

Abstract
Arousal and stress critically regulate memory formation and retention. Increasing levels of stress produce an inverted U-shaped effect on cognitive performance, including the retention of explicit memories, and experiencing a severe stress during a traumatic event may lead to posttraumatic stress disorder (PTSD). The molecular mechanisms underlying the impairing effect of a severe stress on memory and the key contribution of traumatic experiences toward the development of PTSD are still unknown. Here, using increasing footshock intensities in an inhibitory avoidance paradigm, we reproduced the inverted U-shaped curve of memory performance in rats. We then show that the inverted U profile of memory performance correlates with an inverted U profile of corticosterone level in the circulation and of brain-derived neurotrophic factor, phosphorylated tropomyosin-receptor kinase B, and methyl CpG binding protein in the dorsal hippocampus. Furthermore, training with the highest footshock intensity (traumatic experience) led to a significant elevation of hippocampal glucocorticoid receptors. Exposure to an unpredictable, but not to a predictable, highly stressful reminder shock after a first traumatic experience resulted in PTSD-like phenotypes, including increased memory of the trauma, high anxiety, threat generalization, and resistance to extinction. Systemic corticosterone injection immediately after the traumatic experience, but not 3 d later, was sufficient to produce PTSD-like phenotypes. We suggest that, although after a first traumatic experience a suppression of the corticosterone-dependent response protects against the development of an anxiety disorder, experiencing more than one trauma (multiple hits) is a critical contributor to the etiology of PTSD.
SIGNIFICANCE STATEMENT Increasing levels of stress produce an inverted U-shaped effect on memory retention. Humans experiencing an acute trauma may develop posttraumatic stress disorder (PTSD), but the key contributions of trauma to PTSD formation are still unknown. This study in rats shows that a single traumatic experience leads to memory impairment, accompanied by blunted activations of circulating corticosterone and of plasticity molecular changes in the hippocampus. Experiencing a traumatic, unpredictable reminder, but not a repetition of the same trauma (predictable), leads to high anxiety, threat memory generalization, and extinction failure, typical responses of anxiety disorders and PTSD. Thus, although a first trauma elicits inhibiting responses, which may be protective, experiencing more than one unpredictable trauma is a critical contributor of PTSD etiology.
Keywords: hippocampus, memory, molecular mechanisms, PTSD, stress, trauma
Introduction
Although low levels of stress or arousal promote strong and adaptive behavioral responses, high or prolonged stresses can lead to cognitive impairments and may cause or promote mental health disorders (de Kloet et al., 2005; de Quervain et al., 2009). This inverted U curve describing performance under the effect of stress, known as the Yerkes–Dodson law, applies to numerous functions, including memory (Calabrese, 2008). Indeed, increasing levels of arousal or stress are beneficial for strengthening memory up to an optimal level, after which, however, an additional rise in stress leads to a decrease or loss of memory (Cahill and McGaugh, 1996; Diamond et al., 2007; Sandi and Pinelo-Nava, 2007). Notably, this inverted U effect is observed in hippocampal-dependent memories (Okuda et al., 2004; Salehi et al., 2010; Kaouane et al., 2012), whereas simple memories, such as cued threat conditioning or broadly speaking amygdala-dependent tasks, exhibit a proportional increase in memory retention with increasing stress levels (Cordero et al., 2002; Rau et al., 2005). The inverted U effect of stress on memory is mimicked by the stress hormones corticosteroids [corticosterone (cort) in rodents and cortisol in humans], and this effect is mediated by their low-affinity receptors, the glucocorticoid receptors (GRs). In fact, administration of low doses of cortisol or cort facilitates hippocampal-dependent memories in rodents (Okuda et al., 2004; Kaouane et al., 2012) and declarative memories in humans (Schilling et al., 2013), whereas high doses of cort lead to memory impairment (de Quervain et al., 1998; Kaouane et al., 2012).
The formation and strength of long-term memory rely on a process known as consolidation, which leads to a long-lasting memory storage (McGaugh, 2000; Dudai, 2012). The consolidation of hippocampal-dependent memories requires the activation of numerous molecular pathways, among which those leading to cAMP response element-binding protein (CREB)-dependent gene expression play an evolutionarily conserved role (Alberini, 2009). The molecular mechanisms associated with adaptive memory responses have been relatively well investigated: protein kinase signaling patterns are associated with fear memories (Tronson et al., 2012). Stress-induced memory formation and enhancement (adaptive response) requires GR-dependent activation of the mitogen-associated protein kinase (MAPK) pathway, early growth response protein 1 pathways, and synapsin-Ia/Ib. (Revest et al., 2005, 2010). Recently, we showed that the consolidation of rat inhibitory avoidance (IA) memory evoked by a footshock that induces a strong memory formation requires that GRs in the hippocampus activate the brain-derived neurotrophic factor (BDNF) and CREB pathways (Chen et al., 2012a). Furthermore, autoregulation of BDNF expression sustains its own activation for >24 h, thus allowing memory consolidation to occur. This molecular activation returns to baseline by 2 d after training (Bambah-Mukku et al., 2014). Finally, the enhancement of fear memory induced by glucocorticoids requires the BDNF–tropomyosin-receptor kinase B (TrkB) signaling through extracellular signal-regulated kinase 1/2 MAPK phosphorylation (Revest et al., 2014).
However, the molecular mechanisms underlying severe stress-induced memory impairment remain to be understood. In addition, it is not clear whether this memory impairment is attributable to deficit of consolidation or retrieval/expression of the memory. It is also unknown how a traumatic event contributes to the etiology of anxiety pathologies, such as acute stress disorder or posttraumatic stress disorder (PTSD); indeed, although stress and glucocorticoids are known to be critical contributors of the PTSD etiology, only a fraction of ∼10–20% of the individuals exposed to a traumatic event actually develops PTSD (Kessler et al., 2005; Pitman et al., 2012). Understanding the effect and mechanisms underlying memory formation after a traumatic experience is important for understanding the etiology of acute stress disorder and PTSD.
In this study, we sought to determine whether increasingly stressful episodic experiences (i.e., increasing the intensity of the footshock in an IA paradigm) in rats lead to the inverted U-curve effect on memory retention. We then investigated the circulating cort response and the molecular mechanisms in the dorsal hippocampus and medial prefrontal cortex (mPFC) that accompany the stress-memory inverted U effect. Finally, we characterized behavioral effects and mechanisms associated with trauma re-experiencing in predictable and unpredictable conditions.
Materials and Methods
Animals.
Long–Evans male rats weighing between 200 and 250 g at the beginning of the experiments (∼8–9 weeks old) were used. Rats were housed by pair in a 12 h light/dark cycle with ad libitum access to food and water. Experiments were performed during the light cycle. Rats were handled for 3 min/d for 5 consecutive days before any procedure. All protocols complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the New York University Animal Welfare Committee.
IA.
IA was performed as described previously (Chen et al., 2012a). The IA chamber (Med Associates) consisted of a rectangular Perspex box divided into a safe and a shock compartment separated by an automatically operated sliding door. The safe compartment was white and illuminated, and the shock compartment was black and dark. During training, rats were placed into the safe compartment with their heads facing away from the door. After 10 s, the door separating the compartments was opened automatically, allowing the rat access to the shock compartment. The door closed 1 s after the rat entered the dark compartment, and a 2 s/0.6, 0.9, 1.5, or 3 mA intensity footshock was delivered to the grid floor of the shock chamber via a constant current scrambler circuit. After footshock, rats were allowed to stay 10 s in the dark compartment and were then returned to their home cages. Memory retention was measured at the indicated time points after training by placing the rat back into the lit compartment and recording their latency (in seconds) to enter the dark compartment. No footshock was administered during retention tests. Test was terminated once the rat entered the dark compartment or after a 900 s cutoff limit. Activity during retention test was measured as the number of times a rat consecutively broke two of the four infrared beams equally distributed in the light compartment. The unconditioned stimulus (US), which was delivered in the experiment depicted in Figure 1C, was done by placing the rat into the dark compartment, immediately followed by a 2 s electrical footshock. After the footshock, rats were returned immediately to their home cages. The reminder shock (RS) consisted of exposing the rats to a 2 s electrical footshock in a different context, namely a transparent box placed in an illuminated room to avoid any association with the dark compartment. Immediately after the shock, animals were returned to their home cages. In retraining experiments, rats were trained in IA and tested for memory retention 2 d later. After entering into the dark compartment, rats received a footshock. If memory reached the 900 s cutoff limit, the rat was gently forced to enter into the dark compartment for retraining.
Figure 1.

