

Abstract
Mycobacterium tuberculosis (Mtb) responsible for both latent and symptomatic tuberculosis (TB) remains the second leading cause of mortality among infectious diseases worldwide. Mycobacterial biotin protein ligase (MtBPL) is an essential enzyme in Mtb and regulates lipid metabolism through the post-translational biotinylation of acyl coenzyme A carboxylases. We report the synthesis and evaluation of a systematic series of potent nucleoside-based inhibitors of MtBPL that contain modifications to the ribofuranosyl ring of the nucleoside. All compounds were characterized by isothermal titration calorimetry (ITC) and shown to bind potently with KD's below 2 nM. Additionally, we obtained high-resolution co-crystal structures for a majority of the compounds. Despite fairly uniform biochemical potency, the whole-cell Mtb activity varied greatly with minimum inhibitory concentrations (MIC) ranging from 0.78 to >100 μM. Cellular accumulation studies showed a nearly 10-fold enhanced accumulation of a C-2′-α analog over the corresponding C-2′-β analog, consistent with their differential whole-cell activity.
Keywords: Biotin protein ligase, Mycobacterium tuberculosis, tuberculosis, bisubstrate adenylation inhibitors
Introduction
Tuberculosis (TB) is a leading cause of bacterial infectious disease mortality and morbidity. The bacillus Mycobacterium tuberculosis (Mtb) is responsible for both latent and symptomatic TB and belongs to the greater Mycobacterium tuberculosis complex, which includes M. bovis, M. africanum, M. canettii, and M. microti.1 The current treatment for drug sensitive TB requires combination therapy with the first line drugs isoniazid, rifampin, pyrazinamide and ethambutol for 6–9 months.2 Successful treatment of TB necessitates (1) stringent supervision by well-trained care givers, (2) an ample and consistent supply of high quality medicines and (3) drug intake fidelity by patients to minimize the risks of recrudescence and drug resistance. These are already tall obstacles to surmount in remote and under developed regions where TB is endemic. Consequently, the emergence of multidrug- and extensively drug-resistant TB (MDR-TB and XDR-TB) strains which are minimally resistant to the two most effective drugs, isoniazid and rifampin, has alarmed clinicians and threatens the positive gains made in combatting TB.3 Hence, it is imperative to develop new classes of anti-tubercular agents for this worldwide clinical need that are effective against drug-resistant TB and ideally possess a novel mode of action.
From the onset Mtb has both intrigued and frustrated researchers. Its cellular envelope contains a panoply of structurally diverse lipids that provide a protective barrier and modulate host-pathogen interactions.4,5 The fortified waxy cell envelope in turn makes TB a particularly challenging and intransigent infection to treat. The mycolic acids (1) are the most abundant lipids in Mtb comprising approximately 30% of its dry cellular weight and also the largest lipids known in nature containing up to 90 carbon atoms (Figure 1).6,7 These essential lipids are covalently attached to the arabinogalactan core of the cell wall and anchor the outer membrane. Other lipids and lipidated metabolites such as the phenolic glycolipids (PGLs, 2), phthiocerol dimycocerosate A (PDIM-A, 3), sulfolipids (SLs, 4) and mycobactins (MBTs, 5) are non-covalently associated with the cell envelope and critical for virulence.8–10
Figure 1.

Unique lipids and lipidated metabolites found in cell envelope of Mycobacterium tuberculosis. All of the molecules shown exist as a suite of related isomers that vary in the lipid chain length. If reported, the major isomer is shown otherwise a representative molecule is depicted. The mycolic acids are represented by the most abundant a mycolic acid (α-MA, 1), the phenolic glycolipids are represented by 2, the phthiocerol dimycocerosates are exemplified by PDIM-A (3), the sulfolipids are represented by SL-1 (4) and the mycobactins by 5.
All mycobacterial lipids are derived from simple malonyl coenzyme A (CoA) building blocks that are synthesized by acyl CoA carboxylases (ACCs). Distinct from most bacteria, Mtb possesses multiple ACCs for preparation of malonyl CoA, methylmalonyl CoA, and long-chained malonyl CoAs needed for synthesis of simple fatty acids, methyl brached lipids, and the mycolic acids, respectively.11–13 The multimeric ACCs require post-translational biotinylation in order to become functionally active, which is catalyzed by biotin protein ligase (BPL) encoded by birA (Rv3273c) in Mtb.14 The biotin cofactor mediates carboxy group transfer onto the acyl CoA substrates (Figure 2B). Consequently, MtBPL universally regulates lipid metabolism in Mtb and therefore represents an attractive biochemical target for therapeutic development.15
Figure 2.

(A) MtBPL catalyzed biotinylation of ACC, a biotin dependent enzyme proceeds in two steps by sequential adenylation of biotin (6) to form Bio-AMP (7) followed by acylation of the biotin carboxylase carrier protein domain of ACCs to furnish holo-ACC (8). (B) Biotinylated ACC proteins mediate carboxyl group transfer onto acyl-CoAs forming malonyl-CoAs for lipid biosynthesis. The N-1 atom of biotin is carboxylated in an ATP-dependent manner by the biotin-carboxylase domain of ACCs to furnish 9. In the second half-reaction, ACCs transfer the carboxy group onto an acyl-CoA, mediated by the biotin carboxylase domain of ACCs.
MtBPL catalyzes a two-step reaction initiated when the substrate biotin (6) and ATP bind to form an acyl-adenylate intermediate (7, Bio-AMP) and pyrophosphate (Figure 2A).16 In the succeeding step, MtBPL binds the ACC and catalyzes the transfer of biotin from 7 onto the terminal amine of a conserved lysine residue to afford an amide bound biotinylated-ACC complex (8). We have reported the design of a bisubstrate inhibitor of MtBPL termed Bio-AMS (14) as well as several related analogs including 15, 16, and 17.15, 17 These analogs mimic 7 through substitution of the labile acyl-phosphate linkage in 7 with stabile acyl-sulfamide moieties (Figure 3). Compound 14 is exceptionally potent, binding MtBPL with a KD of approximately 0.5 nM or more than 1,700 times more tightly than biotin, and derives its potency through interactions with the biotin and adenosine substrate binding pockets.17 The sulfamide is a near perfect phosphate isostere and is also negatively charged at pH 7 (the estimated pKa of the acyl-sulfamide NH group that is N-acylated is ~3), which ensures preservation of the critical electrostatic interactions with R69, R72, and K138 of MtBPL. Several other related bisubstrate biotin-adenosine inhibitors of BPLs from other organisms have also been described including: alkylphosphate 10,18 β-ketophosphonate 11,19 1,2,3-triazole 12,20 and acylsulfamate 13. More recently, Tieu, Abell and coworkers identified a novel series of Staphylococcus aureus BPL inhibitors containing a heterocyclic benzoxazoline moiety expemplifid by 18 wherein the adenosine nucleobase has been replaced altogether.21,22 Furthermore, biotin analogs (19 and 20) with selective S. aureus BPL inhibitory activity have also been reported.23
Figure 3.

(A) Representative examples of biotin-AMP nucleoside-based bisubstrate inhibitors of BPL including: alkylphosphate 10, β-ketophosphonate 11, 1,2,3 triazole 12, acylsulfamate 13, acylsulfamide 14, acylsulfonamide 15, 3-deaza acylsulfamate 16, and 2-tert-butylethynyl-acylsulfamate 17. (B) Benzoxazoline moiety 18, a selective inhibitor of S. aureus BPL. (C) Biotin based inhibitors 19 and 20 of S. aureus BPL.
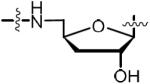
In addition to its potent biochemical activity, 14 was shown to possess promising antimycobacterial activity against ten MDR-TB and XDR-TB strains with minimum inhibitory concentrations (MICs) ranging from 0.16 to 0.625 μM.15 Proteomic analysis demonstrated 14 inhibited biotinylation in whole-cell Mtb while overexpression of MtBPL conferred resistance in Mtb and M. smegmatis. Collectively, these results provide strong evidence for the designed mechanism of action.15 The structure of 14 and 7 bound to MtBPL reveals a large number of electrostatic interactions with the biotin, adenine, and acylsulfamate linkage as well as limited space in the active site to further extend the ligand at these positions.15,24 By contrast, the ribose moiety of the nucleoside has relatively few interactions with the protein and an adjacent accessible crevice to grow the ligand. As a result, we believe modification of the ribose offers the greatest opportunity to further increase potency and improve drug disposition properties. Based on these considerations, we describe herein the design, synthesis and evaluation of a systematic series of rationally designed bisubstrate inhibitors of MtBPL that probe the consequences of modifications to the ribofuranosyl ring of 14.
RESULTS AND DISCUSSION
Chemistry
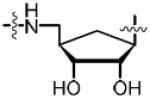
The 4′-ribofuranosyl oxygen of the lead compound 14 does not appear to have any interactions with MtBPL.15 To investigate the impact of deleting the 4′-ribofuranosyl oxygen we proposed the synthesis of a trio of analogs that include carbocyclic Bio-AMS 28, carbocyclic 2′,3′-dideoxy-Bio-AMS 36 and carbocyclic 2′,3′-dideoxy-2′,3′-dehydro-Bio-AMS 34. Analogs 28, 34 and 36 are all derived from the common intermediate 23. Scheme 1 illustrates the synthesis of 28. The aristeromycin derivative 24 was synthesized from Vince lactam (21),25,26 which was first elaborated to the cyclopentenylamine intermediate 22 in three steps following methodology developed by Jung and Rhee.27,28 The installation of the adenine moiety was accomplished via a palladium catalyzed allylic amination to afford adenine intermediate 23. The use of [PdCl(C3H5)]2/triisopropylphosphite as the catalyst and Cs2CO3 as the base was found to be superior to previous reported conditions for related compounds.28 Pivalate hydrolysis of 23 followed by dihydroxylation and acetonide protection afforded a mixture of diasteromeric carbocyclic nucleosides 24α and 24β in an approximately 1:1 ratio. The relative stereochemistry was confirmed by NOESY studies, comparison of 1H NMR with literature reported values, and co-injection with an authentic standard of 24α obtained from the natural product aristeromycin.29–31 The aristeromycin derivative 24α was then converted into the bis-Boc-protected intermediate 25 in two steps, followed by desilylation to afford 26. A Mitsunobu reaction with tert-butyl sulfamoylcarbamate was employed for direct installation of the sulfamide moiety at C-5′. It was discovered that excess triphenylphosphine and DIAD resulted in a significant generation of an iminophosphorane byproduct of 27 (not shown). This was avoided using three equivalents of tert-butyl sulfamoylcarbamate (3.0 equiv) and slow addition via syringe pump of a slight excess of DIAD (1.05 equiv). Biotinylation employing d-(+)-biotin N-hydroxysuccinimide ester (Bio-NHS) mediated by Cs2CO3 followed by global deprotection with 50% aqueous TFA, reverse-phase HPLC purification and lyophilization afforded the carbocylic Bio-AMS 28 in 56% over two steps as the triethylammonium salt.
Scheme 1.

Synthesis of Carbocyclic-Bio-AMS (28).
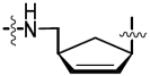
The retention of the C2′-C3′ olefinic bond in analog 34 and saturation of the olefin in cyclopentyl analog 36, were designed to explore the impact of deletion of the corresponding hydroxyl groups and examine the subtle conformational implications on binding that result from saturation of the olefin. Continuing from intermediate 23, hydrolysis of the C-5′ pivaloyl ester proceeded with facility to afford 29 (Scheme 2). Reprotection of the C-5′ alcohol as the silyl ether 30 followed by bis-Boc protection of the N6 amine provided 31. Desilylation of the primary alcohol yielded 32 that was utilized in the succeeding Mitsunobu reaction to give sulfamide intermediate 33. Biotinylation provided the penultimate crude material, which was followed by global deprotection, reverse-phase HPLC purification, and lyophilization to achieve analog 34 in 52% over two steps. Analog 36 was synthesized over three synthetic steps from previously synthesized unsaturated intermediate 33. Hydrogenation using 10 wt% Pd/C in EtOAc supplied the saturated carbocyclic Boc-sulfamide intermediate 35. Biotinylation followed by direct global deprotection of the crude material with 80% aqueous TFA, reverse-phase HPLC purification and lyophilization furnished final analog 36 in 58% over two steps.
Scheme 2.

Synthesis of Cyclopentenyl Bio-AMS (34) and Cyclopentyl Bio-AMS (36).
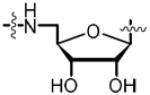
To directly target and manipulate the interactions that exist between the 2′-hydroxyl moiety of our lead compound 14 and an Asp167 residue in the binding pocket and provide a functional handle to grow ligands into the adjacent crevice, we envisioned the introduction of alternate functional groups at the C-2′ carbon. Analogs 46, 47, 57, 63 and 69 are representative archetypal 2′-deoxy-molecules that contain α-azido, α-amino, β-azido, α-fluoro and β-fluoro substituents, respectively. The synthesis of 2′-α-azido (46) and 2′-α-amino (47) analogs is shown in Scheme 3. Vidarabine (37) was protected simultaneously at the C-3′ and C-5′ alcohols using the Markiewicz protecting group yielding cyclic siloxane 38.32 Conversion of the C-2′ alcohol into the corresponding triflate 39 occurred smoothly. SN2 displacement of the triflate by NaN3 in DMF at 65 °C proceeded with inversion of configuration to provide 40.33 bis-Boc protection of the N6 amine furnished 41.34 The selective mono-deprotection of either the C-3′ or C-5′ alcohol nucleosides protected by the Markiewicz group is extensively described in the literature.35 Utilizing a TFA, H2O and THF (1:1:4) solution it is possible to exclusively deprotect the C-5′ alcohol while leaving the C-3′ alcohol protected.36 We were successful in the aforementioned deprotection only to discover that any subsequent chemistry on the C-5′ alcohol was hampered by the steric bulk of the Markiewicz group remnants still attached to C-3′. It was decided that the complete removal of the Markiewicz protecting group with TBAF and 1% AcOH followed by reprotection was the best avenue to pursue. Thus, removal of the Markiewicz group afforded 42 followed by TBS reprotection of the C-3′ and the C-5′ alcohols using standard protocols achieved 43. Regioselective removal of the C-5′ TBS group employing a 3:1:1 AcOH–THF–H2O mixture provided the free C-5′ alcohol 44.37 Despite the formation of inseparable side products, formation of sulfamide intermediate 45 via the Mitsunobu reaction proceeded smoothly. The standard biotinylation reaction described previously yielded the biotinylated product that was immediately followed by global deprotection in 80% aqueous TFA. The crude product was purified via reverse phase HPLC in a yield of 62% over two steps to furnish 46. Further elaboration of the 2′-α-azide to the 2′-α-amine, an isostere of the C2′-hydroxyl on 14, was affected by the Staudinger reduction to provide 47.38,39
Scheme 3.
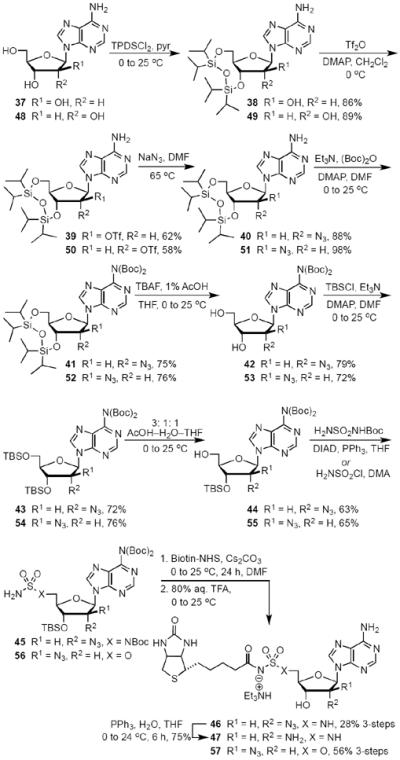
Synthesis of 2′-α-Azido-2′-deoxy-Bio-AMS (46), 2′-α-Amino-2′-deoxy-Bio-AMS (47) and 2′-β-azido-2′-deoxy-Bio-AMS (57).
The only difference between 2′-α-azido (46) described above and 2′-β-azido (57) is the β-facing appendage at the C-2′ carbon. This minor alteration precluded us from utilizing the more sterically demanding Mitsunobu reaction to install the sulfamide arm. Therefore, we were forced to pursue sulfamate analogs for our β-C-2′ analogs. The sulfamates were installed using the Okada protocol.40 Sulfamoyl chloride is sterically smaller and vastly more reactive than tert-butyl sulfamoylcarbamate. Towards the synthesis of analog 57 shown in Scheme 3, adenosine (48) was analogously elaborated to 55, which was treated with sulfamoyl chloride in dimethylacetamide (DMA) to afford sulfamate 56. Biotinylation of the sulfamate intermediate was followed by acidic global deprotection, HPLC purification and lyophilization to yield 57 in 76% over two steps. Our previous experiences with a biotinylated acyl-sulfamate analog had been marred with rapid decomposition via an intra-molecular displacement reaction to form the biologically inactive 3′,5′-cyclo-5′-deoxyadenosine and N-(biotinyl)sulfamic acid.15 Remarkably in the cases of analogs 57 as well as 69 and 73 (vide infra), we were pleasantly surprised to discover that they resisted any cyclonucleoside formation. Their stability can be attributed to the β-facing C-2′ moieties, which preclude the adenine base from adopting the syn-conformation around the glycosidic linkage from which the intramolecular displacement reaction occurs.
Introduction of a fluorine atom at the C-2′ position of nucleosides is well known to increase anomeric stability, modulate ring pucker and binding affinity as well as enhance pharmacokinetic properties.41,42 Scheme 4 illustrates our synthesis of the 2′-deoxy-2′-α-fluoro analog 63 starting with commercially available 2′-deoxy-2′-α-fluoro-adenosine 58. TBS protection of the C-3′ and C-5′ alcohols furnished 59. Again, bis-Boc protection of the N6 amine cleanly afforded 60 and mono-desilylation, using our previously described conditions, achieved the desired product 61. Formation of the sulfamide intermediate 62 using our optimized Mitsunobu procedure gave slightly impure product, which was used in the next reaction. Biotinylation, followed by global deprotection in 80% aqueous TFA, furnished the final analog 63 in 56% over the final two steps. We also pursued the synthesis of the 2′-deoxy-2′-β-fluoro biotinylated analog 69 (Scheme 4). Compound 64 was analogously elaborated to 67. Sulfamate formation (68) was followed by biotinylation and then global deprotection with 80% aqueous TFA. Reverse phase HPLC purification and lyophilization furnished the final analog 69 in a three-step yield of 51%.
Scheme 4.

Synthesis of 2′-Deoxy-2′-αfluoro-Bio-AMS (63) and C2′-deoxy-2′-β-fluoro-Bio-AMS (69).
Further modifications of the glycoside unit steered us towards the synthesis of analogs 73 in Scheme 5 and 82 in Scheme 6. Addition of the β-methyl to C-2′ facilitated our investigation into whether the application of a small lipophilic arm would have any effect on binding. Analog 73 was synthesized over 9-steps from the tribenzoyl protected ribose 70. Compound 71 was attained over six arduous chemical steps.43 Sulfamate formation in DMA afforded 72 in good yield.28,30 Subsequent biotinylation was followed by global deprotection to afford analog 73 in 45% yield over three steps.
Scheme 5.
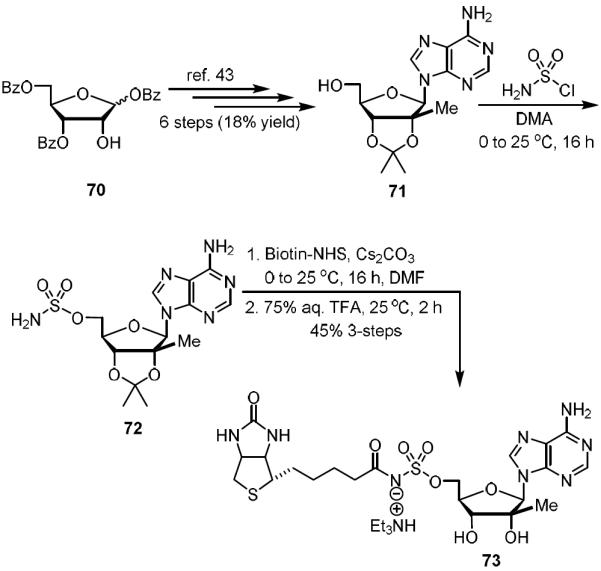
Synthesis of C-2′-β-methyl-Bio-AMS (73).
Scheme 6.

Synthesis of Morpholinyl-Bio-AMS (82).
