

Abstract
Amyloid β peptide (Aβ) is causatively associated with Alzheimer’s disease (AD), and N-terminally truncated and pyroglutamylated Aβ peptides (AβpE) exert hypertoxic effect by an unknown mechanism. Recent evidence has identified the prefibrillar oligomers of Aβ, not the fibrils, as the prevalent cytotoxic species. Structural characterization of Aβ and AβpE oligomers is therefore important for better understanding of their toxic effect. Here we have used isotope-edited Fourier transform infrared (FTIR) spectroscopy to identify the conformational changes in Aβ1-42 and AβpE3-42 upon aggregation, individually and in 1:1 molar combination. During the first two hours of exposure to aqueous buffer, the peptides undergo transition from mostly α-helical to mostly β-sheet structure. Data on peptides 13C,15N-labeled at K16L17V18 or V36G37G38V39 allowed construction of structural models for the monomer and early oligomers. The peptide monomer comprises a β-hairpin that involves residues upstream of the K16L17V18 sequence and an N-terminal α-helix. The oligomers form by non-H-bonding interactions between the β-strands of neighboring β-hairpins, in lateral or staggered manner, with the strands running parallel or antiparallel. Relative α-helical and β-sheet propensities of Aβ1-42 and AβpE3-42 depend on the ionic strength of the buffer, emphasizing the importance of ionic interactions in Aβ peptide structure and aggregation. It is inferred that N-terminal modification of AβpE3-42 affects the helix stability and thereby modulates β-sheet oligomer formation. The data thus provide new insight into the molecular mechanism of Aβ oligomerization by emphasizing the role of the N-terminal transient α-helical structure and by identifying structural constraints for molecular organization of the oligomers.
1. Introduction
Extracellular fibrillar deposits of Aβ peptide constitute a typical histopathological feature of AD.1 Earlier studies suggested a causative role of the Aβ plaques in the disease.2-4 However, recent evidence has identified the prefibrillar soluble oligomers of Aβ as the main neurotoxic entities.5-13
Although the mechanism underlying Aβ toxicity has not been determined, studies have led to various plausible pathways. The channel hypothesis suggests that Aβ forms pores and/or ion channels in the neuronal plasma membrane resulting in dysregulation of cellular homeostasis and cell death.14-16 The endoplasmic reticulum (ER) pathway suggests that binding of Aβ to ER causes Ca2+ release ensued by abnormal activation of certain cytosolic enzymes and apoptotic cell death.7,17-19 Altered mitochondrial dynamics is an early event in AD,20 and is paralleled with recruitment of Aβ to mitochondria and subsequent apoptosis.18,21-24 Finally, Aβ is known to bind to a range of receptors, entailing aberrant cell signaling events and leading to either necrosis or apoptosis.10,25,26
Aβ is derived from the amyloid precursor protein (APP), a bitopic protein in neuronal membranes, by proteolytic cleavage at the transmembrane region and the juxtamembrane extracellular region. The most prevalent forms are the 40- and 42-residue peptides. The fraction of Aβ1-42 (DAEFRHDSGY10EVHHQKLVFF20AEDVGSNKGA30IIGLMVGGVV40IA) increases during AD,27 suggesting its involvement in AD pathogenesis. Further processing of the peptide by amino- or carboxy-peptidases and enzymatic modifications result in an array of shorter peptides.1,27,28 Among them, N-terminally truncated and pyroglutamylated (at Glu3 or Glu11) Aβ peptides (AβpE) have attracted much attention as they have been found in AD brains at significant quantities, reaching 50% of total Aβ peptide.29-33 AβpE peptides have been shown to aggregate at increased rates34-38 and to be hypertoxic as compared to the unmodified peptide.29,34,39,40 Moreover, the presence of AβpE at low molar fractions (5% and below) substantially augmented the neurotoxicity of Aβ peptide.29
Solution NMR studies on peptides corresponding to the APP region that comprised the whole or part of Aβ1-42 sequence, reconstituted in detergent micelles, revealed a disordered N-terminus and a flexible α-helix starting at K16L17V18, with a bend involving the V36G37G38V39 segment.41,42 These data implied that the Aβ sequence was mostly α-helical within the context of APP, before cleavage and release into the aqueous medium. On the other hand, the extracellular amyloid plaques are composed of fibrils of parallel, in-register cross β-sheet structure, where the β-strands are perpendicular and the intermolecular H-bonding is parallel to the fibrillar axis.43-49 Obviously, the fibrillogenesis process involves a dramatic α-helix to β-sheet structural transition. Given that the prefibrillar oligomers exert the main cytotoxic effect,5-13 analysis of the structural transitions during fibrillogenesis and characterization of the intermediate Aβ species are of paramount importance in understanding the structural basis of Aβ toxicity.