Inverted U function between IA memory retention and increasing footshock intensity; anxiety and context generalization. Experimental schedules are shown above each panel. A, Data are shown as mean ± SEM latency expressed in seconds. Latency was measured at 2 d (T1) and 7 d (T2) after TR that was elicited with a 0.6, 0.9, 1.5, or 3 mA footshock. B, Data are expressed as the number ± SEM of beam breaks per minute. Animals' activity was tested at 2 d (T1) and 7 d (T2) after training. C, Latency was tested 2 d after the presentation of the 0.6, 1.5, or 3 mA footshock as an US (T1) and compared with the latency of a different group at TR. D, Latency was measured in context B (ctx B) 2 d after TR. E–H, Open-field test (OFT) after TR and an RS. Data are expressed as the mean ± SEM of total activity, center activity, center time, and center/total activity. Total activity in the arena (E), activity in the center of the arena (F), time spent in the center of the arena (G), and ratio of center activity over total activity (H) were measured in naive and trained animals with a 0.6, 1.5, or 3 mA footshock 2 d after training. *p < 0.05, **p < 0.01, ***p < 0.001.
IA extinction.
To elicit IA extinction learning, memory retention was tested once a day for 5 consecutive days. In each session, after entering the shock (dark) compartment, rats were confined there for 5 min. Animals that exhibited the cutoff latency were gently forced into the dark compartment. At the end of the extinction protocol, rats received an RS to elicit the reinstatement of extinguished memories. An alternative protocol of extinction used for experiments shown in Figure 5C consisted in conducting IA testing twice a day with 30 min intervals between sessions for 7 consecutive days.
Figure 5.
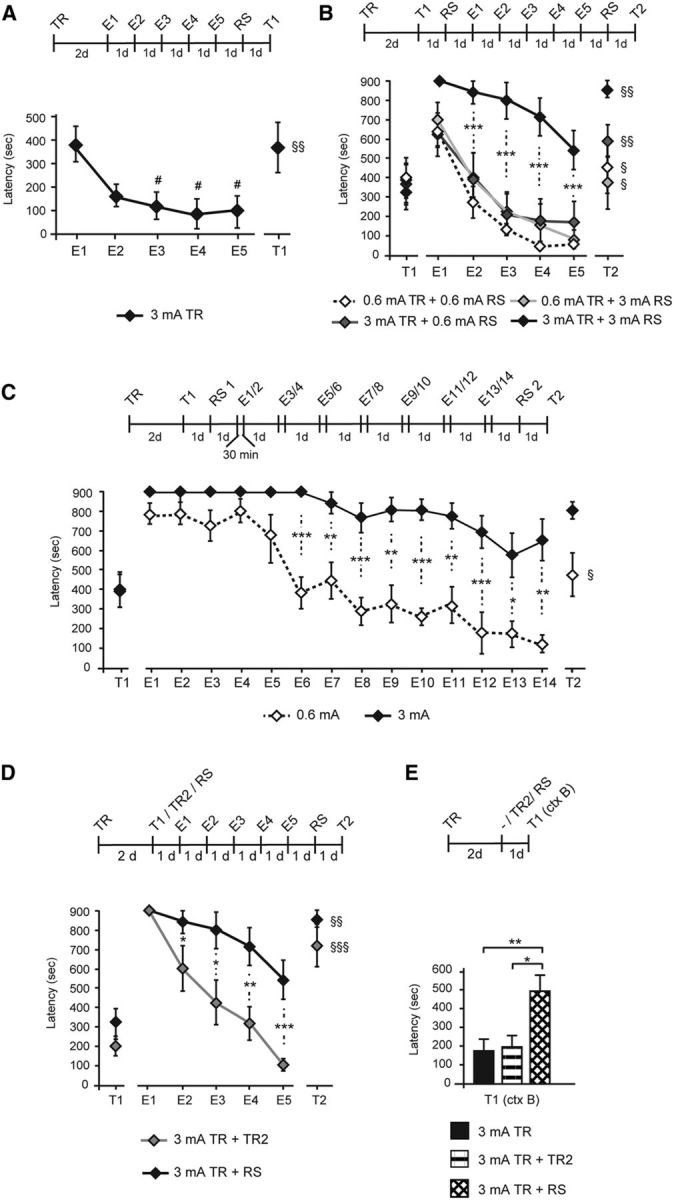
Training with a strong footshock followed by a strong RS elicits extinction-resistant memories. Experimental schedule is shown above each panel. Data are expressed as the mean ± SEM of latency (in seconds) to enter the dark compartment. A, Memory was tested for 5 consecutive days from day 2 after 3 mA-induced training (E1–E5). At the end of each testing session, animals were enclosed for 5 min into the dark compartment of the IA chamber to promote memory extinction. An RS of the same intensity as that received at training was delivered 1 d after E5 and memory was tested for reinstatement 1 d later (T1). #p < 0.05 (vs T1); §§p < 0.01 (E5 vs T1). B, Memory was tested 2 d after training elicited with 0.6 or 3 mA intensity. One day after T1, an RS of 0.6 or 3 mA intensity was delivered, and memory was tested for 5 consecutive days (E1–E5) with the same protocol described in A. An RS of the same intensity as that received at training was delivered 1 d after E5, and memory was tested 1 d later for reinstatement (T2). Data are expressed as the mean ± SEM of latency (in seconds) to enter the dark compartment. ***p < 0.001; §p < 0.05 and §§p < 0.01 (E5 vs T2). C, Rats trained with 0.6 or 3 mA intensity footshock were tested 2 d after training (T1). An RS of the same intensity as during training was delivered 1 d later, and rats were tested twice a day with an intra-testing interval of 30 min for 7 consecutive days (E1–E14). One day after E14, an RS (RS2) was delivered, and memory was tested 1 d later (T2). *p < 0.05, **p < 0.01, ***p < 0.001; §p < 0.05 (T15 vs T16). D, Re-experiencing the same traumatic experience does not elicit a traumatic memory phenotype. Memory was tested at 2 d after a 3 mA training (T1). At the end of T1, rats were either retrained at 3 mA intensity (TR2) or were exposed to a 3 mA RS. Extinction protocol was given for 5 consecutive days (E1–E5). One day after E5, an 3 mA RS was delivered, and memory was tested 1 d later (T2) for reinstatement. *p < 0.05, **p < 0.01, ***p < 0.001; §§p < 0.01 and §§§p < 0.001 (E5 vs T2). E, Two days after training elicited with a 3 mA footshock, rats were left in their home cages (−), retrained at 3 mA, or exposed to a 3 mA RS, and memory was tested for IA generalization in a modified context (ctx B) 1 d later (T1). *p < 0.05, **p < 0.01.
IA context generalization.
Rats were trained in IA, and memory was tested in a different shuttle box (context B). This modified IA chamber consisted of a rectangular Perspex box divided into a light and a dark compartment with modified shape, wall colors and patterns, and a colored plastic board in place of the floor grid.
Open-field test.
The open-field test was conducted in a brightly illuminated (210 lux) square arena (1 × 1 × 0.5 m). Rats were placed in a corner of the arena, and activity was monitored for 10 min using a video camera mounted above the center of the arena. Activity was analyzed by an experimenter blind to experimental conditions. The open-field arena was virtually divided into 16 identical squares. Animals' activity was measured by scoring the number of times they moved from one square to another. Activity in the entire arena (total activity), activity in the four squares composing the center of the arena (center activity), time spent in the four squares composing the center of the arena (center time), and the ratio of total activity over center activity were quantified.
Whole protein extracts, synaptoneurosomal preparations, and Western blots were performed as described previously (Chen et al., 2012a). Dorsal hippocampi or mPFC were dissected rapidly in cold dissection buffer (in mm: 2.6 KCl, 1.23 sodium phosphate monobasic, 26 sodium bicarbonate, 5 kynurenic acid, 212 sucrose, 10 dextrose, 0.5 CaCl2, and 1 MgCl2), followed by homogenization in buffer containing 10 mm HEPES, 2 mm EDTA, 2 mm EGTA, 0.5 mm DTT, 2 mm NaF, 1 μm microcystin, 1 mm benzamidine, and phosphatase and protease inhibitor mixtures (Sigma-Aldrich). A glass–Teflon homogenizer was used, and homogenates were either used directly for obtaining whole extracts or filtered sequentially through a 100 μm nylon mesh filter and a 5 μm nitrocellulose filter to generate synaptoneurosome preparations. Synaptoneurosome fractions were next obtained by resuspending pellets in homogenization buffer after centrifugation at 1000 × g for 10 min. For whole protein extracts, 0.2 m NaCl was added to lysates before Polytron homogenization. Protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad). For Western blot analyses, equal amounts of protein (10–20 μg) to which 10% β-mercaptoethanol was added were resolved using 10–15% denaturing SDS-PAGE and transferred to Immunobilon-FL PVDF membranes (Millipore) by electroblotting. The following primary antibodies were used: actin (1:2000; Santa Cruz Biotechnology), activity-regualted cytoskeleton-associated protein (Arc; 1:1000; Synaptic Systems), BDNF (1:200; Santa Cruz Biotechnology), phospho(Ser3)–cofilin (1:1000; Cell Signaling Technology), phospho(Ser133)–CREB (1:1000; Millipore), CREB (1:1000; Cell Signaling Technology), methyl CpG binding protein (MeCP2; 1:1000; Cell Signaling Technology), phospho(Y817)–TrkB (1:1000; Abcam), and TrkB (1:1000; Cell Signaling Technology). The following secondary antibodies were used: anti-mouse IRDye 680 (1:10,000; LI-COR) or anti-rabbit IRDye 800 (1:10,000; LI-COR). Membranes were scanned using the Odyssey Infrared Imaging System (LI-COR), and images were analyzed with Image Studio software (LI-COR). Integrated intensities were normalized to corresponding values for actin expression and expressed as fold changes relative to the control value.