An even more extreme alteration can be seen in derivative 82 in which the ribosyl ring has been altogether removed in favor of a morpholine ring. Analog 82 was designed primarily as a platform for preparation of derivatives based upon modification of the morpholine nitrogen. The synthetic route to morpholine analog 82 (Scheme 9) began with commercially available N6-benzoyl adenosine (74). After selective TBDPS protection of the C-5′ hydroxyl to afford 75, reaction with sodium meta-periodate in MeOH resulted in oxidative cleavage of the C-2′–C-3′ bond to reveal the nonisolable dihemiacetal intermediate 76, which underwent in situ reaction with ammonium biborate to afford dihemiaminal 77. Reduction with sodium cyanoborohydride in the presence of glacial acetic acid provided crude morpholine 78.44,45 N-Tritylation furnished 79 in 43% yield over four synthetic steps.44,46 Desilylation followed by sulfamoylation of 80 in DME–MeCN afforded sulfamate 81 that was biotinylated using our standard protocol. The synthesis was completed by ammonolysis to remove the N6-benzoyl group and by subsequent treatment with 80% aqueous acetic acid to deprotect the N-trityl group to give 82 in 36% yield over three steps from 81.
To examine the importance of the 2′ and 3′-hydroxyls for biological activity, we attempted to prepare 2′-deoxy- and 3′-deoxy-Bio-AMS analogs. We initially synthesized 2′-deoxy-Bio-AMS (not shown), but discovered the molecule was innately unstable and rapidly decomposed at room temperature. We attribute this instability to a vastly less pronounced anomeric effect which is derived from poor orbital overlap between C-1′ and C-2′ resulting in a high propensity to depurinate.47 However, 3′-deoxy-Bio-AMS 87 was successfully synthesized over six steps from commercially available 3′-deoxy-adenosine (83, Scheme 7). TBS protection of the C-2′ and C-5′ alcohols was followed by bis-Boc protection of N6 furnished 84 in 82% over two steps. Mono-desilylation yielded the desired product 85. Formation of the sulfamide intermediate using our optimized Mitsunobu protocol afforded slightly impure product 86, which was used directly in next reaction. Biotinylation was followed by global deprotection in 80% aqueous TFA to supply the final analog 87 in 62% over the last three steps.
Scheme 7.

Synthesis of 3′-Deoxy-Bio-AMS (87).
To complete our narrative we examined the consequence of altogether eliminating the ribose moiety with acyclo-Bio-AMS analog 90 (Scheme 8). Acyclo-sulfamide analog 90 was prepared from the reported acycloadenosine (88),48 that was converted to sulfamide intermediate 89, which was biotinylated and deprotected to provide 90 in 52% over three steps.
Scheme 8.
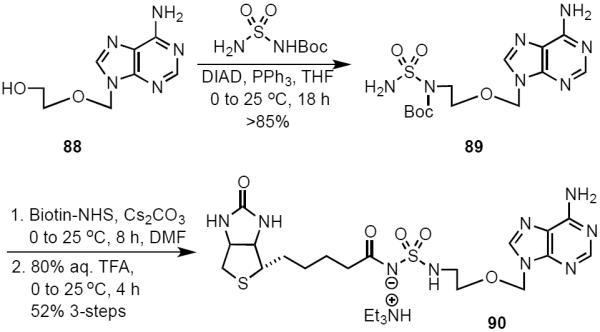
Synthesis of Acyclo-Bio-AMS (90).
Ligand characterization by co-crystallization with MtBPL and isothermal titration calorimetry
The previously described co-crystal structures with 1415 and 724 revealed a binding site with two deep and largely enclosed subsites for the biotinyl and adenosyl groups, and the sidechain of the Trp74 stacked between them. The ribose lies across the open mouth of this pocket. The 2′-hydroxyl of 14 is engaged in a hydrogen bond with the carboxylate of Asp167, while the 3′-hydroxyl makes a water mediated contact with Gly73 (Figure 4A). In order to characterize possible structural consequences of glycosyl substitutions, co-crystal structures with compounds 36, 46, 57, 63, 69, 73, 82, 87, and 90 have been determined at high resolution (Diffraction data and summary refinement statistics are provided in Table S2). The various complexes with bisubstrate inhibitors are remarkably similar. The presence of an acylsulfamate with the 5′ oxygen in 57, 69, 73, and 82 instead of an acylsulfamide with the 5′ nitrogen in 14, 36, 46, 63, 87, and 90 seems structurally inconsequential; a hydrogen bond between the deprotonated acylsulfamate/acylsulfamide nitrogen and the amide NH of Arg69 is conserved in all structures.
Figure 4.
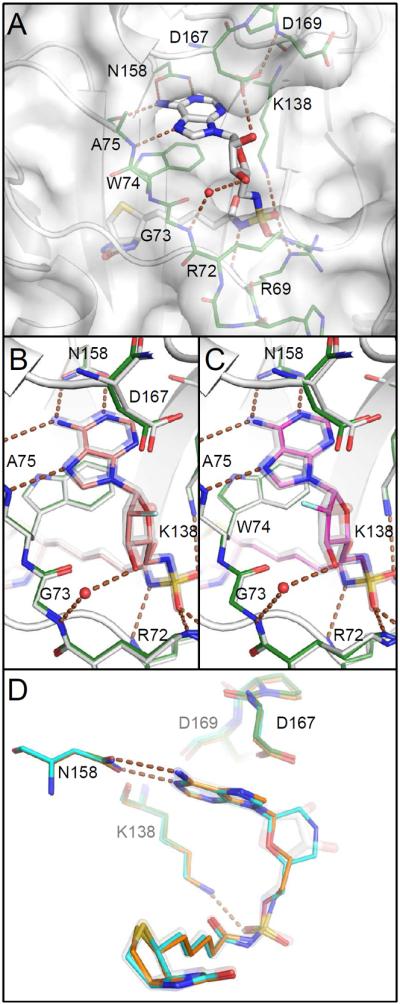
Comparison of co-crystal structures. A) Bio-AMS (14, white) binds with adenosyl and biotinyl groups in deep pockets with Trp74 stacked between them. Protein carbon atoms of this complex are green included in other panels for comparison. Hydrogen bonds are illustrated with brown dashed lines. The ribose is positioned across the open mouth of a larger unoccupied space. B and C) Binding site and ribose ring pucker are unchanged upon binding of the 2′ fluoro analogs with standard (63, salmon) or reversed (69, magenta) stereochemistry, despite the loss of the hydrogen bond to Asp167. Azido analogs (46, 57) bind similarly (not shown). D) Morpholino (82, cyan) and acyclo (90, orange) analogs adopt conformations that mimic the ribose.
A rotameric shift of Asp167 might have been anticipated upon loss of the 2′-hydroxyl, but no change is observed. A hydrogen bond from OD265 to the amide NH of Asp169 holds the carboxylate in position in all structures (Figure 4A). No shift occurs even in the case of analogs missing the 2′-hydroxyl (36 or 90). Altered stereochemistry at the 2′-position is also well tolerated. The 2′-α-fluoro and 2′-β-fluoro compounds (63 and 69, respectively) bind so similarly that all other atoms in the vicinity of the ligands almost perfectly align (Figure 4B, C). Both azido analogs 46 and 57 also compare similarly. In cases where a 2′-substituent is aliphatic, as in the 2′-β-methyl of 73 or the morpholine ring of 82, the Asp167 side chain shifts just far enough (without changing rotamers) to extend the contact distance between OD165 and the carbon to the sum of their van der Waals radii (3.4 Å). The similarity of the structures is reflected in the flat SAR in binding affinity observed by ITC for these compounds.
Analogs 82 and 90 are interesting in that they represent the most diverse departures from the ribofuranosyl ring, yet are well tolerated. Even the acyclic analog (90) follows a trajectory through C-1′, O, and C-5′ adopted by the ribose in Bio-AMS (14) (Figure 4D). The morpholine of 82 also superimposes well upon this triad. The conformation is likely necessary to allow the adenosine and biotin moieties to retain the same relative orientations upon binding.
Little density is observed for the azido functional group in complexes with 46 and 57. Sufficient density confirms the stereochemistry of the 2′ substitution, but the remainder of the azido group appears disordered in rotation about the C-2′-N bond. In each complex, this group has been modeled in the conformation that gives rise to at least (−) density in a Fo-Fc difference maps, but any assignment of a preferred rotamer based on diffraction data is uncertain at best.
Because of the exceedingly tight-binding nature (KD values ≤ 2 nM) of our family of analogs to MtBPL we were unable to determine their potency using a steady-state kinetic assay. Thus we enlisted a technique that has gained popularity to determine ligand protein binding affinities. Originally described in 1965 by Christensen and coworkers, isothermal titration calorimetry (ITC) was first used to concurrently determine the Keq and ΔH of various weak acid–base equilibria and metal ion complexation reactions.49 Today, ITC has become a powerful and commonplace tool to directly characterize the thermodynamics of ligand-protein interactions. ITC enables determination of the dissociation constant (KD), Gibbs free energy (ΔG), enthalpy (ΔH) and entropy (ΔS) of ligand-protein interactions allowing researchers to tease out nuanced information of ligand binding.50 Using 14 as our benchmark we evaluated a series of analogs for their activity against MtBPL using ITC (Table 1). Unfortunately, the remarkably tight-binding nature of our analogs exceeded the limit (KD ≤ 10−8 M) of accurate determination for binding constants by ITC.51 Therefore, we employed a more reliable approach for determining the dissociation constants less than 10−8 M by using displacement ITC, in which our high affinity ligands are titrated into a solution of protein that is bound to a weaker ligand (in this case, biotin). Enthalpies (ΔH) were determined by direct titration for all ligands while dissociation constants (KD) and Gibbs free energies (ΔG) were measured by displacement ITC experiments (see methods, Figures S1–S14, and Table S1).
Table 1.
MIC90 and thermodynamic data of biotinylated nucleosides. 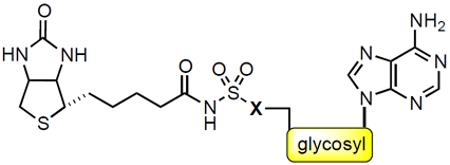
| Compound | MIC90, μMb | KD, nMa | ΔG, kcal/mola | ΔH, kcal/mola | -TΔS, kcal/mol | ΔTm, °C | PDB | |
|---|---|---|---|---|---|---|---|---|
| 14, Bio-AMS |
|
0.78 | 0.865 ± 0.040 | −12.4 ± 0.5 | −18.9 ± 0.1 | 6.6 ± 0.1 | 14.5 ± 1.0 | 3rux |
| 28 |
|
>100 | 0.753 ± 0.002 | −12.4 ± 0.0 | −20.2 ± 0.2 | 7.8 ± 0.2 | 12.2 ± 1.0 | n.d.c |
| 34 |
|
3.12 | 2.14 ± 0.08 | −11.8 ± 0.2 | −22.1 ± 0.2 | 10.3 ± 0.2 | 12.5 ± 0.5 | n.d.c |
| 36 |
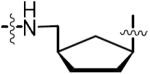
|
1.56 | 0.540 ± 0.007 | −12.6 ± 0.2 | −23.5 ± 0.4 | 10.7 ± 0.4 | 14.5 ± 1.0 | 4xtv |
| 46 |
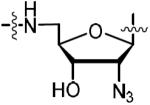
|
3.12 | 0.933 ± 0.045 | −12.3 ± 0.6 | −23.2 ± 0.2 | 10.9 ± 0.3 | 13.0 ± 0.5 | 4xyw |
| 47 |
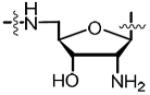
|
1.25 | 0.627 ± 0.016 | −12.6 ± 1.0 | −19.1 ± 0.1 | 6.5 ± 0.2 | 19.2 ± 0.6 | n.d.c |
| 57 |
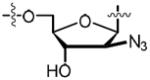
|
>100 | 0.027 ± 0.003 | −14.4 ± 0.2 | –29.3 ± 0.1 | 14.9 ± 0.1 | 17.0 ± 1.0 | 4xtx |
| 63 |
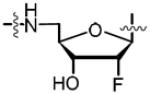
|
1.25 | 0.614 ± 0.018 | −12.6 ± 0.3 | −24.3 ± 0.5 | 11.8 ± 0.5 | 15.2 ± 0.8 | 4xty |
| 69 |
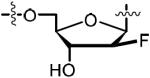
|
50.0 | 0.153 ± 0.000 | −13.4 ± 0.0 | −28.4 ± 0.6 | 14.7 ± 0.5 | 17.7 ± 0.8 | 4xtz |
| 73 |
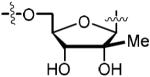
|
50.0 | 0.769 ± 0.006 | −12.4 ± 0.4 | −20.3 ± 0.4 | 8.1 ± 0.5 | 14.2 ± 0.3 | 4xu0 |
| 82 |
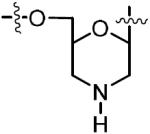
|
3.12 | 0.324 ± 0.023 | −12.9 ± 0.9 | −18.8 ± 0.0 | 5.9 ± 0.1 | 13.8 ± 0.8 | 4xu1 |
| 87 |
|
0.78 | 0.592 ± 0.009 | −12.6 ± 0.1 | −21.4 ± 0.5 | 8.8 ± 0.6 | 15.0 ± 1.0 | 4xu2 |
| 90 |
|
3.12 | 0.315 ± 0.003 | −13.0 ± 0.1 | −25.5 ± 0.7 | 12.6 ± 0.7 | 14.7 ± 0.3 | 4xu3 |
Competitive ITC experiments to determine KD and ΔG were performed at least in triplicate with 2 different enzyme batches while direct titration experiments were done in triplicate. All analogs showed n values of 1 ± 0.2.
Minimum inhibitory concentrations (MIC) that resulted in >90% growth inhibition of M. tuberculosis H37Rv were determined by a broth microdilution assay in GAS medium. Experiments were performed twice independently in triplicate. The MIC is defined as the lowest concentration of inhibitors that prevented growth, as determined by measuring the endpoint OD600 values.
Not determined
All ligands bound tightly to MtBPL with KD's ranging from 27 pM to 2.14 nM. Binding was ethalpically driven and the ΔH's ranged from −18.8 to −29.3 kcal/mol (Table 1). Carbocycle 28 that varies by replacement of the ribofuranosyl ring oxygen with a CH2 moiety was nearly equipotent to 14 with a KD of 0.75 nM. 28 had a 1.2 kcal/mol decrease in ΔH that was compensated by an equal increase in the entropic term, resulting in an identical ΔG of −12.4 kcal/mol. Remarkably, cyclopentenyl 34, cyclopentanyl 36 and acyclo 90 all are missing key hydrogen bond donors and acceptors (C-2′ and C-3′ hydroxyls), yet still bound tightly with KD's of 2.14, 0.54, and 0.315 nM, respectively. These analogs confirmed our notion that the ribofuranosyl oxygen alone played a limited role in the binding pocket. The results also suggest that deletion of the C-2′ and C-3′ hydroxyls in addition to the removal of the 4′-ribofuranosyl oxygen is also well tolerated inferring that the numerous other electrostatic hydrogen bonding interactions between the ligand and protein are sufficient to maintain affinity. Interestingly, compound 90 has improved enthalpic contributions, which could be attributed to improved interactions with MtBPL as a result of its increased flexibility. Cyclopentyl 34 is the least potent of all ligands evaluated. The presumed constrained Cs envelope conformation of the cyclopentene ring likely does not allow optimal interactions within the binding pocket. Compounds 46, 47, and 57 all replace the hydrogen bond donating 2′-hydroxyl with a 2′-amino or 2′-azido group. Both 2′-α substituted compounds 46 and 47 were nearly equipotent to 14, with 47 being slightly better than 46. Surprisingly, the 2′-β-azido analog (57) proved to be the tightest binder, with a KD of 27 pM, which is 32-fold more potent than 14. Additionally, 57 bound with the highest enthalpy of −29.3 kcal/mol, which cannot be easily reconciled. We speculate the azide may directly interact with MtBPL, however, it is not clear from the crystallographic data since there is little electron density for the azide. Both compounds 63 and 69 are epimeric with respect to their C-2′-fluoro substituents and bind with KD's of 0.614 nM and 0.153 nM. Although 63 and 69 are similar in potency to 14, they exhibit unique thermodynamic binding signatures driven by large enthalpic contributions of −24.3 kcal/mol and −28.4 kcal/mol. This could be attributed to the improved dipole-dipole interactions with MtBPL by the incorporation of the fluorine atom into the structure. Compounds 73 and 87 both had slight ring modifications, incorporating a 2′-β-methyl into the ring or removal of the 3′-hydroxyl, respectively. Both compounds exhibited similar binding profiles in terms of KD with 87 displaying slightly tighter binding affinities. The improved enthalpic contribution of 73 can most likely be attributed to van der Waals interactions from the 2′-β-methyl group. Finally, the morpholine 82 analog with the most significant structural change to the ring structure was well tolerated and bound with a KD of 0.324 nM as well as an identical thermodynamic binding signature compared to 14. This result indicates MtBPL is quite tolerant to the perturbations of the glycosyl ring.
We also investigated how well our analogs stabilize MtBPL through a general binding experiment using differential scanning fluorimetry (DSF).52 They all had significant stabilization properties as evident by their change in melting temperature ranging from a change of 12.2 to 19.2 °C (Table 1). There seems to be limited correlation between the change in melting temperature and binding affinity, and these data could only be interpreted as a qualitative measure of ligand stabilization to MtBPL.
Anti-tubercular Activity and Cytotoxicity
We next determined the minimum inhibitory concentration that inhibited greater than 90% of cell growth (MIC) of our analogs against Mtb H37Rv. We previously published the MIC for 14 against H37Rv as 0.78 μM.15 The MIC values of our Bio-AMS derivatives are as diverse as the molecules themselves (Table 1). The parent 14 still reigns supreme as the most potent inhibitor as determined by its whole-cell activity. The carbocyclic molecules 28, 34 and 36 are all deprived of the 4′-ribofuranosyl oxygen. Analog 34 incorporates an olefin while 36 is a saturated cyclopentane. Interestingly, analog 28 does not show strong activity against Mtb, even at 100 μM, but to our amazement both analogs 34 and 36 displayed MIC values that were comparable to 14.
Analogs 46 and 57 along with compounds 63 and 69 are respective C-2′ epimers, but differ in their substituents. Yet this minor modification in the stereochemistry had significant implications on the anti-tubercular activity of the molecules. The C-2′-α substituted compounds 46, 47 and 63 are roughly as active as 14 against Mtb H37Rv, most likely due to the linearity and directionality of the new appendages which mimic that of the C-2′ hydroxyl in Bio-AMS. This can be further attested by comparing the subtle changes in analogs 46 and 47; 2′-α-amino analog 47 is slightly more potent than 2′-α-azido analog 46, likely due to amino groups being significantly superior bioisosteres of hydroxyls than azides. Alternatively, compounds 57 with the 2′-β-azido and 69 with the 2′-β-fluoro handles are void of any biological activity. The poor anti-tubercular activity of compounds 57 and 69 is mirrored by analog 73 which also contains a β-facing attachment (CH3) at the C-2′ position. These results suggest a steric clash between the β-facing C-2′ appendages and a putative nucleoside transporting protein responsible for uptake or association.
Of the current set of Bio-AMS analogs, the morpholine analog 82 and acyclo analog 90, which exhibited the greatest structural changes, also exhibited potent antitubercular activity. The increased flexibility of the acyclo moiety in 90 and 6-membered ring in 82,53 as opposed to a 5-membered ribofuranosyl ring, could allow for perturbations without sacrificing whole-cell activity or binding affinity. We were also confident that deletion of the C-3′ hydroxyl would result in a marginal change to the MIC when juxtaposed to Bio-AMS. As expected, compound 87 showed an equipotent MIC value of 0.78 μM to that of 14. Moreover, the synthesis of 87 from 3′-deoxy-adenosine was facile in comparison by circumventing the obligatory C-3′ and C-5′ hydroxyl protection with the Markiewicz protecting group which rendered the syntheses of 46, 47 and 57 cumbersome.
To assess the potential cytotoxicity of our compounds, Bio-AMS and a subset of the most active analogs (14, 36, 63, 82, and 90) were evaluated against HepG2 cells using an MTT assay. No inhibition of growth was observed (IC50 > 100 μM) for any of the tested compounds.
Cellular Uptake of C-2′-fluoro-Bio-AMS Analogs
The discordant whole-cell MIC data compared to binding data for C-2′ epimers 63 and 69 prompted investigation of their cellular accumulation. Utilizing the uptake methodology developed by Rhee and coworkers,54,55 the amounts of compounds 63 and 69 associated with the cells were quantified by LC-MS/MS. The amount of cell-associated compound 63 was determined to be approximately 7-fold higher than compound 69 when Mtb H37Rv was incubated with 25 μM of each analog. This method, however, does not discern between the amounts of compound that is intracellular versus cell-associated. Nevertheless, there is still a substantial difference between the amounts of compound 63 and 69 that are accumulated giving credence to the hypothesis that the disparate MIC and binding data of the C-2′ fluoro analogs can be potentially explained by differential cellular accumulation.
