

Abstract
Key points
Cardiac myocytes are subjected to fluid shear stress during the cardiac cycle and haemodynamic disturbance.
A longitudinally propagating, regenerative Ca2+ wave is initiated in atrial myocytes under shear stress.
Here we determine the cellular mechanism for this shear‐induced Ca2+ wave using two‐dimensional confocal Ca2+ imaging combined with pressurized fluid flow.
Our data suggest that shear stress triggers the Ca2+ wave through ryanodine receptors via P2Y1 purinoceptor–phospholipase C‐type 2 inositol 1,4,5‐trisphosphate receptor signal transduction in atrial myocytes, and that this mechanotransduction is activated by gap junction hemichannel‐mediated ATP release.
Shear‐specific mechanotransduction and the subsequent regenerative Ca2+ wave may be one way for atrial myocytes to assess mechanical stimuli directly and alter their Ca2+ signalling accordingly.
Abstract
Atrial myocytes are exposed to shear stress during the cardiac cycle and haemodynamic disturbance. In response, they generate a longitudinally propagating global Ca2+ wave. Here, we investigated the cellular mechanisms underlying the shear stress‐mediated Ca2+ wave, using two‐dimensional confocal Ca2+ imaging combined with a pressurized microflow system in single rat atrial myocytes. Shear stress of ∼16 dyn cm−2 for 8 s induced ∼1.2 aperiodic longitudinal Ca2+ waves (∼79 μm s−1) with a delay of 0.2−3 s. Pharmacological blockade of ryanodine receptors (RyRs) or inositol 1,4,5‐trisphosphate receptors (IP3Rs) abolished shear stress‐induced Ca2+ wave generation. Furthermore, in atrial myocytes from type 2 IP3R (IP3R2) knock‐out mice, shear stress failed to induce longitudinal Ca2+ waves. The phospholipase C (PLC) inhibitor U73122, but not its inactive analogue U73343, abolished the shear‐induced longitudinal Ca2+ wave. However, pretreating atrial cells with blockers for stretch‐activated channels, Na+−Ca2+ exchanger, transient receptor potential melastatin subfamily 4, or nicotinamide adenine dinucleotide phosphate oxidase did not suppress wave generation under shear stress. The P2 purinoceptor inhibitor suramin, and the potent P2Y1 receptor antagonist MRS 2179, both suppressed the Ca2+ wave, whereas the P2X receptor antagonist, iso‐PPADS, did not alter it. Suppression of gap junction hemichannels permeable to ATP or extracellular application of ATP‐metabolizing apyrase inhibited the wave. Removal of external Ca2+ to enhance hemichannel opening facilitated the wave generation. Our data suggest that longitudinally propagating, regenerative Ca2+ release through RyRs is triggered by P2Y1–PLC–IP3R2 signalling that is activated by gap junction hemichannel‐mediated ATP release in atrial myocytes under shear stress.
Abbreviations
- 9‐AC
9‐anthracenecarboxylic acid
- 2‐APB
2‐aminoethoxydiphenyl borate
- CICR
Ca2+‐induced Ca2+ release
- DPI
diphenyleneiodonium
- FDHM
full duration at half‐maximum
- IP3R2
type 2 inositol 1,4,5‐trisphosphate receptor
- KO
knock‐out
- NCX
Na+−Ca2+ exchanger
- NOX
nicotinamide adenine dinucleotide phosphate oxidase
- PLC
phospholipase C
- ROI
region‐of‐interest
- RyR
ryanodine receptor
- SAC
stretch‐activated channel
- SR
sarcoplasmic reticulum
- Tp
time‐to‐peak
- TRPM4
transient receptor potential melastatin subfamily 4
- Vp
propagation velocity
- WT
wild‐type
Introduction
Changes in the mechanical environment of the heart, caused by each cardiac cycle, alter cardiac excitation and contraction (Nazir & Lab, 1996). An increase in atrial pressure and volume under pathological conditions, such as valve disease, hypertension or heart failure, is thought to be an important cause of altered atrial excitability (Nazir & Lab, 1996; Nattel, 2002). During each contraction and haemodynamic disturbance, cardiac myocytes are subjected to fluid shear stress caused by blood flow and the relative movement of myocyte sheets (LeGrice et al. 1995; Costa et al. 1999). There is also intriguing clinical evidence that a regurgitant blood‐jet during mitral valve incompetence causes atrial arrhythmia (Nazir & Lab, 1996), and that a direct irritation due to a catheter whip on the intra‐atrial wall elicits ectopic atrial tachycardia (Conwell et al. 1993). Atria often become enlarged and dilated under pathological conditions, and the responses of atrial myocytes to stretch, including the opening of stretch‐activated ion channels (SACs), are well documented (Hagiwara et al. 1992; Sato & Koumi, 1998; Tavi et al. 1998; Zhang et al. 2000; Kamkin et al. 2003). However, atrial responses to shear stimulus remain poorly understood.
Recent evidence indicates that shear stress significantly modulates the functions of cardiac myocytes. Shear stress causes a propagation of action potential in cultured ventricular myocytes (Kong et al. 2005), increases the occurrence of atrial Ca2+ sparks (focal Ca2+ releases through a single RyR cluster; Cheng et al. 1993) (Woo et al. 2007), induces global Ca2+ waves (Woo et al. 2007) and whole‐cell Ca2+ transients (Belmonte & Morad, 2008) in atrial myocytes, and upregulates atrial ultra‐rapid outward K+ currents (Boycott et al. 2013). Furthermore, shear stress enhances ventricular Ca2+ transients (Lee et al. 2008) and suppresses ventricular L‐type Ca2+ currents (Lee et al. 2008; Rosa et al. 2013). These responses to shear stress in cardiac myocytes occur even in the presence of SAC inhibitors (Lee et al. 2008; Boycott et al. 2013; Rosa et al. 2013). The effect of shear stress on Ca2+ sparks is larger in the periphery than in the interior of atrial myocytes lacking transverse tubules (Carl et al. 1995; Woo et al. 2007). At high shear stresses, a longitudinally propagating global Ca2+ wave develops from a local Ca2+ release site in atrial myocytes (Woo et al. 2007). The inhibition of L‐type Ca2+ current and enhancement of Kv1.5 current under shear stress are suppressed by cytosolic Ca2+ buffering (Lee et al. 2008; Boycott et al. 2013), indicating a role of Ca2+ signalling in the modulation of these channel proteins. To date, it remains unknown which cellular mechanisms cause the enhancement in Ca2+ sparks and trigger the Ca2+ waves in atrial cells under shear stress.
In the present study, we investigated the cellular mechanisms underlying the generation of the longitudinal global Ca2+ wave in atrial myocytes subjected to shear stress using two‐dimensional confocal Ca2+ imaging. We applied shear stress of approximately 16 dyn cm−2 to single atrial myocytes using pressurized fluid flow to elicit a global Ca2+ wave as previously reported (Woo et al. 2007). We found that the shear stress‐induced longitudinal Ca2+ wave is generated by the activation of the type 2 inositol 1,4,5‐trisphosphate receptor (IP3R2) via P2Y1 purinoceptor–phospholipase C (PLC) signalling and subsequent Ca2+‐induced Ca2+ release (CICR) through RyRs; this shear response is associated with ATP release from atrial myocytes via gap junction hemichannels.
Methods
Cell isolation
Atrial myocytes were enzymatically isolated (Lee et al. 2008) from male Sprague–Dawley rats (250–350 g) and from wild‐type (WT) and IP3R2 knock‐out (KO) mice (Li et al. 2005) (C57/B6 background, 3–5 months of age, 24–28 g). This study conforms with the Guiding Principles for the Care and Use of Experimental Animals published by the Korean Food and Drug Administration and Animal and Plant Quarantine Agency in South Korea. The experiments were carried out according to the guidelines laid down by the Chungnam National University Animal Care and Use Committee (Approval No. CNU‐00368), and conform to the principles of UK regulations, as described in Drummond (2009). Rats or mice were deeply anaesthetized with pentobarbital sodium (150 mg kg−1, i.p.), the chest cavity was opened and hearts were excised. Then the animals were killed with anaesthetic overdose. The excised hearts were retrogradely perfused at 7 ml min−1 for rat heart and at 1.9 ml min−1 for mouse heart through the aorta (at 36.5°C), first for 3 min with Ca2+‐free Tyrode solution composed of (in mm) 137 NaCl, 5.4 KCl, 10 Hepes, 1 MgCl2 and 10 glucose, pH 7.3, and then with Ca2+‐free Tyrode solution containing collagenase (1.4 mg ml−1 for rat; 1 mg ml−1 for mouse; Type A (EC 3.4.24.3), Roche, Grenzacherstrasse, Basel, Switzerland) and protease (0.14 mg ml−1 for rat, 0.08 mg ml−1 for mouse, Type XIV (EC 3.4.24.31), Sigma, St Louis, MO, USA) for 12 min, and finally with Tyrode solution containing 0.2 mm CaCl2 for 5 min. The atria of the digested heart were then cut into several sections and subjected to gentle agitation to dissociate the cells. The freshly dissociated cells were stored at room temperature in Tyrode solution containing 0.2 mm CaCl2.
Application of shear stress
Pressurized flows of solutions were applied onto the single myocytes through a microbarrel (internal diameter, 250 μm), the tip of which was placed ∼150 μm from the cell. The microbarrel was connectedto a fluid reservoir with a height of 40 cm (Woo et al. 2007; Lee et al. 2008). The tip of the microbarrel, touching the chamber bottom, was tilted to one side at an angle of 45 deg. An electronically controllable solenoid valve was installed in the middle of tubing connecting the fluid reservoir and the microbarrel. The shear stress (dyn cm−2) was calculated for flow in cylindrical tubes according to the equation (Olesen et al. 1988):
where μ represents the fluid viscosity (1.002 × 10‒2 dyn s cm−2 for water), Q is the flow rate (cm3 s−1) and r is the internal radius (cm) of the microbarrel. The micro‐flow system generated shear stress of ∼16 dyn cm−2 (equal to 0.16 N m−2) at 40 cm reservoir height. The positioning of the microbarrel was performed under a microscope using a micromanipulator (48260; Prior Scientific Ltd., Cambridge, UK). The experimental cells were attached to the bottom of the chamber without a coating material. Using a microscope and video monitor it was confirmed that no movement of the cell occurred during the fluid puffing before the start of the experiments. All experiments were carried out at room temperature (22−24°C).