Solution and solid-state NMR studies have been conducted to determine the structure of Aβ1-40 and Aβ1-42 oligomers, where the oligomeric state has been stabilized by lyophilization,11,49,50 sodium dodecyl sulfate,11,51 co-solvents such as 10% hexafluoroisopropanol (HFIP),52 or antibodies.53 Despite the considerable differences in the structural details, the prefibrillar oligomers of Aβ1-42 were found to form loosely packed antiparallel β-structures or mixed parallel/antiparallel β-sheets, as opposed to tightly packed parallel in-register β-sheets of the fibrils. Secondary structural differences between Aβ1-42 fibrils and oligomers have also been detected by other spectroscopic methods. Teplow and co-workers prepared low molecular weight Aβ1-40 samples and used circular dichroism (CD) to monitor structural transitions upon aggregation.54,55 The sum of α-helix + β-sheet fractions increased from monomers to tetramers and correlated with cytotoxicity, while the fibrils had zero α-helical content and showed low cytotoxicity.55 Molecular dynamics (MD) studies on α-helix to β-sheet transition upon exposure of Aβ1-42 or Aβ1-40 to aqueous media predicted mixed α-helix/β-sheet intermediates as well.56-58 Consistent with this, combined CD, solution NMR, and MD studies unveiled α-helix to β-sheet transition of Aβ1-42 with increasing fraction of water in HFIP/H2O.59 Ono et al.55 and Abedini and Raleigh60 considered a mechanism where the initial aggregation occurs between the transient α-helical structures, which then convert to β-sheets. FTIR amide I bands of Aβ1-42 oligomers showed a peak around 1645 cm−1, suggesting irregular structure, which shifted to the β-sheet region of 1630 cm−1 upon induction of fibrillogenesis by heating.49 Another FTIR analysis of Aβ1-42 indicated formation of antiparallel β-sheets by the oligomers and parallel β-sheets by the fibrils.61 Thus, the intermediate, prefibrillar Aβ species assume complex structures, involving α-helix, β-sheet, and irregular structures, which evidently contribute to Aβ toxicity.
Structural data on AβpE are scarce. Studies using CD, electron microscopy, and proteolysis-resistance showed that AβpE formed intermolecular β-sheets and fibrils that were shorter and thicker compared to fibrils of unmodified Aβ.34-37 Solution NMR showed that AβpE3-40 in trifluoroethanol/water (2:3) had a reduced α-helical propensity compared to Aβ1-40.62 This echoes with the findings of higher β-sheet propensity and faster aggregation of AβpE.35,36 However, other studies opposed this notion by reporting that AβpE had similar content of β-sheet structure and aggregated slower than unmodified Aβ.63,64 The structural distinctions between Aβ and AβpE, as well as the structural basis for hypertoxicity of AβpE remain poorly characterized.