CONCLUSION
We have successfully designed and synthesized a focused series of MtBPL bisubstrate inhibitors by strategically modifying the ribofuranosyl ring. All analogs bound tightly to MtBPL, with KD values ranging from 27 pM to 2.14 nM. Aside from the weakest binding 34 and strongest binding 57, all compounds exhibited a narrow range of potency from 0.153 to 0.933 nM. ITC also illustrated the subtleties in the thermodynamic binding signatures of each analog that cannot be easily reconciled. These data, along with X-ray co-crystal structures, demonstrate modification or substitution of the ribofuranosyl ring does not substantially impact the binding of the analogs to MtBPL. However, the relatively flat biochemical potencies sharply contrast with whole-cell antitubercular activity. Notably, all of the analogs containing a C-2′-β substituent including 57, 69 and 73 had substantially reduced whole-cell activities with MIC ranging from 50 to >100 μM despite potent binding affinities. This can only be attributed to differences in cellular accumulation through enhanced efflux or reduced uptake, and when examining the differences in cellular accumulation between 63 and 69, there was substantial preference for cellular association of compound 63 over 69. The other analogs in the series, except 28, possessed whole-cell activity commensurate with 14 with MIC values ranging from 0.78–3.12 μM. Both 47 and 82 represent promising ligands for further optimization since they can be readily functionalized through `click chemistry' or substitution of the endocyclic nitrogen, respectively.
EXPERIMENTALS
General materials and methods
Chemicals and solvents were purchased from Acros Organics, Alfa Aesar, Sigma-Aldrich, and TCI America and were used as received. The nucleosides 9-β-d-arabinofuranosyladenine 37 and 3-deoxyadenosine 83 were obtained from Berry & Associates (Dexter, MI) while 2′-deoxy-2′-fluoroadenosine 58 and 9-(2′-deoxy-2′-fluoro-β-d-arabinofuranosyl)adenine 64 were obtained from Metkinen Chemistry (Kuopio, Finland), and N6-benzoyladenosine 74 was obtained from Carbosynth (Berkshire, UK). tert-Butyl sulfamoylcarbamate,56 sulfamoyl chloride,57 d-(+)-biotin N-hydroxysuccinimide ester,58 5′-[N-(d-biotinoyl)sulfamoyl]amino-5′-deoxyadenosine triethylammonium salt (Bio-AMS) 14,15 {(1S,4R)-4-[N-(phenylsulfonyl)pivalamido]cyclo-pent-2-enyl}methyl pivalate 22,28 2′,3′-O-isoproylidene-β-2′-methyladenosine 71,59 and 9-[(2-hydroxyethoxy)methyl]adenine 88,48 were prepared as described. An anhydrous solvent dispensing system using two packed columns of neutral alumina was used for drying THF and CH2Cl2, while two packed columns of molecular sieves were used to dry DMF, and the solvents were dispensed under argon gas (Ar). Anhydrous grade MeOH, MeCN, pyridine, and DMA were purchased from Aldrich. EtOAc and hexanes were purchased from Fisher Scientific. All reactions were performed under an inert atmosphere of dry argon gas (Ar) in oven-dried (180 °C) glassware. TLC analyses were performed on TLC silica gel plates 60F254 from EMD Chemical Inc. and were visualized with UV light. Optical rotations values were obtained on a polarimeter using a 1 dm cell. Purification by flash chromatography was performed using a medium-pressure flash chromatography system equipped with flash column silica cartridges with the indicated solvent system. Preparative reversed-phase HPLC purification was performed on a Phenomenex Gemini 10 μm C18 250 × 20 mm column operating at 30.0 mL/min with detection at 254 nm with the indicated solvent system. Analytical reversed-phase HPLC purification was performed on a Phenomenex Gemini 5 μm C18 250 × 4.6 mm column operating at 1 mL/min with detection at 254 nm employing a linear gradient from 5% to 50% MeCN in 50 mM aqueous triethylammonium bicarbonate (TEAB) at pH 7.5 for 30 min (Method A). 1H and 13C spectra were recorded on 400, 600 or 900 MHz NMR spectrometers. Proton chemical shifts are reported in ppm from an internal standard of residual chloroform (7.26), methanol (3.31), dimethyl sulfoxide (2.50), or mono-deuterated water (HDO, 4.79); carbon chemical shifts are reported in ppm from an internal standard of residual chloroform (77.0), methanol (49.1), or dimethyl sulfoxide (39.5). Proton chemical data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, dt = doublet of triplets, t = triplet, q = quartet, pentet = pent, m = multiplet, ap = apparent, br = broad, ovlp = overlapping), coupling constant(s), integration. High-resolution mass spectra were obtained on an LTQ Orbitrap Velos (Thermo Scientific, Waltham, MA). All compounds were determined to be >95 % by analytical reverse-phase HPLC (purities for each final compound are given in the experimental section below).
General Procedure for Mitsunobu Reaction
To a solution of nucleoside (1.0 equiv), tert-butyl sulfamoylcarbamate (3.0 equiv) and PPh3 (1.5 equiv) in THF (0.05 M for the limiting reagent) at 0 °C was added DIAD (1.05 equiv) dropwise over 15 min. The solution was stirred at 0 °C for 15 min, then gradually warmed to 23 °C over 3 h and stirred for 12 h at 23 °C. The reaction was concentrated in vacuo to afford the crude sulfamide, which was purified by flash chromatography using the indicated solvent system. The product was typically contaminated with triphenylphosphine oxide (TPPO). In this case the calculated yield was obtained by a simple linear equation relating the total isolated weight to the ratio of product:TPPO and formula weights for each compound.
General Procedure for Okada Sulfamoylation Protocol
To a solution of nucleoside (1.0 equiv) in DMA (0.4 M) was added freshly prepared sulfamoyl chloride (4.0 equiv) at 0 °C. The reaction was slowly warmed to 23 °C over 6 h and then stirred an additional 18 h at 23 °C. The reaction was diluted with EtOAc (200 mL/mmol) and washed successively with H2O (5 × 100 mL/mmol), saturated aqueous NaCl (200 mL/mmol), then dried (Na2SO4) and concentrated under vacuum to afford the crude sulfamate that was typically greater than 90% pure contaminated only with residual DMA and used directly in the biotinylation step.
General Procedure for Biotinylation
To a solution of the intermediate nucleoside sulfamate/sulfamide (1.0 equiv) and Cs2CO3 (2.5 equiv) in DMF (0.5 M for the limiting reagent) at 0 °C was added d-(+)-biotin N-hydroxysuccinimide ester (1.5 equiv). The reaction mixture was stirred for 24 h at 23 °C during which time all starting material was consumed as monitored by electrospray mass spectrometry in the negative mode. DMF was removed by rotary evaporation under high vacuum (P = 0.01 torr) with mild heating (water bath ≤ 35 °C) to afford the crude biotinylated nucleoside. The crude product was used in the next global deprotection reaction directly without further purification.
General Procedure for TFA Global Deprotection
To a solution of the crude biotinylated nucleoside prepared above in H2O (10 mL/mmol) at 0 °C was added TFA (40 mL/mmol) dropwise to achieve a final concentration of 0.02 M. The reaction mixture was stirred for 8 h at 23 °C during which time all starting material was consumed as monitored by electrospray mass spectrometry in the negative mode. The solvent was removed under vacuum. The crude material was re-dissolved in 1:1 MeCN–50 mM TEAB (10–20 mg/mL) and filtered to remove insoluble solids. The resulting solution was purified by preparative reverse phase HPLC with a Phenomenx Gemini C18 (250 × 20 mm) column at a flow rate of 30.0 mL/min employing a linear gradient of 5–15% acetonitrile (solvent B) in 50 mM aqueous triethylammonium bicarbonate (TEAB) at pH 7.5 (solvent A) for 30 min. The appropriate fractions were pooled and lyophilized to afford the final compound as the triethylammonium salt as a white foam.
General Procedure for TBS Protection
To a solution of nucleoside (1.0 equiv), imidazole (6.6 equiv), and DMAP (0.1 equiv) in DMF (0.2 M of limiting nucleoside substrate) at 0 °C was added TBSCl (3.3 equiv). The mixture was warmed to 23 °C over 3 h then stirred at 23 °C for an additional 13 h. The reaction was quenched with a saturated aqueous NH4Cl (40 mL/mmol). The crude mixture was extracted with EtOAc (5 × 100 mL/mmol). The combined organic layers were washed with H2O (100 mL/mmol), saturated aqueous NaCl (100 mL/mmol) and then concentrated in vacuo. Purification by flash chromatography (EtOAc–hexanes) afforded the title compound.
General Procedure for bis-Boc Protection
To a solution of nucleoside (1.0 equiv), Et3N (3.0 equiv) and DMAP (0.5 equiv) in DMF (0.2 M in the limiting reagent) at 0 °C was added Boc2O (3.0 equiv). The reaction was warmed to 23 °C over 3 h then stirred at 23 °C for an additional 10 h. The reaction was quenched with ice cold 1 N aqueous HCl (50 mL/mmol) and extracted with EtOAc (5 × 40 mL mL/mmol). The combined organic extracts were washed with saturated aqueous NaCl (60 mL mL/mmol), dried (MgSO4), and concentrated in vacuo. Purification by flash chromatography (EtOAc–hexanes) afforded the title compound.
(1′R,4′S)-9-{(4′-Pivaloyloxy)methyl]cyclopent-2′-en-1′-yl}adenine (23)
To a round bottom flask charged with adenine (65 mg, 0.50 mmol, 1.2 equiv) and Cs2CO3 (156 mg, 0.50 mmol, 1.2 equiv) was added DMF (2.0 mL) and the mixture stirred at 22 °C for 30 min. To another flask charged with 22 (169 mg, 0.40 mmol, 1.0 equiv), [PdCl(C3H5)]2 (1.5 mg, 0.0040 mmol, 0.010 equiv) and P(OiPr)3 (8.3 mg, 10.0 μL, 0.040 mmol, 0.10 equiv) was added DMF (2.0 mL) and the mixture stirred at 23 °C for 30 min. The solution containing the substrate 22 was transferred via cannula over 5 min at 0 °C into the flask containing the cesium salt of adenine. The reaction mixture was stirred for another 5 h at 23 °C then quenched with saturated aqueous NH4Cl (5 mL). The crude mixture was extracted with EtOAc (5 × 15 mL) and the combined organic layers were washed with saturated aqueous NaCl (20 mL) and then concentrated under vacuum. Purification by flash chromatography (10:1 CH2Cl2–MeOH) afforded the title compound (116 mg, 92%) as white foam: Rf = 0.45 (9:1 CH2Cl2–MeOH); (c 0.50, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 1.19 (s, 9H), 1.64–1.68 (m, 1H), 2.87–2.92 (m, 1H), 3.17–3.19 (m, 1H), 4.09–4.15 (m, 2H), 5.73–5.76 (m, 1H), 5.92 (br s, 2H), 5.96–5.97 (m, 1H), 6.16–6.17 (m, 1H), 7.82 (s, 1H), 8.36 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 27.3, 35.9, 38.9, 44.7, 59.4, 66.5, 120.0, 130.4, 137.6, 138.3, 149.9, 153.0, 155.9, 178.4; HRMS (ESI+) calcd for C16H22N5O2 [M + H]+ 316.1768, found 316.1774 (error 1.9 ppm).
(1′R,2′S,3′S,5′S)-9-{1′,2′-Dihydroxy-1′,2′-O-isopropylidene-5′-[(pivaloyloxy)methyl]cyclopent-3-yl}adenine (24a) or 2′,3′-O-(Isopropylidene)aristeromycin (24α) and (1′S,2′R,3′S,5′S)-9-{1,2-Dihydroxy-1′,2′-O-isopropylidene-5′-[(pivaloyloxy)methyl]cyclopent-3-yl}adenine (24β)
To a solution of 23 (315 mg, 1.0 mmol, 1.0 equiv) in MeOH (5 mL) at 0 °C was added K2CO3 (414 mg, 3.0 mmol, 3.0 equiv) in one portion. The suspension was stirred at 0 °C for 30 min then heated at 60 °C for 4 h. The solution was concentrated in vacuo and re-suspended in 10:1 CH2Cl2–MeOH (20 mL). Filtration over Celite and concentration afforded the crude product. Purification by flash chromatography (CH2Cl2 to 10:90 MeOH–EtOAc, linear gradient) afforded (1′R,4′S)-9-[(4′-hydroxymethyl)cyclopent-2′-en-1′-yl]adenine (215 mg, 89%) as colorless oil: Rf = 0.10 (CH2Cl2–MeOH); 1H NMR, 13C NMR, and HRMS data matched literature values.2
To a solution of (1′R,4′S)-9-[(4′-hydroxymethyl)cyclopent-2′-en-1′-yl]adenine prepared above (92.4 mg, 0.40 mmol, 1.0 equiv) in 1:1 THF–H2O (8 mL) at 0 °C was added OsO4 (250 μL of 4 wt% in tBuOH, 10 mol%) and NMO (93.7 mg, 0.80 mmol, 2.0 equiv). The dark brown mixture was stirred at 0 °C for 48 h then quenched with 10% aqueous Na2S2O5 (5.0 mL). The resulting mixture was extracted with EtOAc (5 × 15 mL) and the combined organic extracts were concentrated in vacuo to afford the crude diol, which was used in next reaction directly.
To a solution of the crude diol prepared above in THF (4.0 mL) was added 2,2-dimethoxypropane (2.0 mL, 16.3 mmol, 41 equiv) and p-TsOH•H2O (76.1 mg, 0.40 mmol, 1.0 equiv) at 0 °C. The mixture was allowed to gradually warm up to 23 °C and stirred for 15 h, then quenched with solid NaHCO3 (3.4 g, 40 mmol, 100 equiv) and the suspension stirred for 30 min at 23 °C. The mixture was filtered and the solid washed with EtOAc (20 mL). The filtrate was concentrated in vacuo to afford the crude product. Purification by flash chromatography (CH2Cl2 to 10:90 MeOH–CH2Cl2, linear gradient) afforded the title compound 24α and the undesired diastereomer 24β as an inseparable 1:1 mixture (65 mg, 53% combined yield over 2 steps): Rf = 0.25 (1:9 MeOH–CH2Cl2). The diastereomers (24α/β) were separated by reverse-phase HPLC (22:77 CH3CN–H2O, isocratic condition, 1.5 mL/min, tR(24β) = 18 min, tR(24α) = 21 min) with a Phenomenx Gemini 5 μm C18 (250 × 20 mm) column. 1H NMR, 13C NMR, HRMS data for 24α are consistent with previously reported values.3 Data for 24β: 1H NMR (400 MHz, MeOH-d4) δ 1.27 (s, 3H), 1.47 (s, 3H), 2.12–2.22 (m, 3H), 3.71 (dd, J = 10.8, 5.6 Hz, 1H), 3.88 (dd, J = 10.8, 5.6 Hz, 1H), 4.76–4.83 (m, 3H), 8.21 (br s, 2H); 13C NMR (100 MHz, MeOH-d4) δ 24.0, 25.8, 30.4, 44.3, 57.0, 61.7, 79.5, 80.9, 111.9, 119.7, 142.2, 150.8, 153.6, 157.2. All other characterization data are identical to reported data.
N6,N6-bis(tert-Butoxycarbonyl)-5′-O-tert-butyldimethylsilyl-2′,3′-O-(isopropylidene)aristeromycin (25)
Compound 24α (47.0 mg, 0.154 mmol) was converted to 5′-O-tert-butyldimethylsilyl-2′,3′-O-(isopropylidene)aristeromycin using the general procedure for TBS protection, which was used in the next step without further purification: Rf = 0.3 (1:9 MeOH–CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 0.06 (s, 6H), 0.88 (s, 9H), 1.28 (s, 3H), 1.56 (s, 3H), 2.35–2.51 (m, 3H), 3.77 (d, J = 4.4 Hz, 2H), 4.65 (dd, J = 7.2, 4.4 Hz, 1H), 4.76–4.80 (m, 1H), 5.01 (t, J = 6.0 Hz, 1H), 5.68 (br s, 2H), 7.85 (s, 1H), 8.33 (s, 1H); 13C NMR (100 MHz, CDCl3) δ −5.45, −5.43, 18.3, 25.1, 25.8, 29.7, 33.4, 45.7, 61.9, 63.0, 80.7, 83.8, 113.5, 121.9, 139.6, 150.2, 152.8, 155.; All other characterization data are identical to reported data.
Crude 5′-O-tert-butyldimethylsilyl-2′,3′-O-(isopropylidene)aristeromycin prepared above (60.5 mg, 0.14 mmol, 1.0 equiv) was converted to the title compound using the general procedure for bis-Boc protection. Purification by flash chromatography (3:1 to 1:1 hexane–EtOAc, linear gradient) afforded the title compound (82 mg, 92% over 2 steps) as a colorless oil: Rf = 0.4 (1:1 EtOAc–hexanes); 1H NMR (400 MHz, CDCl3) δ 0.07 (s, 3H), 0.08 (s, 3H), 0.92 (s, 9H), 1.32 (s, 3H), 1.47 (s, 18H), 1.57 (s, 3H), 2.39–2.42 (m, 2H), 2.55–2.60 (m, 1H), 3.76–3.79 (m, 2H), 4.68 (dd, J = 6.8, 4.4 Hz, 1H), 4.83–4.85 (m, 1H), 5.03 (t, J = 6.4 Hz, 1H), 8.11 (s, 1H), 8.83 (s, 1H); 13C NMR (100 MHz, CDCl3) δ −5.47, −5.45, 18.3, 25.1, 25.8, 27.6, 27.8, 33.1, 45.6, 62.3, 62.7, 80.6, 83.7, 83.8, 113.6, 129.5, 143.9, 150.4, 150.6, 151.8, 153.3; HRMS calcd for C30H50N5O7Si [M + H]+ 620.3474, found 620.3452 (error 3.5 ppm).
N6,N6-bis(tert-Butoxycarbonyl)-2′,3′-O-(isopropylidene)aristeromycin (26)
To a solution of 25 (124 mg, 0.20 mmol, 1.0 equiv) in THF (4 mL) at 0 °C was added a 1.0 M TBAF solution in THF (0.30 mL, 0.30 mmol, 1.5 equiv) dropwise. The solution was warmed to 23 °C and stirred for 1 h then quenched with saturated aqueous NaHCO3 (20 mL). The aqueous layer was extracted with EtOAc (5 × 15 mL) and the combined organic extracts were washed with H2O (30 mL), saturated aqueous NaCl (30 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash chromatography (2:1 to 1:1 hexane–EtOAc, linear gradient) afforded the title compound (94 mg, 93%) as a white foam: Rf = 0.25 (1:1 EtOAc–hexanes); 1H NMR (400 MHz, CDCl3) δ 1.33 (s, 3H), 1.46 (s, 18H), 1.51 (s, 3H), 2.44–2.49 (m, 3H), 3.82–3.86 (m, 2H), 4.74 (dd, J = 6.8, 4.0 Hz, 1H), 4.83–4.86 (m, 1H), 5.05 (t, J = 6.4 Hz, 1H), 8.13 (s, 1H), 8.84 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 25.1, 27.6, 27.8, 33.0, 45.5, 62.9, 63.8, 82.0, 83.8, 84.0, 113.7, 129.7, 144.3, 150.6 (2 C), 151.8, 153.1; HRMS calcd for C24H36N5O7 [M + H]+ 506.2609, found 506.2588 (error 4.1 ppm).
N6,N6-bis(tert-Butoxycarbonyl)-5′-[N-(tert-butoxycarbonyl)-N-(sulfamoyl)amino]-5′-deoxy-2′,3′-O-(isopropylidene)aristeromycin (27)
Compound 26 (50.5 mg, 0.10 mmol, 1.0 equiv) was converted to the title compound using the general procedure for the Mitsunobu reaction. Purification by flash chromatography (3:1 to 1:1 hexane–EtOAc, linear gradient) afforded the title compound (62 mg, 83% based on 1H NMR analysis) partially contaminated with triphenylphosphine oxide: Rf = 0.28 (1:1 EtOAc–hexanes); 1H NMR (400 MHz, CDCl3) δ 1.30 (s, 3H), 1.48 (s, 9H), 1.54 (s, 3H), 1.58 (s, 18H), 2.39–2.51 (m, 3H), 3.87 (dd, J = 14.4, 4.4 Hz, 1H), 4.00 (dd, J = 14.4, 9.2 Hz, 1H), 4.11 (q, J = 7.2 Hz, 2H), 4.83–4.87 (m, 1H), 4.92 (t, J = 7.2 Hz, 1H), 5.05–5.09 (m, 1H), 5.66 (br s, 2H), 8.07 (s, 1H), 8.84 (s, 1H).
