil dysfunction might contribute to risk of infection in sickle cell disease, and measurement of neutrophil oxidative burst might be used to identify patients at greatest risk of infection, who might benefit from enhanced prophylaxis.
Introduction
Sickle cell disease (SCD) is an autosomal recessive disorder caused by point mutation of the β-globin gene, resulting in abnormal forms of hemoglobin that cause increased red blood cell rigidity and hemolysis. Over the next 40 years, it is estimated that 14 million births will be affected by SCD, with over 80% in Africa.1 SCD causes considerable morbidity and premature death2 and is estimated to be one of the top 10 causes of life-years lost in West Africa.3 Among its many complications, individuals with SCD demonstrate dramatically increased susceptibility to specific bacterial infections,4 including non-typhoid Salmonella (NTS), Streptococcus pneumoniae and Haemophilus influenzae.5,6 Infection is a major cause of SCD-associated mortality worldwide,5,7 and splenic dysfunction is thought to be the major cause of this susceptibility.8 By 12 months of age, three-quarters of individuals with SCD exhibit either asplenism or hyposplenism, leaving them less able to mount an effective immune response to encapsulated bacteria.9 Prior to the introduction of Pneumococcal conjugate vaccines and widespread penicillin prophylaxis, S. pneumoniae was the most common pathogen isolated from children with SCD presenting with invasive bacterial disease in higher income countries.10 In Africa, S. pneumoniae is also frequently isolated in bacteremic children with SCD,6 but other studies have found NTS, S. aureus and Klebsiella are common,11–13 perhaps indicating that splenic dysfunction is not the only cause of susceptibility to infection in SCD.
The severity of hemolysis in HbSS SCD is partially determined by the relative proportions of HbS and HbF.14 Hydroxyurea treatment increases the proportion of HbF, decreases HbS and hemolysis,15,16 and reduces the risk of infections,17 suggesting that hemolysis may contribute to susceptibility to infection in SCD. We have recently shown that both acute and progressive hemolysis can cause impairment of neutrophil oxidative burst capacity, which results in susceptibility to bacteremia in a mouse model.18 This was true whether hemolysis was induced by a chemical hemolytic agent or malaria infection, and was dependent on both hemolysis-derived heme and induction of the heme catabolizing enzyme, heme oxygenase-1 (HO-1), during granulopoiesis in the bone marrow. A similar defect in neutrophil oxidative burst activity, which correlated with the extent of hemolysis, was demonstrated in children with malaria.19 The extent of hemolysis and HO-1 induction in SCD can be greater than that seen in malaria,20 and so we hypothesized that hemolysis-related neutrophil dysfunction might occur in SCD.18 If this is the case, it may provide an additional explanation for susceptibility to infection. In the current study, we investigated whether neutrophil function is impaired in treatment-naïve children with SCD, and whether any such impairment is related to hemolysis and HO-1 activity.
Methods
Patients and controls
Ethical approval was granted by the NRES Committee London- City & East (Ref. 136415). Samples were obtained after written informed consent from the parent or legal guardian of children (aged 1 to 15 years inclusive) attending clinics at St Mary’s Hospital, Imperial College Healthcare NHS Trust. Eligible SCD patients had previously confirmed HbSS, were not on hydroxyurea therapy, no blood transfusion within 120 days, no evidence of infection, and had a clinical requirement for blood tests. Control subjects having routine blood tests (e.g. before elective surgery), without hemoglobinopathy, significant comorbidity, or symptoms of infection or inflammation, were frequency matched to SCD patients by age, gender and ethnicity (in respective order of priority). Up to 2.5 mL venous blood was collected for co-oximetry (GEM4000 analyzer, Instrumentation Laboratory), whole blood oxidative burst, plasma separation, and HO-1 expression (sodium heparin Vacutainer, BD). Bone marrow (< 2 mL) was collected from a limited number of subjects during “back-up” (SCD) or donation (controls) harvests.
Flow cytometry assays
Neutrophil oxidative burst and degranulation assays were performed, as described previously19 with the following antibodies: Brilliant Violet-421 conjugated anti-CD11b (Biolegend) and allophycocyanin conjugated anti-CD15 (eBioscience). Samples were analyzed using a BD LSRFortessa flow cytometer and FlowJo v. X.07 (Tree Star). The oxidative burst was assessed by the rhodamine fluorescence intensity [(calibrated between samples using rainbow fluorescent particles (Biolegend)], which we have previously shown to be unimpaired by the presence of heme.21 Degranulation was assessed by fold change in CD11b expression. HO-1 expression in blood was determined as described previously,19 with the following antibodies: Alexa647 conjugated anti-CD16, phycoerythrin-Cy7 conjugated anti-CD14 and anti-CD49d, Alexa700 conjugated anti-CD15, Brilliant Violet421 conjugated anti-CD24, anti-CD11b conjugated to each of these fluorophores (all Biolegend except Alexa700 anti-CD11b from AbDSerotec), polyclonal rabbit anti-HO-1 (Enzo LifeSciences), and FITC conjugated goat anti-rabbit IgG (Santa Cruz).
ELISAs
Human hemopexin SimpleStep ELISA and haptoglobin ELISA kits (both Abcam) were used according to the manufacturer’s instructions.