Confocal Ca2+ imaging and image analysis
Myocytes were loaded with 3 μm fluo‐4 acetoxymethyl (AM) ester (Invitrogen) for 10 min. The dye‐loaded cells were continuously superfused with 2 mm Ca2+‐containing normal Tyrode solution. Intracellular Ca2+ fluorescence was imaged in two dimensions using a laser scanning confocal imaging system (A1, Nikon, Japan) attached to an inverted microscope (Eclipse Ti, Nikon) fitted with a ×60 oil‐immersion objective lens (Plan Apo, NA 1.4) (Subedi et al. 2011). Dyes were excited at 488 nm using an Ar ion laser (Ommichrome) and fluorescence emission at > 510 nm was detected. Images were acquired and analysed with workstation software (NIS Elements AR, v3.2, Nikon). To record the whole‐cell Ca2+ wave, Ca2+ imaging was performed at 60 Hz at the expense of the time resolution. In some experiments, Ca2+ images were acquired at 120 Hz to detect Ca2+ releases on field stimulations (Fig. 13). The recording of shear stress‐induced Ca2+ change was normally preceded by a train of electrical pulses at 1 Hz using a pair of Pt electrodes connected to a stimulator (D‐7806, Hugo Sachs Elektronik, March‐Hugstetten, Germany) to maintain stable sarcoplasmic reticulum (SR) Ca2+ loading during the experimental period.
Figure 13. Shear‐induced longitudinal Ca2+ wave in field‐stimulated atrial myocytes and its effect on depolarization‐induced Ca2+ releases .
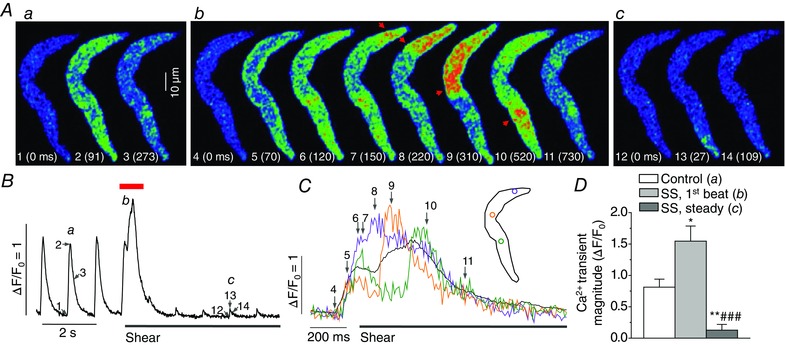
A, confocal Ca2+ images recorded in a field‐stimulated (1 Hz) atrial cell before and after application of shear stress (16 dyn cm−2). Images were selected at the time points marked by corresponding numbers in Ca2+ transient traces a, b and c in B. 0 ms (‘1’) indicates onset of depolarization. Confocal Ca2+ images of depolarization‐induced Ca2+ release with immediate shear stimulation (b) show development of longitudinal Ca2+ wave (arrows) by shear right after depolarization‐induced transverse Ca2+ wave. B, averaged Ca2+ signal from whole area of the Ca2+ images showing larger and prolonged Ca2+ increases (b) immediately after the onset of shear stimulation and significant attenuation of the Ca2+ transients (c) after such enhancement. C, local Ca2+ signals measured from coloured ROIs during the period marked by red bar above the Ca2+ trace in B, representing longitudinal Ca2+ propagation. D, average Ca2+ transient magnitudes measured in field‐stimulated atrial cells at 1 Hz before (‘Control’) and after shear stimulation (‘SS’). The magnitudes of the first Ca2+ transient after the onset of shear stimulation (‘SS, 1st beat’) and those of Ca2+ transients when shear effect was stabilized (‘SS, steady’) were assessed. *P < 0.05, **P < 0.01 vs. Control. ### P < 0.01 vs. ‘SS, 1st beat’ (paired t test; n = 8).
In order to estimate Ca2+ changes, the average resting fluorescence intensity (F 0) was calculated from several frames measured before Ca2+ increase. Tracings of Ca2+ changes were shown as the average fluorescence of each region‐of‐interest (ROI) normalized relative to the F 0 (F/F 0) (Woo et al. 2007; Subedi et al. 2011). Shear‐mediated Ca2+ increase sometimes induced slight contraction, and in such cases only stationary areas were included for the image analysis. The propagation velocity (V p) of the Ca2+ wave was measured using the equation:
where L is the distance (μm) between the core (S0) of the Ca2+ wave and a site (S1) to which the Ca2+ wave significantly moved, and Δt is the delay (ms), calculated as the difference between the time‐to‐peaks of the Ca2+ transients measured from S0 and S1. We counted Ca2+ waves that showed longitudinal movement to a distance of more than approximately 40% of the cell length.
Chemicals and treatment
Reagents used to make Tyrode solutions, and tetracaine, 2‐aminoethoxydiphenyl borate (2‐APB), suramin, apyrase, carbenoxolone, diphenyleneiodonium (DPI), GdCl3, and 9‐anthracenecarboxylic acid (9‐AC) were purchased from Sigma. U73122, U73343 and CGP‐37157 were from Calbiochem (Merck Millipore Corporation, Darmstadt, Germany). GsMTx‐4, a peptide toxin from Grammostola spatulata spider venom, was purchased from Alomone Labs (Jerusalem, Israel). KB‐R7943, 9‐phenanthrol, pyridoxalphosphate‐6‐azophenyl‐2′,5′‐disulfonic acid (iso‐PPADS) and MRS 2179 were obtained from Tocris Bioscience (Bristol, UK). EGTA (1 mm) was added to the external solutions to make Ca2+‐free solutions (Fig. 8). Short pre‐exposure to a drug (e.g. tetracaine and GsMTx‐4) was done by rapid puffing with low reservoir height (4–5 cm) having no shear effect on the Ca2+ level. Long‐term exposures (>30 s) to drug solutions (e.g. 2‐APB and U73122, etc.) were done with superfusion.
Figure 8. Gap junction hemichannels play a role in the induction of longitudinal Ca2+ waves under shear stress .
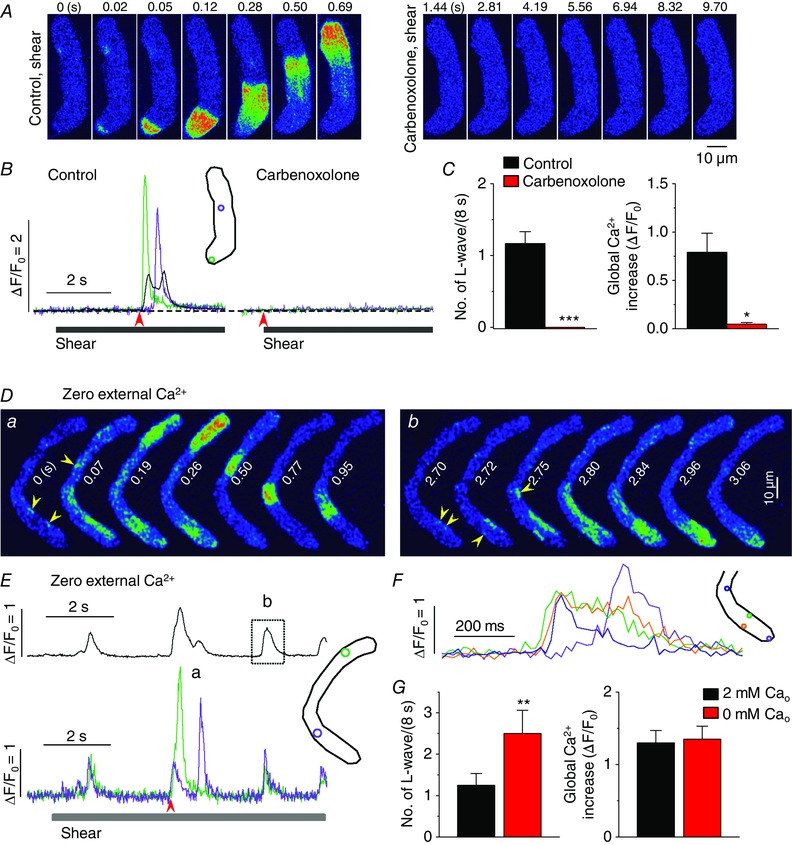
A, representative confocal Ca2+ images, recorded during the applications of shear (16 dyn cm−2) in the absence (‘Control, shear’) and presence of the inhibitor of gap junction hemichannels carbenoxolone (50 μm; ‘carbenoxolone, shear’) show the blockade of the occurrence of longitudinal Ca2+ waves under shear stress by this drug. B, changes in local (green and violet) and global (black) Ca2+ levels measured from the ROIs (inset) on the series of confocal Ca2+ images recorded from the cell shown in the A. The time marked by arrowheads matches with 0 s shown in the confocal images. C, summary of the effects of carbenoxolone on the occurrence of longitudinal Ca2+ waves and on global Ca2+ changes during the application of shear (8 s). *P < 0.05, ***P < 0.001 vs. Control (n = 6, paired Student's t test). D, representative confocal Ca2+ images showing shear‐induced longitudinal Ca2+ wave (a) and local Ca2+ propagations (b) induced by shear stress in the absence of external Ca2+. Arrowheads indicate the foci of Ca2+ waves. E, whole‐cell (upper) and local Ca2+ signal traces (lower) measured from ROIs with corresponding colours (inset). Images shown in Da and Db were selected from the 2nd Ca2+ transient and the 3rd Ca2+ transient in the upper (black) trace of E, respectively. The period of shear stress application was indicated by the bar under the local Ca2+ signals. F, local Ca2+ signals measured from different spots (see ROIs in the inset) during the period marked by dotted box in E, showing smaller Ca2+ waves originating from several peripheral sites (see arrowheads in Db). G, summary of the occurrence of global Ca2+ waves and magnitude of whole‐cell Ca2+ transients measured in 2 mm (n = 64) and 0 mm Ca2+‐containing external puffing solutions (n = 6) during 8 s‐long shear stimulus. **P < 0.01 vs. ‘2 mm Cao’ (unpaired t test).
Statistics
The numerical results are presented as means ± standard error of the mean (SEM). n indicates the number of cells tested. Paired or unpaired Student's t tests were used for statistical comparisons depending on the experiments. Differences at P < 0.05 were considered to be statistically significant.
Results
Longitudinal Ca2+ wave is triggered by IP3R2‐mediated Ca2+ release in atrial myocytes under shear stress
Figure 1 A–C shows the generation of the longitudinal Ca2+ wave upon shear stress application (16 dyn cm−2) and the effect of the RyR inhibitor tetracaine on this wave. Shear stress‐induced longitudinal Ca2+ waves developed from one (>80%) or two foci (arrows, Fig. 1 A and D) with a 0.2−3 s delay during an 8‐s shear stimulation, and propagated at 75.7 ± 3.63 μm s−1 (n = 64) on 60 Hz imaging. During the 8‐s shear stress simulation we observed 1.2 ± 0.26 wave events (n = 64), and the occurrence of the wave was not periodic. Thus, wave occurrence was quantified as the number of wave events per 8‐s shear under each experimental condition. After the first 8‐s exposure to shear stress, it took 3–4 min to restitute the shear‐induced Ca2+ wave in the same cells. The increase in local Ca2+ concentration (ΔF/F 0) in the area in which the longitudinal wave was propagated was 3.04 ± 0.25 (n = 64). The whole‐cell Ca2+ signal accompanying the global Ca2+ wave (ΔF/F 0 = 1.25 ± 0.17) showed prolonged and slow Ca2+ increase (time‐to‐peak (T p) = 348 ± 38.1 ms; full duration at half‐maximum (FDHM) = 415 ± 48.0 ms; n = 64).