5′-[N-[(D-Biotinoyl)sulfamoyl]amino-5′-deoxyaristeromycin triethylammonium salt (28)
Compound 27 (34 mg, 0.050 mmol, 1.0 equiv) was converted to the title compound using the general procedure for biotinylation and TFA global deprotection to afford (17 mg, 52% in 2 steps): HPLC purity: 95.0%, tR = 11.9 min, k′ = 2.3 (method A); 1H NMR (CD3OD, 400 MHz) δ 1.28 (t, J = 6.8 Hz, 9H), 1.34–1.48 (m, 3H), 1.54–1.75 (m, 5H), 1.95–2.01 (m, 1H), 2.25 (t, J = 6.8 Hz, 2H), 2.29–2.32 (m, 1H), 2.47–2.50 (m, 1H), 2.65–2.70 (m, 1H), 2.90 (dd, J = 12.8, 4.8 Hz, 1H), 3.14 (q, J = 6.8 Hz, 6H), 3.10–3.25 (m, 1H), 4.03 (t, J = 5.2 Hz, 1H), 4.29 (dd, J = 12.4, 4.4 Hz, 1H), 4.45 (dd, J = 12.4, 4.4 Hz, 1H), 4.57 (dd, J = 8.4, 5.6 Hz, 1H), 4.79–4.86 (m, 1H), 8.17 (s, 1H), 8.24 (s, 1H); 13C NMR (CD3OD, 100 MHz) δ 9.5, 26.7, 29.6, 29.9, 31.2, 38.0, 41.2, 44.4, 47.70, 47.95, 57.2, 61.7, 62.4, 63.4, 74.9, 76.5, 120.8, 142.4, 151.0, 153.7, 157.4, 166.3, 178.2; HRMS (ESI−) calcd for C21H30N9O6S2 [M − H]− 568.1766, found 568.1784 (error 3.2 ppm).
(1′R,4′S)-9-[4′-(Hydroxymethyl)cyclopent-2′-en-1′-yl]adenine (29)
To a solution of 23 (1.43 g, 4.53 mmol, 1.0 equiv) in MeOH (10 mL) was added 1 M aqueous NaOH (10 mL) and the mixture stirred at 80 °C for 4 h. The reaction was concentrated in vacuo and the resulting crude solid residue was purified by flash chromatography (3:7 MeOH–EtOAc) to provide the title compound (0.953 mg, 95%) as a white solid: Rf = 0.40 (3:7 MeOH–EtOAc); 1H NMR, 13C NMR, and HRMS data matched literature values for the compound prepared by an alternate synthetic route.30
(1′R,4′S)-9-[4′-(tert-Butyldimethylsilyloxymethyl)cyclopent-2′-en-1′-yl]adenine (30)
Compound 29 (0.953 g, 4.12 mmol) was converted to the title compound using the general procedure for TBS protection. Purification by flash chromatography (1:5 MeOH–EtOAc) afforded the title compound (1.19 g, 83%) as a white solid: Rf = 0.35 (1:5 MeOH–EtOAc); 1H NMR (600 MHz, CDCl3) δ 0.04 (s, 6H), 0.88 (s, 9H), 1.69 (dt, J = 13.8, 6.0, 1H), 2.80 (dt, J = 13.8, 9.0 Hz, 1H), 2.98–3.05 (m, 1H), 3.61 (dd, J = 9.6, 6.0 Hz, 1H), 3.71 (dd, J = 9.6, 5.4 Hz, 1H), 5.74 (td, J = 5.4, 1.8 Hz, 1H), 5.86–5.88 (m, 2H), 5.89 (br s, 2H), 6.16–6.17 (m, 1H), 7.89 (s, 1H), 8.36 (s, 1H); 13C NMR (150 MHz, CDCl3) δ −5.25, −5.24, 18.5, 26.0, 34.9, 48.0, 59.4, 65.7, 119.8, 129.6, 138.9, 139.2, 149.8, 152.9, 155.9; HRMS (ESI+) calcd for C17H28N5OSi[M + H]+ 346.2058, found 346.2066 (error 2.3 ppm).
(1′R,4′S)-N6,N6-bis(tert-Butoxycarbonyl)-9-[4′-(tert-butyldimethylsilyloxymethyl)cyclopent-2′-en-1′-yl]adenine (31)
Compound 30 (1.25 g, 3.60 mmol) was converted to the title compound using the general procedure for bis-Boc protection. Purification by flash chromatography (2:8 EtOAc–hexanes) afforded the title compound (1.79 g, 91%) as a colorless oil: Rf = 0.50 (3:7 EtOAc–hexanes); 1H NMR (600 MHz, CDCl3) δ 0.03 (s, 6H), 0.86 (s, 9H), 1.43 (s, 18H), 1.71 (dt, J = 13.8, 6.0 Hz, 1H), 2.82 (dt, J = 13.8, 8.4 Hz, 1H), 3.00–3.05 (m, 1H), 3.62 (dd, J = 9.6, 5.4 Hz, 1H), 3.72 (dd, J = 10.2, 4.8 Hz, 1H), 5.82 (t, J = 6.0 Hz, 1H), 5.86–5.87 (m, 1H), 6.18–6.19 (m, 1H), 8.17 (s, 1H), 8.84 (s, 1H); 13C NMR (150 MHz, CDCl3) δ −5.21, −5.19, 18.6, 26.1, 27.9, 34.8, 48.0, 59.9, 65.7, 83.8, 129.2, 129.2, 139.8, 143.4, 150.3, 150.7, 152.0, 153.3; HRMS (ESI+) calcd for C27H44N5O5Si [M + H]+ 546.3106, found 546.3130 (error 4.4 ppm).
(1′R,4′S)-N6,N6-bis(tert-Butoxycarbonyl)-9-[4′ (hydroxymethyl)cyclopent-2′-en-1′-yl]adenine (32)
To a solution of 31 (1.76 g, 3.22 mmol, 1.0 equiv) in THF (15 mL) at 0 °C was added a solution of TBAF (1.0 M in THF, 3.90 mL, 3.86 mmol, 1.2 equiv). The reaction mixture was stirred for 2 h at 0 °C, then quenched with ice cold aqueous 1 N HCl (10 mL). The crude mixture was extracted with EtOAc (5 × 30 mL) and the combined organic extracts were washed with saturated aqueous NaCl (30 mL) and concentrated under vacuum. Purification by flash chromatography (1:9 MeOH–EtOAc) afforded the title compound (1.14 g, 94%) as a colorless oil: Rf = 0.40 (1:9 MeOH–EtOAc); 1H NMR (600 MHz, CDCl3) δ 1.41 (s, 18H), 1.88–1.93 (dt, J = 13.8, 5.4 Hz, 1H), 2.84 (dt, J = 13.8, 9.6 Hz, 1H), 3.08 (br s, 1H), 3.67 (dd, J = 10.8, 4.2 Hz, 1H), 3.81 (dd, J = 10.8, 4.2 Hz,1H), 5.77–5.78 (m, 1H), 5.79 (br s, 1H), 5.83–5.84 (m, 1H), 6.17–6.18 (m, 1H), 8.30 (s, 1H), 8.80 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 27.9, 33.8, 47.7, 60.6, 64.6, 83.8, 129.3, 129.6, 139.3, 144.3, 150.2, 150.6, 151.7, 153.0; HRMS (ESI+) calcd for C21H30N5O5 [M + H]+ 432.2241, found 432.2255 (error 3.2 ppm).
(1′R,4′S)-9[4-({[N-[(D-Biotinoyl)sulfamoyl]amino}methyl)cyclopent-2′-en-1′-yl]adenine triethylammonium salt (34)
Compound 32 (1.11 g, 2.58 mmol, 1.0 equiv), was converted to 33 using the general procedure for the Mitsunobu reaction. Purification by flash chromatography (1:1 to 7:3% EtOAc–hexane, linear gradient) afforded 33 (1.30 g, 70%, ~85% purity by weight, contaminated with PPh3O) that was used in the next step without further purification. Compound 33 (0.378 g, 0.621 mmol) was converted to 34 using the general procedure for biotinylation and TFA deprotection to provide the title compound (0.246 g, 57% over 2 steps) as a white solid as the triethylammonium salt (1.9 equiv of Et3N): HPLC purity: 96.9%, tR = 12.90 min, k′ = 2.4 (method A); (c 1.33, MeOH); 1H NMR (400 MHz, MeOH-d4) δ 1.19 (t, J = 7.3 Hz, 17H, excess Et3N), 1.38–1.48 (m, 2H), 1.51–1.66 (m, 4H), 1.68–1.82 (m, 2H), 2.17 (t, J = 7.8 Hz, 2H), 2.67 (d, J = 12.2 Hz, 1H), 2.91 (q, J = 7.1 Hz, 13H, excess Et3N), 2.98–3.15 (m, 4H), 3.16–3.22 (m, 1H), 4.30 (dd, J = 8.0, 4.2 Hz, 1H), 4.43–4.49 (m, 1H), 5.66–5.73 (m, 1H), 5.95 (d, J = 5.5 Hz, 1H), 6.21 (d, J = 5.5 Hz, 1H), 8.15 (s, 1H), 8.22 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 10.8, 25.0, 28.1, 28.2, 34.9, 36.2, 39.8, 44.4, 45.7, 47.2, 55.4, 59.1, 59.4, 61.1, 119.0, 130.2, 137.6, 138.9, 149.0, 152.3, 156.0, 162.8, 173.5; HRMS (ESI−) calcd for C21H28N9O4S2 [M – Et3NH]− 534.1711, found 534.1711 (error 0 ppm).
(1′R,3′S)-9-[3′-({[N-[(D-Biotinoyl)sulfamoyl]amino}methyl)cyclopentan-1′-yl]adenine triethylammonium salt (36)
To a solution of 33 (0.800 g, 1.31 mmol, 1.0 equiv) in EtOAc (15 mL) was added 10% w/w Pd/C (0.23 g). The nitrogen atmosphere was exchanged for a hydrogen atmosphere (1 atm) and the reaction stirred for 16 h at 23 °C. The reaction was filtered through a pad of Celite washing with MeOH (30 mL). The filtrate was concentrated under reduced pressure. Purification by flash chromatography (30% EtOAc–hexanes) afforded 35 that was used in the next coupling reaction directly without further purification. Compound 35 (0.650 g, 1.06 mmol, 1.0 equiv) was converted to 36 using the general procedure for biotinylation and TFA deprotection to afford the title compound (0.302 g, 58% over 3 steps) as the triethylammonium salt (2.4 equiv of Et3N) as a white solid: HPLC purity: 92.0%, tR = 14.83 min, k′ = 2.9, (method A); (c 1.08, MeOH); 1H NMR (400 MHz, MeOH-d4) δ 1.17 (t, J = 7.2 Hz, 22H, excess Et3N), 1.40–1.49 (m, 2H), 1.54–1.69 (m, 4H), 1.71–1.79 (m, 2H), 1.82–1.91 (m, 1H), 1.94–2.04 (m, 1H), 2.08–2.16 (m, 1H), 2.20 (t, J = 7.4 Hz, 2H), 2.26–2.37 (m, 2H), 2.42–2.51 (m, 1H), 2.67 (d, J = 12.5 Hz, 1H), 2.85 (q, J = 7.0 Hz, 15H, excess Et3N), 2.98–3.05 (m, 2H), 3.16–3.22 (m, 1H), 4.30 (dd, J = 7.8, 4.4 Hz, 1H), 4.43–4.50 (m, 1H) 4.90–4.96 (m, 1H), 8.20 (s, 1H), 8.25 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 11.2, 25.2, 27.5, 28.1, 28.2, 30.9, 36.6, 37.2, 39.9, 45.7, 48.0, 54.9, 55.5, 59.2, 61.1, 119.1, 139.3, 149.3, 152.1, 156.0, 162.8, 174.2 (missing 1 carbon); HRMS (ESI−) calcd for C21H30N9O4S2 [M – Et3NH]− 536.1868, found 536.1880 (error 2.2 ppm).
9-[3′,5-′-O-(1,1,3,3 Tetraisopropyldisiloxyl)-β-D-arabinofuranosyl]adenine (38)
To a solution of 37 (0.200 g, 0.796 mmol, 1.0 equiv) in pyridine (10 mL) at 0 °C was added TIPDSCl2 (0.395 g, 2.63 mmol, 3.3 equiv). The mixture was slowly warmed to 23 °C over 3 h and then stirred for 40 h at 23 °C. The reaction was concentrated in vacuo and the crude oil was taken up in EtOAc (100 mL) and washed successively with ice cold 1 N aqueous HCl (5 × 15 mL) and saturated aqueous NaCl (80 mL). The organic layer was dried (MgSO4) and concentrated under vacuum. Purification by flash chromatography (4:1 EtOAc–hexane to 1:9 MeOH–EtOAc) afforded the title compound (0.35 g, 86%) as a white solid: Rf = 0.40 (10% MeOH–EtOAc); 1H NMR (400 MHz, DMSO-d6) δ 0.92–1.08 (m, 28H), 3.76–3.84 (m, 1H), 3.88–3.96 (m, 1H), 4.10 (dd, J = 12.3, 3.4 Hz, 1H), 4.46–4.61 (m, 2H), 5.77 (d, J = 5.8 Hz, 1H), 6.20 (d, J = 6.4 Hz, 1H), 7.27 (br s, 2H), 8.04 (s, 1H), 8.10 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 12.0, 12.21, 12.23, 12.4, 12.8, 16.80, 16.81, 16.85, 16.9, 17.16, 17.18, 17.3, 61.5, 74.9, 75.1, 79.6, 81.7, 118.5, 139.6, 149.5, 152.3, 156.0; HRMS (ESI+) calcd for C22H40N5O5Si2 [M + H]+ 510.2562, found 510.2539 (error 4.5 ppm).
9-[3′,5′-O-(1,1,3,3-Tetraisopropyldisiloxyl)-2′-O-trifluormethylsulfonyl-β-D-arabinofuranosyl]adenine (39)
To a solution of 38 (0.327 g, 0.685 mmol, 1.0 equiv) and DMAP (42.0 mg, 0.343 mmol, 3.0 equiv) in CH2Cl2 (7 mL) at 0 °C was added Tf2O (0.140 mL, 0.822 mmol, 1.2 equiv). The reaction was stirred for 1.5 h at 0 °C then concentrated in vacuo to provide an oil that was purified directly by flash chromatography (4:1 EtOAc–hexane to EtOAc, linear gradient) to afford the title compound (0.272 g, 62%) as a white foam: Rf = 0.55 (EtOAc); 1H NMR (400 MHz, DMSO-d6) δ 1.00–1.36 (m, 28H), 3.89–4.01 (m, 2H), 4.21 (dd, J = 12.0, 5.4 Hz, 1H), 5.60–5.71 (m, 1H) 6.05 (t, J = 7.6 Hz, 1H), 6.49 (d, J = 7.0 Hz, 1H), 7.39 (br s, 2H), 8.07 (s, 1H), 8.28 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 12.20, 12.22, 12.3, 12.4, 16.61, 16.64, 16.66, 17.06, 17.07, 17.08, 17.2, 62.0, 74.1, 78.4, 78.9, 89.2, 119.1, 119.2 (q, 1JC-F = 310 Hz), 140.4, 149.0, 152.4, 156.1; HRMS (ESI+) calcd for C23H39F3N5O7SSi2 [M + H]+ 642.2055, found 642.2025 (error 4.6 ppm).
2′-Azido-2′-deoxy-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxyl)adenosine (40)
A solution of 39 (0.272 g, 0.424 mmol, 1.0 equiv) and sodium azide (83.0 mg, 1.27 mmol, 3.0 equiv) in DMF (10 mL) was stirred at 60 °C for 15 h. The reaction was concentrated in vacuo and the crude oil was purified by flash chromatography (3:2 to 4:1 EtOAc–hexane) to afford the title compound (0.199 g, 88%) as a white foam: Rf = 0.35 (3:2 EtOAc–hexanes); (c 0.06, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 0.96–1.29 (m, 28H), 3.88–4.08 (m, 3H), 5.01 (d, J = 6.2 Hz, 1H), 5.44 (t, J = 6.5 Hz, 1H) 5.83 (s, 1H), 7.35 (br s, 2H), 8.06 (s, 1H), 8.22 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 12.10, 12.16, 12.4, 12.7, 16.73, 16.78, 16.9, 17.03, 17.09, 17.10, 17.3, 60.8, 64.6, 71.9, 81.0, 86.1, 119.3, 140.0, 148.5, 152.4, 156.1; HRMS (ESI+) calcd for C22H39N8O4Si2 [M + H]+ 535.2627, found 535.2611 (error 3.0 ppm).
2′-Azido-N6,N6-bis(tert-butoxycarbonyl)-2′-deoxy-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxyl)adenosine (41)
Compound 40 (0.199 g, 0.373 mmol) was converted to 41 using the general procedure for bis-Boc protection. Purification by flash chromatography (1:4 EtOAc–hexane) afforded the title compound (0.205 g, 75%) as an off white foam: Rf = 0.65 (3:7 EtOAc–hexane); 1H NMR (400 MHz, DMSO-d6) δ 0.96–1.14 (m, 28H), 1.39 (s, 18H), 3.92–4.06 (m, 3H), 5.19 (d, J = 5.6 Hz, 1H), 5.36 (dd, J = 6.4, 4.8 Hz, 1H), 5.98 (s, 1H), 8.73 (s, 1H), 8.78 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 12.13, 12.18, 12.3, 12.7, 16.71, 16.76, 16.8, 17.03, 17.04, 17.1, 17.2, 27.2, 64.2, 65.2, 71.4, 81.0, 83.5, 86.5, 128.3, 145.8, 149.4, 149.9, 151.4, 151.9; HRMS (ESI+) calcd for C32H55N8O8Si2 [M + H]+ 735.3676, found 735.3662 (error 1.9 ppm).
2′-Azido-N6,N6-bis(tert-butoxycarbonyl)-2′-deoxyadenosine (42)
To a solution of 41 (0.205 g, 0.280 mmol, 1.0 equiv) in THF (5 mL) at 0 °C was added glacial acetic acid (50 μL to provide a 1% v/v solution). The cooling bath was removed and the mixture was stirred at 23 °C for 1.5 h. The reaction was quenched with ice cold 1 N aqueous HCl (15 mL) then extracted with EtOAc (5 × 20 mL). The combined organic extracts were washed with saturated aqueous NaCl (30 mL), dried (MgSO4), and concentrated. Purification by flash chromatography (EtOAc) afforded the title compound (0.099 g, 79%) as a colorless oil: Rf = 0.40 (EtOAc); 1H NMR (400 MHz, CDCl3) δ 1.44 (s, 18H), 3.56 (br s, 1H), 3.79 (d, J = 12.5 Hz, 1H), 3.98 (d, J = 12.5 Hz, 1H), 4.30 (s, 1H), 4.61–4.67 (m, 1H), 4.81–4.88 (m, 1H), 5.54 (br s, 1H), 6.01 (d, J = 7.5 Hz, 1H), 8.26 (s, 1H), 8.85 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 27.7, 62.7, 65.4, 72.6, 84.4, 87.9, 88.9, 130.3, 144.6, 150.2, 151.2, 151.7, 151.9; HRMS (ESI+) calcd for C20H29N8O7 [M + H]+ 493.2154, found 493.2141 (error 2.6 ppm).
2′-Azido-N6,N6-bis(tert-butoxycarbonyl)-3′,5′-O-bis(tert-butyldimethylsilyl)-2′-deoxyadenosine (43)
Compound 42 (0.099 g, 0.202 mmol) was converted to 43 using the general procedure for TBS protection. Purification by flash chromatography (1:4 EtOAc–hexanes) afforded the title compound (0.105 g, 72%) as a yellow oil: Rf = 0.60 (3:7 EtOAc–hexanes); 1H NMR (400 MHz, DMSO-d6) δ −0.11 (s, 3H), −0.03 (s, 3H), 0.20 (s, 6H), 0.79 (s, 9H), 0.95 (s, 9H), 1.38 (s, 18H), 3.74 (dd, J = 11.7, 3.1 Hz, 1H), 3.90 (dd, J = 11.7, 3.6 Hz, 1H), 3.95–4.01 (m, 1H), 4.95–5.02 (m, 2H), 6.16 (d, J = 3.9 Hz, 1H), 8.81 (s, 1H), 8.86 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ −5.64, −5.20, −4.89, 12.8, 17.18, 17.21, 17.7, 17.9, 25.59, 25.62, 27.2, 61.1, 64.1, 71.2, 79.1, 83.4, 84.3, 85.8, 145.4, 149.4, 149.9, 151.7, 152.3; HRMS (ESI+) calcd for C32H57N8O7Si2 [M + H]+ 721.3883, found 721.3858 (error 3.5 ppm).