Plasma heme
Protein-bound plasma heme concentrations were determined by spectrophotometry.22
Cell culture
HL-60 cells (PHE European Collection of Cell Cultures) were maintained at 1−9×105 cells/mL in RPMI with 10% fetal bovine serum (Gibco), penicillin, streptomycin and 2 mM L-glutamine (Sigma) at 37°C and 5% CO. Neutrophilic differentiation with 10−7M all-trans retinoic acid (ATRA, Sigma) for five days23 was confirmed by microscopy of hematoxylin and eosin stained smears using a Nikon Eclipse 50i microscope. Viability was assessed by trypan blue exclusion (Countess automated system, Life Technologies). Ferriprotoporphyrin IX dichloride (hemin) (Frontier Scientific) was prepared and used as described previously.18,19
Statistical analysis
Statistical analysis was undertaken in R (version 3.0.3). Categorical variables were compared using the χ2 test; continuous data were compared using Student’s t-test or the Mann-Witney test. A generalized linear model was used to assess simultaneous effects and interaction of neutrophil count and carboxyhemoglobin on oxidative burst after validation of model assumptions. The relationship was graphically represented by plotting the magnitude of the oxidative burst at every combination of 50 equal divisions in each of neutrophil count and carboxyhemoglobin.
Results
We examined neutrophil oxidative burst activity in 26 children with HbSS-SCD and 26 control subjects, frequency matched for age, gender and ethnicity (in respective order of priority). One subject from the control group was excluded after diagnosis of clinically asymptomatic HbCC disease, a pre-specified exclusion criterion. The groups were similar in terms of age and gender, although the control group had a smaller proportion of Black African/Black British subjects (Table 1). All markers of hemolysis were significantly higher in the subjects with SCD than in the control subjects. White blood cell count and neutrophil count were also significantly higher in subjects with SCD.
Table 1.
Characteristics of study subjects.
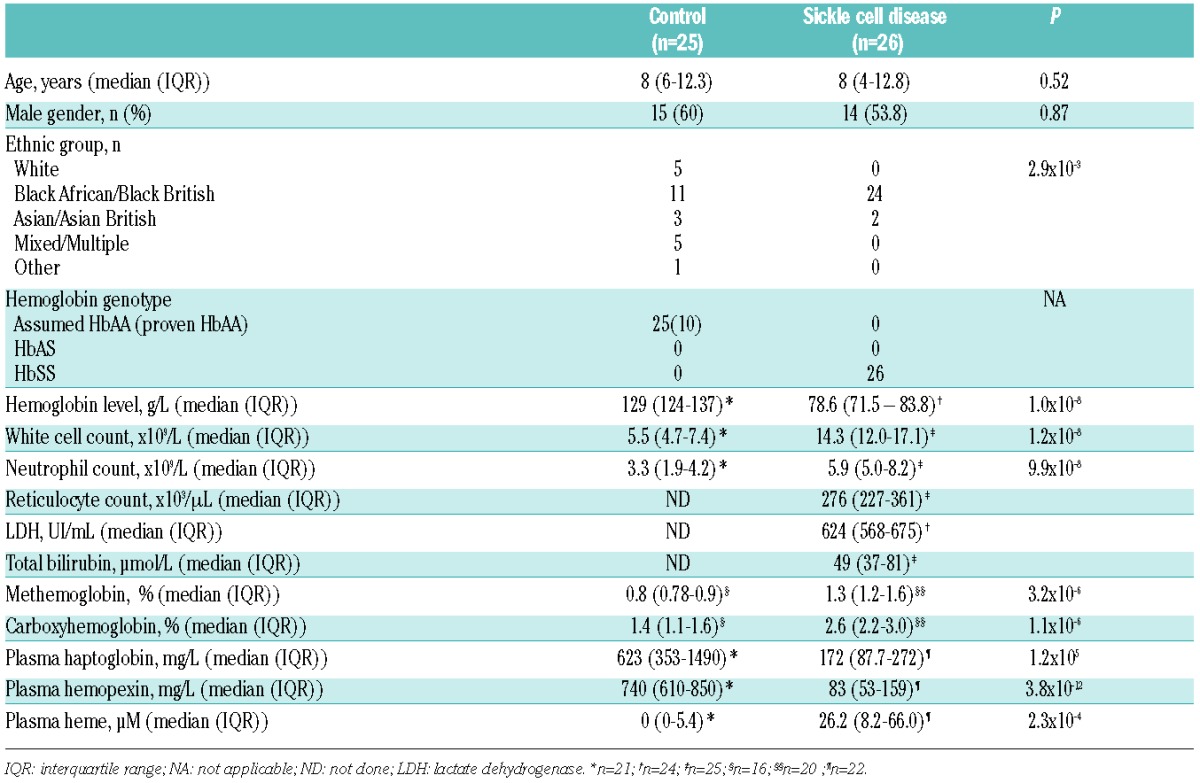
All subjects, whether SCD or controls, showed a unimodal distribution of neutrophil oxidative burst response to PMA-stimulation (Figure 1A). Unstimulated and PMA-stimulated rhodamine fluorescence were both significantly lower in subjects with SCD than in controls, indicating lower basal production of reactive oxygen species, and impairment of the oxidative burst (Figure 1B and C). This defect was specific, because upregulation of surface CD11b expression in response to PMA, a marker of neutrophil activation and degranulation, was similar between groups (Figure 1D). The defect in oxidative burst remained when analysis was restricted to subjects of Black African/Black British ethnicity only (P=0.048 for unstimulated, and P=0.00018 for stimulated rhodamine fluorescence).
Figure 1.
Neutrophil oxidative burst is impaired in children with sickle cell disease. (A) Representative histogram of rhodamine fluorescence intensity of unstimulated (dark fill) and PMA-stimulated (light fill) neutrophils from a child with sickle cell disease. (B–D) Comparison of unstimulated rhodamine fluorescence intensity (B), PMA-stimulated rhodamine fluorescence intensity (C), and PMA-stimulated upregulation of surface CD11b expression (D), in neutrophils of healthy control (n=25) and sickle cell disease subjects (n=26). The Mann-Witney test was used for all comparisons.