Figure 1. Shear stress elicits longitudinal Ca2+ waves by triggering IP3Rs and subsequent CICR through the RyRs .
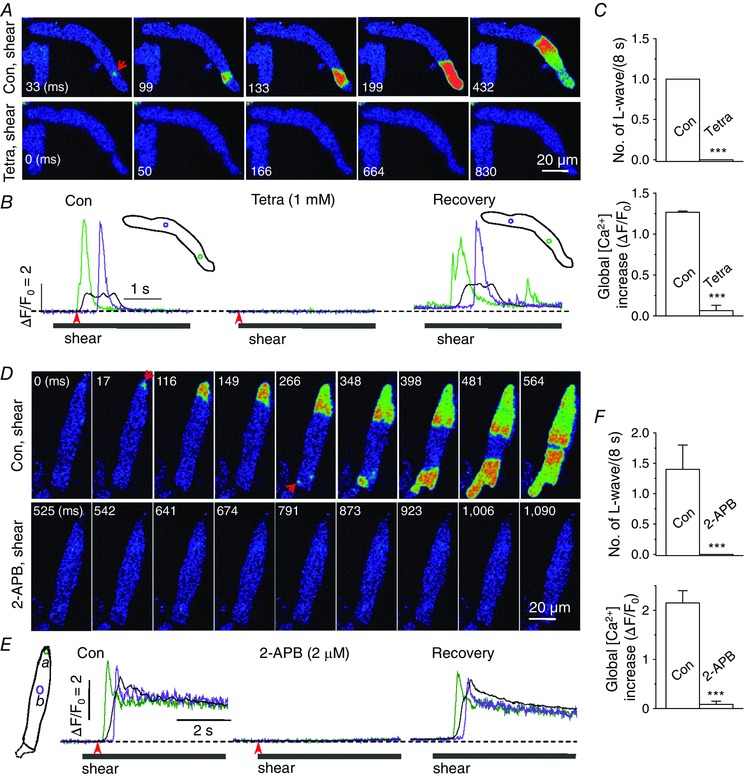
A and D, confocal Ca2+ images recorded during the application of shear stress (∼16 dyn cm−2) in the control solutions (‘Con, shear’) and in the presence of 1 mm tetracaine (10 s; A, ‘Tetra, shear’) or 2 μm 2‐APB (D, ‘2‐APB, shear’) in the representative rat atrial myocytes. Arrows indicate Ca2+ wave core. B and E, changes in Ca2+ fluorescence were measured (at 60 Hz) from the region‐of‐interest (ROI) with corresponding colours (inset) using the Ca2+ images recorded in the cells illustrated in A (B) and D (E). The time marked by arrowheads matches with 0 ms shown in the confocal images. Local Ca2+ signals (green and violet) represent Ca2+ movement during the longitudinal Ca2+ wave. Whole‐cell Ca2+ change during the wave is shown as a black trace. C and F, summary of the number of longitudinal (L‐) waves and whole‐cell Ca2+ increases during 8‐s application of shear stress in the absence (‘Con’) and presence of 1 mm tetracaine (‘Tetra’; n = 9; C) or 2 μm 2‐APB (n = 14; F). ***P < 0.001 vs. ‘Con’ (paired Student's t test).
After a 10‐s pretreatment with 1 mm tetracaine, shear stress failed to induce either the Ca2+ wave or a significant increase in Ca2+ (Fig. 1 A–C). Treatment of tetracaine for 10 s completely abolished resting Ca2+ sparks, confirming RyR blockade (data not shown). The exposure to tetracaine did not alter resting Ca2+ level (in F/F 0: control, 1.00 ± 0 vs. tetracaine, 1.00 ± 0, n = 9, P > 0.05). After tetracaine washout, the shear‐triggered longitudinal Ca2+ wave was observed again (Fig. 1 B, ‘Recovery’). This result indicates that the shear‐induced longitudinal Ca2+ wave is mediated by regenerative Ca2+ release through the RyR, and is consistent with an even distribution of RyRs throughout atrial myocytes (Carl et al. 1995; Lipp et al. 2000; Mackenzie et al. 2002).
Since atrial myocytes also widely express IP3Rs co‐localized with RyRs in the peripheral junctional SR (Lipp et al. 2000; Mackenzie et al. 2002), it is reasonable to postulate that release of Ca2+ through the IP3R induces RyR‐mediated Ca2+ release by CICR (Mackenzie et al. 2002). We therefore examined whether the shear‐induced Ca2+ wave is regulated by IP3R‐mediated Ca2+ release, using its inhibitor 2‐APB. This compound has been successfully used for selective inhibition of IP3Rs in cardiac cells at concentrations of 2–5 μm (Mackenzie et al. 2002; Li et al. 2005). Pretreatment with 2 μm 2‐APB (8–10 min) prevented the generation of longitudinal Ca2+ waves when shear was applied (Fig. 1 D–F). Shear‐induced Ca2+ waves were observed again after 2‐APB washout (Fig. 1 E, ‘Recovery’). The exposure to 2‐APB did not alter resting Ca2+ level (in F/F 0: control, 1.00 ± 0 vs. 2‐APB, 1.01 ± 0.016, n = 14, P > 0.05). These results indicate that IP3Rs are activated under shear stress, and that IP3R‐mediated Ca2+ release may play a role in triggering the RyR via CICR. We did not observe any significant focal Ca2+ signal induced by shear in the presence of tetracaine, suggesting that IP3R‐mediated Ca2+ release may activate co‐localized RyRs to generate a wave core under shear stress.
Although 2‐APB is widely used to study the role of IP3Rs, it has multiple sites of action, including store‐operated channels (Bootman et al. 2002). To confirm the role of IP3R‐mediated Ca2+ release in the generation of longitudinal Ca2+ waves, we examined shear‐mediated Ca2+ signals using IP3R2 KO mice (Li et al. 2005). IP3R2 is the most abundant of the three IP3R subtypes, and they are co‐localized with RyRs in the peripheral junctional SR in atrial myocytes (Lipp et al. 2000; Mackenzie et al. 2002). In WT mouse atrial myocytes, we observed that longitudinal Ca2+ waves were induced by shear stress and were abolished by IP3R blocker 2‐APB application, almost completely suppressing whole‐cell Ca2+ change (Fig. 2 A, C and D). In sharp contrast, in IP3R2‐KO cells, longitudinal Ca2+ waves were not observed at all during shear stimulation (Fig. 2 B, C and D). This result clearly demonstrates that IP3R‐mediated Ca2+ signalling plays a key role in the activation of the longitudinal Ca2+ wave in atrial myocytes under shear stress.
Figure 2. IP3R2 is responsible for triggering longitudinal Ca2+ waves under shear stress .
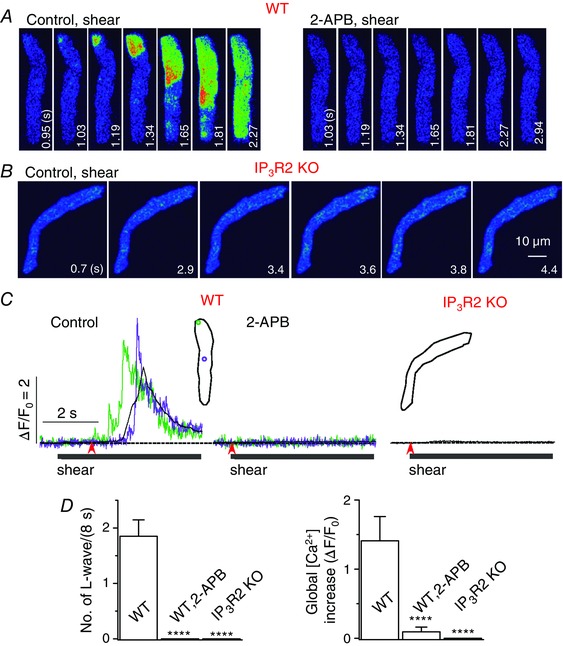
A and B, confocal Ca2+ images recorded in the representative WT (with and without 2‐APB) (A) and IP3R2 KO mouse atrial myocytes (B) during the application of shear stress (∼16 dyn cm−2). Longitudinal Ca2+ waves were not observed during shear application when IP3R2 was deficient. C, Ca2+ fluorescence measured from the ROIs with corresponding colours (inset) from the series of confocal images recorded in the cells illustrated in A and B. The time marked by arrowheads matches with 0 s shown in the confocal images. D, summary of the number of longitudinal Ca2+ waves and whole‐cell Ca2+ increases during 8‐s application of shear stress in WT cells (n = 15) with and without 2‐APB, and in IP3R2 KO cells (n = 12). ****P < 0.0001 vs. WT (unpaired t test).
Role of PLC in the activation of the atrial Ca2+ wave under shear stress
We further examined whether IP3‐generating PLC plays a role in triggering the longitudinal Ca2+ wave under shear stress. In cells in which shear‐dependent longitudinal Ca2+ waves had been recorded under control conditions, shear‐induced waves were no longer observed upon application of the PLC inhibitor U73122 (5 μm, 5 min) (Fig. 3 A–C). In the presence of U73122, although more Ca2+ sparks were detected (Fig. 3 A, right images), there was no significant global Ca2+ change during shear stimulation (Fig. 3 B and C). The exposure to U73122 did not show any effect on resting Ca2+ level (in F/F 0: control, 1.00 ± 0 vs. U73122, 1.06 ± 0.033, n = 22, P > 0.05). Application of U73343 (5 μm, 5 min), the inactive analogue of U73122, did not inhibit formation of shear‐induced Ca2+ waves (Fig. 3 D–F), with no effect on resting Ca2+ level (in F/F 0: control, 1.00 ± 0 vs. U73343, 1.05 ± 0.020, n = 8, P > 0.05), supporting the role of PLC in longitudinal Ca2+ wave generation under shear stress.
Figure 3. PLC plays a role in the generation of longitudinal Ca2+ waves by shear stress in atrial myocytes .