Cort analysis.
Plasma cort levels collected from trunk blood were analyzed using Cayman EIA corticosterone kit (Cayman Chemical Company), according to the instructions of the manufacturer.
Cort injections.
Different doses of cort (0.5, 3.0, and 10.0 mg/kg; Sigma-Aldrich) were dissolved in 5% ethanol and administered subcutaneously. Control animals were injected with vehicle. All preparations of cort were made fresh before injections.
Statistical analyses.
Statistical analyses were performed using one- or two-way ANOVA followed by Tukey's HSD post hoc test for Western blot experiments, Bonferroni's post hoc test for behavioral experiments, or Dunnett's post hoc test when all groups were compared with a single control. Unpaired or paired two-sided Student's t tests were used to compare values from two independent sets of data or data from repeated measures on single animals, respectively.
Results
IA memory retention follows an inverted U function with increasing footshock intensity
To examine the effect of increasing levels of stress on long-term memory, IA training (TR) in rats was elicited using increasing footshock intensities (0.6, 0.9, 1.5, and 3 mA), and memory retention was tested 2 and 7 d after training (T1 and T2, respectively). As shown in Figure 1A, although, compared with acquisition, all groups of rats had significant memory retention, increasing footshock intensity produced retention latencies that followed an inverted U curve at both T1 and T2. Two-way ANOVA comparing latencies across shock intensity (0.6, 0.9, 1.5, and 3 mA) and tests (T1 and T2) showed a significant effect of shock intensity (F(2.72) = 12.60, p < 0.001), no effect of test (F(2.72) = 0.154, p = 0.704), and no shock intensity × test interaction (F(2.72) = 0.54, p = 0.655; n = 10). Bonferroni's post hoc tests revealed that, compared with 0.6 mA, memory evoked by 1.5 mA footshock was significantly increased at T1 (p = 0.005) and T2 (p = 0.001). However, 3 mA footshock led to a significant memory decrease when compared with 1.5 mA at both T1 (p = 0.002) and T2 (p = 0.026).
Locomotor activity did not change with shock intensity and/or day of test (Fig. 1B), as revealed by two-way ANOVA comparing activity across shock intensity (F(2.72) = 1.75, p = 0.164), day of test (F(3.96) = 0.11, p = 0.741), and shock intensity × day of test interaction (F(2.72) = 0.20, p = 0.894; n = 8). Exposure to the footshocks alone (US) did not elicit any IA latency 2 d later (T1; Fig. 1C; one-way ANOVA comparing latency at acquisition and T1, F(2.09) = 1.05, p = 0.411; n = 7–8).
Because strong contextual memories may generalize to different contexts (Vervliet et al., 2013), we tested the retention of the 3 mA-induced memories in a context different from that where training occurred (context B). The 0.6 mA- and 1.5 mA-trained groups were used for comparison. Memory was tested in context B two days after training (T1). As shown in Figure 1D, only rats trained with a 3 mA footshock exhibited significant context generalization after training, as revealed by their significantly higher latency for context B compared with that of rats trained at 0.6 mA.
The open-field test was used to assess anxiety-like behavior revealed by total activity, activity in the center of the arena, time spent in the center of the arena, and the ratio of center/total activity, 2 d after training elicited by 0.6, 1.5, or 3 mA footshock (Fig. 1E–H). One-way ANOVA showed no differences after training among the groups (n = 10–12) in total activity (F(2.79) = 1.20, p = 0.319), center activity (F(2.79) = 2.34, p = 0.085), and time spent in the center of the arena (F(2.79) = 2.56, p = 0.066; Fig. 1E–G), whereas a significant decrease of the ratio of center activity/total activity was observed only in the 3 mA-trained group when compared with the naive or 0.6 mA-trained group [F(2.79) = 4.88, p = 0.005; Bonferroni's post hoc test showed significant differences of 3 mA-trained group compared with the naive or 0.6 mA-trained group (p = 0.025 and p = 0.014, respectively; Fig. 1H)].
Together, these data indicate that the retention of IA memory elicited by increasing footshock intensity follows an inverted U distribution and that only the rats trained with the highest shock intensity, which leads to memory impairment, show significant anxiety and generalization.
Molecular correlations of the inverted U memory retention curve: inverted U concentrations of hippocampal levels of BDNF, pTrkB, and MeCP2
In search for mechanisms underlying the inverted U memory retention curve in response to stress, we examined molecular changes in the dorsal hippocampus and mPFC, brain regions known to be involved in contextual threat and traumatic memories encoding, consolidation, and expression (Squire et al., 2004; Maren et al., 2013). Molecular investigations were performed on the groups of rats that represent the inverted U memory retention curve, i.e., after a 0.6, 1.5, or 3 mA footshock-induced training.
Western blot analyses of dorsal hippocampal extracts were done at 1 h and/or 20 h after training, time points at which previous studies established that IA training elicits significant changes in plasticity markers in the dorsal hippocampus (Alberini, 2009; Chen et al., 2012a). Specifically, we determined the levels of Arc, pCREB, BDNF, pTrkB, and pcofilin, all of which have been reported previously to have a critical role in long-term plasticity and memory formation (Silva et al., 1998; Cunha et al., 2010; Chen et al., 2012a). Furthermore, because MeCP2 plays a key role in the regulation of BDNF expression (Chahrour et al., 2008) and contributes critically to synaptic plasticity, learning, and memory (Na et al., 2013), we also monitored the expression levels of this transcriptional regulator. Finally, we investigated the expression level of GRs, because they play a critical role in stress response and memory consolidation (Chen et al., 2012a; Finsterwald and Alberini, 2014).
As shown in Figure 2A, compared with naive conditions, at 1 h after training, there was a significant induction of Arc [one-way ANOVA (F(2.98) = 4.16, p = 0.016; n = 7–8) followed by Dunnett's post hoc tests at 0.6 mA (p = 0.036), 1.5 mA (p = 0.003), and 3 mA (p = 0.026)] and pCREB [one-way ANOVA (F(2.82) = 4.23, p = 0.01; n = 7–8) followed by Dunnett's post hoc test at 0.6 mA (p = 0.008), 1.5 mA (p = 0.013), and 3 mA (p = 0.014)]. Levels of BDNF, pTrkB, and GR did not change (data not shown). Thus, the initial activation or induction of fundamental hippocampal plasticity mechanisms critical for long-term memory formation occurs in all conditions, irrespectively of the intensity of the threat and memory performance.
Figure 2.
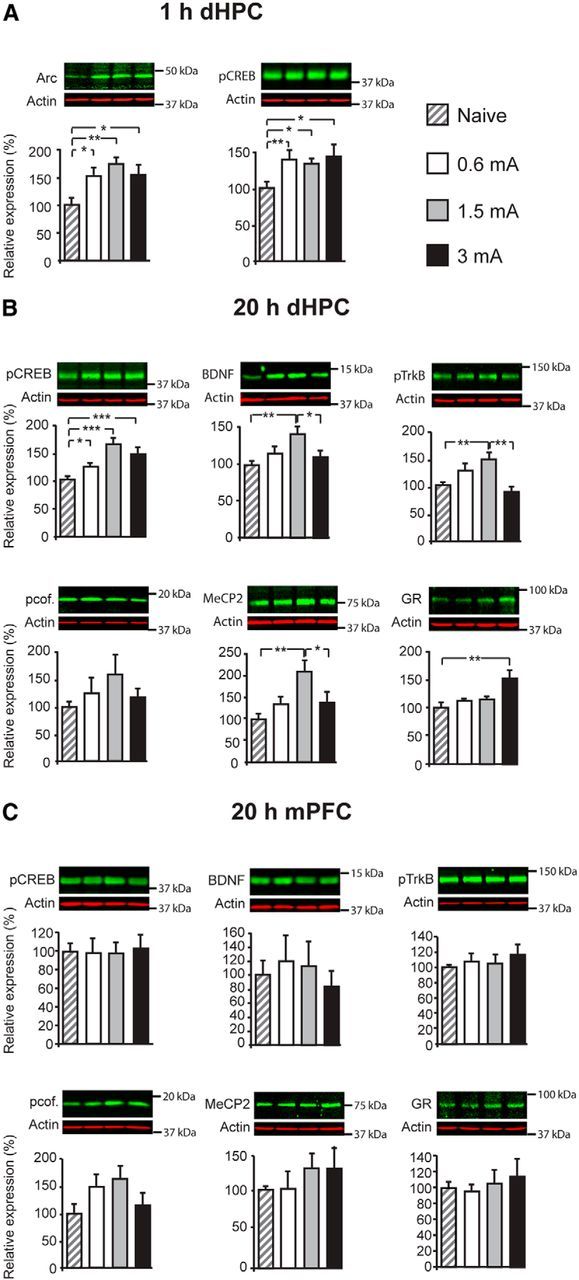
Inverted U in the dorsal hippocampus (dHPC) molecular correlates. Western blot examples are shown above each quantitative panel. A, Levels of Arc and pCREB at 1 h after training elicited with a 0.6, 1.5, or 3 mA footshock. B, Levels of pCREB, BDNF, pcofilin (pcof.), MeCP2, and GR in dHPC total fraction and of pTrkB in synaptoneurosomal fraction at 20 h after training elicited with a 0.6, 1.5, or 3 mA footshock. C, Expression of pCREB, BDNF, pcof., MeCP2, and GR in mPFC total fraction and of pTrkB in mPFC synaptoneurosomal fraction at 20 h after training elicited with a 0.6, 1.5, or 3 mA footshock. *p < 0.05, **p < 0.01, ***p < 0.001.