The extent of hemolysis in SCD is very variable (Table 1), and directly quantifying hemolysis is challenging. For this reason, many indirect measures of hemolysis are used in clinical practice. Carbon monoxide is one of the products of heme catabolism by HO enzymes (along with iron and biliverdin), and because carbon monoxide avidly binds to hemoglobin, carboxyhemoglobin is a systemic marker of HO-mediated heme catabolism and hemolysis.24,25 One way to capture the inter-individual variation in hemolysis is to combine information from multiple indirect measures to generate a hemolytic index (or hemolytic component) using principal component analysis.26,27 Using this approach we derived a hemolytic component based on hemoglobin concentration, reticulocyte count, plasma lactate dehydrogenase concentration, and total plasma bilirubin concentration, which explained 35.5% of the inter-individual variation. Carboxyhemoglobin was significantly correlated with this hemolytic component (r=0.55, P=0.014), providing additional evidence that it is an important marker of hemolysis in our study population, as well as an indicator of the extent of heme degradation by HO-1.
We previously demonstrated in a mouse model that the effects of hemolysis and HO-1 induction on neutrophil oxidative burst are complex, because hemolysis-derived heme can activate the oxidative burst and contributes to the mobilization of neutrophils from the bone marrow, whilst simultaneously inducing HO-1 in neutrophil progenitors during granulopoiesis. It is this induction of HO-1 in bone marrow which impairs the oxidative burst capacity of the subsequently-produced mature neutrophils (Figure 2A).18 As expected from this mechanism, the magnitude of the PMA-stimulated neutrophil oxidative burst did not correlate directly with the hemolytic component (r= −0.0063, P=0.98). We, therefore, assessed the relationship between neutrophil oxidative burst, neutrophil count (as a marker of neutrophil mobilization) and carboxyhemoglobin (as a systemic marker of HO-1 activity) in SCD subjects using a generalized linear model with an interaction term between carboxyhemoglobin and neutrophil count (allowing for the effect of each to be conditional on the value of the other). All terms, including the interaction between neutrophil count and carboxyhemoglobin, were significantly associated with the magnitude of the PMA-stimulated oxidative burst (Table 2). To illustrate this relationship, including the interaction of terms, we plotted the model-predicted effect of joint variation in carboxyhemoglobin and neutrophil count on oxidative burst (Figure 2B and C). Consistent with our mechanistic biological model,18 this statistical model predicts that the greatest impairment of oxidative burst occurs when carboxyhemoglobin and neutrophil count increase simultaneously, as may be the case during an episode of bacterial infection.
Figure 2.
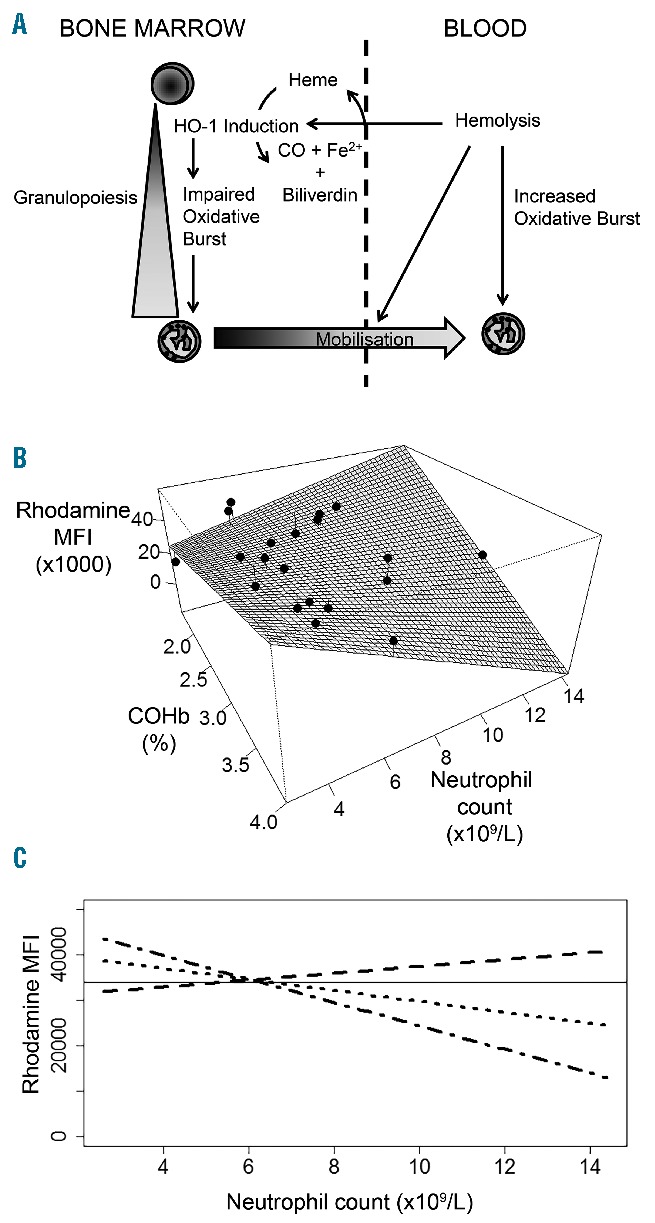
Interacting effects of hemolysis, heme-oxygenase-1 (HO-1) induction, and neutrophil mobilization, on neutrophil oxidative burst. (A) Working model of the interactions between hemolysis, HO-1 induction, and neutrophil oxidative burst. Hemolysis has three interacting effects: stimulating the oxidative burst of mature neutrophils; inducing HO-1 expression in neutrophil progenitors in bone marrow (BM) which leads to impaired oxidative burst capacity during neutrophil maturation; and mobilization of neutrophils from BM to the blood. (B) Interaction between measured carboxyhemoglobin % (COHb, an indicator of hemolysis and HO-1 activity), neutrophil count in peripheral blood, and PMA-stimulated oxidative burst (rhodamine MFI) of neutrophils in patients with sickle cell disease. The fitted surface shows the predicted oxidative burst response (using a generalized linear model), and overlying black dots show individual patient data (n=20). (C) Predicted change in PMA-stimulated oxidative burst with neutrophil count at different levels of COHb [(representing the 25th (dashed line), 50th (dotted line), and 75th centiles (dash-dot line) in subjects with sickle cell disease)]. The horizontal line represents the lowest value observed in control subjects.