Structural analysis of the prefibrillar oligomers of Aβ involved lyophilization, detergents or organic solvents (see above), all of which likely affect the peptide’s structure. On the other hand, few studies have been conducted on AβpE, alone or in combination with the unmodified peptide. Our objective is to analyze the conformational transitions in Aβ and AβpE individually and in combination during aggregation in the absence of biologically impertinent components, and to help structurally characterize the toxic intermediates. Our earlier work indicated that, contrary to the doctrine that AβpE undergoes faster fibrillogenesis and β-sheet formation and thereby elicits a hypertoxic effect, AβpE is actually able to not only delay the aggregation of Aβ but also reverse the cross β-sheet structure of Aβ to α-helix.65 Here we present a biophysical analysis of the structural transitions in Aβ1-42 and AβpE3-42 upon aggregation, individually and in 1:1 molar combination. We have studied unlabeled peptides, as well as peptides 13C,15N-labeled at regions K16L17V18 or V36G37G38V39. The peptides dried from HFIP assume α-helical structure, and transfer to aqueous buffers initiates transition to more complex conformations, including β-sheet, β-turn, α-helix, and irregular structures. Analysis of 12C=O-13C=O and 13C=O-13C=O vibrational couplings provides structural constraints that lead to molecular models of peptide monomers and oligomers.
2. Experimental
2.1. Materials
Unlabeled Aβ1-42 and AβpE3-42 peptides were purchased from rPeptide (Bogart, GA, USA) and Innovagen (Lund, Sweden), respectively, and were > 97% pure. Uniformly 13C,15N-labeled amino acids were purchased from Cambridge Isotope Laboratories (Tewksbury, MA, USA) and sent to EZBiolab (Carmel, IN, USA), where the segmentally labeled peptides were synthesized. The final purity of these peptides was > 95%. Most chemicals including HFIP, salts, buffers were purchased from Fisher Scientific (Hanover Park, IL, USA) and Sigma-Aldrich (St. Louis, MO, USA).
2.2. Experimental procedures
The peptides were obtained from the vendors as lyophilized powder and stored at −20°C in tightly sealed vials. Samples for CD or FTIR experiments were prepared by first dissolving the peptides in HFIP at 50 μM concentration to disperse pre-existing aggregates, as described earlier.66 In CD experiments, the peptide solution was contained in a 4 mm × 4 mm rectangular quartz cuvette and spectra were recorded on a J-810 spectropolarimeter (Jasco, Tokyo, Japan) at 25 °C. Then the solvent was removed under a gentle stream of nitrogen gas followed by desiccation for 30 min, and spectra of dry peptide were measured. Aqueous buffer was then added, accompanied with stirring with a magnetic stir bar, and CD spectra were measured consecutively for two hours to detect structural changes during aggregation. Samples of equimolar combination of Aβ1-42 and AβpE3-42 contained 25 μM of each peptide. Spectra were measured for blank buffers and subtracted from the sample spectra.
In FTIR experiments, 120 μL of 50 μM peptide solution in HFIP were placed on a circular CaF2 FTIR window (32 mm in diameter, 3 mm thick) and dried as described above. The window was mounted on a sample holder and spectra were measured on a Vector-22 FTIR spectrometer (Bruker Optics, Billerica, MA, USA) equipped with a liquid nitrogen-cooled Hg-Cd-Te detector at 2 cm−1 nominal resolution at 25 °C, as described.65 Following measurements of the spectra of dry peptides, the CaF2 window was dismounted, placed horizontally, and 120 μL of D2O-based buffer were added to the dry peptide. A 50 μm Teflon spacer and a second CaF2 window were placed on top of the sample and tightly sealed. Consecutive FTIR spectra in the 4000 cm−1−400 cm−1 region were measured to observe conformational changes in the peptides upon exposure to aqueous buffer.
Two buffers of different ionic compositions were used: 50 mM Na,K-phosphate (pD 7.2) + 50 mM NaCl, and 10 mM Na,K-phosphate (pD 7.2). Both buffers were prepared using D2O, and pD was adjusted using a regular pH-meter, taking into account a 0.4 pH unit isotopic shift.67 Reference spectra were measured using a single CaF2 window for the dry samples and the respective blank buffer between two windows for samples in aqueous buffer. The instrument was purged with dry air throughout the measurements. Absorbance spectra of atmospheric water vapor were measured separately and used to correct the sample spectra when necessary. The final absorbance spectra of the peptide samples were smoothed using a 13-point Savitzky-Golay linear least squares algorithm embedded in the Igor Pro 5.03 software, and baseline corrected. In order to compare spectral line-shapes and calculate spectral differences, all spectra were normalized by dividing each spectrum by its amide I area, using Igor Pro 5.03.