The significant increase in pCREB persisted at 20 h after training in all groups [Fig. 2B; one-way ANOVA (F(2.76) = 9.26, p < 0.001; n = 15–16) followed by Dunnett's post hoc test at 0.6 mA (p = 0.034), 1.5 mA (p < 0.001), and 3 mA (p = 0.001)]. However, at 20 h after training, a time point at which a relatively mild footshock is known to increase the expression of BDNF (Bambah-Mukku et al., 2014), the levels of BDNF and synaptic pTrkB displayed an inverted U profile that paralleled that of memory retentions. One-way ANOVA (F(2.83) = 4.71, p = 0.06; n = 10–12) followed by Tukey's HSD post hoc test revealed that BDNF levels increased significantly after training with 1.5 mA compared with naive (p = 0.005), but that 3 mA training significantly decreased BDNF levels compared with 1.5 mA training (p = 0.049; Fig. 2B). Statistical analysis of pTrkB expression in synaptoneurosomal fractions by one-way ANOVA (F(2.76) = 6.57, p = 0.01; n = 15–16) followed by Tukey's HSD post hoc test revealed a similar significant increase of pTrkB at 1.5 mA compared with naive (p = 0.008) and a significant decrease at 3 mA compared with 1.5 mA (p = 0.001; Fig. 2B). Analysis of pTrkB expression levels also in the whole dorsal hippocampus extract had a similar inverted U profile with increasing shock intensities (data not shown). Furthermore, pcofilin also displayed an inverted U profile, but the changes did not reach any statistical significance [one-way ANOVA (F(2.82) = 0.93, p = 0.436; n = 12)]. The concentration of MeCP2 also followed an inverted U profile at 20 h after training; one-way ANOVA (F(2.87) = 6.46, p = 0.001; n = 10) followed by Tukey's HSD post hoc test revealed a significant increase of MeCP2 at 1.5 mA compared with naive (p = 0.001) and a significant blunting at 3 mA compared with 1.5 mA (p = 0.011). The levels of GRs were significantly higher at 20 h after training only in the 3 mA-trained group compared with naive [one-way ANOVA (F(2.87) = 5.57, p = 0.003; n = 8–12) followed by Dunnett's post hoc test (p < 0.001); Fig. 2B].
In the mPFC, we tested the 20 h posttraining time point. Although it appeared that BDNF and pcofilin displayed a trend toward an inverted U profile, none of the markers changed significantly with increased footshock intensity at training compared with the naive condition (Fig. 2C), as revealed by one-way ANOVA for the expression of pCREB (F(2.95) = 0.04, p = 0.99; n = 8), BDNF (F(3.95) = 0.30, p = 0.827; n = 4), pcofilin (F(2.95) = 1.79, p = 0.173; n = 8), MeCP2 (F(3.49) = 0.55, p = 0.657; n = 4), GR (F(2.95) = 0.32, p = 0.807; n = 8), and pTrkB (F(2.95) = 0.32, p = 0.807; n = 8). Finally, no changes in CREB and TrkB total protein levels were found in the dorsal hippocampus or mPFC (data not shown). Together, these data indicate that some hippocampal markers critical for plasticity and memory consolidation, such as Arc and pCREB, are induced by training independently of the intensity of the footshock—and hence threat—experienced and independently of the inverted U effect on memory retention. However, interestingly, the concentration of BDNF, pTrkB, and MeCP2 in the dorsal hippocampus parallels the inverted U profile of memory retention.
The impaired memory is reinstated by a reminder shock
To determine whether the memory deficit of the 3 mA group results from a disruption of memory consolidation, therefore reflecting loss of memory formation, or a deficit in memory retrieval or expression, we administered a reminder footshock (RS) in a different context that had no associations with the dark compartment, a procedure known to reinstate decreased threat memory responses such as that observed in extinction training (Inda et al., 2011). Hence, rats trained with a 3 mA footshock were given an RS of the same intensity 1 d after T2 and were tested 1 d later (T3). As shown in Figure 3A, one-way ANOVA followed by Bonferroni's post hoc tests revealed that memory was reinstated significantly after RS [(F(3.35) = 16.36, p < 0.001; n = 10; T3 compared with T1 and T2, p < 0.001 for both)], and most rats reached the maximal (cutoff) latency level, indicating that the decrease in retention was likely attributable to impaired memory retrieval or expression rather than impaired consolidation. Administering the 3 mA RS 1 d after T1 (Fig. 3B; paired two-sided Student's t test, p < 0.001; n = 9) or 1 d after training without previous testing (Fig. 3C; n = 8) also increased significantly memory retention and produced latencies that were almost all at the maximal level (cutoff latency). Hence, a 3 mA RS given at 1, 3, or 8 d after training, irrespectively of posttraining testing session, converts a high-stress-induced memory impairment into a very robust memory expression. Furthermore, the 3 mA-induced memory after the RS of the same intensity in a different context significantly increased memory generalization, which was not found in the other groups of rats trained at 3 mA and exposed to an RS of either 0.6 or 1.5 mA (Fig. 3D).
Figure 3.

An RS in a different context produces a very strong memory and high anxiety. A, Latency was tested at 2 d (T1) and 7 d (T2) after TR elicited with a 3 mA footshock; a 3 mA RS was delivered 1 d after T2, and memory was tested 1 d later (T3). B, Latency was measured 2 d after training (T1) and after the RS delivered 1 d after T1 (T2). C, Latency was tested 1 d after exposure to an RS that was delivered 1 d after a 3 mA training (T1). D, Latency was measured in context B (ctx B) 1 d after RS. E–H, Open-field test (OFT). Data are expressed as the mean ± SEM of total activity, center activity, center time, and center/total activity measured 1 d after exposure to a 3 mA RS that was delivered 2 d after a 3 mA-induced training. **p < 0.01, ***p < 0.001.
Finally, the 3 mA RS after 3 mA training promoted significant anxiety-like behavior. All the open-field measures were significantly and dramatically reduced compared with naive controls [Fig. 3E–H; unpaired two-sided Student's t test (n = 17–18) in total activity (p < 0.001), center activity (p < 0.001), center time (p < 0.001), and center/total activity (p = 0.001)].
Together, these results indicate that a highly stressful reminder of the traumatic experience significantly reinstates retention of the memory, which additionally generalizes to other contexts, and dramatically increases anxiety-like behavior.
The blunted induction of BDNF, pTrkB, and MeCP2 after a traumatic event does not change after the RS
We then asked whether molecular changes that occur after training were influenced by the administration of an RS that, as shown in Figure 3A–C, dramatically increases or reinstates memory retention. In particular, we asked whether BDNF and pTrkB concentrations, which exhibit inverted U profiles after training, correlate with the levels of memory retention or the traumatic aspect of the first experience. Rats were trained at 3 mA and 1 d later received an RS of the same shock intensity. Hippocampal and mPFC levels of pCREB, BDNF, pcofilin, MeCP2, and GR in total fractions, as well as synaptic pTrkB, were quantified at 20 h after the RS and compared with levels of naive rats. In the dorsal hippocampus, although pCREB (p = 0.005; n = 15–16) and GR (p = 0.026; n = 8–10) remained significantly elevated after the RS, to a level comparable with that found after training, BDNF (p = 0.17; n = 11–12), pTrkB (p = 0.77; n = 8–9), pcofilin (p = 0.75; n = 8), and MeCP2 (p = 0.096; n = 12) levels remained completely blunted (Fig. 4A). In the mPFC, exposure to an RS did not change the expression of any of the marker tested (Fig. 4B), as revealed by unpaired two-sided Student's t tests for pCREB (p = 0.43), BDNF (p = 0.42), pcofilin (p = 0.44), MeCP2 (p = 0.9), GR (p = 0.42), and pTrkB (p = 0.3; n = 8 per group). The expression levels of total CREB and total synaptic TrkB in the dorsal hippocampus or mPFC did not change among groups (data not shown).
Figure 4.