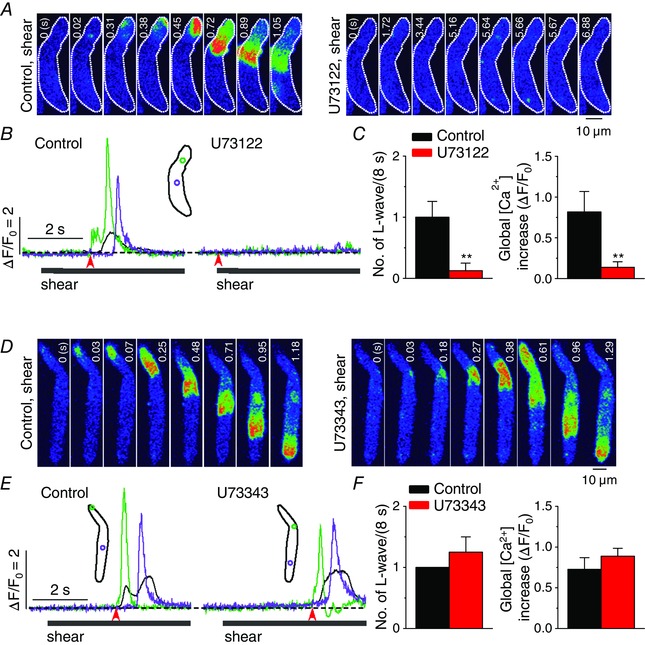
A and D, representative confocal Ca2+ images recorded during the applications of shear (∼16 dyn cm−2) in the absence (‘Control, shear’) and presence of the inhibitor of PLC, U73122 (5 μm; A, ‘U73122, shear’) or its inactive analogue, U73343 (5 μm; D, ‘U73343, shear’), showing no wave under shear stress during the inhibition of PLC. B and E, changes in local (green and violet) and global (black) Ca2+ levels measured from the ROIs (inset) in the series of confocal Ca2+ images recorded from the cells shown in A (B) and D (E). The time marked by arrowheads matches with 0 s shown in the confocal images. C and F, summary of the effects of U73122 (n = 22) or U73343 (n = 8) on the occurrence of longitudinal Ca2+ waves (L‐waves) and on whole‐cell Ca2+ changes (ΔF/F 0) during the application of shear (8 s). **P < 0.01 vs. Control (paired Student's t test).
Shear stress‐mediated Ca2+ wave is not mediated by conventional stretch signalling or transient receptor potential melastatin subfamily 4
Pressurized fluid flow may also elicit local membrane stretch or deterioration. However, shear effects on L‐type Ca2+ channels, Kv1.5 channels, or Ca2+ transients in cardiac myocytes are not inhibited by SAC blockers such as GsMTx‐4, streptomycin, or Gd3+ (Lee et al. 2008; Belmonte & Morad, 2008; Boycott et al. 2013). We therefore tested the effect of GsMTx‐4 (2 μm, 10–20 s) on longitudinal Ca2+ wave occurrence under shear stress in atrial myocytes, and also found that this wave is resistant to this toxin (Fig. 4 A and E). Whole‐cell Ca2+ measurements revealed no significant difference in the magnitude of Ca2+ increase under shear stress in cells pretreated with GsMTx‐4 (Fig. 4 A and E; Table 1). GsMTx‐4 itself did not affect resting Ca2+ level (in F/F 0: control, 1.00 ± 0 vs. GsMTx‐4, 1.00 ± 0, n = 7, P > 0.05).
Figure 4. No role of SAC, NCX, NOX or TRPM4 in the generation of longitudinal Ca2+ waves under shear stress .
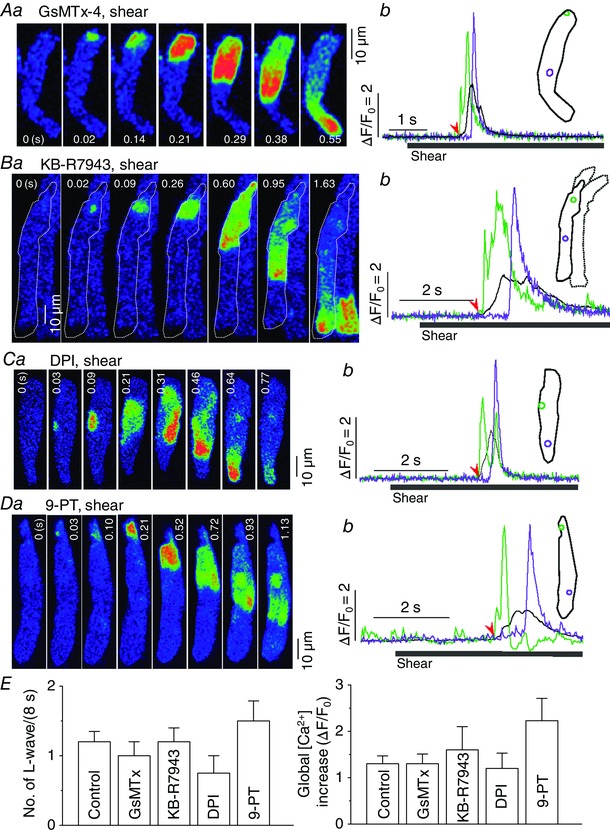
Roles of SAC, NCX, NOX and TRPM4 in the generation of longitudinal Ca2+ wave under shear stress (∼16 dyn cm−2) were tested by the inhibition of each protein using GsMTx‐4 (2 μm; A), KB‐R7943 (0.2 μm; B), diphenyleneiodonium (DPI; 3 μm; C), and 9‐phenanthrol (9‐PT; 10 μm; D), respectively. a and b show series of confocal Ca2+ images, and local and global Ca2+ signals from representative atrial myocytes, respectively. Inset of b illustrates ROIs for the local (green and violet) and global (black) Ca2+ signal traces shown in b. The time marked by the arrowheads matches with 0 s shown in the control image. Note that the inhibition of NCX slowed Ca2+ decay and prolonged Ca2+ signals (Bb; Table 1). E, summary of the occurrence of longitudinal Ca2+ waves (left) and global Ca2+ increase (right) measured on 8 s‐long shear exposure under control conditions and in the presence of each compound. There was no significant change in either parameter by any intervention. Control: n = 64; GsMTx‐4: n = 7; KB‐R7943: n = 6; DPI: n = 5; 9‐PT: n = 7.
Table 1.
Summary of spatiotemporal properties of shear‐induced longitudinal Ca2+ waves
| Local Ca2+ increase (ΔF/F 0) | FDHM of global Ca2+ transient (ms) | Speed of L‐wave (μm s−1) | n | |
|---|---|---|---|---|
| Control | 3.04 ± 0.25 | 415 ± 48.0 | 75.7 ± 3.63 | 64 |
| U73343 (5 μm) | 3.30 ± 0.32 | 607 ± 82.2 | 88.4 ± 12.7 | 7 |
| iso‐PPADS (10 μm) | 2.90 ± 0.68 | 641 ± 83.2 | 70.3 ± 7.32 | 7 |
| KB‐R7943 (0.2 μm) | 4.57 ± 0.54a | 995 ± 91.3b | 93.1 ± 12.2 | 6 |
| GsMTx‐4 (2 μm) | 3.42 ± 2.31 | 400 ± 37.3 | 97.9 ± 11.4 | 7 |
| DPI (3 μm) | 2.72 ± 0.73 | 388 ± 50.8 | 77.1 ± 7.71 | 5 |
| 9‐Phenanthrol (10 μm) | 4.54 ± 0.63a | 751 ± 55.1b | 71.6 ± 5.63 | 7 |
| Zero [Ca2+]o | 2.00 ± 0.42a | 372 ± 43.1 | 73.6 ± 6.68 | 6 |
Values represent mean ± SEM. a P < 0.05 vs. control local Ca2+ increase. b P < 0.05 vs. control FDHM of global Ca2+ transient. L‐wave: longitudinal Ca2+ wave.
Stretch is known to increase intracellular Na+ and Ca2+ concentrations and augment contractility in atrial and ventricular myocytes (Tavi et al. 1998; Luers et al. 2005). We examined whether the Na+−Ca2+ exchanger (NCX) is involved in the activation of the longitudinal Ca2+ wave under shear stress using its specific inhibitor KB‐R7943. After applying 0.2 μm KB‐R7943 for approximately 8 min, shear stress elicited the longitudinal Ca2+ wave with a more prolonged Ca2+ increase (see FDHM in Table 1; Fig. 4 B and E). The exposure to this drug also increased resting Ca2+ level (in F/F 0: control, 1.00 ± 0 vs. KB‐R7943, 1.45 ± 0.08, n = 6, P < 0.05). This result suggests that NCX does not play a role in the development of the Ca2+ wave under shear, but that it is important for the removal of Ca2+ during the longitudinal Ca2+ wave.
Ca2+ sparks are activated by the generation of reactive oxygen species via the activation of subsarcolemmal nicotinamide adenine dinucleotide phosphate oxidase (NOX) in rat ventricular myocytes during whole‐cell stretch (Prosser et al. 2011). We tested whether this mechanism also contributes to the effect of shear on atrial Ca2+ signal using the inhibitor of NOX, DPI. Preincubation of atrial cells in 3 μm DPI‐containing solution for 5 min did not prevent occurrence of the Ca2+ wave or global Ca2+ increase (Fig. 4 C and E; Table 1), indicating that the development of the longitudinal Ca2+ wave under shear is independent of NOX. The drug itself did not affect resting Ca2+ level (in F/F 0: control, 1.00 ± 0 vs. DPI, 1.00 ± 0.004, n = 5, P > 0.05).
We recently observed that transient receptor potential melastatin subfamily 4 (TRPM4) currents are specifically activated by ∼16 dyn cm−2 shear stress (M.‐J. Son, unpublished observations). Because the TRPM4 channel can carry Na+ into the myocytes at resting conditions, it is possible to cause depolarization and cytosolic Ca2+ change (Launay et al. 2002). Therefore, we tested the effect of 9‐phenanthrol, a TRPM4 blocker, on the shear‐induced global Ca2+ wave. This compound has no significant effect on other voltage‐gated ion channels or transient receptor potential channels at concentrations of 10−30 μm (Grand et al. 2008; Simard et al. 2012). The pretreatment of cells with 10 μm 9‐phenanthrol for 4–10 min did not suppress the occurrence of the longitudinal Ca2+ wave during shear stress (Fig. 4 D and E). We observed that the magnitude of local Ca2+ change and FDHM of the global Ca2+ transient during shear‐induced longitudinal Ca2+ wave were significantly increased in the 9‐phenanthrol preincubated cells (Table 1), and that the drug itself increased resting Ca2+ level (in F/F 0: control, 1.00 ± 0 vs. 9‐phenanthrol, 1.08 ± 0.021, n = 7, P < 0.05), suggesting possible contribution of TRPM4 to the shear‐mediated Ca2+ mobilization and to physiological Ca2+ regulation.