2′-Azido-N6,N6-bis(tert-butoxycarbonyl)-3′-O-tert-butyldimethylsilyl-2′-deoxyadenosine (44)
To a solution of 43 (0.105 g, 0.145 mmol, 1.0 equiv) in 1:1 THF–H2O (10 mL) at 0 °C was added glacial AcOH (15 mL) dropwise and the mixture stirred at 0 °C for 36 h. The reaction mixture was extracted with EtOAc (5 × 25 mL) and the combined organic layers were washed with saturated aqueous NaCl (50 mL) and then concentrated. Purification by flash chromatography (3:2 EtOAc–hexane) afforded the title compound (0.056 g, 63%) as a white foam: Rf = 0.45 (1:1 EtOAc–hexane); 1H NMR (400 MHz, MeOH-d4) δ 0.21–0.24 (m, 6H), 1.00 (s, 9H), 1.40 (s, 18H), 3.75 (dd, J = 12.3, 3.1 Hz, 1H), 3.90 (dd, J = 12.3, 3.4 Hz, 1H), 4.17 (q, J = 3.2 Hz, 1H), 4.71 (t, J = 5.6 Hz, 1H), 4.79–4.82 (m, 1H), 6.32 (d, J = 6.0 Hz, 1H), 8.83 (s, 1H), 8.88 (s, 1H); 13C NMR (150 MHz, MeOH-d4) δ −4.63, −4.45, 19.1, 26.4, 28.1, 62.1, 66.8, 74.3, 85.5, 88.2, 88.5, 130.8, 146.8, 151.4, 151.6, 153.3, 154.3; HRMS (ESI+) calcd for C26H43N8O7Si [M + H]+ 607.3018, found 607.2991 (error 4.4 ppm).
2′-Azido-5′-[N-(d-biotinoyl)sulfamoyl]amino-3′,5′-dideoxyadenosine triethylammonium salt (46)
Compound 44 (0.282 g, 0.464 mmol, 1.0 equiv), was converted to 45 using the general procedure for the Mitsunobu reaction to afford (0.312 g, 85%, ~90% purity, contaminated with PPh3O) that used in the next step without further purification. The intermediate nucleoside sulfamide 45 (0.312 g, 0.356 mmol) was converted to 46 using the general procedure for biotinylation and TFA deprotection to afford the title compound (0.230 g, 28% over 3 steps) as the triethylammonium salt (2.7 equiv of Et3N) as a white solid: HPLC purity: 96.5%, tR = 14.25 min, k′ = 2.7 (method A); (c 0.50, MeOH); 1H NMR (400 MHz, MeOH-d4) δ 1.15 (t, J = 7.3 Hz, 25H, excess Et3N), 1.38–1.45 (m, 2H), 1.52–1.65 (m, 3H), 1.66–1.79 (m, 1H), 2.16 (t, J = 7.3 Hz, 2H), 2.66 (d, J = 12.3 Hz, 1H), 2.81 (q, J = 7.3 Hz, 17H, excess Et3N), 3.12–3.28 (m, 4H), 4.18–4.22 (m, 1H), 4.25–4.33 (m, 1H), 4.40–4.45 (m, 1H), 4.68–4.73 (m, 1H), 4.82 (t, J = 5.9 Hz, 1H), 6.02 (d, J = 6.7 Hz, 1H), 8.31 (s, 1H), 8.32 (s, 1H); 13C NMR (100 MHz, MeOH-d4) δ 10.6, 27.7, 29.7, 30.1, 40.0, 41.3, 46.5, 47.5, 57.2, 61.7, 63.5, 66.2, 74.0, 86.3, 88.7, 121.0, 142.0, 150.6, 154.5, 157.7, 166.4, 182.5; HRMS (ESI−) calcd for C20H27N12O6S2 [M − Et3NH]− 595.1623, found 595.1609 (error 2.4 ppm).
2′-Amino-5′-[N-(d-biotinoyl)sulfamoyl]amino-3′,5′-dideoxyadenosine (47)
To a solution of 46 (0.120 g, 0.133 mmol, 1.0 equiv) in 2:1 THF–H2O (6 mL) at 0 °C was added PPh3 (0.140 g, 0.534 mmol, 4 equiv). The ice bath was removed and the reaction was stirred for 6 h at 23 °C during which time all starting material was consumed as monitored by electrospray mass spectrometry in the negative mode. The reaction was concentrated in vacuo. The crude material was re-dissolved in 1:1 MeCN:50 mM TEAB (10–20 mg/mL) and filtered to remove insoluble solids. The resulting solution was purified by preparative reverse phase HPLC with a Phenomenx Gemini C18 (250 × 20 mm) column at a flow rate of 30.0 mL/min employing a linear gradient of 5–15% acetonitrile (solvent B) in 50 mM aqueous triethylammonium bicarbonate (TEAB) at pH 7.5 (solvent A) for 30 min. The appropriate fractions were pooled and lyophilized to afford the title compound (0.090 g, 75%) as the triethylammonium salt (3.0 equiv of Et3N) as a white solid: HPLC purity: 97.5%, tR = 11.53 min, k′ = 2.1, (method A); (c 0.50, MeOH); 1H NMR (400 MHz, MeOH-d4) δ 1.15 (t, J = 7.3 Hz, 27H excess Et3N), 1.38–1.47 (m, 2H), 1.52–1.65 (m, 3H), 1.66–1.74 (m, 1H), 2.19 (t, J = 7.6 Hz, 2H), 2.66 (d, J = 13.2 Hz, 1H), 2.83 (q, J = 7.3 Hz, 19H excess Et3N), 3.12–3.18 (m, 1H), 3.19–3.27 (m, 3H), 4.20–4.29 (m, 4H), 4.41 (dd, J = 7.7, 5.0 Hz, 1H), 5.78 (d, J = 8.3 Hz, 1H), 8.26 (s, 1H), 8.33 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 11.1, 25.0, 28.0, 28.1, 36.3, 39.8, 45.3, 45.7, 55.5, 56.4, 59.1, 61.0, 72.1, 84.6, 89.5, 119.5, 140.6, 149.9, 152.4, 156.2, 162.7, 173.9; HRMS (ESI−) calcd for C20H29N10O6S2 [M − Et3NH]− 569.1718, found 569.1725 (error 1.1 ppm).
3′,5′-O-(1,1,3,3-Tetraisopropyldisiloxyl)adenosine (49)
To a solution of adenosine 48 (0.200 g, 0.748 mmol, 1.0 equiv) in pyridine (5 mL) at 0 °C was added TIPDSCl2 (0.248 g, 0.786 mmol, 1.05 equiv). The mixture slowly warmed to 23 °C over 3 h and was stirred at 23 °C for 45 h. The reaction was concentrated in vacuo and the crude oil was taken up in EtOAc (100 mL) and washed successively with ice cold 1 N aqueous HCl (5 × 25 mL) and saturated aqueous NaCl (100 mL). The organic layer was dried (MgSO4) and concentrated under vacuum. Purification by flash chromatography (4:1 EtOAc–hexane to 1:9 MeOH–EtOAc) afforded the title compound (0.340 g, 89%) as a white solid: Rf = 0.25 (1:19 MeOH–EtOAc); 1H NMR (400 MHz, CDCl3) δ 0.99–1.07 (m, 28H), 3.86–3.94 (m, 1H), 3.98–4.10 (m, 2H) 4.52 (t, J = 4.4 Hz, 1H), 4.79 (dd, J = 8.7, 5.1 Hz, 1H), 5.61 (d, J = 4.4 Hz, 1H), 5.88 (s, 1H), 7.32 (br s, 2H), 8.08 (s, 1H), 8.21 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 12.0, 12.2, 12.4, 12.7, 16.7, 16.83, 16.86, 16.95, 17.12, 17.13, 17.15, 17.3, 60.8, 69.8, 73.6, 80.8, 89.3, 119.3, 139.2, 148.6, 152.5, 156.1; HRMS (ESI+) calcd for C22H40N5O5Si2 [M + H]+ 510.2562, found 510.2543 (error 3.7 ppm).
3′,5′-O-(1,1,3,3-Tetraisopropyldisiloxyl)-2′-O-(trifluormethylsulfonyl)adenosine (50)
To a solution of 49 (5.76 g, 11.3 mmol, 1.0 equiv) and DMAP (4.14 g, 33.9 mmol, 3.0 equiv) in CH2Cl2 (150 mL) at 0 °C was added Tf2O (2.47 mL, 14.7 mmol, 1.3 equiv). The reaction was stirred for 1.5 h at 0 °C then concentrated in vacuo to provide an oil that was purified by flash chromatography (5:95 triethylamine–EtOAc) to afford the title compound (4.20 g, 58% yield) as a white foam: Rf = 0.40 (1:19 MeOH–CH2Cl2); 1H NMR (400 MHz, DMSO-d6) δ 0.90–1.28 (m, 28H), 3.94–4.10 (m, 3H), 5.37 (dd, J = 8.6, 5.0 Hz, 1H), 6.07 (d, J = 4.4 Hz, 1H), 6.46 (s, 1H), 7.43 (br s, 2H), 8.04 (s, 1H), 8.27 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 12.1, 12.2, 12.3, 12.6, 16.53, 16.54, 16.6, 17.00, 17.02, 17.04, 17.2, 59.7, 68.3, 80.3, 85.4, 89.4, 119.2 (q, 1JC-F = 320 Hz), 140.0, 148.5, 152.30, 152. 33, 156.0; HRMS (ESI+) calcd for C23H39F3N5O7SSi2 [M + H]+ 642.2055, found 642.2021 (error 5.6 ppm).
9-[2′-Azido-2′-deoxy-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxyl)-β-d-arabinofuranosyl]adenine (51)
A solution of 50 (0.272 g, 0.424 mmol, 1.0 equiv) and sodium azide (83.0 mg, 1.27 mmol, 3.0 equiv) in DMF (10 mL) was stirred at 60 °C for 15 h. The reaction was concentrated in vacuo and the crude oil was concentrated onto silica gel. Purification by flash chromatography (3:2 to 4:1 EtOAc–hexane, linear gradient) afforded the title compound (0.221 g, 98%) as a light yellow foam: Rf = 0.40 (3:2 EtOAc–hexane); 1H NMR (400 MHz, DMSO-d6) δ 1.00–1.32 (m, 28H), 3.87–3.98 (m, 2H), 4.23 (dd, J = 13.7, 5.4 Hz, 1H), 4.84–4.95 (m, 2H), 6.36–6.43 (m, 1H), 7.36 (br s, 2H), 8.10 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 12.0, 12.1, 12.3, 12.8, 16.6, 16.7, 16.75, 16.77, 17.11, 17.13 (2C), 17.28, 61.4, 67.1, 73.9, 80.6, 81.1, 118.9, 138.9, 149.1, 152.4, 156.1; HRMS (ESI+) calcd for C22H39N8O4Si2 [M + H]+ 535.2627, found 535.2610 (error 5.0 ppm).
9-[2′-Azido-2′-deoxy-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxyl)-β-d-arabinofuranosyl]-N6,N6-bis(tert-butoxycarbonyl)adenine (52)
Compound 51 (0.221 g, 0.413 mmol) was converted to 52 using the general procedure for bis-Boc protection. Purification by flash chromatography (2:8 EtOAc–hexane) afforded the title compound (0.230 g, 76%) as a cloudy oil: Rf = 0.55 (3:7 EtOAc–hexane); 1H NMR (400 MHz, DMSO-d6) δ 0.98–1.08 (m, 22H), 1.12 (t, J = 7.2 Hz, 6H), 1.34 (s, 18H), 3.92–4.04 (m, 2H), 4.21–4.31 (m, 1H), 4.75 (t, J = 8.8 Hz, 1H), 4.98 (t, J = 8.8 Hz, 1H), 6.58 (d, J = 7.1 Hz, 1H), 8.63 (s, 1H), 8.82 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 12.0, 12.30, 12.35, 12.6, 16.4, 16.5, 16.66, 16.69, 17.03 (2C), 17.05, 17.2, 27.1, 27.4, 61.3, 67.0, 73.7, 80.1, 80.8, 81.7, 83.3, 128.2, 144.5, 149.5, 149.6, 151.6, 152.5; HRMS (ESI+) calcd for C32H55N8O8Si2 [M + H]+ 735.3676, found 735.3666 (error 1.4 ppm).
9-(2′-Azido-2′-deoxy-β-d-arabinofuranosyl)-N6,N6-bis(tert-butoxycarbonyl)adenine (53)
To a solution of 52 (0.230 g, 0.313 mmol, 1.0 equiv) in THF (5 mL) was added glacial acetic acid (50 μL to provide a 1% v/v solution). The reaction was warmed to 23 °C and stirred for 6 h then quenched with cold 1 N aqueous HCl (15 mL). The crude mixture was extracted with EtOAc (5 × 20 mL) and the combined organic layers were washed with saturated aqueous NaCl (30 mL), dried (MgSO4) and concentrated under vacuum. Purification by flash chromatography (1:1 EtOAc–hexane) afforded the title compound (0.111 g, 72%) as a light yellow oil: Rf = 0.40 (1:1 EtOAc–hexane); 1H NMR (400 MHz, DMSO-d6) δ 1.36 (s, 18H), 3.66–3.91 (m, 3H), 4.40 (q, J = 8.0 Hz, 1H), 4.71, (t, J = 7.4 Hz, 1H), 5.22 (t, J = 5.1 Hz, 1H), 6.05 (d, J =5.5 Hz, 1H), 6.59 (d, J = 7.1 Hz, 1H), 8.85 (s, 1H), 8.88 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 27.2, 59.5, 67.3, 71.3, 82.1, 83.3, 83.9, 127.8, 144.5, 149.9, 149.3, 149.8, 151.8, 152.7; HRMS (ESI+) calcd for C20H29N8O7 [M + H]+ 493.2154, found 493.2149 (error 1.0 ppm).
9-[2′-Azido-3′,5′-O-bis(tert-butyldimethylsilyl)-2′-deoxy-β-d-arabinofuranosyl]-N6,N6-bis(tert-butoxycarbonyl)adenine (54)
Compound 53 (0.804 g, 1.63 mmol) was converted to 54 using the general procedure for TBS protection. Purification by flash chromatography (2:8 EtOAc–hexane) afforded the title compound (0.893 g, 76%) as a white solid: Rf = 0.55 (3:7 EtOAc–hexane); 1H NMR (400 MHz, DMSO-d6) δ 0.05 (s, 3H), 0.07 (s, 3H), 0.12 (s, 6H), 0.90 (s, 18H), 1.35 (s, 18H), 3.78–3.89 (m, 1H), 3.90–4.02 (m, 2H), 4.47 (t, J = 8.0 Hz, 1H), 4.86 (t, J = 7.6 Hz, 1H), 6.64 (d, J = 6.8 Hz, 1H), 8.73 (s, 1H), 8.90 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ −5.57, −5.53, −4.93, −4.92, 17.5, 18.1, 25.4, 25.8, 27.1, 61.1, 67.9, 72.1, 82.1, 82.5, 83.3, 127.6, 144.2, 149.4, 149.7, 151.9, 152.6; HRMS (ESI+) calcd for C32H57N8O7Si2 [M + H]+ 721.3883, found 721.3867 (error 2.2 ppm).
9-(2′-Azido-3′-O-tert-butyldimethylsilyl-2′-deoxy-β-d-arabinofuranosyl)-N6,N6-bis(tert-butoxycarbonyl)adenine (55)
To a solution of 54 (0.893 g, 1.24 mmol, 1.0 equiv) in 1:1 H2O–THF (20 mL) at 0 °C was added glacial AcOH (30 mL) dropwise. The reaction was stirred at 0 °C for 46 h then extracted with EtOAc (5 × 60 mL). The combined organic layers were washed with saturated aqueous NaCl (100 mL), dried (MgSO4), and concentrated under vacuum. Purification by flash chromatography (3:7 EtOAc–hexane) afforded the title compound (0.489 g, 65%) as a yellow foam: Rf = 0.25 (3:7 EtOAc–hexane); 1H NMR (400 MHz, CDCl3) δ 0.13 (s, 3H), 0.39 (s, 3H) 0.89 (s, 9H), 1.36 (s, 18H), 3.62–3.69 (m, 1H), 3.76–3.83 (m, 1H), 3.85–3.94 (m, 1H), 4.49 (t, J = 7.98 Hz, 1H), 4.82 (t, J = 7.28 Hz, 1H), 5.29–5.35 (m, 1H), 6.63 (d, J = 6.7 Hz, 1H) 8.86 (s, 1H), 8.89 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ −5.03, −4.82, 17.6, 25.5, 27.2, 59.1, 68.1, 72.4, 82.1, 82.5, 83.2, 127.7, 144.7, 149.3, 149.8, 151.8, 152.7; HRMS (ESI+) calcd for C26H43N8O7Si [M + H]+ 607.3018, found 607.3000 (error 3.0 ppm).
9-{2′-Azido-5′-O-[N-(d-biotinoyl)sulfamoyl]-2′-deoxy-β-d-arabinofuranosyl}adenine triethylammonium salt (57)
Compound 55 (0.267 g, 0.440 mmol) was converted to 56 using the general procedure for sulfamylation using the Okada protocol that was greater than 90% pure contaminated with residual DMA and used directly in the next step. The intermediate sulfamate 56 was converted to 57 using the general procedure for biotinylation and TFA deprotection to afford the title compound (0.193 g, 55% over 3 steps) as the triethylammonium salt (4.4 equiv of Et3N) as a white solid: HPLC purity: 97.2%, tR = 14.66 min, k′ = 2.9 (method A); (c 1.08, MeOH); 1H NMR (400 MHz, MeOH-d4) δ 1.11 (t, J = 7.2 Hz, 40H, excess Et3N), 1.39–1.47 (m, 2H), 1.53–1.67 (m, 3H), 1.68–1.77 (m, 1H), 2.22 (t, J = 7.2 Hz, 2H), 2.63–2.67 (m, 1H), 2.86 (dd, J = 12.8, 5.1 Hz, 1H), 2.71 (q, J = 7.3 Hz, 28H, excess Et3N), 3.11–3.17 (m, 1H), 4.09–4.14 (m, 1H), 4.28 (dd, J = 7.8, 4.2 Hz, 1H), 4.36–4.40 (m, 2H), 4.41–4.46 (m, 1H), 4.47–4.52 (m, 1H), 4.54–4.59 (m, 1H), 6.47 (d, J = 6.0 Hz, 1H), 8.21 (s, 1H), 8.34 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 9.35, 25.5, 25.9, 28.1, 28.4, 39.9, 45.7, 55.5, 59.1, 61.1, 66.3, 66.9, 72.7, 80.7, 81.6, 118.5, 139,5, 149.1, 152.5, 155.9, 162.8, 177.9; HRMS (ESI−) calcd for C20H26N11O7S2 [M − Et3NH]− 596.1464, found 596.1472 (error 1.3 ppm).
3′,5′-O-bis(tert-Butyldimethylsilyl)-2′-deoxy-2′-fluoroadenosine (59)
Compound 58 (2.05 g, 7.62 mmol) was converted to the title compound using the general procedure for TBS protection. Purification by flash chromatography (1:19 MeOH–EtOAc) afforded the title compound (3.23 g, 85%) as a white solid: Rf = 0.45 (1:19 MeOH–EtOAc); 1H NMR (600 MHz, CDCl3) δ −0.11 (s, 3H), −0.02 (s, 3H), 0.14 (s, 3H), 0.15 (s, 3H), 0.77 (s, 9H), 0.91 (s, 9H), 3.71 (d, J = 11.2 Hz, 1H), 3.91 (d, J = 11.7 Hz, 1H), 3.94–3.98 (m, 1H), 4.86–4.94 (m, 1H), 5.63 (d, 2JC-F = 53.5 Hz, 1H), 6.22 (d, 3JC-F = 19.2 Hz, 1H), 7.33 (br s, 2H), 8.12 (s, 1H), 8.25 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ −5.71, −5.68, −5.23, −4.89, 17.7, 18.8, 25.54, 25.57, 60.8, 68.8 (d, 2JC-F = 15.6 Hz), 82.5, 85.9 (2JC-F = 33.6 Hz), 92.5 (1JC-F = 187 Hz), 119.0, 139.6, 148.7, 152.6, 156.1; HRMS (ESI+) calcd for C22H41FN5O3Si2 [M + H]+ 498.2726, found 498.2709 (error 3.4 ppm).