Curve-fitting was performed using GRAMS spectroscopic software to estimate the fractions of various secondary structures in the peptides. The locations of the amide I components were determined based on the second derivatives. The result of the curve-fitting procedure was considered satisfactory when the peak wavenumbers of the components agreed with those predicted by the second derivatives, the “curvefit,” i.e. the sum of all components, reasonably fitted the actual spectrum , and the widths of the components were within meaningful limits.
3. Results and discussion
FTIR and CD have been used to assess the structural transitions in the unmodified and pyroglutamylated Aβ peptides during aggregation in aqueous buffers. The peptides were dissolved in HFIP to disperse pre-existing aggregates66 and dried by desiccation. The secondary structure of the dehydrated peptides was determined, followed by addition of aqueous buffers of various ionic strengths and monitoring conformational transitions upon aggregation. Since Aβ is derived from APP, involving part of its transmembrane domain, the dehydrated peptide may mimic the peptide sequestered from the aqueous phase into the cell plasma membrane. The observed structural changes initiated by addition of the aqueous buffer mimic the conformational transitions of the peptides upon transfer to the cytosol or the extracellular milieu. It has been demonstrated that Aβ1-42 or AβpE3-42 form oligomers upon incubation in aqueous media at concentrations 10-100 μM and temperatures 25-37°C for 2 hours or less, and the onset of fibrillogenesis occurs beyond the first two hours.66,68-70 Hence, we have employed a biophysical approach to characterize the structural transitions from the α-helical monomeric structure to the initial oligomers of mostly β-sheet structure within this time frame.
3.1. Dehydrated peptides adopt α-helical conformation
3.1.1. Unlabeled peptides
To monitor the structural changes in the peptides during aggregation in an aqueous medium, their secondary structure was determined in HFIP, after solvent removal by desiccation, and during the first 2 hours of exposure to an aqueous buffer. The CD spectra of Aβ1-42, AβpE3-42, and their equimolar combination dissolved in HFIP all showed a minimum around 203 nm and a prominent shoulder around 220 nm (Figure 1a), indicative of irregular and α-helical conformations, respectively.71-73 This is consistent with earlier CD and NMR data indicating predominantly α-helical structure for Aβ1-42 in pure HFIP66 or HFIP/H2O media at high HFIP content59 and for AβpE3-40 in 40% trifluoroethanol/60% water.62 Upon removal of the solvent, the dry peptides assume mostly α-helical structure with CD minima at 208-209 nm and 221-224 nm (Figure 1b-d). These data imply an intrinsic α-helical propensity of both peptides in a desolvated state. Addition of aqueous buffer to the dry peptides induces α-helix to β-sheet structural transition. The spectra of Aβ1-42 acquire a well-defined β-sheet line-shape with a minimum at 215 nm upon exposure to water for 10 min, which undergoes little change during incubation for 2 hours (Figure 1b). The behavior of AβpE3-42 is different; at 10 min of incubation in the aqueous buffer it retains significant α-helical structure, as evidenced by double minima around 206 and 221 nm, followed by gradual transition to β-sheet upon further exposure to water (Figure 1c). The equal molar combination of the two peptides displays a structural behavior similar to that of AβpE3-42, i.e., a slower conversion to β-sheet and longer retention of the intermediate α-helical structure (cf. gray spectra in Figure 1c and 1d). (CD spectra have been measured every 10 min; the intermediate spectra are omitted for clarity).
Figure 1.