Role of the P2Y1 purinergic receptor in the generation of the longitudinal Ca2+ wave under shear stress
A similar type of Ca2+ wave (starting from a focal site) has been reported in vascular endothelial cells under shear stress and has been associated with local release of ATP (Yamamoto et al. 2011). ATP activates PLC via P2Y signalling. We therefore investigated whether the same signalling is responsible for the generation of this atrial Ca2+ wave. We first used the non‐selective P2Y purinoceptor antagonist, suramin. Preincubation of cells with this chemical for 10 min at 10 μm completely eliminated the Ca2+ wave during shear stimulation (Fig. 5 A–C). Suramin did not alter resting Ca2+ level (in F/F 0: control, 1.00 ± 0 vs. suramin, 1.08 ± 0.044, n = 6, P > 0.05). P2Y1 is one of the main subtypes of the P2Y receptor in adult cardiac myocytes (Webb et al. 1996) and plays a major role in atrial pacemaker cells (Ju et al. 2003). We examined the effect of a potent P2Y1‐specific antagonist, 2’‐deoxy‐N 6‐methyladenosine‐3’,5’‐bisphosphate (MRS 2179; 0.2 μm, 10 min) (von Kugelgen & Wetter, 2000), on the induction of the longitudinal Ca2+ wave by shear stress (Fig. 5 D–F). The exposure to MRS 2179 did not alter resting Ca2+ level (in F/F 0: control, 1.00 ± 0 vs. MRS 2179, 1.06 ± 0.025, n = 8, P > 0.05). Although shear‐induced longitudinal Ca2+ waves were clearly observed under control conditions, they were not recorded at all after exposure to MRS 2179.
Figure 5. Role of P2Y1 purinoceptor in the development of longitudinal Ca2+ waves under shear stress .
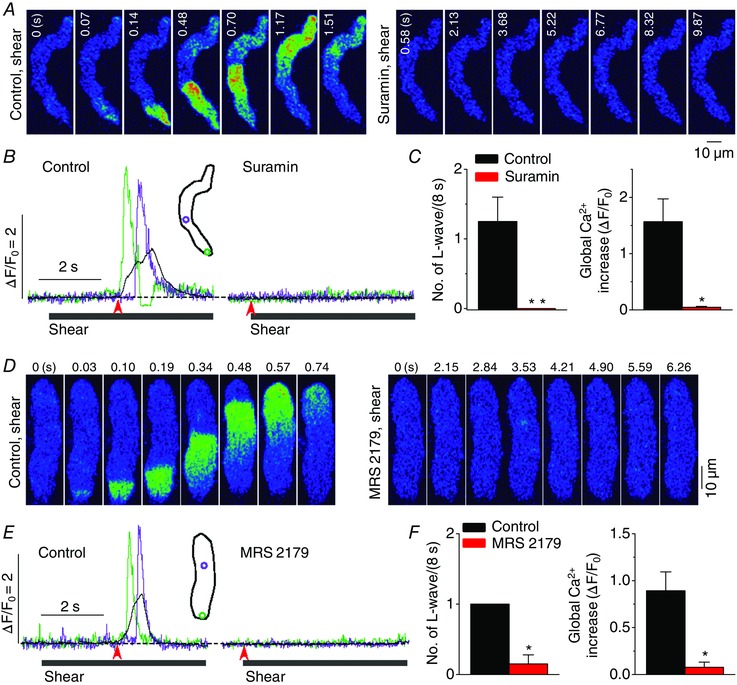
A and D, representative confocal Ca2+ images recorded during the applications of shear stress (∼16 dyn cm−2) in the absence (‘Control, shear’) and presence of the non‐selective inhibitor of P2 purinoceptor, suramin (10 μm; A, ‘Suramin, shear’), or the selective antagonist of P2Y1 receptor, MRS 2179 (0.2 μm; D, ‘MRS 2179, shear’). Both chemicals inhibited the occurrence of longitudinal Ca2+ wave under shear stress. B and E, changes in local (green and violet) and global (black) Ca2+ levels measured from the ROIs (inset) on the series of confocal Ca2+ images recorded in the cells shown in A (B) and D (E). The time marked by arrowheads matches with 0 s shown in the confocal images. C and F, summary of the effects of suramin (n = 6) and MRS 2179 (n = 8) on the occurrence of longitudinal Ca2+ waves and on global Ca2+ changes during the application of shear (8 s). *P < 0.05, **P < 0.01 vs. Control (paired Student's t test).
We examined whether P2X purinoceptors also contribute to the generation of the shear‐induced longitudinal Ca2+ wave. We used the P2X receptor‐selective antagonist, iso‐PPADS, at 1, 10 and 50 μm (10 min). No effect was observed on the Ca2+ wave or global Ca2+ signal at any concentration tested during shear stimulation (Fig. 6). The treatment of iso‐PPADS, at 1, 10 and 50 μm, did not change resting Ca2+ level (in F/F 0: control, 1.00 ± 0 vs. iso‐PPADS at 1 μm, 1.00 ± 0, n = 4, P > 0.05; 10 μm, 1.02 ± 0.05, n = 7, P > 0.05; 50 μm, 1.03 ± 0.03, n = 4, P > 0.05). These findings indicate that the P2X receptor is not involved in the generation of the longitudinal Ca2+ wave under shear stress.
Figure 6. No role for P2X receptor in the activation of longitudinal Ca2+ waves under shear stress .
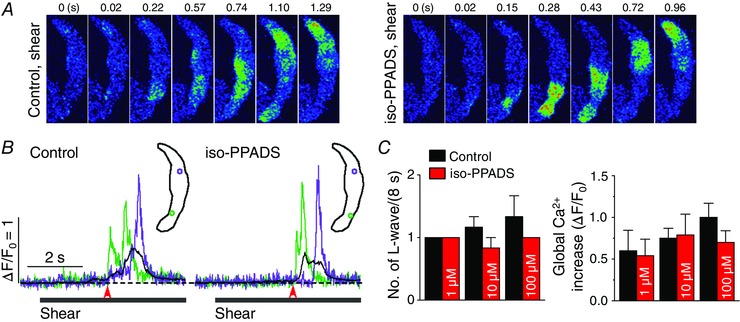
A, representative confocal Ca2+ images recorded during the applications of shear (∼16 dyn cm−2) in the absence (‘Control, shear’) and presence of the selective antagonist of P2X purinoceptor, iso‐PPADS (10 μm; ‘iso‐PPADS, shear’). This chemical did not suppress the occurrence of longitudinal Ca2+ wave under shear stress. B, changes in local (green and violet) and global (black) Ca2+ levels measured from the ROIs (inset) on the series of confocal Ca2+ images recorded in the cell shown in A. The time marked by arrowheads matches with 0 s shown in the confocal images. C, summary of the effects of iso‐PPADS (1 μm, n = 4; 10 μm, n = 7; 50 μm, n = 4) on the occurrence of longitudinal Ca2+ waves and on global Ca2+ changes during the application of shear (8 s). P > 0.05 vs. Control (paired Student's t test).
Role of ATP release through gap junction hemichannels in shear‐mediated longitudinal Ca2+ wave propagation
The P2 purinoceptor is activated by extracellular ATP. In several other cell types, including cardiac myocytes and vascular endothelial cells, mechanical stimulus is implicated in ATP release from the cell to the extracellular space (Cotrina et al. 1998; Nishida et al. 2008; Yamamoto et al. 2011; Oishi et al. 2012). To examine whether ATP released from atrial myocytes elicits the longitudinal Ca2+ wave via purinoceptor–PLC signalling under shear stimulation, we used apyrase, which metabolizes extracellular ATP to AMP and reduces its concentration near the cell membrane. After incubating the cells in solutions containing apyrase (2 U ml−1) for 30 min, shear stress no longer induced Ca2+ waves (Fig. 7), indicating that ATP release from atrial myocytes is involved in the generation of the longitudinal Ca2+ wave under shear stress. The application of apyrase did not alter resting Ca2+ level (in F/F 0: control, 1.00 ± 0 vs. apyrase, 0.986 ± 0.063, n = 8, P > 0.05).
Figure 7. ATP released to extracellular space elicits longitudinal Ca2+ waves during shear stress .
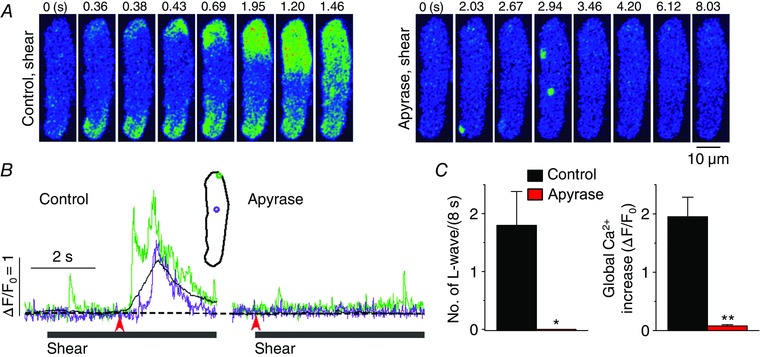
A, representative confocal Ca2+ images recorded during the applications of shear (∼16 dyn cm−2) in the absence (‘Control, shear’) and presence of the ATP metabolizing enzyme, apyrase (2 U ml‒1; ‘Apyrase, shear’). This enzyme did suppress the occurrence of longitudinal Ca2+ waves under shear stress. B, changes in local (green and violet) and global (black) Ca2+ levels measured from the ROIs (inset) on the series of confocal Ca2+ images recorded from the cell shown in A. The time marked by arrowheads matches with 0 s shown in the confocal images. C, summary of the effects of apyrase (n = 8) on the occurrence of longitudinal Ca2+ waves and on global Ca2+ changes during the application of shear (8 s). *P < 0.05, **P < 0.01 vs. Control (paired Student's t test).
One way by which ATP is released in excitable cells is via gap junction hemichannels (Cotrina et al. 1998; Nishida et al. 2008). In addition, the expression of connexin hemichannels in other cell types is regulated by mechanical stress, including shear stress, suggesting that this protein may be an effector that responds to flow shear stress (DePaola et al. 1999; Meens et al. 2013). To further examine the hypothesis that ATP efflux plays a role in the activation of the longitudinal Ca2+ wave through the purinoceptor in atrial cells under shear, we examined the effect of the gap junction hemichannel inhibitor carbenoxolone on shear‐induced Ca2+ waves. Shear stress applied to cells preincubated with carbenoxolone (50 μm, 10 min) never induced the longitudinal Ca2+ wave or any significant Ca2+ rise, although it triggered the Ca2+ wave in the same myocytes under control conditions (Fig. 8 A–C). Carbenoxolone itself had no effect on the resting Ca2+ level (in F/F 0: control, 1.00 ± 0 vs. carbenoxolone, 1.05 ± 0.03, n = 6, P > 0.05). These results suggest that ATP release through gap junction hemichannels may trigger the longitudinal Ca2+ wave in atrial myocytes during shear stimulation.