Table 2.
Generalized linear model of the effect of carboxyhemoglobin concentration and blood neutrophil count on neutrophil oxidative burst in subjects with sickle cell disease.
In order to confirm the relevance of our mechanistic model of HO-1-dependent impairment of the oxidative burst in human neutrophils, we examined HO-1 expression directly. In healthy donor blood, incubation with hemin results in robust upregulation of HO-1 protein expression in monocytes, but not in neutrophils (Figure 3A). Similarly, there was no evidence that chronic exposure to hemolysis in subjects with SCD resulted in a marked increase in HO-1 expression in circulating neutrophils when compared to healthy controls (Figure 3B). However, HO-1 expression was clearly increased in a CD16intermediate cell population in bone marrow from subjects with SCD when compared with healthy child donors (Figure 4A). During granulopoiesis there is a continuous gradient of increasing CD16 expression,28 suggesting that these CD16intermediate cells may be neutrophil progenitors or immature neutrophils. Additional surface staining demonstrated that these cells had high expression of CD49d, low expression of CD24, intermediate expression of CD15, and variable expression of CD11b (Figure 4B), an expression pattern which would be consistent with the transition stage between proliferating and non-proliferating neutrophil progenitors, and unlikely to include monocytes or erythroid lineage cells.28–30 Together these findings support our mechanistic model in which SCD hemolysis induces HO-1 in neutrophil progenitors, but not in mature neutrophils, similar to our previous findings in mice and in children with malarial hemolysis.18,19
Figure 3.
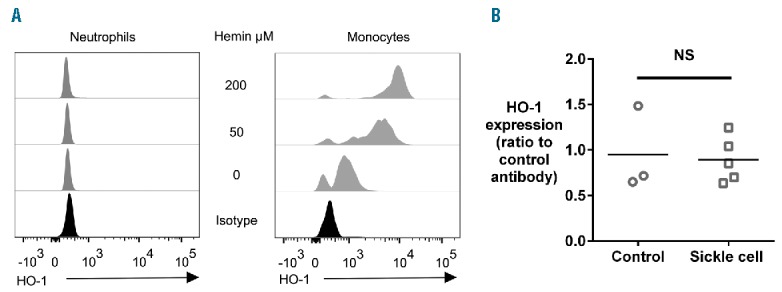
Heme oxygenase-1 (HO-1) is not induced in mature human neutrophils by hemin treatment or hemolysis (A) Illustrative flow cytometry analysis of the effect of incubation of human whole blood with varying concentrations of hemin for 18 h on expression of HO-1 in monocytes and neutrophils. Results shown from one healthy adult donor, representative of 5 independent experiments. (B) HO-1 expression in neutrophils of healthy control subjects (n=3) compared with subjects with sickle cell disease (n=5). The Mann-Witney test was used for comparison.
Figure 4.

Flow-cytometry analysis of heme oxygenase-1 (HO-1) expression in bone marrow (BM) from subjects with sickle cell disease and healthy donors. (A) Comparison of staining with anti-HO-1 antibody and control antibody in subjects with sickle cell disease (n=2) and healthy child BM donors (n=2, one HbAS, one HbAA), plotted against CD16 expression as a marker of neutrophil maturation. The gated population is CD16 intermediate, and is proposed to represent developing neutrophils. (B) Expression of surface markers which define the maturational stage of developing neutrophils in BM. For each subject with sickle cell disease, the expression of surface markers on the HO-1+CD16intermediate population (gated in A) is presented in comparison with the expression of the same markers in total BM (upper panels). The HO-1+ cells express high levels of CD49d, low levels of CD24, intermediate-to-high levels of CD11b, and intermediate levels of CD15, suggesting they are around the point of transition from proliferating to non-proliferating neutrophil progenitors.
To test whether HO-1 induction during human neutrophil granulopoiesis could be responsible for impairment of the oxidative burst, we used the HL-60 promyelocytic leukemia cell line as a model system.31 Incubation of HL-60 cells with 50 μM hemin (a concentration exceeded in the plasma of one-third of our SCD subjects) during their differentiation into neutrophil-like cells resulted in increased expression of HO-1 (Figure 5A), but did not alter morphological maturation (Figure 5B) or cell viability (which was consistently greater than 90%). Hemin treatment impaired the PMA-stimulated oxidative burst of differentiated HL-60 cells, indicating that HO-1 induction during maturation of neutrophil-like HL-60 cells was associated with impairment of the oxidative burst. These findings support a causal role for the hemolysis-induced HO-1 expression in the bone marrow of SCD subjects in impairment of the oxidative burst of circulating neutrophils.
Figure 5.