N6,N6-bis(tert-Butoxycarbonyl)-3′,5′-O-bis(tert-butyldimethylsilyl)-2′-deoxy-2′-fluoroadenosine (60)
Compound 59 (3.20 g, 6.43 mmol) was converted to the title compound using the general procedure for bis-Boc protection. Purification by flash chromatography (1:3 EtOAc–hexane) afforded the title compound (4.13 g, 92%) as a clear oil: Rf = 0.60 (3:7 EtOAc–hexane); 1H NMR (600 MHz, CDCl3) δ −0.15 (s, 3H), −0.03 (s, 3H), 0.15 (s, 6H), 0.77 (s, 9H), 0.92, (s, 9H), 1.38 (s, 18H), 3.75 (dd, J = 12.2, 3.1 Hz, 1H), 3.94 (dd, J = 12.0, 2.4 Hz, 1H), 3.98–4.03 (m, 1H), 4.94 (ddd, J = 22.0, 8.1, 4.4 Hz, 1H), 5.69 (dd, J = 52.8, 3.3 Hz, 1H), 6.39 (d, J = 19.2 Hz, 1H) 8.76 (s, 1H), 8.83 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ −5.73, −5.70, −5.25, −4.91, 17.7, 17.8, 25.51, 25.54, 27.2, 60.5, 68.6 (d, 2JC-F = 15.3 Hz), 82.6, 83.4, 86.3 (d, 2JC-F = 34.4 Hz), 92.5 (d, 1JC-F = 187 Hz), 128.0, 145.3, 149.3, 149.9, 151.5, 152.0; HRMS (ESI+) calcd for C32H57FN5O7Si2 [M + H]+ 698.3775, found 698.3760 (error 2.1 ppm).
N6,N6-bis(tert-Butoxycarbonyl)-3′-O-tert-butyldimethylsilyl-2′-deoxy-2′-fluoroadenosine (61)
To a solution of 60 (3.90 g, 5.60 mmol, 1.0 equiv) in 1:1 H2O–THF (20 mL) at 0 °C was added glacial AcOH (30 mL). The mixture was stirred at 0 °C for 13 h then quenched with ice cold 1 N aqueous HCl (50 mL). The mixture was extracted with EtOAc (5 × 50 mL) and the combined organic layers were washed with saturated aqueous NaCl (50 mL) and concentrated under vacuum. Purification by flash chromatography (1:1 EtOAc–hexane) afforded the title compound (2.09 g, 66% yield) as a white foam: Rf = 0.40 (1:1 EtOAc–hexane); 1H NMR (600 MHz, CDCl3) δ 0.13 (s, 3H), 0.15 (s, 3H), 0.91 (s, 9H), 1.41 (s, 18H), 3.55–3.60 (m, 1H), 3.74–3.79 (m, 1H), 4.00–4.04 (m 1H), 4.70–4.77 (m, 1H), 5.15–5.19 (m, 1H), 5.65 (d, J = 52.8 Hz, 1H), 6.40 (d, J = 16.6 Hz, 1H), 8.86 (s, 1H), 8.87 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ −5.16, −4.95, 17.8, 25.6, 27.3, 59.7, 69.6 (d, 2JC-F = 15.4 Hz), 83.5, 84.5, 85.9 (d, 2JC-F = 33.6 Hz), 92.3 (d, 1JC-F = 189 Hz), 128.1, 145.1, 149.3, 150.0, 151.7, 152.3; HRMS (ESI+) calcd for C26H43FN5O7Si [M + H]+ 584.2910, found 584.2898 (error 2.1 ppm).
5′-[N-(d-Biotinoyl)sulfamoyl]amino-2′,5′-dideoxy-2′-fluoroadenosine triethylammonium salt (63)
Compound 61 (2.13 g, 3.65 mmol) was converted to 62 using the general procedure for the Mitsunobu reaction. Purification by flash chromatography (30% EtOAc–hexanes) afforded 62 (>85% purity, contaminated with PPh3O) that used in the next step without further purification. Compound 62 (2.78 g, 3.65 mmol) was converted to 63 using the general procedures for biotinylation and TFA deprotection to afford the title compound (0.861 g, 35% over 3 steps) as the triethylammonium salt (1.7 equiv of Et3N) as a white solid: HPLC purity: 96.9%, tR = 12.90 min, k′ = 2.4 (method A); (c 0.49, MeOH); 1H NMR (400 MHz, MeOH-d4) δ 1.18 (t, J = 7.4 Hz, 16H, excess Et3N), 1.37–1.45 (m, 2H), 1.52–1.60 (m, 3H), 1.66–1.75 (m, 1H), 2.16 (t, J = 7.7 Hz, 2H), 2.67 (d, J = 12.6 Hz, 1H), 2.89 (q, J = 7.7 Hz, 12H, excess Et3N), 3.14–3.24 (m, 3H), 4.18–4.23 (m, 1H), 4.28 (dd, J = 8.5, 4.6 Hz, 1H), 4.42–4.46 (m, 1H), 4.71 (dt, J = 15.8, 5.8 Hz, 2H), 5.53 (dt, J = 53.1, 4.1 Hz, 1H), 6.23 (dd, J = 17.0, 3.4 Hz, 1H), 8.28 (s, 1H), 8.29 (s, 1H); 13C NMR (150 MHz, MeOH-d4) δ 9.5, 26.7, 29.6, 29.9, 38.0, 40.5, 41.2, 46.1, 48.0, 57.0, 61.7, 63.4, 71.7 (d, 2JC-F = 15.8 Hz), 84.2, 89.2 (d, 2JC-F = 33.3 Hz), 94.2 (d, 1JC-F = 190 Hz), 121.0, 142.0, 150.2, 154.5, 157.7, 166.3, 178.1; HRMS (ESI−) calcd for C20H26FN8O7S2 [M − H]− 573.1355, found 573.1524 (error 3.0 ppm).
9-[3′,5′-O-bis(tert-Butyldimethylsilyl)-2′-deoxy-2′-fluoro-β-d-arabinofuranosyl]adenine (65)
Compound 64 (0.230 g, 0.853 mmol) was converted to the title compound using the general procedure for TBS protection. Purification by flash chromatography (1:19 MeOH–EtOAc) afforded the title compound (0.39 g, 92%) as a white foam: Rf = 0.45 (1:19 MeOH–EtOAc); 1H NMR (600 MHz, DMSO-d6) δ 0.06 (s, 3H), 0.07 (s, 3H), 0.13 (s, 6H), 0.89 (s, 18H), 3.79–3.91 (m, 3H), 4.67–4.71 (m, 1H), 5.27–5.40 (m, 1H), 6.44 (dd, J = 12.4, 4.7 Hz, 1H), 7.36 (br s, 2H), 8.15 (s, 1H), 8.16 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ −5.52 (2C), −5.26, −4.89, 17.6, 18.0, 25.5, 25.7, 61.4, 73.5 (d, 2JC-F = 23.3 Hz), 80.6 (d, 2JC-F = 17.2 Hz), 82.0 (d, 2JC-F = 7.0 Hz), 95.2 (d, 1JC-F = 193 Hz), 118.1, 138.90, 149.2, 152.9, 156.0; HRMS (ESI+) calcd for C22H41FN5O3Si2 [M + H]+ 498.2726, found 498.2716 (error 2.0 ppm).
N6,N6-bis(tert-Butoxycarbonyl)-9-[3′,5′-O-bis(tert-butyldimethylsilyl)-2′-deoxy-2′-fluoro-β-d-arabinofuranosyl]adenine (66)
Compound 65 (0.390 g, 0.784 mmol) was converted to the title compound using the general procedure for bis-Boc protection. Purification by flash chromatography (1:3 EtOAc–hexane) afforded the title compound (0.443 g, 81%) as a colorless oil: Rf = 0.55 (3:7 EtOAc–hexane); 1H NMR (600 MHz, DMSO-d6) δ 0.05 (s, 3H), 0.06, (s, 3H), 0.12 (s, 6H), 0.89 (s, 18H), 1.36 (s, 18H), 3.85 (dd, J = 12.0, 4.0 Hz, 1H), 3.89–3.97 (m, 2H), 4.72 (dt, J = 18.7, 5.5 Hz, 1H), 5.47 (dt, J = 59.2, 4.9 Hz, 1H), 6.62 (dd, J = 10.1, 5.4 Hz, 1H), 8.71 (s, 1H), 8.88 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ −5.59, −5.58, −5.23, −4.95, 17.6, 18.0, 25.4, 25.7, 27.1, 63.4, 73.0 (d, 2JC-F = 23.0 Hz), 81.1 (d, 2JC-F = 17.1 Hz), 82.1 (d, 2JC-F = 7.6 Hz), 83.3, 95.2 (d, 1JC-F = 194 Hz), 127.5, 144.60, 144.62, 149.4, 149.7, 151.9, 152.4; HRMS (ESI+) calcd for C32H57FN5O7Si2 [M + H]+ 698.3775, found 698.3765 (error 1.4 ppm).
N6,N6-bis(tert-Butoxycarbonyl)-9-(3′O-tert-butyldimethylsilyl-2′-deoxy-2′-fluoro-β-d-arabinofuranosyl)adenine (67)
To a solution of 66 (0.443 g, 0.635 mmol, 1.0 equiv) in H2O–THF (10 mL) at 0 °C was added glacial AcOH (15 mL). The reaction was stirred at 0 °C for 23 h then extracted with EtOAc (5 × 50 mL). The combined organic layers were washed with saturated aqueous NaCl (50 mL) and concentrated under vacuum. Purification by flash chromatography (1:1 EtOAc–hexane) afforded the title compound (0.245 g, 66%) as a white foam: Rf = 0.25 (1:1 EtOAc–hexane); 1H NMR (600 MHz, DMSO-d6) δ 0.06 (s, 6H), 0.88 (s, 9H), 1.36 (s, 18H), 3.86–3.96 (m, 3H), 4.51 (dt, J = 19.7, 4.5 Hz, 1H), 5.39 (dt, J = 59.2, 4.7 Hz, 1H), 6.08 (s, 1H), 6.61 (dd, J = 10.5, 5.0 Hz, 1H), 8.72 (s, 1H), 8.89 (s, 1H); 13C NMR (226 MHz, MeOH-d4) δ −4.71, −4.56, 19.0, 26.3, 28.1, 61.5, 75.6 (d, 2JC-F = 24.3 Hz), 84.4 (d, 3JC-F = 18.2 Hz), 85.5, 85.8 (d, 2JC-F = 5.3 Hz), 97.2 (d, 1JC-F = 194.0 Hz), 130.1, 147.0, 151.2, 151.5, 153.5, 154.5; HRMS (ESI+) calcd for C26H43FN5O7Si [M + H]+ 584.2910, found 584.2902 (error 1.4 ppm).
9-{5′-O-[N-(d-Biotinoyl)sulfamoyl]-2′-deoxy-2′-fluoro-β-d-arabinofuranosyl}adenine triethylammonium salt (69)
Compound 67 (0.245 g, 0.419 mmol) was converted to 68 using the general procedure for sulfamoylation employing the Okada protocol. Purification by flash chromatography (30% EtOAc–hexanes) afforded 68 that was greater than 90% pure contaminated with residual DMA and used directly in the next step. The intermediate sulfamate was converted to 69 using the general procedure for biotinylation and TFA deprotection to afford the title compound (0.182 g, 56% over 3 steps) as the triethylammonium salt (4.0 equiv of Et3N) as a white solid: HPLC purity >99%, tR = 14.23 min, k′ = 2.8 (method A); (c 1.43, MeOH); 1H NMR (400 MHz, MeOH-d4) δ 1.12 (t, J = 7.2 Hz, 36H, excess Et3N), 1.40–1.48 (m, 2H), 1.53–1.67 (m, 3H), 1.68–1.77 (m, 1H), 2.21 (t, J = 7.3 Hz, 2H), 2.66 (d, J = 12.5 Hz, 1H), 2.74 (q, J = 7.3 Hz, 24H, excess Et3N), 2.87 (dd, J = 12.8, 4.9 Hz, 1H), 3.13–3.19 (m, 1H), 4.18–4.23 (m, 1H), 4.28 (dd, J = 8.0, 4.5 Hz, 1H), 4.30–4.34 (m, 2H), 4.41–4.46 (m, 1H), 4.61 (dt, J = 16.9, 3.2 Hz, 1H), 5.12 (dt, J = 52.3, 3.5 Hz, 1H), 6.51 (dd, J = 16.5, 3.8 Hz, 1H), 8.21 (s, 1H), 8.33 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 9.35, 25.2, 25.9, 28.1, 28.4, 39.8, 45.6, 55.5, 59.1, 61.0, 66.4, 73.3 (d, 2JC-F = 23.5 Hz), 81.0, 81.6 94.8 (d, 1JC-F = 191 Hz), 118.1, 139.5, 149.1, 152.8, 156.0, 162.8, 177.7; HRMS (ESI−) calcd for C20H26FN8O7S2 [M − Et3NH]− 573.1355, found 573.1364 (error 1.5 ppm).
5′-O-[N-(d-Biotinoyl)sulfamoyl]-β-2'-methyladenosine triethylammonium salt (73)
Compound 71 (100 mg, 0.311 mmol) was converted to 72 using the general procedure for sulfamoylation employing the Okada protocol that was used directly without further purification. The intermediate sulfamate 72 was converted to 73 using the general procedures for biotinylation and TFA deprotection to afford the title compound (101 mg, 45% over 3 steps) as the triethylammonium salt (6.2 equiv of Et3N) as a white solid: HPLC purity: 89.3%, tR = 13.04 min, k′ = 2.5 (method A); 1H NMR (400 MHz, MeOH-d4) δ 0.92 (s, 3H), 1.10 (t, J = 7.6 Hz, 56H, excess Et3N), 1.38–1.46 (m, 2H), 1.59–1.66 (m, 3H), 1.69–1.76 (m, 1H), 2.22 (t, J = 7.0 Hz, 2H), 2.71 (q, J = 7.0 Hz, 40H, excess Et3N), 2.83–2.89 (m, 2H), 3.11–3.16 (m, 1H), 4.19–4.29 (m, 3H), 4.41–4.51 (m, 3H), 6.10 (s, 1H), 8.19 (s, 1H), 8.44 (s, 1H); NOE 1H NMR (600 MHz, DMSO-d6) (Irr, H-1'), enhancement at 0.75 (CH3) and 8.29 (H-2); 13C NMR (150 MHz, DMSO-d6) δ 11.1 (Et3NH), 20.03 (CH3), 26.3 (C-9”), 28.4 (C-7”), 28.8 (C-8”), 39.1 (C-10”), 40.4 (Et3NH), 46.4 (C-4”), 56.1 (C-6”), 59.2 (C-3'), 61.4 (C-4'), 66.6 (C-5'), 72.8 (C-6a), 80.4 (C-3a), 87.4 (C-2'), 91.3 (C-1'), 119.1 (C-5), 139.5 (C-2), 149.3 (C-8), 153.0 (C-4), 156.1 (C-6), 163.4 (C-2”), 179.1 (C-11”); HRMS (ESI−) calcd for C21H29N8O8S2 [M − Et3NH]− 585.1555, found 585.1512 (error 7.3 ppm).
N6-Benzoyl-5′-O-(tert-butyldiphenylsilyl)adenosine (75)
To a suspension of N6-benzoyladenosine 74 (2.50 g, 6.73 mmol, 1.0 equiv) and DMAP (82 mg, 0.67 mmol, 0.1 equiv) in pyridine (50 mL) at 23 °C was added tert-butylchlorodiphenylsilane (1.93 mL, 7.41 mmol, 1.1 equiv) dropwise. The reaction was stirred for 19 h at 23 °C. Solvent was removed in vacuo and the residue was re-dissolved in EtOH (10 mL). A white precipitate started to form, and further precipitation was induced by the addition of Et2O (200 mL). The gummy solid was filtered, washed with H2O (2 × 50 mL) and recrystallized from MeOH (50 mL) to afford the title compound (4.03 g, 98%) as a white solid: mp: 113–116 °C; Rf = 0.31 (1:19 MeOH–CH2Cl2); 1H NMR (400 MHz, CDCl3) 0.94 (s, 9H), 3.78–3.96 (m, 2H), 4.33–4.43 (m, 1H), 4.51–4.59 (m, 1H), 4.78 (t, J = 5.5 Hz, 1H), 6.06 (d, J = 5.9 Hz, 1H), 7.29–7.46 (m, 6H), 7.48–7.66 (m 7H), 8.02 (d, J = 7.4 Hz, 2H), 8.26 (s, 1H), 8.71 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 19.1, 26.7, 63.9, 72.2, 75.9, 86.9, 90.3, 122.9, 127.81, 127.83, 127.9, 128.9 (2C), 129.9, 130.0, 132.5, 132.6, 133.5, 135.4, 135.5 (2C), 141.3, 149.5, 150.9, 152.2; HRMS (ESI+) calcd for C33H36N5O5Si [M + H]+ 610.2480, found 610.2473 (error 1.1 ppm).
(2′R,6′S)-N6-Benzoy-9-{6′-[(tert-butyldiphenylsilyloxy)methyl]-N-tritylmorpholin-2′-yl}adenine (79)
To a suspension of 75 (1.47 g, 2.41 mmol, 1.0 equiv) in methanol (25 mL) at 23 °C was added sodium meta-periodate (570 mg, 2.66 mmol, 1.1 equiv) and ammonium biborate tetrahydrate (700 mg, 2.66 mmol, 1.1 equiv). The reaction mixture was stirred at 23 °C for 1.5 h during which time the starting material was consumed and a cluster of closely-spaced ninhydrin-active spots (TLC) was generated. The reaction mixture was filtered and to the filtrate was added successively activated powdered 4 Å molecular sieves (300 mg/mmol of 75), sodium cyanoborohydride (330 mg, 5.3 mmol, 2.2 equiv) and glacial acetic acid (0.30 mL, 5.30 mmol, 2.2 equiv) dropwise. The reaction mixture was stirred for another 5 h at 23 °C. The mixture was then filtered through Celite and evaporated to dryness. The residue was dissolved in CHCl3 (50 mL) and washed with H2O (30 mL), saturated aqueous NaCl (30 mL), dried (Na2SO4) and concentrated in vacuo to yield crude 78 as a white solid, which was used in the next step directly without purification.
The crude 78 prepared above was co-evaporated with DMF (2 × 20 mL). Crude 78 was suspended in DMF (10 mL) and to the suspension was added triethylamine (0.50 mL, 3.62 mmol, 1.5 equiv), and trityl chloride (810 mg, 2.89 mmol, 1.2 equiv) portion-wise at 0 °C. After 2 h, the reaction mixture was quenched with MeOH (1 mL), and evaporated to dryness. The residue was then dissolved in EtOAc (50 mL) and washed with H2O (30 mL), saturated aqueous NaCl (30 mL), dried (Na2SO4) and concentrated under vacuum. Purification by flash chromatography (0:1 to 1:9 MeOH–CH2Cl2, linear gradient) afforded the title compound (0.86 g, 43% over 4 steps) as a white foam: Rf = 0.49 (1:19 MeOH–CH2Cl2); 1H NMR (400 MHz, CDCl3) 0.95 (s, 9H), 1.62 (t, J = 11.0 Hz, 1H), 1.75 (t, J = 11.0 Hz, 1H), 3.36 (d, J = 11.7 Hz, 1H), 3.47 (d, J = 11.4 Hz, 1H), 3.61 (dd, J = 10.5, 5.9 Hz, 1H), 3.77 (dd, J = 10.6, 4.3 Hz, 1H), 4.29–4.41 (m, 1H), 6.38 (d, J = 8.6 Hz, 1H), 7.16–7.63 (m, 28H), 7.84 (s, 1H), 8.00 (d, J = 7.4 Hz, 2H), 8.81 (s, 1H), 8.96 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 19.2, 26.7, 49.9, 53.3, 64.5, 77.2, 80.2, 122.6, 126.5, 127.6, 127.8, 127.9, 128.8, 129.2, 129.68, 129.73, 132.7, 133.1, 133.2, 133.6, 135.5, 140.6, 149.3, 151.2, 152.8, 164.5; HRMS (ESI+) calcd for C52H51N6O3Si [M + H]+ 835.3786, found 835.3785 (error 0.1 ppm).