To confirm whether gap junction hemichannels are involved in the induction of the longitudinal Ca2+ wave, we applied shear stress with zero external Ca2+ solutions to enhance gap junction channel opening (John et al. 1999). Under external Ca2+‐free conditions, shear stress generated more longitudinal Ca2+ waves and smaller Ca2+ waves from several foci during the 8‐s exposure to shear (Fig. 8 D and E). Laterally propagating Ca2+ waves along the cell periphery were often observed during shear exposure under these conditions (Fig. 8 Db and F), manifesting as smaller Ca2+ transients in the whole‐cell Ca2+ measurement (see Fig. 8 Eb). In the absence of external Ca2+, more longitudinal global waves were observed, while the magnitude of global Ca2+ transients for each longitudinal wave was not significantly altered (Fig. 8 G; Table 1). This result further supports our hypothesis that gap junction hemichannels play a role in ATP release that elicits the longitudinal Ca2+ wave via P2Y1 signalling in atrial myocytes under shear stress.
Discussion
In the present study, using two‐dimensional confocal Ca2+ imaging in atrial myocytes in combination with pharmacological and genetic interventions, we have demonstrated for the first time, to our knowledge, that shear stress elicits a longitudinal global Ca2+ wave via IP3R2‐mediated Ca2+ release accompanied by CICR in adult atrial myocytes. We also demonstrated that ATP autocrine action on the P2Y1 purinoceptor coupled with PLC signalling is responsible for this specific Ca2+ signalling. Preincubation of atrial cells with specific inhibitors for IP3R, RyR, PLC, P2 receptor, or P2Y1 receptor eliminated the shear‐induced longitudinal Ca2+ waves. Using type 2 IP3R KO mice we clearly showed that IP3R2‐mediated Ca2+ release plays a role in triggering the Ca2+ wave under shear stress. Apyrase, an enzyme that metabolizes extracellular ATP, and blockade of gap junction hemichannels, also completely abolished the Ca2+ wave under shear stress. Consistent with these observations, removal of extracellular Ca2+, which enhances hemichannel activity, enhanced the occurrence of the wave under shear. These findings further suggest that ATP released from atrial cells through hemichannels plays a role in triggering the longitudinal Ca2+ wave under shear, and explains the activation of P2Y1 purinergic signalling under shear stress. However, generation of the wave under shear stress was not altered by the inhibition of NCX, SAC, NOX, TRPM4, or P2X receptor, providing evidence of mechanotransduction specific to shear stress in cardiac myocytes, which is distinct from the mechanical signalling associated with stretch.
Shear‐specific mechanotransduction in atrial myocytes
Our data demonstrate that shear stress activates purinergic signalling and a subsequent Ca2+ wave in atrial myocytes. Purinoceptor activation by ATP release in atrial cells under shear stress was confirmed by our finding that the ATP metabolizing enzyme apyrase or the respective P2 and P2Y1 purinoceptor antagonists, suramin and MRS 2179, abolished wave occurrence (Figs 5 and 7). Generation of the Ca2+ wave from a focus within a cell during shear stress has already been shown in cultured vascular endothelial cells (Yamamoto et al. 2011). The generation of the Ca2+ wave in the endothelial cells under shear stress is also mediated by purinoceptors activated by ATP release from endothelial cells (Yamamoto et al. 2003, 2011). These endothelial cell responses appear to be similar to our observations in atrial cells. Interestingly, the pathway involved in ATP release under shear stress and the purinoceptor subtype involved in the Ca2+ response seem to differ between the two types of cells. In vascular endothelial cells, shear‐mediated ATP release was associated with caveolae (Yamamoto et al. 2011), whereas in atrial myocytes, ATP release appears to be mediated by gap junction hemichannels (Fig. 8). We did not observe any reliable effect of methyl‐β‐cyclodextrin, which disrupts caveolae and lipid rafts by depleting plasma membrane cholesterol, on longitudinal wave occurrence in atrial cells under shear. The shear‐mediated atrial Ca2+ wave was observed more often in the absence of external Ca2+ (Fig. 8 D–G), when the activity of gap junction hemichannels is increased (John et al. 1999). Blockade of gap junction hemichannels eliminated longitudinal Ca2+ wave generation under shear stress (Fig. 8 A–C). In contrast, the Ca2+ response in endothelial cells under shear is thought to be caused by Ca2+ influx through P2X4 purinergic receptors due to ATP release (Yamamoto et al. 2000, 2003). In fact, ATP release through gap junction hemichannels has been recognized in cardiac myocytes under other mechanical stimuli, such as direct touch (Suadicani et al. 2000) and stretch (Nishida et al. 2008; Oishi et al. 2012). However, it should also be noted that the specific shear/P2Y1‐mediated Ca2+ response in atrial myocytes was not affected by inhibitors of SAC or NOX, important mediators of stretch‐induced Ca2+ responses (Fig. 4 A, C and E). Moreover, it is distinct from stretch‐induced ventricular fibrosis that involves P2Y6 signalling by released ATP (Nishida et al. 2008).
There are other ATP release pathways such as ATP‐permeable anion channels (e.g. maxi anion channels, volume‐regulated Cl− channels), ATP‐binding cassette transporters (e.g. the cystic fibrosis transmembrane conductance regulator) and exocytotic secretion (Cotrina et al. 1998; Schneider et al. 1999; Bodin & Burnstock, 2001; Bell et al. 2003). Depending on the cell type and stimulus, ATP release occurs via one or two such pathways. We used 50 μm Gd3+, which blocks maxi anion channels with little effect on gap junction channels (Bell et al. 2003), and 9‐AC (1 mm), which blocks most Cl− channels, including cystic fibrosis transmembrane conductance regulator and volume‐regulated Cl− channels, to test the possible role of the ATP‐permeable anion channels in longitudinal Ca2+ wave propagation in atrial cells under shear. However, we found no suppression of the Ca2+ wave by these chemicals (Figs 9 and 10). Because no significant shear‐induced Ca2+ increase was observed during IP3R blockade, the ATP release that initiates the P2Y1 receptor–IP3 signalling required for Ca2+ wave generation does not seem to involve Ca2+‐dependent vesicular secretion.
Figure 9. No role for Cl − channels in the generation of longitudinal Ca2+ waves in atrial myocytes under shear stress .
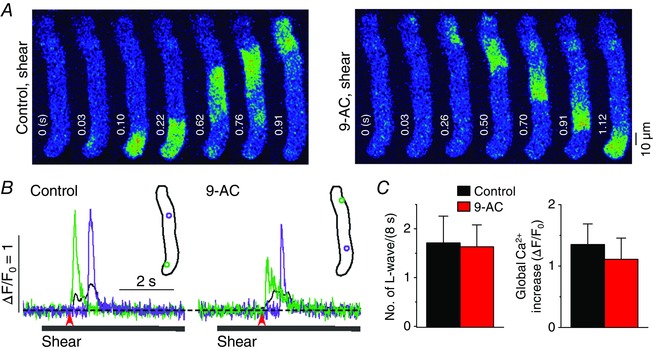
A, series of confocal Ca2+ images recorded in a representative rat atrial myocyte during shear stress treatment of ∼16 dyn cm−2, showing longitudinal Ca2+ wave propagation in the absence (‘Control, shear’) and presence of 1 mm 9‐AC (6 min; ‘9‐AC, shear’), the inhibitor of Cl− channels. B, Ca2+ changes from the correspondingly coloured ROIs, illustrated in the inset, show local (green and violet) and global (black) Ca2+ transients during the shear stimulation. The period of shear application was indicated by the grey bar below the Ca2+ traces. The time point marked by the red arrow matches with 0 s at the confocal images in A. C, comparison of mean values of the occurrence of longitudinal Ca2+ waves (L‐waves) and the magnitude of global Ca2+ transient induced by 8 s‐long shear stimulation before and after application of 1 mm 9‐AC. There was no significant change in either parameter produced by this chemical (4 cells, P > 0.05, paired Student's t test).
Figure 10. No contribution of maxi anion channels to shear‐mediated longitudinal Ca2+ waves .
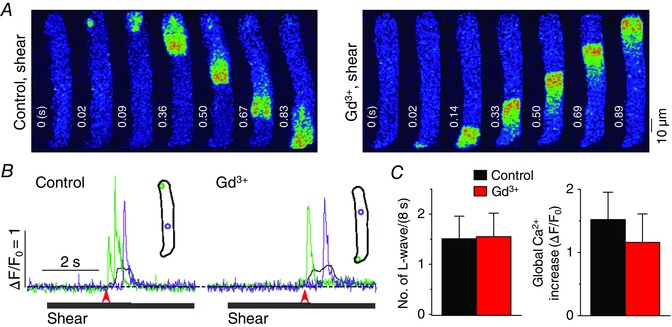
A, a series of confocal Ca2+ images recorded in a representative rat atrial myocyte during the treatment of shear stress of ∼16 dyn cm−2, showing longitudinal Ca2+ wave propagation in the absence and presence of 50 μm Gd3+ (5 min), the inhibitor of maxi anion channels. It should also be noted that higher concentration of Gd3+ is a mild inhibitor of gap junction channels (John et al. 1999). B, Ca2+ changes from the correspondingly coloured ROIs, illustrated in the inset, show local (green and violet) and global (black) Ca2+ transients during the shear stimulation. The period of shear application was indicated by the grey bar below the Ca2+ traces. The time point marked by the red arrow matches with 0 s at the confocal images in A. C, comparison of mean values of the occurrence of longitudinal Ca2+ waves (L‐waves) and the magnitude of global Ca2+ transient induced by 8 s‐long shear stimulation before and after application of Gd3+. There was no significant change in either parameter in the presence of Gd3+ (5 cells, P > 0.05, paired Student's t test).
The mechanism of hemichannel activation as an early response to shear stress remains to be uncovered.
Longitudinal Ca2+ waves originated mostly from focal sites located in the upper and lower parts, and ends of atrial myocytes (see Figs 1 A and D, 2 A, 3 A and D, 4 A, 5 A and D, 7 A and 8 A). This observation suggests that mechanosensors connected to hemichannels, or the hemichannels themselves, are located in the vicinity of the Ca2+ wave core. Gap junctions predominate at intercalated discs under normal conditions, but gap junction hemichannels are also found at lateral sites (Uzzaman et al. 2000). These hemichannels have been suggested to play a role in ventricular ATP release under pathological conditions such as pressure overload (Nishida et al. 2008). In zero external Ca2+ solution, where hemichannels open more frequently, shear stress‐mediated Ca2+ release started from more peripheral sites (Fig. 8 D–G), consistent with the role of laterally localized hemichannels in conducting ATP to generate atrial Ca2+ waves. Shear stress can deform the surface membrane and its proteins, and also affect cytoskeletal proteins linked to these membrane proteins. The hot spots of cytoskeletal strain have been suggested to coincide with the locations of ATP release because such sites are repositioned by shear stress (Helmke et al. 2003). In fact, gap junction hemichannels are connected to cytoskeletal proteins (Meens et al. 2013), and the hemichannels themselves are also considered to be mechanosensitive (Bao et al. 2004). The spatiotemporal characteristics of gap junction hemichannel‐mediated ATP release, and mechanosensing associated with hemichannel opening, need to be examined in atrial cells under shear stress in the future.