Molecular changes in the dorsal hippocampus (dHPC) and mPFC after training followed by an RS. Data are expressed as the percentage mean ± SEM relative to the naive condition. Western blot examples are shown above each quantitative panel. A, Levels of pCREB, BDNF, pcofilin (pcof.), MeCP2, and GR in the total fraction and of pTrkB in the synaptoneurosomal fraction of the dHPC after 3 mA training followed by an RS compared with levels in naive animals. B, Levels of pCREB, BDNF, pcof., MeCP2, and GR in the total fraction and of pTrkB in the synaptoneurosomal fraction of the mPFC after 3 mA training followed by an RS compared with the levels in naive animals. *p < 0.05, **p < 0.01.
These data indicate that, although memory expression is increased significantly by the RS, the levels of hippocampal BDNF, pTrkB, and MeCP2 remain blunted, suggesting that these hippocampal changes are not targets of the RS effect and that the decreased activation of these hippocampal markers correlates with the experience of a severe episodic stress rather than the levels of memory expression or performance.
The unpredictability of the RS is key for eliciting PTSD-like phenotypes, including extinction failure
Because failure of extinction is another characteristic phenotype of traumatic memories associated with PTSD (Cain et al., 2012; Steckler and Risbrough, 2012; Parsons and Ressler, 2013), we assessed extinction learning in rats that received a 3 mA footshock training followed by a 3 mA RS and compared it with extinction curves of memories induced by lower shock intensities (i.e., 0.6 mA).
Using a protocol that rapidly produces IA extinction as it confines the animal to the dark side of the IA chamber for a prolonged time (5 min) in the absence of footshock, we first determined the extinction profile of a traumatic memory formed after 3 mA footshock training. As shown in Figure 5A, IA memory extinguished rapidly. Latencies were analyzed throughout the extinction protocol (E1–E5) using one-way ANOVA (F(2.59) = 3.29, p = 0.014; n = 8) followed by Dunnett's post hoc test [E1 vs E2 (p = 0.084), E1 vs E3 (p = 0.036), E1 vs E4 (p = 0.016), and E1 vs E5 (p = 0.019)]. An RS delivered after the last extinction trial reinstated the memory (paired two-sided Student's t test comparing E5 and T1 p = 0.002), hence validating the extinction protocol.
We then tested extinction learning of the memory elicited after the RS and asked whether/how the intensity of the initial training and/or of the RS influences extinction. Hence, we compared the extinction curve of memories elicited by 0.6 or 3 mA TR followed by either a 0.6 or a 3 mA RS. As shown in Figure 5B, the memories elicited with a 0.6 mA TR followed by either a 0.6 or 3 mA RS, as well as memories elicited with a 3 mA TR followed by a 0.6 mA RS, were all significantly and similarly sensitive to extinction. In contrast, compared with these, the memory induced by a 3 mA TR followed by a 3 mA RS was significantly resistant to extinction as shown by one-way ANOVA at E1 (F(2.99) = 1.83, p = 0.167), E2 (F(2.99) = 5.26, p = 0.006), E3 (F(2.99) = 10.04, p < 0.001), E4 (F(2.99) = 9.04, p < 0.001), and E5 (F(2.99) = 7.14, p = 0.001) followed by Bonferroni's post hoc tests (p < 0.001 from E1 to E5 for the 3 mA TR plus 3 mA RS group compared with the three other groups; n = 6–8). A second RS after the last trial significantly reinstated the extinguished memories in all groups, thus validating the extinction protocol [paired two-sided Student's t test revealed significant memory reinstatement after an RS (E5 vs T2): 0.6 mA TR plus 0.6 mA RS (p = 0.01), 0.6 mA TR plus 3 mA RS (p = 0.027), 3 mA TR plus 0.6 mA RS (p = 0.009), and 3 mA TR plus 3 mA RS (p = 0.007)]. These results were confirmed using a similar extinction paradigm that excluded the testing before exposure to the RS (data not shown), as well as another extinction protocol that consisted of exposing the rats to repeated testing trials for 7 consecutive days [Fig. 5C; mean latency between 0.6 mA training plus RS and 3 mA training plus RS were compared using unpaired two-sided Student's t tests: E1 (p = 0.073), E2 (p = 0.083), E3 (p = 0.058), E4 (p = 0.15), E5 (p = 0.07), E6 (p < 0.001), E7 (p = 0.007), E8 (p < 0.001), E9 (p = 0.001), E10 (p < 0.001), E11 (p = 0.003), E12 (p < 0.001), E13 (p = 0.022), and E14 (p = 0.009); the mean latencies at E14 and T2 were compared using paired two-sided Student's t tests for the 0.6 mA group (p = 0.011) and the 3 mA group (p = 0.26); n = 8].
Together, these results indicate that two severely stressful experiences (traumatic experiences) are critical for generating memories that are resistant to extinction, a hallmark of traumatic memories associated with PTSD (Cain et al., 2012; Steckler and Risbrough, 2012).
The unpredictability in experiencing a stressor appears to critically contribute to the development of trauma-induced psychopathologies in humans (Grupe and Nitschke, 2013) and animal models (Foa et al., 1992). To investigate the contribution of the unpredictability of the second traumatic experience to the behavioral phenotypes, we compared the extinction curves and memory generalization of rats that underwent a 3 mA-induced TR followed by either a 3 mA RS (given in a context different from training) or a second identical training trial with 3 mA footshock (2TRs). As shown in Figure 5D, memories elicited by 2TRs exhibited rapid extinction, in marked contrast to extinction-resistant memories elicited by TR followed by an RS [unpaired two-sided Student's t tests comparing latencies between groups that were exposed to either 2TRs (n = 8) or TR followed by an RS (n = 7) at E2 (p = 0.048), E3 (p = 0.015), E4 (p = 0.005), and E5 (p < 0.001)]. An RS after the last extinction test significantly reinstated extinguished memories, thus validating the extinction protocol [paired two-sided Student's t tests comparing latencies between E5 and T2 (p < 0.001) and (p = 0.007)]. In addition, memory generalization of rats that received 2TRs at 3 mA was similar to that of rats that had a single TR, whereas rats that received a TR plus RS exhibited a significantly increased memory generalization compared with rats exposed to one or two training trials [Fig. 5E; one-way ANOVA (F(3.47) = 6.845, p = 0.005; n = 8) followed by Bonferroni's post hoc tests; 3 mA TR vs 3 mA TR plus RS (p = 0.01) and 3 mA 2TRs vs 3 mA TR plus RS (p = 0.016)].
Thus, despite the fact that the rats were exposed to two traumatic experiences of the same physical intensity, only those who received a second traumatic event in an unpredictable context developed behavioral phenotypes similar to PTSD.
Cort response is blunted with high shock; cort administration at training with high shock, but not at later times, is sufficient to induce PTSD-like phenotypes
The levels of circulating cort after training with different shock intensity were determined. As shown in Figure 6A, an inverted U profile of cort plasma levels was found 30–45 min after training with the different shock intensities. One-way ANOVA (F(3.71) = 6.90, p = 0.008; n = 7–8) followed by Tukey's HSD post hoc tests revealed that, compared with naive animals trained with 0.6 mA, showed a trend toward an increase in cort plasma level (p = 0.19) and animals trained with 1.5 mA had a robust and significant elevation of plasma cort (p = 0.001), which was also significantly higher than that found with 0.6 mA (p = 0.01). Conversely, training with 3 mA significantly decreased the cort plasma level compared with 1.5 mA training (p = 0.042). A similar experiment done 10 min after training also showed a significant decrease in cort plasma level in 3 mA-trained rats compared with 1.5 mA trained rats (data not shown). Thus, we hypothesized that the decrease in circulating cort after high threat may result from a blunted cort synthesis. If that were the case, administering cort at training with 1.5 or 3 mA shock would produce opposite outcomes. Indeed, in line with this hypothesis, one-way ANOVA (F(3.29) = 34.42, p < 0.0001; n = 7–8) followed by Tukey's HSD post hoc tests revealed that, compared with vehicle, a subcutaneous injection of cort (10 mg/kg) immediately after training significantly impaired a 1.5 mA memory (p < 0.001) and significantly enhanced a 3 mA memory (p < 0.001) tested 2 d later (Fig. 6B).
Figure 6.

Systemic cort administration immediately after a strong footshock training is sufficient to produce PTSD-like phenotype. A, Plasma cort levels at 30–45 min after training (TR) elicited with a 0.6, 1.5, or 3 mA footshock. B, Systemic injection of cort, compared with vehicle (Veh), immediately after training with 1.5 or 3 mA footshock leads to opposite effects on IA latency (in seconds) tested 2 d later. C, Latency of the groups of rats described in B exposed to a new context (ctx) 2 d after TR. D–G, Open-field test (OFT) performed on the groups of rats described in B and compared with naive rats. Data are expressed as the mean ± SEM of total activity, center activity, center time, and center/total activity. Total activity in the arena (D), activity in the center of the arena (E), time spent in the center of the arena (F), and ratio of center activity over total activity (G). H, Extinction curves after TR with 1.5 or 3 mA shock and vehicle or cort injection Data are expressed in latency (in seconds). I, Extinction curves of memory first tested 2 d after training (T1) with either 1.5 or 3 mA footshock, injected 1 d later with cort (0.5, 3.0, or 10.0 mg/kg) or vehicle (Veh), and, starting 1 d later, undergone extinction for 5 consecutive days (E1–E5). One day after E5, an RS was given and memory was tested 24 h later (T2). Data are expressed in latency (in seconds). p < 0.001, E5 versus T2 for all groups except for cort at 0.5 mg/kg in the 3 mA group at p < 0.01. J, Plasma cort levels were measured at 45–60 min after RS or a second training trial (TR2) delivered 1 d after the original training and compared with those of naive animals. *p < 0.05, **p < 0.01, ***p < 0.001.