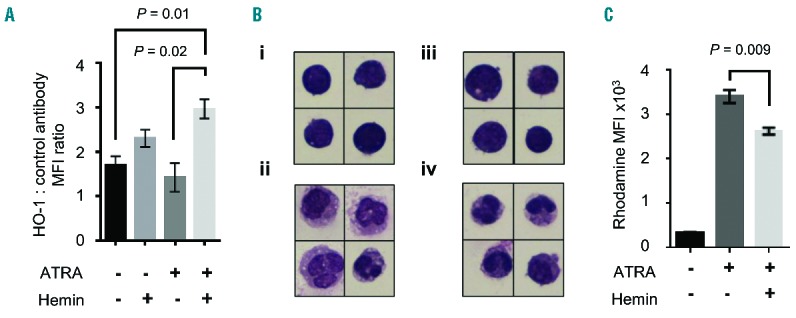
Heme oxygenase-1 (HO-1) induction during maturation impairs the oxidative burst of neutrophil-like differentiated HL60 cells (A) Comparison of HO-1 expression in the promyelocytic HL-60 cell line incubated with or without the HO-1 substrate and inducer, hemin (50 μM), for five days in the presence or absence of 10−7 M all-trans retinoic acid (ATRA) (n=3 per group), which stimulates differentiation into neutrophil-like cells. (B) Representative morphology of hematoxylin and eosin stained HL-60 cells: (i) undifferentiated; (ii) differentiated cells following five days with 10−7 M ATRA; (iii) undifferentiated, hemin treated; (iv) differentiated, hemin treated. (C) Comparison of PMA-stimulated oxidative burst of HL-60 cells, undifferentiated or differentiated into neutrophil-like cells in the presence or absence of hemin (50 μM) (n=3 per group). Results representative of 3 independent experiments. Images were obtained at ×60 magnification using a Nikon Eclipse 50i microscope, all cells shown at the same scale. Statistical comparison was performed using unpaired t-test.
Discussion
Children with SCD are prone to bacteremia and overwhelming sepsis, risks that have been reduced, but not entirely eradicated, by antibiotic prophylaxis and modern conjugate vaccines.2,32 In African children with SCD, infection is thought to be the most common cause of death, and neither penicillin prophylaxis nor current vaccination regimes offer protection against one of the most common causes of NTS bacteremia.6 Splenic dysfunction has long been considered the most important cause of susceptibility to encapsulated bacterial infection in SCD.5 However, children with SCD are also susceptible to infections with other organisms, including S. aureus and Klebsiella,11–13 suggesting that splenic dysfunction may not be the only cause of susceptibility in this population. NTS bacteremia is strongly associated with other diseases which also cause severe hemolysis, particularly malaria and Oroya fever.33 Hydroxyurea treatment of children with SCD reduces levels of hemolysis,15 all-cause mortality,34 and infection-related mortality,17 suggesting a role for hemolysis in promoting susceptibility to infection in this population. We, and others, have previously shown that hemolysis increases susceptibility to NTS sepsis in a mouse model,18,35,36 whereas anemia due to blood loss alone does not.35 We demonstrated that this susceptibility to infection was dependent on hemolysis-derived heme causing induction of HO-1 during granulopoiesis, which resulted in the production of neutrophils with reduced oxidative burst capacity.18 This neutrophil defect was found in the acute and progressive hemolysis that occurs in malaria infections in both mice and humans,18,19 and we predicted that a similar defect might be seen in SCD.18
The results of our current study demonstrate that impairment of the neutrophil oxidative burst occurs in children with SCD. Importantly, we assessed this in children who had never received hydroxyurea therapy, who had not received a blood transfusion within the last four months, and who did not have intercurrent infection. We found that the extent of impairment of neutrophil function was extremely variable, with many children with SCD having values within the “normal” range, but some showing less than 50% of the average in healthy controls. Whilst the magnitude of this defect is substantially less than that seen in chronic granulomatous disease,37 when combined with hyposplenism and poor humoral immunity in young children it may still be sufficient to increase susceptibility to some pathogens such as NTS.38,39 Unlike S. pneumoniae, NTS are well adapted to intracellular survival through virulence mechanisms which specifically perturb the phagocyte oxidative burst,40,41 and may have an additive effect with any defect induced by hemolysis. Thus, impairment of neutrophil oxidative burst capacity may be a key factor explaining the well recognized, but poorly understood, phenomenon of susceptibility to NTS bacteremia and osteomyelitis in SCD.
We found evidence to support our working model (Figure 2A) of the interacting effects of hemolysis, HO-1 activity, neutrophil mobilization and oxidative burst. Cell- free heme, released during hemolysis, promotes neutrophil mobilization from the bone marrow and can prime the oxidative burst, but also induces HO-1 in developing neutrophils which subsequently have reduced oxidative burst capacity.18,19,42 The net effect on the oxidative burst of circulating neutrophils depends on the relative balance of these influences, such that the oxidative burst is most impaired when there is both high HO-1 activity (indicated by high carboxyhemoglobin concentration) and mobilization of neutrophils from the bone marrow (indicated by a high circulating neutrophil count). Others have demonstrated that immature neutrophils are increased in the peripheral circulation in SCD, indicating that there is indeed increased mobilization from the bone marrow.43 The same authors also found that neutrophils from SCD patients exhibited enhanced resistance to apoptosis, a characteristic which could be a consequence of the cytoprotective functions of HO-1 during granulopoiesis.44 Our model may also be influenced by the systemic inflammation of SCD, which contributes to neutrophil mobilization,45 and may promote HO-1 induction and priming of the neutrophil oxidative burst. Interestingly, we found a unimodal distribution of the neutrophil oxidative burst in all subjects with SCD, which is in contrast to our findings in children with malaria, in whom we saw a bimodal response of each individual’s neutrophils following PMA stimulation.19 We speculate that the chronic hemolysis of steady-state SCD results in relatively uniform impairment of neutrophil function, whereas the acute and cyclical pattern of hemolysis in malaria results in a mixture of newly mobilized neutrophils with impaired oxidative burst and circulating neutrophils with normal or enhanced oxidative burst.