PLC–IP3R signalling‐specific longitudinal Ca2+ wave in atrial myocytes under shear stress
We demonstrated that the shear‐induced longitudinal Ca2+ wave is mediated by CICR via the RyR, which is triggered by Ca2+ release through the IP3R2 (Figs 1 and 2). Application of 8‐s long shear stress sometimes induced a much faster transverse Ca2+ propagation from the entire cell periphery to the cell interior (T p of whole‐cell Ca2+ transient = 10–30 ms) with preceding increase in background Ca2+ level. The shear‐induced rapid transverse Ca2+ wave with the background Ca2+ increases was maintained in IP3R2 KO atrial cells and in 2‐APB‐pretreated cells (J.‐C. Kim, unpublished observations), although we never observed longitudinal Ca2+ waves in those myocytes, suggesting that there may be an IP3R signalling‐independent pathway under shear stress, which generates a rapid global Ca2+ transient in atrial cells. In this regard, it was previously reported, using Ca2+ epifluorescence measurements from whole atrial myocytes loaded with fura‐2 (Belmonte & Morad, 2008), that 2 s‐long shear stimulation (25 dyn cm‒2) induces slow Ca2+ transients of which T p (∼130 ms) and magnitude (similar to depolarization‐induced Ca2+ transient) are smaller than those of global Ca2+ signals during the shear (8 s)‐induced longitudinal Ca2+ waves observed in the present study (T p: ∼350 ms; magnitude: ∼2‐fold larger than depolarization‐induced Ca2+ transient; Fig. 13 C). The Ca2+ transients, measured under 2 s‐long shear in the epifluorescence system, develop with a shorter and uniform latency period (0.25−0.3 s), and increase monophasically (Belmonte & Morad, 2008). In sharp contrast, the shear‐induced global Ca2+ transients associated with longitudinal Ca2+ waves showed longer and varied latency (0.2−3 s) and multiphasic increases (e.g. plateau peak, multiple peaks or single peak with humps), depending on the propensity (see Fig. 1 A, B, D and E, and Table 1 for examples) and the speed of wave. The same report suggested that the subcellular source of the shear‐induced fura‐2 transient is not IP3R‐mediated Ca2+ release but mitochondria that are separate from the Ca2+ release pool activated by depolarization, and that Ca2+ flux via the mitochondrial NCX is responsible for the mitochondrial Ca2+ mobilization under shear (Belmonte & Morad, 2008). However, the shear‐mediated longitudinal Ca2+ waves were not affected by the mitochondrial uncoupling and inhibitor of mitochondrial NCX (Figs 11 and 12). In addition, the magnitude of the global Ca2+ signal, associated with the shear‐induced longitudinal Ca2+ wave, was similar to that of depolarization‐induced global Ca2+ transient recorded immediately after shear stimulation (compare Fig. 13 Bb to ‘Global [Ca2+] increase’ in all figures). Although the reasons for the observations of global Ca2+ transient signals with different kinetics and mechanism using a similar fluid puffing system are not clear, differences in the detailed experimental conditions such as shear duration (8 s vs. 2 s) and/or strength (16 vs. 25 dyn cm−2) and superfusion/puffing flow rate that can affect ATP washing might cause more prominent activation of one of the shear‐mediated signalling mechanisms. Together, these results suggest that shear stress activates multiple Ca2+ signalling pathways that have distinct spatiotemporal patterns and mechanisms in atrial myocytes. In the present study, we provide clear evidence for shear‐mediated PLC–IP3R–Ca2+ signalling, specifically associated with a slow longitudinal Ca2+ propagation wave (V p = ∼75 μm s−1) in atrial myocytes.
Figure 11. No role of mitochondrial Na+Ca2+ exchanger on the generation of longitudinal Ca2+ waves in atrial myocytes under shear stress .
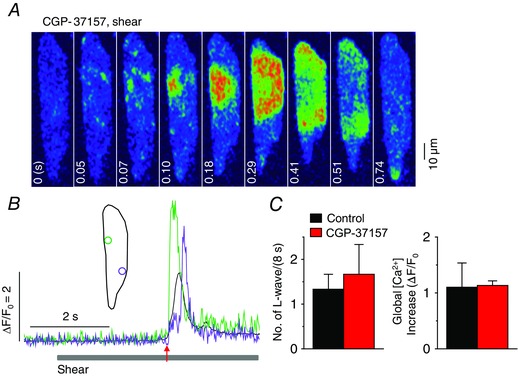
A, a series of confocal Ca2+ images recorded in a representative rat atrial myocyte pretreated with 1 μm CGP‐37157 (1 min), a blocker of the mitochondrial Na+−Ca2+ exchanger, during shear stress treatment of ∼16 dyn cm−2, showing longitudinal Ca2+ wave propagation. B, Ca2+ changes from the correspondingly coloured ROIs, illustrated in the inset, show local (green and violet) and global (black) Ca2+ transients during the shear stimulation. The period of shear application is indicated by the grey bar below the Ca2+ traces. The time point marked by the red arrow matches with 0 s on the confocal images in A. C, comparison of mean values of the occurrence of longitudinal Ca2+ waves (L‐waves) and the magnitude of global Ca2+ transient induced by 8 s‐long shear stimulation before and after application of 1 μm CGP‐37157. There was no significant change in either parameters in the presence of CGP‐37157 (6 cells, P > 0.05, paired Student's t test).
Figure 12. No effect of carbonyl cyanide m‐chlorophenylhydrazone (CCCP) on shear‐induced longitudinal Ca2+ waves in atrial myocytes .
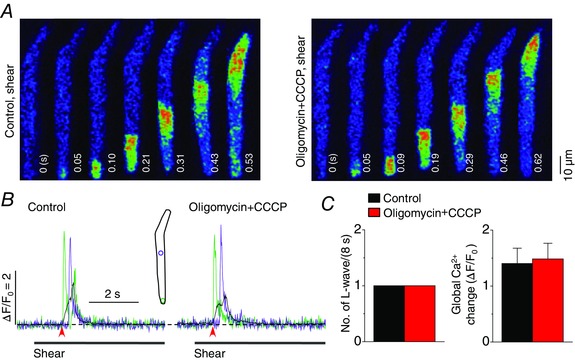
A, representative confocal Ca2+ images recorded during the applications of shear (∼16 dyn cm−2) in the absence (‘Control, shear’) and presence of the mitochondrial uncoupler CCCP (1 μm) together with pretreated oligomycin (1 μg ml−1), the inhibitor of F1/FO‐ATP synthase (‘Oligomycin+CCCP, shear’). Oligomycin was pretreated for 5–6 min to prevent ATP depletion. B, changes in local (green and violet) and global (black) Ca2+ levels measured from the ROIs (inset) on the series of confocal Ca2+ images recorded from the cell shown in A. The time marked by arrowheads matches with 0 s shown in the confocal images. C, summary of the effects of oligomycin + CCCP (n = 4) on the occurrence of longitudinal Ca2+ waves and on global Ca2+ changes during the application of shear (8 s), showing no significant changes in either parameter as a result of the drug treatments.
Pathophysiological implications
The shear stress‐mediated longitudinal Ca2+ wave may significantly alter atrial Ca2+ signalling that normally involves a transverse global Ca2+ wave. We observed the shear stress (16 dyn cm‒2)‐induced longitudinal global Ca2+ propagation during physiological Ca2+ cycling in atrial myocytes (Fig. 13 Ab and C). In addition, the depolarization‐induced Ca2+ transient was significantly increased and prolonged under shear stimulation, and this was soon followed by a dramatic decrease in the Ca2+ transient (Fig. 13 A, B and D). Our observations and previous reports indicate that single cells, including cardiac myocytes and endothelial cells, show significant responses to shear stress in the range of approximately 0.3−30 dyn cm−2 (Olesen et al. 1988; Woo et al. 2007; Belmonte & Morad, 2008; Boycott et al. 2013). The level of interlaminar shear stress in the normal adult rat atria, roughly estimated using the Couette flow model, is ∼0.43 dyn cm−2 at resting heart rate (Boycott et al. 2013). Shear stress increases during regurgitant blood‐jet and volume/pressure overload due to conditions such as valvular heart disease (stenosis), congestive heart failure and hypertension. Considering that the shear stress generated during mitral regurgitation is dependent on the atrioventricular valve orifice and the pressure gradient across the valve during systole, it may be even more difficult to estimate the fluid‐jet force on single myocytes in vivo. The fact that the shear stress used to elicit the longitudinal Ca2+ waves in single cells is ∼35 times higher than the estimated interlaminar shear stress in the adult rat atrium, combined with the rather dramatic decrease in the Ca2+ transient after the initial Ca2+ wave (Fig. 13) suggests pathological relevance of this response such as depression of contraction. Although the level of shear stress that each myocyte would receive in an intact atrial chamber wall during such haemodynamic disturbances is unclear, the consequences of such regurgitation (e.g. mitral regurgitation) or volume overload are atrial arrhythmias and remodelling (Nazir & Lab, 1996; Nattel, 2002). The evidence regarding the absence of after‐transients or spontaneous Ca2+ transients between electrically induced Ca2+ transients in the paced myocytes under shear stress (Fig. 13) supports the view that the shear response involving longitudinal Ca2+ waves does not act as a trigger of arrhythmia in these myocytes. Possible remodelling of Ca2+ signalling tool‐kit proteins and voltage‐dependent ionic currents by physiologically or pathologically relevant shear stress needs to be investigated to fully understand the alterations of atrial Ca2+ signalling under shear stress.
Additional information
Competing interests
None declared.
Author contributions
J.‐C.K. and S.‐H.W. contributed to the conception and design of experiments and were involved in the experiments and collection, analysis and interpretation of data. Experiments were conducted in the lab of S.‐H.W. J.‐C.K. and S.‐H.W. drafted the manuscript. Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean Government (MEST) (2012‐0005369, 2015R1A2A2A01002625).
Acknowledgements
We thank Dr J. Chen at University of California at San Diego (USA) for the IP3R2 knock‐out mice.