(2′R,6′S)-N6-Benzoyl-9-[6′-(hydroxymethyl)-N-tritylmorpholin-2′-yl]adenine (80)
To a solution of 79 (1.67 g, 2.00 mmol, 1.0 equiv) in THF (20 mL) at 23 °C was added a solution of TBAF (1.0 M in THF, 3.00 mL, 3.00 mmol, 1.5 equiv). The reaction mixture was stirred for 2 h at 23 °C then concentrated and diluted with EtOAc (30 mL), washed with H2O (25 mL), saturated aqueous NaCl (25 mL), dried (Na2SO4), and concentrated in vacuo. Purification by flash chromatography (0:1 to 1:19 MeOH–CH2Cl2, linear gradient) afforded the title compound (1.15 g, 96%) as a white solid: Rf = 0.29 (5% MeOH–CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 1.59 (t, J = 11.2 Hz, 1H), 1.79 (t, J = 10.8 Hz, 1H), 3.10 (d, J = 11.7 Hz, 1H), 3.50 (d, J = 11.7 Hz, 1H), 3.53–3.64 (m, 2H), 3.81 (br s, 1H), 4.29 (br s, 1H), 6.34 (d, J = 9.4 Hz, 1H), 7.01–7.23 (m, 3H), 7.29 (t, J = 7.4 Hz, 7H), 7.38–7.53 (m, 8H), 7.53–7.65 (m, 1H), 7.83 (s, 1H), 8.03 (d, J = 7.4 Hz, 1H), 8.77 (s, 1H), 9.60 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 45.8, 48.7, 53.0, 63.4, 76.9, 77.2, 77.6, 80.4, 122.1, 126.6, 127.9, 128.0, 128.7, 129.1, 132.6, 133.8, 140.1, 149.6, 150.8, 152.9, 164.8; HRMS (ESI+) calcd for C36H33N6O3 [M + H]+ 597.2609, found 597.2622 (error 2.2 ppm).
(2′R,6′S)-N6-Benzoyl-9-(6′-{[(sulfamoyl)oxy]methyl}-N-tritylmorpholin-2′-yl)adenine (81)
Compound 80 (200 mg, 0.335 mmol) was converted to the title compound using the Okada general procedure for sulfamoylation. Purification by flash chromatography (0:1 to 3:2 EtOAc–hexane, linear gradient) afforded the title compound (180 mg, 78%) as a white solid: Rf = 0.49 (5% MeOH–CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 1.60 (t, J = 11.4 Hz, 1H), 1.80 (t, J = 10.6 Hz, 1H), 3.20 (d, J = 11.7 Hz, 1H), 3.43 (d, J = 11.7 Hz, 1H), 4.08–4.23 (m, 2H), 4.55 (br s, 1H), 5.85 (br s, 2H), 6.28 (d, J = 8.2 Hz, 1H), 7.13–7.56 (m, 18H), 7.81 (s, 1H), 7.88 (d, J = 7.8 Hz, 2H), 8.68 (s, 1H), 9.02 (br s, 1H); 13C NMR (100 MHz, CDCl3) 48.6, 52.7, 56.8, 70.0, 74.3, 80.1, 122.2, 126.8, 126.9, 127.8, 128.1, 128.7, 128.8, 129.1, 132.8, 133.3, 140.7, 148.9, 150.8, 152.7, 164.8; HRMS (ESI−) calcd for C36H32N7O5S [M − H]− 674.2191, found 674.2192 (error 0.1 ppm).
(2′R,6′S)-9-[6′-({[N-(d-Biotinoyl)sulfamoyl]oxy}methyl)morpholin-2′-yl]adenine triethylammonium salt (82)
Compound 81 (50 mg, 0.073 mmol) was biotinylated using the general procedure for biotinylation. To a solution of this crude biotinylated nucleoside in pyridine (1 mL) at 23 °C was added concentrated aqueous ammonia (1 mL). The reaction mixture was stirred for 25 h at 23 °C during which time all starting material was consumed as monitored by electrospray mass spectrometry in the negative mode. The solvent was removed under vacuum. The crude debenzoylated material was deprotected using the general procedure for TFA deprotection to afford the title compound (18 mg, 36% over 2 steps) as the triethylammonium salt (1.6 equiv of Et3N) as a white solid: HPLC purity: 92.0%, tR = 13.2 min (method A), k′ = 1.4 (method A); 1H NMR (600 MHz, MeOH-d4) δ 1.14 (t, J = 7.2 Hz, 15H, excess Et3N), 1.24 (t, J = 7.0 Hz, 2H), 1.32–1.46 (m, 2H), 1.46–1.75 (m, 5H), 2.15 (t, J = 7.2 Hz, 2H), 2.67 (d, J = 12.5 Hz, 1H), 2.79 (q, J = 7.0 Hz, 11H, excess Et3N), 2.89 (dd, J = 12.9, 5.1 Hz, 2H), 3.02 (d, J = 12.5 Hz, 1H), 3.18 (d, J = 6.7 Hz, 2H), 4.04–4.16 (m, 2H), 4.28 (dd, J = 7.6, 4.5 Hz, 1H), 4.39–4.51 (m, 1H), 5.85 (t, J = 6.3 Hz, 1H), 7.46 (d, J = 7.4 Hz, 1H), 7.54 (s, 1H), 7.86 (d, J = 7.8 Hz, 1H), 8.20 (s, 1H), 8.34 (s, 1H); 13C NMR (100 MHz, MeOH-d4) 25.8, 28.1, 28.4, 38.4, 39.7, 45.2, 45.9, 46.8, 47.9, 48.1, 55.6, 60.2, 61.9, 68.3, 75.8, 80.1, 127.1, 128.1, 131.5, 139.2, 148.7, 152.5; HRMS (ESI−) calcd for C20H28N9O6S2 [M – Et3NH]− 554.1609, found 554.1612 (error 0.5 ppm).
N6,N6-bis(tert-Butoxycarbonyl)-2′,5′-O-bis(tert-butyldimethylsilyl)-3′-deoxyadenosine (84)
Compound 83 (0.200 g, 0.796 mmol) was converted to 2′,5′-O-bis(tert-butyldimethylsilyl)-3′-deoxyadenosine using the general procedure for TBS protection. Purification by flash chromatography (5% MeOH–EtOAc) afforded 2′,5′-O-bis(tert-butyldimethylsilyl)-3′-deoxyadenosine (0.327 g, 86%), which was used in the next step without further purification. 2′,5′-O-bis(tert-Butyldimethylsilyl)-3′-deoxyadenosine (0.327 g, 0.685 mmol) was converted to the title compound using the general procedure for bis-Boc protection. Purification by flash chromatography (2:3 EtOAc–hexane) afforded the title compound (0.466 g, 82%, 2 steps) as a colorless oil: Rf = 0.50 (3:7 EtOAc–hexane); (c 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 0.00 (s, 3H), 0.01 (s, 3H), 0,04 (s, 6H), 0.83 (s, 9H), 0.88 (s, 9H), 1.36 (s, 18H), 1.95–2.03 (m, 1H), 2.27–2.37 (m, 1H), 3.79 (d, J = 9.5 Hz, 1H), 3.97 (d, J = 10.9 Hz, 1H), 4.41–4.48 (m, 1H), 4.80–4.85 (m, 1H), 6.04 (s, 1H), 8.81 (s, 1H), 8.86 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ −5.52, −5.49, −5.10, −5.08, 17.6, 18.0, 25.5, 25.8, 27.2, 33.9, 63.9, 76.3, 80.5, 83.3, 90.7, 128.1, 144.2, 149.2, 149.8, 151.7, 152.3; HRMS (ESI+) calcd for C32H58N5O7Si2 [M + H]+ 680.3869, found 680.3852 (error 2.5 ppm).
N6,N6-bis(tert-Butoxycarbonyl)-2′-O-tert-butyldimethylsilyl-3′-deoxyadenosine (85)
To a solution of 84 (0.466 g, 0.685 mmol, 1.0 equiv) in 1:1 H2O–THF (10 mL) at 0 °C was added glacial AcOH (15 mL). The mixture was stirred at 0 °C for 7 h then extracted with EtOAc (5 × 50 mL). The combined organic layers were washed with saturated aqueous NaCl (50 mL) and then concentrated under vacuum. Purification by flash chromatography (2:3 EtOAc–hexane) afforded the title compound (0.244 g, 63%) as a white foam: Rf = 0.50 (1:1 EtOAc–hexane); (c 0.79, MeOH); 1H NMR (400 MHz, DMSO-d6) δ −0.02 (s, 3H), 0.01 (s, 3H), 0.82 (s, 9H), 1.38 (s, 18H), 1.90–2.01 (m, 1H), 2.24–2.36 (m, 1H), 3.51–3.63 (m, 1H), 3.69–3.79 (m, 1H), 4.35–4.46 (m, 1H), 4.82 (br s, 1H), 5.11 (t, J = 5.1 Hz, 1H), 6.03 (d, J = 1.7 Hz, 1H), 8.87 (s, 1H), 8.91 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ −5.10, −5.08, 17.6, 25.5, 27.2, 34.1, 62.1, 76.2, 80.5, 83.4, 90.7, 128.1, 144.6, 149.2, 149.9, 151.6, 152.4; HRMS (ESI+) calcd for C26H44N5O7Si2 [M + H]+ 566.3005, found 566.2991 (error 2.5 ppm).
5′-[N-(d-Biotinoyl)sulfamoyl]amino-3′,5′-dideoxyadenosine triethylammonium salt (87)
Compound 85 (0.238 g, 0.421 mmol, 1.0 equiv) was converted to 86 using the general procedure for the Mitsunobu reaction. Purification by flash chromatography (45% EtOAc–hexanes) afforded 86 (>85% purity, contaminated with PPh3O) that used in the next step without further purification. The intermediate sulfamide 86 was converted to 87 using the general procedures for biotinylation and TFA deprotection to afford the title compound (171 mg, 62% over 3 steps) as the triethylammonium salt (3.3 equiv of Et3N) as a white solid: HPLC purity: 96.3%, tR = 12.44 min, k′ = 2.3 (method A); (c 0.56, MeOH); 1H NMR (400 MHz, MeOH-d4) δ 1.13 (t, J = 7.3 Hz, 30H, excess Et3N), 1.19–1.30 (m, 2H), 1.39–1.48 (m, 2H), 1.53–1.66 (m, 3H), 1.68–1.77 (m, 1H), 2.18 (t, J = 7.1 Hz, 2H), 2.33–2.42 (m, 1H), 2.66 (d, J = 12.2 Hz, 1H), 2.75 (q, J = 7.1 Hz, 20H, excess Et3N), 2.87 (dd, J = 13.0, 5.0, 1H), 3.11–3.16 (m, 1H), 3.23–3.26 (m, 1H), 4.28 (dd, J = 8.4, 4.9 Hz, 1H), 4.41–4.46 (m, 1H), 4.54–4.61 (m, 1H), 4.74–4.75 (m, 1H), 5.91 (d, J = 2.3 Hz, 1H), 8.26 (s, 1H), 8.28 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 9.01, 21.9, 24.4, 27.98, 35.0, 35.7, 45.7, 46.7, 55.3 59.1, 61.0, 67.8, 73.2, 77.3, 91.4, 119.4, 139.8, 148.5, 152.5, 156.2, 162.7, 171.7; HRMS (ESI−) calcd for C20H28N9O6S2 [M – Et3NH]− 554.1609, found 554.1618 (error 1.7 ppm).
9-[({N-[d-Biotinoyl))sulfamoyl]amino}ethoxy)methyl]adenine triethylammonium salt (90)
Compound 88 (0.213 g, 1.02 mmol) was converted to 89 using the general procedure for the Mitsunobu reaction. Purification by flash chromatography (60% EtOAc–hexanes) afforded 89 (>85% purity, contaminated with PPh3O) that used in the next step without further purification. The intermediate sulfamide 89 was converted to 90 using the general procedures for biotinylation and TFA deprotection to afford the title compound (438 mg, 52% over 3 steps) as the triethylammonium salt (3.5 equiv of Et3N) as a white solid: HPLC purity: 97.2%, tR = 12.37 min, k′ = 2.3 (method A); (c 1.06, MeOH); 1H NMR (400 MHz, MeOH-d4) δ 1.11 (t, J = 7.3 Hz, 46H, excess Et3N), 1.39–1.47 (m, 2H), 1.53–1.66 (m, 3H), 1.68–1.78 (m, 1H), 2.14 (t, J = 7.4 Hz, 2H), 2.70 (q, J = 7.3 Hz, 32H, excess Et3N), 2.89 (dd, J = 12.9, 5.3 Hz, 1H), 3.06 (t, J = 5.6 Hz, 2H), 3.16–3.22 (m, 1H), 3.66 (t, J = 5.4 Hz, 2H), 4.30 (dd, J = 7.7, 4.4 Hz, 1H), 4.47 (dd, J = 7.7, 4.9 Hz, 1H), 5.64 (s, 2H), 8.23 (s, 1H), 8.27 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 10.9, 25.1, 28.0, 28.2, 36.8, 39.8, 42.3, 45.7, 55.4, 59.2, 61.0, 67.6, 71.9, 118.4, 141.1, 149.7, 152.9, 156.0, 162.7, 175.1; HRMS (ESI−) calcd for C18H26N9O5S2 [M − Et3NH]− 512.1504, found 512.1512 (error 1.6 ppm).
MtBPL cloning, expression and purification
MtBPL was cloned, expressed and purified as previously described.15
Isothermal titration calorimetry
All ITC experiments were conducted on an automated microcalorimeter (Malvern Instruments). The experiments were performed at 25 °C in ITC buffer (10 mM Tris pH 7.5, 200 mM KCl, 2.5 mM MgCl). MtBPL was exchanged (2 × 10 mL) into ITC buffer using an Amicon Ultra concentrator, and the final filtrate was used to prepare a solution of the analogs from a 10 mM stock in DMSO. Protein concentrations were 10 μM for MtBPL (determined by active site titration with 14), 100 μM analog and 100 μM biotin (determined by weighing sample on an ultramicrobalance [Mettler Toledo] accurate to 0.001 mg). In the direct titration experiments, the analog was injected into a solution of the enzyme. In the competitive titration experiments, the analog was injected into a solution of the enzyme and biotin. Titrations were carried out with a stirring speed of 750 rpm and 200 s interval between 4 μL injections. The first injection for each sample was excluded from data fitting. Titrations were run past the point of enzyme saturation to correct for heats of dilution. The experimental data were fitted to a theoretical titration curve using the Origin software package (version 7.0) provided with the instrument to afford values of KAapp (the apparent binding constant of the ligands in the presence of biotin in M−1), n (stoichiometry of binding) and ΔH (the binding enthalpy change in kcal/mol). The KA values for each ligand was obtained from the KAapp value using equation 1:
| (1) |
where [B] is the concentration of biotin, and KAB is the association constant for biotin experimentally determined to be 7.29 × 105. The thermodynamic parameters (ΔG and ΔS) were calculated from K determined from the displacement titration and ΔH from the direct binding titration using equation 2:
| (2) |
where ΔG, ΔH, and ΔS are the changes in free energy, enthalpy, and entropy of binding, respectively; R = 1.98 cal mol−1 K−1; and T is the absolute temperature (298 K). The affinity of the ligands for the protein is given as the dissociation constant (KD = 1/KA). ITC experiments were run in triplicate and analyzed independently, and the thermodynamic values were averaged.
Differential Scanning Fluorimetry
Differential scanning fluorimetry was performed using a Bio-Rad CFX96 according to established protocols.52 In brief, purified MtBPL was diluted to give 40 μL solution consisting of: 17.3 μM (0.5 mg/mL) MtBPL, 43 μM analog (diluted from a 2 mM stock in DMSO), 10 mM Tris pH 8.0, 200 mM KCl, 2.5 mM MgCl, 10% (v/v) glycerol, and 5 × SYPRO Orange (Life Technologies). Each analog was run in triplicate. The fluorescence response (melting curve) was measured across a temperature range between 10 and 85 °C with 1 min incubation per 0.5 °C temperature increase. The melting temperature (Tm) was determined from the peak of the first derivatives of the melting curve; calculations were made using the Bio-Rad CFX Manager software and plots were produced using the program Plot (http://plot.micw.eu/).
M. tuberculosis MIC Assay
All compounds MICs were experimentally determined as previously described.15 Minimum inhibitory concentrations (MICs) were determined in quadruplicate in standard GAST medium according to the broth micro dilution method using compounds from DMSO stock solutions or with control wells treated with an equivalent amount of DMSO. Plates were incubated at 37 °C (100 μL/well) and growth was recorded by measurement of optical density at 580 nm. All measurements reported herein used an initial cell density of 105 cells/assay and growth monitored at 10–14 days, with the untreated and DMSO-treated control cultures reaching an OD620 ~0.8–1.0.
Co-Crystallization of Ligands with MtBPL
MtBPL at 1.5 mg/ml (10 mM HEPES pH 7.5, 50 mM NaCl, 1 mM DTT) was combined with inhibitors (200 μM in DMSO) at 1:30 molar ratio, incubated at 37°C for 30 min, and then concentrated using Amicon centrifugal filters (EMD Millipore) to 10 mg/ml. The concentrated protein-compound solution was utilized to obtain crystals via vapor-diffusion using apo crystals as micro-seeds in hanging drops as previously described.15 Crystals grew within 1 week. Mother liquor included 17–24% PEG MME 2000, 100 mM Tris pH 8.5 and 100 mM trimethylamine N-oxide. Crystals were frozen in liquid nitrogen after a quick soak in cryo-protectant solution containing mother liquor supplemented with 20% PEG 400.
Crystallographic Data collection and structure solution
All diffraction data was collected at synchrotron X-ray sources under cryogenic conditions of 100 K (−173 °C). Data for the complex with compound 82 was collected at beamline 23-ID-B (GM-CA CAT) of the Advanced Photon Source (APS; Argonne, IL) equipped with a MAR 300 CCD detector. Data was integrated and scaled with HKL3000 (HKL Research, Inc). Data for the complex with compound 90 were collected at beamline 4.2.2 of the Advanced Light Source (ALS) in Berkeley, CA equipped with a NOIR-1 CCD detector. HKL-2000 was used to index, integrate and scale the data.60 All other data were collected at APS beamline 17-ID (IMCA-CAT) equipped with a Dectris PILATUS 6M detector. The program autoPROC was utilized to index, integrate and scale the data.61 The complex with compound 36 was first to be phased by molecular replacement,62 using the MtBPL complex with 14 (PDB-id 3RUX)15 stripped of inhibitor and solvent as a search model. The refined structure with 36 without inhibitor was subsequently used as the search model in molecular replacement model to phase complexes with the remaining compounds. All structures were subjected to subsequent rounds of model building using COOT63 and refinement using phenix.refine64 to acquire the final refined structures. Topology and parameter files for all ligands were generated using JLigand.65
Cell Cytotoxicity-MTT Assay
Human liver cells (HepG2, ATCC) cells were plated in 96-well plates at (2.5–5.0) × 104 cells per well (200 μL). HepG2 cells were maintained in Eagle's minimum essential medium (EMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. Compounds were prepared as 10 mM stock solutions in DMSO, and 1 μL of the compound stock solution was added to each well in 200 μL of EMEM, yielding a final compound concentration of 100 μM. Control wells contained either 1% DMSO (negative control) or 40 μM phenol (positive control), and all reactions were done in triplicate. The plate was incubated for 48 h at 37 °C in a 5% CO2/95% air humidified atmosphere. Measurement of cell viability was carried out using a modified method of Mosmann based on 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT).66 The MTT solution was prepared fresh at 1 mg/mL in serum-free, phenol red free DMEM. MTT solution (200 μL) was added to each well, and the plate was incubated as described above for 3 h. The MTT solution was removed, and the formazan crystals were solubilized with 200 μL of 2 N KOH. The plate was read on a M5e spectrophotometer (Molecular Devices) at 570 nm for formazan and 650 nm for background subtraction. Cell viability was estimated as the percentage absorbance of sample relative to the DMSO control.
LC-MS/MS Quantitation of Compound Accumulation
Compounds 63 and 69 were evaluated for cellular accumulation using the method reported by Rhee and coworkers.54,55 Briefly, M. tuberculosis H37Rv cultures were grown in GAST medium containing tween 80 for 5 days, then transferred to 7H10 agar plates containing 0, 5, and 25 μM 63 and 69 for 24 hours. Samples were isolated using the method previously reported.54 Samples were then analyzed by LC-MS/MS (Shimadzu UFLC XR-AB SCIEX QTRAP 5500) and quantified by reverse-phase LC on a Kinetix C18 column (50 mm × 2.1 mm, 2.6 μm particle size; Phenomenex, Torrance, CA) at a flow rate of 0.5 mL/min. Initial conditions were 5% acetonitrile containing 0.1% formic acid (solvent B) and 95% water containing 0.1% formic acid (solvent A) from 0 to 0.5 min, after which solvent B was increased to 95% from 0.5 to 3 min. The column was washed in 95% solvent B for 2 min, returned to 5% solvent B over 0.2 min, and allowed to re-equilibrate for 2.8 min in 5% solvent B to provide a total run time of 8 min. The column oven was maintained at 40 °C and the injection volume was 10 μL. All analytes were analyzed by MS in positive ionization mode by Multiple Reaction Monitoring (MRM). To determine the optimum MRM settings, compounds 63 and 69 were infused at a concentration of 10 μM (in 1:1 water/acetonitrile containing 0.1% formic acid) onto the MS by a syringe pump at a flow of 10 μL/min. The MS parameters are listed in table S3.