In keeping with our previous observations in malaria,18,19 we found HO-1 protein expression in circulating neutrophils was essentially undetectable by flow cytometry, and was not substantially increased by hemin treatment or in SCD. Lanaro et al. have also shown that HMOX1 mRNA expression is very low in neutrophils compared to monocytes, although HMOX1 mRNA was significantly higher in SCD neutrophils than in controls.46 Whether this indicates a limit to the sensitivity of the flow cytometry assay, or that very low-level HMOX1 mRNA and protein expression do not correlate well in neutrophils, is unclear. Interestingly, Lanaro et al. also found HMOX1 mRNA remained elevated in neutrophils of SCD patients treated with hydroxurea,46 although it is not known whether this represents a direct effect of hydroxyurea, or perhaps more severe underlying disease in patients selected for hyroxyurea therapy.
In contrast to our observations in circulating neutrophils, we found that HO-1 was robustly induced in a neutrophil progenitor population in the bone marrow of children with SCD. The small number of bone marrow samples is a limitation of our study, but we supplemented this with an in vitro model of neutrophilic maturation to determine whether heme-induced HO-1 expression could impair the development of normal oxidative burst capacity. The fact that HO-1 could be induced during neutrophilic maturation and resulted in an impairment of the oxidative burst is consistent with our proposed mechanism. Whilst neutrophilic differentiation and maturation of HL-60 cells is not identical to natural granulopoiesis in bone marrow, the observations of this study together with evidence from our previous work, make a compelling case that the impairment of the neutrophil oxidative burst in SCD is dependent on induction of HO-1 in bone marrow. Some important questions about the underlying mechanisms remain unanswered. Why does heme exert differential effects on neutrophil progenitors and circulating neutrophils¿ Does this perhaps relate to differences in the expression of receptors which might mediate the uptake of heme, for example CD91, which binds heme-hemopexin complexes¿47 And how does increased HO-1 expression lead to impairment of oxidative burst¿ Does it degrade heme which is needed for the NADPH oxidase, or do heme degradation products such as carbon monoxide drive more global changes in gene expression through oxidative conditioning¿48 In a mouse model, we found that non-heme induction of HO-1 did not cause susceptibility to NTS, whereas inhibition of HO-1 abrogated the susceptibility to NTS caused by hemolysis, together suggesting that increases in both heme uptake and degradation are necessary to produce neutrophil dysfunction.18 Further studies will be needed to dissect the molecular mechanisms.
The suggestion that poor bactericidal activity of neutrophils may contribute to infection susceptibility in SCD is not entirely new,49 and defects have been demonstrated in the oxidative burst response in adults with SCD,50 although the role of hemolysis has not previously been explored. Our study confirms that this defect is seen in a very well defined group of children with HbSS SCD, and is not confounded by the effect of blood transfusion or disease modifying therapy. To our knowledge, no studies have yet investigated whether the oxidative burst response in SCD is predictive of subsequent risk or outcome of infection. However, in mice, a similar reduction in the magnitude of the oxidative burst prior to infection resulted in greatly diminished resistance to NTS.18 A prospective study will be required to determine whether the extent of impairment of the oxidative burst is clinically significant. Neutrophil oxidative burst can be determined by flow cytometry relatively easily, but as we have shown it might also be predicted from routine measurement of neutrophil count and carboxyhemoglobin concentration, the latter being measurable non-invasively.51
HO-1-dependent suppression of neutrophil reactive oxygen species (ROS) may represent an appropriate adaptive response to hemolysis.18 Increased expression of HO-1 is associated with activation of multiple anti-oxidative response pathways,48,52 although the exact mechanisms by which HO-1 induction leads to impaired neutrophil oxidative burst remain unknown. In the presence of cell-free heme, ROS produced by neutrophils may cause significant tissue damage,47,52 and a mechanism which senses hemolysis-derived heme and constrains the production of neutrophil ROS would be protective. In support of this, genetic polymorphisms in the promoter region of HMOX1 have been shown to influence HO-1 expression,21 and are important modifiers of non-infectious complications of SCD.53 However, a consequence of this adaptive response is impairment of an important mechanism of defence against bacteria, and it would be interesting to investigate whether HMOX1 promoter polymorphisms also influence susceptibility to bacterial infection in SCD. There is considerable interest in harnessing the therapeutic potential of hemoglobin- and heme-scavengers, HO-1 and carbon monoxide in SCD.47,54,55 Our results might be interpreted to suggest that inhibition of HO-1 could be beneficial during infections in SCD, but this would be hazardous because toxic heme would undoubtedly accumulate. A better strategy may be to direct hemolysis-derived heme away from the bone marrow and into specific cell types, such as monocytes and macrophages (which do not show impaired oxidative burst following HO-1 induction19) via the haptoglobin receptor CD163,47 reducing the circulating heme that is available to induce HO-1 during granulopoiesis.
Acknowledgments
The authors would like to thank Dr Elizabeth Jones and Dr Nick Fordham for assistance with collection of clinical information, Meena Paul for assistance with co-oximetry measurements, the staff of the flow cytometry core facility, and the Paediatric phlebotomy, haematology, and administrative staff at Imperial College Healthcare NHS Trust for assistance with identification of eligible subjects and sample collection.