Furthermore, the cort-injected rats after 3 mA footshock training, but not after 1.5 mA footshock training, had significantly increased memory generalization compared with the 1.5 mA–vehicle (p < 0.001), 1.5 mA–cort (p < 0.001), and 3 mA–vehicle (p < 0.05), as revealed by one-way ANOVA (F(3.29) = 10.44, p < 0.001) followed by Tukey's HSD post hoc test. There were no other significant differences between the other groups (Fig. 6C).
Cort administration after a 3 mA footshock training also augmented anxiety-like behaviors: all open-field measures were reduced significantly compared with naive controls [Fig. 6D–G; unpaired two-sided Student's t test (n = 8) in total activity (p < 0.001), center activity (p < 0.001), center time (p < 0.001), and center/total activity (p < 0.001)]. There was also a significant decrease in center/total activity for the 3 mA–vehicle-treated group compared with naive animals (p < 0.001).
Cort administration after 3 mA but not 1.5 mA training also resulted in a memory resistant to extinction (Fig. 6H). Extinction training began 3 d after training. A one-way ANOVA across extinction sessions, at E1 (F(3.23) = 22.37, p < 0.001), E2 (F(3.23) = 40.94, p < 0.001), E3 (F(3.23) = 100.2, p < 0.001), E4 (F(3.23) = 58.75, p < 0.001), and E5 (F(3.23) = 31.65, p < 0.001) followed by Bonferroni's post hoc tests revealed a significant slower extinction for the 3 mA cort-injected group compared with vehicle injected and both 1.5 mA trained rats (p < 0.001 on E1–E5 for 3 mA with cort compared with 1.5 mA–vehicle, 1.5 mA–cort, and 3 mA–vehicle groups; p < 0.05 on E1 for 1.5 vehicle group compared with 1.5 mA–cort and 3 mA–vehicle groups). All of the other groups had comparable extinction. These extinction curves did not change if an RS was added 2 d after training, except for the 3 mA trained rats injected with vehicle, which with RS showed PTSD-like phenotypes (data not shown).
However, cort administered 3 d after training, thus given in the place of the RS, failed to elicit both latency increase and extinction resistance. cort (0.5, 3.0, or 10 mg/kg) injected subcutaneously 24 h after the first test (T1) to rats trained with 1.5 or 3 mA footshock did not change latencies or the extinction curve in either group (Fig. 6I), as determined by a one-way ANOVA for the 1.5 mA group (n = 6) at E1 (F(3.27) = 0.93, p = 0.442), E2 (F(3.27) = 0.40, p = 0.756), E3 (F(3.27) = 0.31, p = 0.814), E4 (F(3.27) = 0.93, p = 0.442), and E5 (F(3.27) = 0.35, p = 0.791), and for the 3 mA group (n = 6) at E1 (F(3.27) = 0.56, p = 0.648), E2 (F(3.27) = 0.35, p = 0.787), E3 (F(3.27) = 0.29, p = 0.834), E4 (F(3.27) = 0.31, p = 0.815), and E5 (F(3.27) = 0.30, p = 0.824). An RS given after the last extinction test significantly reinstated the extinguished memories, hence validating the extinction protocol (E5 vs T2, p < 0.001 for all groups except 0.5 mg/kg cort in the 3 mA group at p < 0.001).
Together, these results show that cort administered immediately after 3 mA training but not days later is sufficient to elicit the PTSD-like phenotypes.
Analyses of plasma cort levels 45–60 min after either the 3 mA TR plus RS or 2TRs revealed that, compared with naive rats, both groups had a significant elevation of cort concentrations. The increase after RS was significantly higher than that detected after a second training trial [Fig. 6J; one-way ANOVA (F(2,19) = 23.08, p < 0.001; n = 6–8) followed by Tukey's HSD post hoc tests; naive vs 2TRs (p < 0.001), naive vs TR plus RS (p < 0.001), and 2TRs vs TR plus RS (p < 0.05)].
Together, these data indicate that a first traumatic experience blunts both cort surge and memory expression; this memory is readily extinguishable. However, a second unpredictable, but not a predictable, traumatic experience converts the blunted response in PTSD-like phenotypes. Notably, increasing cort during the first traumatic experience, but not at later times, in lieu of a second unpredictable trauma is sufficient to elicit the PTSD-like phenotypes.
Discussion
We showed that, in rats, increasing the severity of stress in a contextual experience produces an inverted U effect on memory retention, which is a characteristic effect of complex cognitive performance in humans (Diamond et al., 2007). The memory decrease elicited by a strong footshock during a first traumatic experience correlates with blunted circulating cort response and blunted hippocampal activation of BDNF, pTrkB, and MeCP2, but not of pCREB and Arc. A reminder of the traumatic experience in a different context, but not a repetition of the experience in the same context, elicits PTSD-like behavioral phenotypes that include strong memory of the trauma, high anxiety, threat context generalization, and extinction failure. Hence, although a first traumatic experience induces a correlative molecular signature in the hippocampus with memory impairment, it does not produce a PTSD-like behavior. The induction of a PTSD-like phenotype requires that at least a second severe and unpredictable stressful episode be experienced. The unpredictability of the second traumatic experience is key to produce a PTSD-like phenotype.
Our data, in line with those of Diamond et al. (1996, 1999) and Salehi et al. (2010), indicate that animal models, including rat IA, can be used to reproduce the inverted U effect of stress on hippocampal-dependent memories and underscores the important role of hippocampal systems in the development and regulation of stress-induced human pathologies, including anxiety disorders and PTSD (Kim and Diamond, 2002; Miller and McEwen, 2006). These models, rather than milder (and hence adaptive) threat conditioning paradigms, should be used for preclinical investigations aimed at identifying effective mechanisms and selective therapeutic approaches for neuropsychiatric disorders in humans.
In fact, as our data indicate, both behavioral and molecular responses underlying a threat memory evoked by a highly stressful (traumatic) experience (e.g., 3 mA) are different from those evoked by a strong but not traumatic stress (e.g., 1.5 mA). First, as revealed by training with increasing shock intensities, the inverted U memory retention correlates with inverted U level of circulating cort, indicating that a traumatic experience may differentially regulate the stress responses. In agreement with this idea, a significant increase in GR levels in the dorsal hippocampus only occurs after the traumatic experience, which is associated with a decrease in memory performance and circulating cort concentration. In several species and paradigms, including chicks (Sandi and Rose, 1997), ground squirrels (Mateo, 2008), rats (Roozendaal et al., 1999; Okuda et al., 2004), and humans (Andreano and Cahill, 2006), either very low or high glucocorticoid levels have been reported to be associated with poor performance in a variety of learning and memory tasks (Park et al., 2006). Although other paradigms report a linear increase in cort plasma level with increasing shock intensities (Conrad, 2008), the very strong footshock that was used here may have been accompanied by a distinct regulation of circulating cort and of the hypothalamic–pituitary–adrenal (HPA) axis. The blunting of circulating cort response is intriguing and should be understood in detail. Because none of the two time points investigated (10 and 30 min after training) revealed a plasma cort surge at 3 mA intensity, we speculate that the decrease in plasma cort compared with milder intensity shock (i.e., 1.5 mA) may be attributable to a very rapid or differential tissues sequestration or to a dampened synthesis or release. In both cases, the end result appears to be protective against the development of anxiety disorders. Interesting regulatory mechanisms that may be involved in blunting cort and hippocampal responses in the high-shock intensity condition is the regulation of corticotropin-releasing hormone and its receptor, and the regulation of GR expression, both of which thus far have been mostly studied in prolonged and chronic stress (Reichardt et al., 2000; Chen et al., 2012b).
Second, the correlation with an inverted U curve of hippocampal levels of BDNF–pTrkB and MeCP2, but not pCREB and Arc, indicate that some but not all critical pathways associated with memory consolidation and persistence are activated after a traumatic experience. Alternatively, the severe stress of 3 mA footshock may produce a negative regulation of the BDNF pathway, perhaps attributable to cort blunting. Although the activation of pCREB and induction of Arc indicate that memory consolidation has been initiated, the blunting of the BDNF/pTrkB activation at 20 h after training suggests that the persistence of the consolidation process is potentially compromised. Therefore, this inhibition, which also remains after the RS, appears to be a signature of exposure to a traumatic experience and might occur to limit or prevent excessively strong or prolonged activation, which could lead to an overconsolidated memory of a trauma. In other words, we suggest that the blunting of the cort response, BDNF expression and activation of pTrkB may represent a protective mechanism against PTSD development after experiencing a trauma. In agreement with this suggestion, cort injection immediately after 3 mA training leads to PTSD-like phenotypes. Furthermore, the hippocampal inverted U profile of MeCP2 suggests that this factor may play a positive regulatory role in memory consolidation and learning-dependent BDNF expression in the hippocampus. Alternatively, the regulation of BDNF release and expression after traumatic experiences may follow very different rules. This interesting correlation requires additional studies to be better understood.