Analyte and internal standard peak areas were calculated (MultiQuant, version 2.0.2, AB SCIEX). Analyte peak areas were normalized to internal standard peak areas and the analyte concentrations were determined with an appropriate standard curve (Figure S15). The protein concentration of the lysate in each of the samples were calculated by Bradford assay using Bovine Serum Albumin (BSA) as a standard. The average protein concentration in the lysate samples was determined to be 5.74 ± 1.35 μg/mL. The normalized concentration is given as a ratio of analyte concentration over protein concentration and were found to be 0.035 ± 0.014 μg analyte/μg protein for 63 at 25 μM and 0.005 ± 0.003 μg analyte/μg protein for 69 at 25 μM.
Supplementary Material
Acknowledgements
This work was supported by a grant (AI091790 to D.S.) from the National Institutes of Health. Isothermal titration calorimetry was carried out using an ITC-200 microcalorimeter, funded by the NIH Shared Instrumentation Grant S10-OD017982. Mass spectrometry was carried out in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, University of Minnesota, funded in part by Cancer Center Support Grant CA-77598. We also gratefully acknowledge resources from the University of Minnesota Supercomputing Institute. Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute.
Abbreviations
- ACCs
acyl CoA carboxylases
- AMP
adenosine monophosphate
- Bio-AMP
biotinyl-5′-adenosine monophosphate
- Bio-AMS
biotinyl-5′-adenosine monosulfamide
- Bio-NHS
D-(+)-biotin N-hydroxysuccinimide ester
- Boc
tert-butyloxycarbonyl
- BPL
biotin protein ligase
- DIAD
diisopropyl azodicarboxylate
- DMA
dimethylacetamide
- DSF
differential scanning fluorimetry
- HPLC
high performance liquid chromatography
- ITC
isothermal titration calorimetry
- MtBPL
Biotin protein ligase
- MIC
minimum inhibitory concentration
- Mtb
Mycobacterium tuberculosis
- MtBPL
Mycobacterium tuberculosis biotin protein ligase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide TB, tuberculosis
- TBDPS
tert-butyldiphenylsilyl
- TBS
tert-butyl dimethylsilyl
- TFA
trifluoroacetic acid
- MDR-TB
multi drug resistant tuberculosis
- XDR-TB
extensively drug resistant tuberculosis
Footnotes
Supporting Information. Full 1H and 13C NMR spectra of all described compounds, as well as direct and indirect raw ITC data for all final analogs included as Figures S1–14 and Table S1. A summary of observed diffraction and refinement statistics, along with figures depicting ligand omit difference density is included as Table S2.
Author Contributions The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Ancillary Information: Atomic coordinates and diffraction data for MtBPL complexes with compounds 36, 46, 57, 63, 69, 73, 82, 87 and 90 have been deposited with the Protein Data Bank with accession codes 4xtv, 4xtw, 4xtx, 4xty, 4xtz, 4xu0, 4xu1, 4xu2 and 4xu3, respectively.
References
- (1).Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, Garnier T, Gutierrez C, Hewinson G, Kremer K, Parsons LM, Pym AS, Samper S, van Soolingen D, Cole ST. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc. Natl. Acad. Sci. USA. 2002;99:3684–3689. doi: 10.1073/pnas.052548299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Treatment of tuberculosis. Guidelines. 4th ed. World Health Organization; Geneva, Switzerland: 2010. [PubMed] [Google Scholar]
- (3).Stoffels K, Allix-Beguec C, Groenen G, Wanlin M, Berkvens D, Mathys V, Supply P, Fauville-Dufaux M. From multidrug- to extensively drug-resistant tuberculosis: upward trends as seen from a 15-year nationwide study. PLoS One. 2013;8:e63128. doi: 10.1371/journal.pone.0063128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bansal-Mutalik R, Nikaido H. Mycobacterial outer membrane is a lipid bilayer and the inner membrane is unusually rich in diacyl phosphatidylinositol dimannosides. Proc. Natl. Acad. Sci. USA. 2014;111:4958–4963. doi: 10.1073/pnas.1403078111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Brennan PJ, Nikaido H. The envelope of mycobacteria. Annu. Rev. Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- (6).Barry CE, III, Lee RE, Mdluli K, Sampson AE, Schroeder BG, Slayden RA, Yuan Y. Mycolic acids: structure, biosynthesis and physiological functions. Prog. Lipid. Res. 1998;37:143–179. doi: 10.1016/s0163-7827(98)00008-3. [DOI] [PubMed] [Google Scholar]
- (7).Verschoor JA, Baird MS, Grooten J. Towards understanding the functional diversity of cell wall mycolic acids of Mycobacterium tuberculosis. Prog. Lipid Res. 2012;51:325–339. doi: 10.1016/j.plipres.2012.05.002. [DOI] [PubMed] [Google Scholar]
- (8).Onwueme KC, Vos CJ, Zurita J, Ferreras JA, Quadri LE. The dimycocerosate ester polyketide virulence factors of mycobacteria. Prog. Lipid Res. 2005;44:259–302. doi: 10.1016/j.plipres.2005.07.001. [DOI] [PubMed] [Google Scholar]
- (9).Jackson M, Stadthagen G, Gicquel B. Long-chain multiple methyl-branched fatty acid-containing lipids of Mycobacterium tuberculosis: biosynthesis, transport, regulation and biological activities. Tuberculosis (Edinb) 2007;87:78–86. doi: 10.1016/j.tube.2006.05.003. [DOI] [PubMed] [Google Scholar]
- (10).Ratledge C. Iron, mycobacteria and tuberculosis. Tuberculosis (Edinb) 2004;84:110–130. doi: 10.1016/j.tube.2003.08.012. [DOI] [PubMed] [Google Scholar]
- (11).Portevin D, de Sousa-D'Auria C, Montrozier H, Houssin C, Stella A, Laneelle MA, Bardou F, Guilhot C, Daffe M. The acyl-AMP ligase FadD32 and AccD4-containing acyl-CoA carboxylase are required for the synthesis of mycolic acids and essential for mycobacterial growth: identification of the carboxylation product and determination of the acyl-CoA carboxylase components. J. Biol. Chem. 2005;280:8862–8874. doi: 10.1074/jbc.M408578200. [DOI] [PubMed] [Google Scholar]
- (12).Gago G, Kurth D, Diacovich L, Tsai SC, Gramajo H. Biochemical and structural characterization of an essential acyl coenzyme A carboxylase from Mycobacterium tuberculosis. J. Bacteriol. 2006;188:477–486. doi: 10.1128/JB.188.2.477-486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kurth DG, Gago GM, de la Iglesia A, Bazet Lyonnet B, Lin TW, Morbidoni HR, Tsai SC, Gramajo H. ACCase 6 is the essential acetyl-CoA carboxylase involved in fatty acid and mycolic acid biosynthesis in mycobacteria. Microbiology. 2009;155:2664–2675. doi: 10.1099/mic.0.027714-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Purushothaman S, Gupta G, Srivastava R, Ramu VG, Surolia A. Ligand specificity of group I biotin protein ligase of Mycobacterium tuberculosis. PLoS One. 2008;3:e2320. doi: 10.1371/journal.pone.0002320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Duckworth BP, Geders TW, Tiwari D, Boshoff HI, Sibbald PA, Barry CE, III, Schnappinger D, Finzel BC, Aldrich CC. Bisubstrate adenylation inhibitors of biotin protein ligase from Mycobacterium tuberculosis. Chem. Biol. 2011;18:1432–1441. doi: 10.1016/j.chembiol.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Paparella AS, Soares da Costa TP, Yap MY, Tieu W, Wilce MC, Booker GW, Abell AD, Polyak SW. Structure guided design of biotin protein ligase inhibitors for antibiotic discovery. Curr. Top. Med. Chem. 2014;14:4–20. doi: 10.2174/1568026613666131111103149. [DOI] [PubMed] [Google Scholar]
- (17).Shi C, Tiwari D, Wilson DJ, Seiler CL, Schnappinger D, Aldrich CC. Bisubstrate inhibitors of biotin protein ligase in Mycobacterium tuberculosis resistant to cyclonucleoside formation. ACS Med. Chem. Lett. 2013;4:1213–1217. doi: 10.1021/ml400328a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Brown PH, Cronan JE, Grøtli M, Beckett D. The biotin repressor: modulation of allostery by corepressor analogs. J. Mol. Biol. 2004;337:857–869. doi: 10.1016/j.jmb.2004.01.041. [DOI] [PubMed] [Google Scholar]
- (19).Sittiwong W, Cordonier EL, Zempleni J, Dussault PH. β-Keto and β-hydroxyphosphonate analogs of biotin-5′-AMP are inhibitors of holocarboxylase synthetase. Bioorg. Med. Chem. Lett. 2014;24:5568–5571. doi: 10.1016/j.bmcl.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Soares da Costa TP, Tieu W, Yap MY, Pendini NR, Polyak SW, Sejer Pedersen D, Morona R, Turnidge JD, Wallace JC, Wilce MCJ, Booker GW, Abell AD. Selective inhibition of biotin protein ligase from Staphylococcus aureus. J. Biol. Chem. 2012;287:17823–17832. doi: 10.1074/jbc.M112.356576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tieu W, Soares da Costa TP, Yap MY, Keeling KL, Wilce MCJ, Wallace JC, Booker GW, Polyak SW, Abell AD. Optimising in situ click chemistry: the screening and identification of biotin protein ligase inhibitors. Chem. Sci. 2013;4:3533–3537. [Google Scholar]
- (22).Tieu W, Jarrad AM, Paparella AS, Keeling KA, Soares da Costa TP, Wallace JC, Booker GW, Polyak SW, Abell AD. Heterocyclic acyl-phosphate bioisostere-based inhibitors of Staphylococcus aureus biotin protein ligase. Bioorg. Med. Chem. Lett. 2014;24:4689–4693. doi: 10.1016/j.bmcl.2014.08.030. [DOI] [PubMed] [Google Scholar]
- (23).Soares da Costa TP, Tieu W, Yap MY, Zvarec O, Bell JM, Turnidge JD, Wallace JC, Booker GW, Wilce MCJ, Abell AD, Polyak SW. Biotin analogues with antibacterial activity are potent inhibitors of biotin protein ligase. ACS Med. Chem. Lett. 2012;3:509–514. doi: 10.1021/ml300106p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ma Q, Akhter Y, Wilmanns M, Ehebauer MT. Active site conformational changes upon reaction intermediate biotinyl-5'-AMP binding in biotin protein ligase from Mycobacterium tuberculosis. Protein Sci. 2014;23:932–939. doi: 10.1002/pro.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Daluge S, Vince R. Synthesis of carbocyclic aminonucleosides. J. Org. Chem. 1978;43:2311–2320. [Google Scholar]
- (26).Singh R, Vince R. 2-Azabicyclo[2.2.1]hept-5-en-3-one: chemical profile of a versatile synthetic building block and its impact on the development of therapeutics. Chem. Rev. 2012;112:4642–4686. doi: 10.1021/cr2004822. [DOI] [PubMed] [Google Scholar]
- (27).Jung ME, Rhee H. π-Allylpalladium formation from allylic amines via N,N-Ditosylimides and N-Tosylamides: Efficient synthesis of the antiviral agent carbovir. J. Org. Chem. 1994;59:4719–4720. [Google Scholar]
- (28).Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE, III, Bennett EM, Aldrich CC. Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: Structure–activity relationships of the nucleobase domain of 5′-O-[N-(Salicyl)sulfamoyl]adenosine. J. Med. Chem. 2008;51:5349–5370. doi: 10.1021/jm800567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yang M, Ye W, Schneller SW. Preparation of carbocyclic S-adenosylazamethionine accompanied by a practical synthesis of (−)-aristeromycin. J. Org. Chem. 2004;69:3993–3996. doi: 10.1021/jo040119g. [DOI] [PubMed] [Google Scholar]
- (30).Somu RV, Wilson DJ, Bennett EM, Boshoff HI, Celia L, Beck BJ, Barry CE, III, Aldrich CC. Antitubercular nucleosides that inhibit siderophore biosynthesis: SAR of the glycosyl domain. J. Med. Chem. 2006;49:7623–7635. doi: 10.1021/jm061068d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ainai T, Wang YG, Tokoro Y, Kobayashi Y. Highly stereoselective synthesis of aristeromycin through dihydroxylation of 4-aryl-1-azido-2-cyclopentenes. J. Org. Chem. 2004;69:655–659. doi: 10.1021/jo034672u. [DOI] [PubMed] [Google Scholar]
- (32).Markiewicz WT, Wiewiórowski M. A new type of silyl protecting groups in nucleoside chemistry. Nucleic Acids Res. 1978;1:s185–s190. [Google Scholar]
- (33).O'Mahony G, Svensson S, Sundgren A, Grøtli M. Synthesis of 2-([1,2,3]triazol-1-yl)-2-deoxyadenosine. Nucleosides, Nucleotides Nucleic Acids. 2008;27:449–459. doi: 10.1080/15257770802086880. [DOI] [PubMed] [Google Scholar]
- (34).Ikeuchi H, Meyer ME, Ding Y, Hiratake J, Richards NG. A critical electrostatic interaction mediates inhibitor recognition by human asparagine synthetase. Bioorg. Med. Chem. 2009;17:6641–6650. doi: 10.1016/j.bmc.2009.07.071. [DOI] [PubMed] [Google Scholar]
- (35).Zhu XF, Williams HJ, Scott AI. Aqueous trifluoroacetic acid—an efficient reagent for exclusively cleaving the 5′ end of 3′, 5′ -TIPDS protected ribonucleosides. Tetrahedron Lett. 2000;41:9541–9545. [Google Scholar]
- (36).Dowden J, Brown RS, Moreau C. Chemical synthesis of the novel Ca2+ messenger NAADP. Nucleosides, Nucleotides Nucleic Acids. 2005;24:513–518. doi: 10.1081/ncn-200061786. [DOI] [PubMed] [Google Scholar]
- (37).Kim SE, Kim SY, Kim S, Kang T, Lee J. Deoxyribosyl analogues of methionyl and isoleucyl sulfamate adenylates as inhibitors of methionyl-tRNA and isoleucyl-tRNA synthetases. Bioorg. Med. Chem. Lett. 2005;15:3389–3393. doi: 10.1016/j.bmcl.2005.05.035. [DOI] [PubMed] [Google Scholar]
- (38).Staudinger H, Meyer J. Über neue organische Phosphorverbindungen III. Phosphinmethylenderivate und Phosphinimine. Helv. Chim. Acta. 1919;2:635–646. [Google Scholar]
- (39).Gololobov YG, Zhmurova IN, Kasukhin LF. Sixty years of Staudinger reaction. Tetrahedron. 1981;37:437–472. [Google Scholar]
- (40).Okada M, Iwashita S, Koizumi N. Efficient general method for sulfamoylation of a hydroxyl group. Tetrahedron Lett. 2000;41:7047–7051. [Google Scholar]
- (41).Pankiewicz KW. Fluorinated nucleosides. Carbohydr. Res. 2000;327:87–105. doi: 10.1016/s0008-6215(00)00089-6. [DOI] [PubMed] [Google Scholar]
- (42).Qiu XL, Xu XH, Qing FL. Recent advances in the synthesis of fluorinated nucleosides. Tetrahedron. 2010;66:789–843. [Google Scholar]
- (43).Cappellacci L, Franchetti P, Vita P, Petrelli R, Lavecchia A, Costa B, Spinetti F, Martini C, Klotz KN, Grifantini M. 50-Carbamoyl derivatives of 20-C-methyl-purine nucleosides as selective A1 adenosine receptor agonists: Affinity, efficacy, and selectivity for A1 receptor from different species. Bioorg. Med. Chem. 2008;16:336–353. doi: 10.1016/j.bmc.2007.09.035. [DOI] [PubMed] [Google Scholar]
- (44).Pattanayak S, Paul S, Nandi B, Sinha S. Improved protocol for the synthesis of flexibly protected morpholino monomers from unprotected ribonucleosides. Nucleosides, Nucleotides Nucleic Acids. 2012;31:763–782. doi: 10.1080/15257770.2012.724491. [DOI] [PubMed] [Google Scholar]
- (45).Bell NM, Wong R, Micklefield J. A non-enzymatic, DNA template-directed morpholino primer extension approach. Chem. Eur. J. 2010;16:2026–2030. doi: 10.1002/chem.200902237. [DOI] [PubMed] [Google Scholar]
- (46).Abramova TV, Kassakin MF, Lomzov AA, Pyshnyi DV, Silnikov VN. New oligonucleotide analogues based on morpholine subunits joined by oxalyl diamide tether. Bioorg. Chem. 2007;35:258–275. doi: 10.1016/j.bioorg.2006.12.003. [DOI] [PubMed] [Google Scholar]
- (47).Angyal SJ. Conformational analysis in carbohydrate chemistry I. Conformational free fnergies. The conformations and α : β ratios of aldopyranoses in aqueous solutions. Aust. J. Chem. 1968;21:2737–2746. [Google Scholar]
- (48).Schaeffer HJ, Gurwara S, Vince R, Bittner S. Novel substrate of adenosine deaminase. J. Med. Chem. 1971;14:367–369. doi: 10.1021/jm00286a024. [DOI] [PubMed] [Google Scholar]
- (49).Christensen JJ, Johnston HD, Izatt RM. An Isothermal titration calorimeter. Rev. Sci. Instrum. 1968;39:1356–1359. [Google Scholar]
- (50).Schön A, Madani N, Smith AB, Lalonde JM, Freire E. Some binding related drug properties are dependent on thermodynamic signature. Chem. Biol. Drug Des. 2011;77:161–165. doi: 10.1111/j.1747-0285.2010.01075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Velazquez-Campoy A, Freire E. Isothermal titration calorimetry to determine association constants for high affinity ligands. Nat. Protoc. 2006;1:186–191. doi: 10.1038/nprot.2006.28. [DOI] [PubMed] [Google Scholar]
- (52).Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- (53).Summerton J, Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997;7:187–195. doi: 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]
- (54).de Carvalho LP, Fischer SM, Marrero J, Nathan C, Ehrt S, Rhee KY. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem. Biol. 2010;17:1122–1131. doi: 10.1016/j.chembiol.2010.08.009. [DOI] [PubMed] [Google Scholar]
- (55).Bryk R, Arango N, Maksymiuk C, Balakrishnan A, Wu YT, Wong CH, Masquelin T, Hipskind P, Lima CD, Nathan C. Lipoamide channel binding sulfonamides selectively inhibit mycobacterial lipoamide dehydrogenase. Biochemistry. 2013;52:9375–9384. doi: 10.1021/bi401077f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Kurokawa T, Kim M, Du Bois J. Synthesis of 1,3-diamines through rhodium-catalyzed C-H insertion. Angew. Chem. Int. Ed. 2009;48:2777–2779. doi: 10.1002/anie.200806192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Heacock D, Forsyth CJ, Shiba K, Musier-Forsyth K. Synthesis and aminoacyl-tRNA synthetase inhibitory activity of prolyl adenylate analogs. Bioorg. Chem. 1996;24:273–289. [Google Scholar]
- (58).Susumu K, Uyeda HT, Medintz IL, Pons T, Delehanty JB, Mattoussi H. Enhancing the stability and biological functionalities of quantum dots via compact multifunctional ligands. J. Am. Chem. Soc. 2007;129:13987–13996. doi: 10.1021/ja0749744. [DOI] [PubMed] [Google Scholar]
- (59).Franchetti P, Cappellacci L, Marchetti S, Trincavelli L, Martini C, Mazzoni MR, Lucacchini A, Grifantini M. 2'-C-Methyl analogues of selective adenosine receptor agonists: synthesis and binding studies. J. Med. Chem. 1998;41:1708–1715. doi: 10.1021/jm9707737. [DOI] [PubMed] [Google Scholar]
- (60).Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Vol. 276. Academic Press; New York: 1997. [DOI] [PubMed] [Google Scholar]
- (61).Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T, Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011;67:293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).McCoy AJ, Grosse Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (64).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Lebedev AA, Young P, Isupov MN, Moroz OV, Vagin AA, Murshudov GN. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012;68:431–440. doi: 10.1107/S090744491200251X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.