Footnotes
Funding
This study was supported by the Rosetrees Trust, the National Institute for Health Research (NIHR) Biomedical Research Centre based at Imperial College Healthcare NHS Trust and Imperial College London, an Institutional Wellcome Trust Value in People Award (to AJC), and a MRC Clinician Scientist Fellowship (MR/L006529/1; to AJC). The views expressed are those of the authors and not necessarily those of the funders.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Piel FB, Hay SI, Gupta S, et al. Global burden of sickle cell anaemia in children under five, 2010–2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10(7):e1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chakravorty S, Williams TN. Sickle cell disease: a neglected chronic disease of increasing global health importance. Arch Dis Child. 2015;100(1):48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385(9963):117–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramakrishnan M, Moisi JC, Klugman KP, et al. Increased risk of invasive bacterial infections in African people with sickle-cell disease: a systematic review and meta-analysis. Lancet Infect Dis. 2010;10(5):329–337. [DOI] [PubMed] [Google Scholar]
- 5.Booth C, Inusa B, Obaro SK. Infection in sickle cell disease: a review. Int J Infect Dis. 2010;14(1):e2–e12. [DOI] [PubMed] [Google Scholar]
- 6.Williams TN, Uyoga S, Macharia A, et al. Bacteraemia in Kenyan children with sickle-cell anaemia: a retrospective cohort and case-control study. Lancet. 2009;374(9698): 1364–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quinn CT, Rogers ZR, Buchanan GR. Survival of children with sickle cell disease. Blood. 2004;103(11):4023–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bohnsack JF, Brown EJ. The role of the spleen in resistance to infection. Annu Rev Med. 1986;37:49–59. [DOI] [PubMed] [Google Scholar]
- 9.Rogers ZR, Wang WC, Luo Z, et al. Biomarkers of splenic function in infants with sickle cell anemia: baseline data from the BABY HUG Trial. Blood. 2011;117(9): 2614–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Norris CF, Smith-Whitley K, McGowan KL. Positive blood cultures in sickle cell disease: time to positivity and clinical outcome. J Pediatr Hematol Oncol. 2003;25(5):390–395. [DOI] [PubMed] [Google Scholar]
- 11.Akuse RM. Variation in the pattern of bacterial infection in patients with sickle cell disease requiring admission. J Trop Pediatr. 1996;42(6):318–323. [DOI] [PubMed] [Google Scholar]
- 12.Kizito ME, Mworozi E, Ndugwa C, Serjeant GR. Bacteraemia in homozygous sickle cell disease in Africa: is pneumococcal prophylax is justified¿ Arch Dis Child. 2007;92(1): 21–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aken’ova YA, Bakare RA, Okunade MA. Septicaemia in sickle cell anaemia patients: the Ibadan experience. Cent Afr J Med. 1998;44(4):102–104. [PubMed] [Google Scholar]
- 14.Akinsheye I, Alsultan A, Solovieff N, et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118(1):19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kinney TR, Helms RW, O’Branski EE, et al. Safety of hydroxyurea in children with sickle cell anemia: results of the HUG-KIDS study, a phase I/II trial. Pediatric Hydroxyurea Group. Blood. 1999;94(5): 1550–1554. [PubMed] [Google Scholar]
- 16.Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377(9778):1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lobo CL, Pinto JF, Nascimento EM, et al. The effect of hydroxcarbamide therapy on survival of children with sickle cell disease. Br J Haematol. 2013;161(6):852–860. [DOI] [PubMed] [Google Scholar]
- 18.Cunnington AJ, de Souza JB, Walther M, Riley EM. Malaria impairs resistance to Salmonella through heme- and heme oxygenase-dependent dysfunctional granulocyte mobilization. Nat Med. 2011;18(1):120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cunnington AJ, Njie M, Correa S, et al. Prolonged neutrophil dysfunction after Plasmodium falciparum malaria is related to hemolysis and heme oxygenase-1 induction. J Immunol. 2012;189(11):5336–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cunnington AJ, Kendrick SF, Wamola B, et al. Carboxyhemoglobin levels in Kenyan children with Plasmodium falciparum malaria. Am J Trop Med Hyg. 2004;71(1):43–47. [PubMed] [Google Scholar]
- 21.Walther M, De Caul A, Aka P, et al. HMOX1 gene promoter alleles and high HO-1 levels are associated with severe malaria in Gambian children. PLoS Pathog. 2012;8(3): e1002579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shinowara GY, Walters MI. Hematin–studies on protein complexes and determination in human plasma. Am J Clin Pathol. 1963;40:113–122. [DOI] [PubMed] [Google Scholar]
- 23.Breitman TR, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc Natl Acad Sci USA. 1980;77(5):2936–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sears DA, Udden MM, Thomas LJ. Carboxyhemoglobin levels in patients with sickle-cell anemia: relationship to hemolytic and vasoocclusive severity. Am J Med Sci. 2001;322(6):345–348. [DOI] [PubMed] [Google Scholar]