BDNF and pTrkB activation has been shown previously to be impaired in both acute and chronic stress paradigms (Duman and Monteggia, 2006; Andero and Ressler, 2012). Given the feedbacks, homeostatic and allostatic regulations, as well as the temporal differences between acute and chronic stresses (McEwen, 2000), the effects in the two conditions may not be comparable. Hence, the decrease of BDNF after an acute severe trauma may not necessarily be accompanied by the same mechanisms as those elicited by chronic stress.
Although it is known that mPFC plays an important role in PTSD pathophysiology (Liberzon and Sripada, 2008), we did not find in this brain region any of the molecular changes occurring in the dorsal hippocampus at 20 h after training. Hence, increases in hippocampal pCREB and GR levels and the inverted U effect of stress on hippocampal MeCP2, BDNF, and pTrkB do not occur at the same extent or similar temporal profile in the mPFC. Additional studies after more extensive temporal molecular investigations in the mPFC and other cortical regions are needed to better understand the role of the cortex in processing traumas.
Our data also revealed that the encounter of a second traumatic event that reminds of the original traumatic experience, but is not a predictable repetition of the first experience (e.g., an RS), unleashes severe behavioral responses that are similar to those described in PTSD. To produce the PTSD-like phenotype, the second event needs to be highly stressful (e.g., 3 mA RS) and to follow a previous severely stressful (traumatic) experience (e.g., 3 mA footshock training). An initial or second less intense stressful episode (e.g., 0.6 mA footshock) is not sufficient. The resulting PTSD-like phenotypes included a remerging of a very strong IA response that was also accompanied by an elevated anxiety, increased context generalization, and finally extinction failure. All these behavioral responses are similar to those of patients experiencing anxiety disorder and PTSD. It is possible that the extrinsic nature of the stress, which occurs with the RS because it is triggered by conditions unrelated to the first trauma, is key to generate PTSD-like phenotypes; conversely, an intrinsic stress such as one originated by contextual elements of the first trauma (second training trial) is not able to elicit pathological responses (Sandi and Pinelo-Nava, 2007).
A context generalization response was reported previously in mice after an injection of cort given after a high-shock intensity pavlovian threat conditioning training (Kaouane et al., 2012), which is in agreement with our data showing that the RS leads to a significant increase in plasma cort concentration. However, we found that cort injection given in place of the RS is not sufficient to produce extinction failure, a typical behavioral response in PTSD. Thus, we suggest that, in addition to cort surge, additional mechanisms (yet to be identified) activated by a second, severe and unpredictable stressful event interact with the changes occurred after the first trauma to produce PTSD-like behavioral phenotypes.
Furthermore, although a second predictable exposure to the same trauma (retraining) produces a significant increase in circulating cort levels, a second unpredictable traumatic experience (RS) causes a significantly higher cort surge. Only the unpredictable but not the predictable second traumatic experience generated PTSD-like behavioral phenotypes. We suggest that re-experiencing a trauma is a necessary condition for inducing the psychopathology and that the unpredictability (and hence uncontrollability) of the traumatic re-experience critically contributes to the development of the psychopathology (Foa et al., 1992; Grillon et al., 2009; Bowirrat et al., 2010; Koolhaas et al., 2011). In agreement with our suggestion, unpredictability of a stressor is known to evoke a high-stress response, whereas repeated, similar exposures to the same stressor lead to a dampened stress response (Muir and Pfister, 1987; Grissom and Bhatnagar, 2009). As proposed by Grissom and Bhatnagar (2009), the decline of the HPA response with repeated exposures to stressors may reflect a higher-order adaptive process that results from the increased predictability and control over the stressful situation. This could explain why a second training event, which exposes the animals to the same high footshock intensity as an RS, does not produce PTSD-like phenotypes.
The temporal window of exposure to the reminder is rather prolonged, as shown by our results indicating that the two traumatic events given 1, 3, or 8 d apart do evoke the PTSD-like phenotypes. Future experiments shall address whether there is a temporal limit and other boundary conditions for evoking PTSD-like phenotypes after two traumatic events.
We conclude that, although a first traumatic experience elicits a blunted stress and memory expression, perhaps as protective measure, an unpredictable second traumatic experience, therefore multiple traumatic hits, critically contributes to generating behavioral responses typical of psychopathologies such as PTSD. These understandings can be used to reproduce in animal models PTSD-like phenotypes for research and screening in preclinical studies and can be informative for reviewing clinical data.
Footnotes
This work was supported by National Institute of Mental Health Grants R01 MH065635 and R01 MH074736 (C.M.A.) and Swiss National Science Foundation Fellowship PBLAP3_140173 (C.F.). We thank Nelson Humala and Gabriella Pollonini for technical help.
The authors declare no competing financial interests.
References
- Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiol Rev. 2009;89:121–145. doi: 10.1152/physrev.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andero R, Ressler KJ. Fear extinction and BDNF: translating animal models of PTSD to the clinic. Genes Brain Behav. 2012;11:503–512. doi: 10.1111/j.1601-183X.2012.00801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreano JM, Cahill L. Glucocorticoid release and memory consolidation in men and women. Psychol Sci. 2006;17:466–470. doi: 10.1111/j.1467-9280.2006.01729.x. [DOI] [PubMed] [Google Scholar]
- Bambah-Mukku D, Travaglia A, Chen DY, Pollonini G, Alberini CM. A positive autoregulatory BDNF feedback loop via C/EBPbeta mediates hippocampal memory consolidation. J Neurosci. 2014;34:12547–12559. doi: 10.1523/JNEUROSCI.0324-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowirrat A, Chen TJ, Blum K, Madigan M, Bailey JA, Chuan Chen AL, Downs BW, Braverman ER, Radi S, Waite RL, Kerner M, Giordano J, Morse S, Oscar-Berman M, Gold M. Neuro-psychopharmacogenetics and neurological antecedents of posttraumatic stress disorder: unlocking the mysteries of resilience and vulnerability. Curr Neuropharmacol. 2010;8:335–358. doi: 10.2174/157015910793358123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill L, McGaugh JL. Modulation of memory storage. Curr Opin Neurobiol. 1996;6:237–242. doi: 10.1016/S0959-4388(96)80078-X. [DOI] [PubMed] [Google Scholar]
- Cain CK, Maynard GD, Kehne JH. Targeting memory processes with drugs to prevent or cure PTSD. Expert Opin Investig Drugs. 2012;21:1323–1350. doi: 10.1517/13543784.2012.704020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese EJ. Stress biology and hormesis: the Yerkes-Dodson law in psychology—a special case of the hormesis dose response. Crit Rev Toxicol. 2008;38:453–462. doi: 10.1080/10408440802004007. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DY, Bambah-Mukku D, Pollonini G, Alberini CM. Glucocorticoid receptors recruit the CaMKIIalpha-BDNF-CREB pathways to mediate memory consolidation. Nat Neurosci. 2012a;15:1707–1714. doi: 10.1038/nn.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Andres AL, Frotscher M, Baram TZ. Tuning synaptic transmission in the hippocampus by stress: the CRH system. Front Cell Neurosci. 2012b;6:13. doi: 10.3389/fncel.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad CD. Chronic stress-induced hippocampal vulnerability: the glucocorticoid vulnerability hypothesis. Rev Neurosci. 2008;19:395–411. doi: 10.1515/revneuro.2008.19.6.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero MI, Kruyt ND, Merino JJ, Sandi C. Glucocorticoid involvement in memory formation in a rat model for traumatic memory. Stress. 2002;5:73–79. doi: 10.1080/1025389029000124404. [DOI] [PubMed] [Google Scholar]
- Cunha C, Brambilla R, Thomas KL. A simple role for BDNF in learning and memory? Front Mol Neurosci. 2010;3:1. doi: 10.3389/neuro.02.001.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- de Quervain DJ, Roozendaal B, McGaugh JL. Stress and glucocorticoids impair retrieval of long-term spatial memory. Nature. 1998;394:787–790. doi: 10.1038/29542. [DOI] [PubMed] [Google Scholar]