References
- Bao L, Sachs F & Dahl G (2004). Connexins are mechanosensitive. Am J Physiol Cell Physiol 287, C1389–C1395. [DOI] [PubMed] [Google Scholar]
- Bell PD, Lapointe JY, Sabirov R, Hayashi S, Peti‐Peterdi J, Manabe K, Kovacs G & Okada Y (2003). Macular densa cell signaling involves ATP release through a maxi anion channel. Proc Natl Acad Sci USA 100, 4322–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte S & Morad M (2008). ‘Pressure–flow’‐triggered intracellular Ca2+ transients in rat cardiac myocytes: possible mechanisms and role of mitochondria. J Physiol 586, 1379–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodin P & Burnstock G (2001). Evidence that release of adenosine trisphosphate from endothelial cells during increased shear stress is vesicular. J Cardiovasc Pharmacol 38, 900–908. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Collins TJ, Mackenzie L, Roderick HL, Berridge MJ & Peppiatt CM (2002). 2‐Aminoethoxydiphenyl borate (2‐APB) is a reliable blocker of store‐operated Ca2+ entry but an inconsistent inhibitor of InsP3‐induced Ca2+ release. FASEB J 16, 1145–1150. [DOI] [PubMed] [Google Scholar]
- Boycott HE, Barbier CSM, Eichel CA, Costa KD, Martins RP, Louault F, Dilanian G, Coulombe A, Hatem SN & Balse E (2013). Shear stress triggers insertion of voltage‐gated potassium channels from intracellular compartments in atrial myocytes. Proc Natl Acad Sci USA 110, E3955–E3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carl SL, Felix K, Caswell AH, Brandt NR, Ball WJ, Vaghy PL, Meissner G & Ferguson DG (1995). Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium. J Cell Biol 129, 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ & Cannell MB (1993). Calcium sparks: elementary events underlying excitation‐contraction coupling in heart muscle. Science 262, 740–744. [DOI] [PubMed] [Google Scholar]
- Conwell JA, Cocalis MW & Erickson LC (1993). EAT to the beat: ‘ectopic’ atrial tachycardia caused by catheter whip. Lancet 342, 8873. [DOI] [PubMed] [Google Scholar]
- Costa KD, Takayama Y, McCuloch AD & Covell JW (1999). Laminar fiber architecture and three‐dimensional systolic mechanics in canine ventricular myocardium. Am J Physiol 276, H595–H607. [DOI] [PubMed] [Google Scholar]
- Cotrina ML, Lin JHC, Alves‐Rodrigues A, Liu S, Li J, Azmi‐Ghadimi H, Kang J, Naus CCG & Nedergaard M (1998). Connexins regulate calcium signaling by controlling ATP release. Proc Natl Acad Sci USA 95, 15735–15740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePaola N, Davier PF, Pritchard WF, Florez L, Harbeck N & Polacek DC (1999). Spatial and temporal regulation of gap junction connexin43 in vascular endothelial cells exposed to controlled disturbed flows in vitro . Proc Natl Acad Sci USA 96, 3154–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB (2009). Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol 587, 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grand T, Demion M, Norez C, Mettey Y, Launay P, Becq F, Bois P & Guinamard R (2008). 9‐Phenanthrol inhibits human TRPM4 but not TRPM5 cationic channels. Br J Pharmacol 153, 1697–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara N, Masuda H, Shoda M & Irisawa H (1992). Stretch‐activated anion currents of rabbit cardiac myocytes. J Physiol 456, 285–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmke BP, Rosen AB & Davies PF (2003). Mapping mechanical strain of an endogenous cytoskeletal network in living endothelial cells. Biophys J 84, 2691–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John SA, Kondo R, Wang SY, Goldhaber JI & Weiss JN (1999). Connexin‐43 hemichannels opened by metabolic inhibition. J Biol Chem 274, 236–240. [DOI] [PubMed] [Google Scholar]
- Ju YK, Huang W, Jiang L, Barden JA & Allen DG (2003). ATP modulates intracellular Ca2+ and firing rate through a P2Y1 purinoceptor in cane toad pacemaker cells. J Physiol 552, 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamkin A, Kiseleva I, Wagner KD, Bohm J, Theres H, Günther J & Scholz H (2003). Characteristics of stretch‐activated ion currents in isolated atrial myocytes from human hearts. Pflugers Arch 446, 339–346. [DOI] [PubMed] [Google Scholar]
- Kong CR, Bursac N & Tung L (2005). Mechanical excitation by fluid jets in monolayers of cultured cardiac myocytes. J Appl Physiol (1985) 98, 2328–2336. [DOI] [PubMed] [Google Scholar]
- Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R & Kinet JP (2002). TRPM4 is a Ca2+‐activated nonselective cation channel mediating cell membrane depolarization. Cell 109, 397–407. [DOI] [PubMed] [Google Scholar]
- Lee S, Kim JC, Li Y, Son MJ & Woo SH (2008). Fluid pressure modulates L‐type Ca2+ channel via enhancement of Ca2+‐induced Ca2+ release in rat ventricular myocytes. Am J Physiol Cell Physiol 294, C966–C976 [DOI] [PubMed] [Google Scholar]
- LeGrice IJ, Takayama Y & Covell JW (1995). Transverse shear along myocardiac cleavage planes provides a mechanism for normal systolic wall thickening. Circ Res 77, 182–193. [DOI] [PubMed] [Google Scholar]
- Li X, Zima AV, Sheikh F, Blatter LA & Chen J (2005). Endothelin‐1‐induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol‐1,4,5‐trisphosphate (IP3)‐receptor type 2‐deficient mice. Circ Res 96, 1274–1281. [DOI] [PubMed] [Google Scholar]
- Lipp P, Laine M, Tovey SC, Burrell KM, Berridge MJ, Li W & Bootman MD (2000). Functional InsP3 receptors that may modulate excitation–contraction coupling in the heart. Curr Biol 10, 939–942. [DOI] [PubMed] [Google Scholar]
- Luers C, Fialka F, Elgner A, Zhu D, Kockskämper J, Lewinski DV & Pieske B (2005). Stretch‐dependent modulation of [Na+]i, [Ca2+]i, and pHi in rabbit myocardium – a mechanism for the slow force response. Cardiovasc Res 68, 454–463. [DOI] [PubMed] [Google Scholar]
- Mackenzie L, Bootman MD, Laine M, Berridge MJ, Thuring J, Holmes A, Li WH & Lipp P (2002). The role of inositol 1,4,5‐trisphosphate receptors in Ca2+ signalling and the generation of arrhythmias in rat atrial myocytes. J Physiol 541, 395–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meens MJ, Pfenniger A, Kwak BR & Delmar M (2013). Regulation of cardiovascular connexins by mechanical forces and junctions. Cardiovasc Res 99, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattel S (2002). New ideas about atrial fibrillation 50 years on. Nature 415, 219–226. [DOI] [PubMed] [Google Scholar]
- Nazir SA & Lab MJ (1996). Mechanoelectric feedback and atrial arrhythmias. Cardiovasc Res 32, 52–61. [PubMed] [Google Scholar]
- Nishida M, Sato Y, Uemura A, Narita Y, Tozaki‐Saitoh H, Nakaya M, Ide T, Suzuki K, Inoue K, Nagao T & Kurose H (2008). P2Y6 receptor‐Gα12/13 signalling in cardiomyocytes triggers pressure overload‐induced cardiac fibrosis. EMBO J 27, 3104–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi S, Sasano T, Tateishi Y, Tamura N, Isobe M & Furukawa T (2012). Stretch of atrial myocytes stimulates recruitment of macrophages via ATP released through gap‐junction channels. J Pharmacol Sci 120, 296–304. [DOI] [PubMed] [Google Scholar]
- Olesen S‐P, Clapham DE & Davies PF (1988). Hemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature 331, 168–170. [DOI] [PubMed] [Google Scholar]
- Prosser BL, Ward CW & Lederer WJ (2011). X‐ROS signaling: rapid mechano‐chemo transduction in heart. Science 333, 1440–1445. [DOI] [PubMed] [Google Scholar]
- Rosa AO, Yamaguchi N & Morad M (2013). Mechanical regulation of native and the recombinant calcium channel. Cell Calcium 53, 264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato R & Koumi S‐I (1998). Characterization of the stretch‐activated chloride channel in isolated human atrial myocytes. J Membr Biol 163, 67–76. [DOI] [PubMed] [Google Scholar]
- Schneider SW, Egan ME, Jena BP, Guggino WB, Oberleithner H & Geibel JP (1999). Continuous detection of extracellular ATP on living cells by using atomic force microscopy. Proc Natl Acad Sci USA 96, 12180–12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard C, Salle L, Rouet R & Guinamard R (2012). Transient receptor potential melastatin 4 inhibitor 9‐phenanthrol abolishes arrhythmias induced by hypoxia and re‐oxygenation in mouse ventricle. Br J Pharmacol 165, 2354–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suadicani SO, Vink MJ & Spray DC (2000). Slow intercellular Ca2+ signaling in wild‐type and Cx43‐null neonatal mouse cardiac myocytes. Am J Physiol Heart Circ Physiol 279, H3076–H3088. [DOI] [PubMed] [Google Scholar]
- Subedi KP, Kim JC, Kang M, Son MJ, Kim YS & Woo SH (2011). Voltage‐dependent anion channel 2 modulates resting Ca2+ sparks, but not action potential‐induced Ca2+ signaling in cardiac myocytes. Cell Calcium 49, 136–143. [DOI] [PubMed] [Google Scholar]
- Tavi P, Han C & Weckström M (1998). Mechanisms of stretch‐induced changes in [Ca2+]i in rat atrial myocytes. Circ Res 83, 1165–1177. [DOI] [PubMed] [Google Scholar]
- Uzzaman M, Honjo H, Takagishi Y, Emdad L, Magee AI, Severs NJ & Kodama I (2000). Remodeling of gap junctional coupling in hypertrophied right ventricles of rats with monocrotaline‐induced pulmonary hypertension. Circ Res 86, 871–878. [DOI] [PubMed] [Google Scholar]
- von Kugelgen I & Wetter A (2000). Molecular pharmacology of P2Y‐receptors. Naunyn Schmiedebergs Arch Pharmacol 362, 310–323. [DOI] [PubMed] [Google Scholar]
- Webb TE, Boluyt MO & Barnard EA (1996). Molecular biology of P2Y purinoceptors: expression in rat heart. J Auton Pharmacol 16, 303–307. [DOI] [PubMed] [Google Scholar]
- Woo SH, Risius T & Morad M (2007). Modulation of local Ca2+ release sites by rapid fluid puffing in rat atrial myocytes. Cell Calcium 41, 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Furuya K, Nakamura M, Kobatake E, Sokabe M & Ando J (2011). Visualization of flow‐induced ATP release and triggering of Ca2+ waves at caveolae in vascular endothelial cells. J Cell Sci 124, 3477–3483. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Korenaga R, Kamiya A & Ando J (2000). Fluid shear stress activates Ca2+ influx into human endothelial cells via P2X4 purinoceptors. Circ Res 87, 385–391. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sokabe T, Ohura N, Nakatsuka H, Kamiya A & Ando J (2003). Endogenously released ATP mediates shear stress‐induced Ca2+ influx into pulmonary artery endothelial cells. Am J Physiol Heart Circ Physiol 285, H793–H803. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Youm JB, Sung HK, Lee SH, Ryu SY, Ho WK & Earm YE (2000). Stretch‐activated and background non‐selective cation channels in rat atrial myocytes. J Physiol 523, 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]