- 25.Coburn RF, Williams WJ, Kahn SB. Endogenous carbon monoxide production in patients with hemolytic anemia. J Clin Invest. 1966;45(4):460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Minniti CP, Sable C, Campbell A, et al. Elevated tricuspid regurgitant jet velocity in children and adolescents with sickle cell disease: association with hemolysis and hemoglobin oxygen desaturation. Haematologica. 2009;94(3):340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nouraie M, Lee JS, Zhang Y, et al. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica. 2013; 98(3):464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elghetany MT, Ge Y, Patel J, et al. Flow cytometric study of neutrophilic granulopoiesis in normal bone marrow using an expanded panel of antibodies: correlation with morphologic assessments. J Clin Lab Anal. 2004;18(1):36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lund-Johansen F, Terstappen LW. Differential surface expression of cell adhesion molecules during granulocyte maturation. J Leukoc Biol. 1993;54(1):47–55. [DOI] [PubMed] [Google Scholar]
- 30.Behbehani GK, Bendall SC, Clutter MR, et al. Single-cell mass cytometry adapted to measurements of the cell cycle. Cytometry A. 2012;81(7):552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fleck RA, Romero-Steiner S, Nahm MH. Use of HL-60 cell line to measure opsonic capacity of pneumococcal antibodies. Clin Diagn Lab Immunol. 2005;12(1):19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baskin MN, Goh XL, Heeney MM, Harper MB. Bacteremia risk and outpatient management of febrile patients with sickle cell disease. Pediatrics. 2013;131(6):1035–41. [DOI] [PubMed] [Google Scholar]
- 33.Orf K, Cunnington AJ. Infection-related hemolysis and susceptibility to Gram-negative bacterial co-infection. Front Microbiol. 2015;6:666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thornburg CD, Files BA, Luo Z, et al. Impact of hydroxyurea on clinical events in the BABY HUG trial. Blood. 2012; 120(22):4304–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaye D, Hook EW. The Influence of Hemolysis or Blood Loss on Susceptibility to Infection. J Immunol. 1963;91:65–75. [PubMed] [Google Scholar]
- 36.Kaye D, Hook EW. The Influence of Hemolysis on Susceptibility to Salmonella Infection: Additional Observations. J Immunol. 1963;91:518–527. [PubMed] [Google Scholar]
- 37.Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363(27):2600–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacLennan CA, Gondwe EN, Msefula CL, et al. The neglected role of antibody in protection against bacteremia caused by nontyphoidal strains of Salmonella in African children. J Clin Invest. 2008;118(4):1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gondwe EN, Molyneux ME, Goodall M, et al. Importance of antibody and complement for oxidative burst and killing of invasive nontyphoidal Salmonella by blood cells in Africans. Proc Natl Acad Sci USA. 2010;107(7):3070–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vazquez-Torres A, Xu Y, Jones-Carson J, et al. Salmonella pathogenicity island 2-dependent evasion of the phagocyte NADPH oxidase. Science. 2000; 287(5458):1655–1658. [DOI] [PubMed] [Google Scholar]
- 41.de Jong HK, Parry CM, van der Poll T, Wiersinga WJ. Host-pathogen interaction in invasive Salmonellosis. PLoS Pathog. 2012;8(10):e1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porto BN, Alves LS, Fernandez PL, et al. Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J Biol Chem. 2007;282(33):24430–24436. [DOI] [PubMed] [Google Scholar]
- 43.Almeida CB, Favero ME, Pereira-Cunha FG, et al. Alterations in cell maturity and serum survival factors may modulate neutrophil numbers in sickle cell disease. Exp Biol Med (Maywood). 2011;236(11):1239–1246. [DOI] [PubMed] [Google Scholar]
- 44.Rushworth SA, MacEwan DJ. HO-1 underlies resistance of AML cells to TNF-induced apoptosis. Blood. 2008;111(7):3793–3801. [DOI] [PubMed] [Google Scholar]
- 45.Beckman JD, Belcher JD, Vineyard JV, et al. Inhaled carbon monoxide reduces leukocytosis in a murine model of sickle cell disease. Am J Physiol Heart Circ Physiol. 2009;297(4):H1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lanaro C, Franco-Penteado CF, Albuqueque DM, et al. Altered levels of cytokines and inflammatory mediators in plasma and leukocytes of sickle cell anemia patients and effects of hydroxyurea therapy. J Leukoc Biol. 2009;85(2):235–242. [DOI] [PubMed] [Google Scholar]
- 47.Schaer DJ, Buehler PW, Alayash AI, et al. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood. 2013;121(8):1276–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bilban M, Haschemi A, Wegiel B, et al. Heme oxygenase and carbon monoxide initiate homeostatic signaling. J Mol Med. 2008;86(3):267–279. [DOI] [PubMed] [Google Scholar]
- 49.Humbert JR, Winsor EL, Githens JM, Schmitz JB. Neutrophil dysfunctions in sickle cell disease. Biomed Pharmacother. 1990;44(3):153–158. [DOI] [PubMed] [Google Scholar]
- 50.Qari MH, Zaki WA. Flow cytometric assessment of leukocyte function in sickle cell anemia. Hemoglobin. 2011;35(4):367–381. [DOI] [PubMed] [Google Scholar]
- 51.Caboot JB, Jawad AF, McDonough JM, et al. Non-invasive measurements of carboxyhemoglobin and methemoglobin in children with sickle cell disease. Pediatr Pulmonol. 2012;47(8):808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. 2010;50:323–354. [DOI] [PubMed] [Google Scholar]
- 53.Bean CJ, Boulet SL, Ellingsen D, et al. Heme oxygenase-1 gene promoter polymorphism is associated with reduced incidence of acute chest syndrome among children with sickle cell disease. Blood. 2012;120(18): 3822–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Belcher JD, Young M, Chen C, et al. MP4CO, a pegylated hemoglobin saturated with carbon monoxide, is a modulator of HO-1, inflammation, and vaso-occlusion in transgenic sickle mice. Blood. 2013; 122(15):2757–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Belcher JD, Mahaseth H, Welch TE, et al. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J Clin Invest. 2006; 116(3):808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]