- de Quervain DJ, Aerni A, Schelling G, Roozendaal B. Glucocorticoids and the regulation of memory in health and disease. Front Neuroendocrinol. 2009;30:358–370. doi: 10.1016/j.yfrne.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Fleshner M, Ingersoll N, Rose GM. Psychological stress impairs spatial working memory: relevance to electrophysiological studies of hippocampal function. Behav Neurosci. 1996;110:661–672. doi: 10.1037/0735-7044.110.4.661. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Park CR, Heman KL, Rose GM. Exposing rats to a predator impairs spatial working memory in the radial arm water maze. Hippocampus. 1999;9:542–552. doi: 10.1002/(SICI)1098-1063(1999)9:5<542::AID-HIPO8>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Campbell AM, Park CR, Halonen J, Zoladz PR. The temporal dynamics model of emotional memory processing: a synthesis on the neurobiological basis of stress-induced amnesia, flashbulb and traumatic memories, and the Yerkes-Dodson law. Neural Plast. 2007;2007:60803. doi: 10.1155/2007/60803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudai Y. The restless engram: consolidations never end. Annu Rev Neurosci. 2012;35:227–247. doi: 10.1146/annurev-neuro-062111-150500. [DOI] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Finsterwald C, Alberini CM. Stress and glucocorticoid receptor-dependent mechanisms in long-term memory: from adaptive responses to psychopathologies. Neurobiol Learn Mem. 2014;112C:17–29. doi: 10.1016/j.nlm.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foa EB, Zinbarg R, Rothbaum BO. Uncontrollability and unpredictability in post-traumatic stress disorder: an animal model. Psychol Bull. 1992;112:218–238. doi: 10.1037/0033-2909.112.2.218. [DOI] [PubMed] [Google Scholar]
- Grillon C, Pine DS, Lissek S, Rabin S, Bonne O, Vythilingam M. Increased anxiety during anticipation of unpredictable aversive stimuli in posttraumatic stress disorder but not in generalized anxiety disorder. Biol Psychiatry. 2009;66:47–53. doi: 10.1016/j.biopsych.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissom N, Bhatnagar S. Habituation to repeated stress: get used to it. Neurobiol Learn Mem. 2009;92:215–224. doi: 10.1016/j.nlm.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupe DW, Nitschke JB. Uncertainty and anticipation in anxiety: an integrated neurobiological and psychological perspective. Nat Rev Neurosci. 2013;14:488–501. doi: 10.1038/nrn3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inda MC, Muravieva EV, Alberini CM. Memory retrieval and the passage of time: from reconsolidation and strengthening to extinction. J Neurosci. 2011;31:1635–1643. doi: 10.1523/JNEUROSCI.4736-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaouane N, Porte Y, Vallée M, Brayda-Bruno L, Mons N, Calandreau L, Marighetto A, Piazza PV, Desmedt A. Glucocorticoids can induce PTSD-like memory impairments in mice. Science. 2012;335:1510–1513. doi: 10.1126/science.1207615. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3:453–462. doi: 10.1038/nrn849. [DOI] [PubMed] [Google Scholar]
- Koolhaas JM, Bartolomucci A, Buwalda B, de Boer SF, Flügge G, Korte SM, Meerlo P, Murison R, Olivier B, Palanza P, Richter-Levin G, Sgoifo A, Steimer T, Stiedl O, van Dijk G, Wöhr M, Fuchs E. Stress revisited: a critical evaluation of the stress concept. Neurosci Biobehav Rev. 2011;35:1291–1301. doi: 10.1016/j.neubiorev.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Liberzon I, Sripada CS. The functional neuroanatomy of PTSD: a critical review. Prog Brain Res. 2008;167:151–169. doi: 10.1016/S0079-6123(07)67011-3. [DOI] [PubMed] [Google Scholar]
- Maren S, Phan KL, Liberzon I. The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat Rev Neurosci. 2013;14:417–428. doi: 10.1038/nrn3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo JM. Inverted-U shape relationship between cortisol and learning in ground squirrels. Neurobiol Learn Mem. 2008;89:582–590. doi: 10.1016/j.nlm.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS. Allostasis and allostatic load: implications for neuropsychopharmacology. Neuropsychopharmacology. 2000;22:108–124. doi: 10.1016/S0893-133X(99)00129-3. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Memory—a century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- Miller MM, McEwen BS. Establishing an agenda for translational research on PTSD. Ann NY Acad Sci. 2006;1071:294–312. doi: 10.1196/annals.1364.023. [DOI] [PubMed] [Google Scholar]
- Muir JL, Pfister HP. Time course of the corticosterone and prolactin response following predictable and unpredictable novelty stress in Rattus norvegicus. Physiol Behav. 1987;40:103–107. doi: 10.1016/0031-9384(87)90191-0. [DOI] [PubMed] [Google Scholar]
- Na ES, Nelson ED, Kavalali ET, Monteggia LM. The impact of MeCP2 loss- or gain-of-function on synaptic plasticity. Neuropsychopharmacology. 2013;38:212–219. doi: 10.1038/npp.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S, Roozendaal B, McGaugh JL. Glucocorticoid effects on object recognition memory require training-associated emotional arousal. Proc Natl Acad Sci U S A. 2004;101:853–858. doi: 10.1073/pnas.0307803100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CR, Campbell AM, Woodson JC, Smith TP, Fleshner M, Diamond DM. Permissive influence of stress in the expression of a U-shaped relationship between serum corticosterone levels and spatial memory errors in rats. Dose Response. 2006;4:55–74. doi: 10.2203/dose-response.004.01.005.Park. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons RG, Ressler KJ. Implications of memory modulation for post-traumatic stress and fear disorders. Nat Neurosci. 2013;16:146–153. doi: 10.1038/nn.3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, Milad MR, Liberzon I. Biological studies of post-traumatic stress disorder. Nat Rev Neurosci. 2012;13:769–787. doi: 10.1038/nrn3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau V, DeCola JP, Fanselow MS. Stress-induced enhancement of fear learning: an animal model of posttraumatic stress disorder. Neurosci Biobehav Rev. 2005;29:1207–1223. doi: 10.1016/j.neubiorev.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Reichardt HM, Tronche F, Bauer A, Schütz G. Molecular genetic analysis of glucocorticoid signaling using the Cre/loxP system. Biol Chem. 2000;381:961–964. doi: 10.1515/BC.2000.118. [DOI] [PubMed] [Google Scholar]
- Revest JM, Di Blasi F, Kitchener P, Rougé-Pont F, Desmedt A, Turiault M, Tronche F, Piazza PV. The MAPK pathway and Egr-1 mediate stress-related behavioral effects of glucocorticoids. Nat Neurosci. 2005;8:664–672. doi: 10.1038/nn1441. [DOI] [PubMed] [Google Scholar]
- Revest JM, Kaouane N, Mondin M, Le Roux A, Rougé-Pont F, Vallée M, Barik J, Tronche F, Desmedt A, Piazza PV. The enhancement of stress-related memory by glucocorticoids depends on synapsin-Ia/Ib. Mol Psychiatry. 2010;15:1125, 1140–1151. doi: 10.1038/mp.2010.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revest JM, Le Roux A, Roullot-Lacarrière V, Kaouane N, Vallée M, Kasanetz F, Rougé-Pont F, Tronche F, Desmedt A, Piazza PV. BDNF-TrkB signaling through Erk1/2 MAPK phosphorylation mediates the enhancement of fear memory induced by glucocorticoids. Mol Psychiatry. 2014;19:1001–1009. doi: 10.1038/mp.2013.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, Nguyen BT, Power AE, McGaugh JL. Basolateral amygdala noradrenergic influence enables enhancement of memory consolidation induced by hippocampal glucocorticoid receptor activation. Proc Natl Acad Sci U S A. 1999;96:11642–11647. doi: 10.1073/pnas.96.20.11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi B, Cordero MI, Sandi C. Learning under stress: the inverted-U-shape function revisited. Learn Mem. 2010;17:522–530. doi: 10.1101/lm.1914110. [DOI] [PubMed] [Google Scholar]
- Sandi C, Pinelo-Nava MT. Stress and memory: behavioral effects and neurobiological mechanisms. Neural Plast. 2007;2007:78970. doi: 10.1155/2007/78970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandi C, Rose SP. Training-dependent biphasic effects of corticosterone in memory formation for a passive avoidance task in chicks. Psychopharmacology. 1997;133:152–160. doi: 10.1007/s002130050385. [DOI] [PubMed] [Google Scholar]
- Schilling TM, Kölsch M, Larra MF, Zech CM, Blumenthal TD, Frings C, Schächinger H. For whom the bell (curve) tolls: cortisol rapidly affects memory retrieval by an inverted U-shaped dose-response relationship. Psychoneuroendocrinology. 2013;38:1565–1572. doi: 10.1016/j.psyneuen.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- Squire LR, Stark CE, Clark RE. The medial temporal lobe. Annu Rev Neurosci. 2004;27:279–306. doi: 10.1146/annurev.neuro.27.070203.144130. [DOI] [PubMed] [Google Scholar]
- Steckler T, Risbrough V. Pharmacological treatment of PTSD—established and new approaches. Neuropharmacology. 2012;62:617–627. doi: 10.1016/j.neuropharm.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson NC, Corcoran KA, Jovasevic V, Radulovic J. Fear conditioning and extinction: emotional states encoded by distinct signaling pathways. Trends Neurosci. 2012;35:145–155. doi: 10.1016/j.tins.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervliet B, Baeyens F, Van den Bergh O, Hermans D. Extinction, generalization, and return of fear: a critical review of renewal research in humans. Biol Psychol. 2013;92:51–58. doi: 10.1016/j.biopsycho.2012.01.006. [DOI] [PubMed] [Google Scholar]