(a) CD spectra of 50 μM peptide solutions in HFIP. Black solid, dotted, and gray solid lines correspond to Aβ1-42, AβpE3-42, and their 1:1 combination, respectively. Panels b), c), and d) present spectra of Aβ1-42, AβpE3-42, and their 1:1 combination, respectively, in dry state (black solid lines), 10 min in aqueous buffer (50 mM NaCl, 50 mM Na,K-phosphate, pH 7.2) (gray solid lines), and 2 h in buffer (dotted lines).
FTIR spectroscopy was used to further analyze the structural transitions in the peptides. The peptides dried from HFIP on an FTIR CaF2 window demonstrate mostly α-helical structure with prominent peaks at 1658-1662 cm−1 (Figure 2). These peak wavenumbers are close to those measured for other α-helical peptides corresponding to phospholamban74,75 or the transmembrane domain of influenza hemagglutinin.76 Fractions of α-helix in the peptide samples were determined by curve-fitting and ranged from 57% to 86% (not shown). Other amide I components were assigned to turns or irregular structures.
Figure 2.
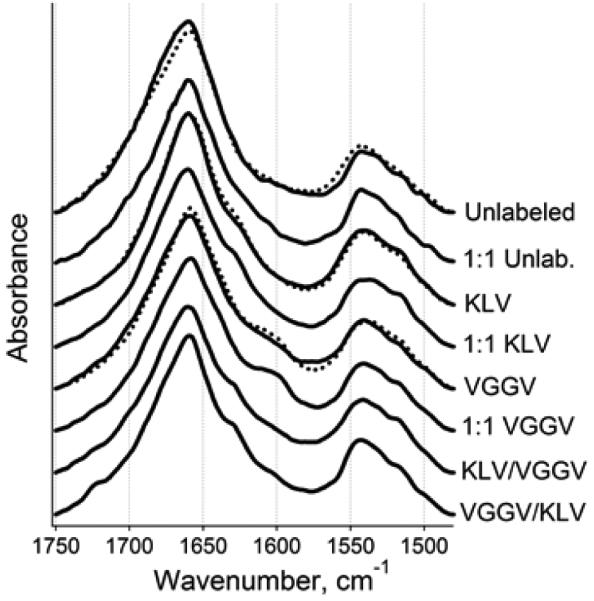
FTIR spectra of dry peptides in amide I and amide II regions. Spectra for unlabeled and isotopically labeled Aβ1-42 and AβpE3-42 peptides and their 1:1 combinations are presented. Dotted lines correspond to AβpE3-42, and solid lines correspond either to Aβ1-42 or to combined samples, as indicated. KLV or VGGV imply the peptides have been labeled at K16L17V18 or V36G37G38V39, respectively. In KLV/VGGV or VGGV/KLV samples, the first stretch applies to Aβ1-42 and the second to AβpE3-42. All spectra are normalized to a total area of 1.0.
3.1.2. Isotopically labeled peptides
In addition to the unlabeled peptides, four segmentally 13C,15N-labeled peptides were studied. Each of the Aβ1-42 and AβpE3-42 peptides was labeled either at K16L17V18 or V36G37G38V39 stretches. Site-directed labeling of proteins or peptides with stable isotopes such as 13C or both 13C and 15N generates a characteristic, downshifted FTIR amide I signal and thereby allows one to probe the local secondary structure.67,77-80 Moreover, vibrational couplings between amino acid residues of similar or different isotopic contents result in distinct spectral features which provide additional structural details.80-83 Spectra were analyzed using a resolution enhancement procedure, i.e., 2nd derivatives, where the spectral components appear as downward peaks. Despite the inherent noise in the 2nd derivative spectra, they show well defined features like the major α-helical component at 1658-1662 cm−1 (Figure S1 in Electronic Supplementary Information). The spectra of the samples containing peptides 13C,15N-labeled at K16L17V18 consistently display an additional component located at 1624-1628 cm−1. This ~32 cm−1 downshift is less than expected based on an isolated harmonic oscillator model but is consistent with FTIR data on α-helical synthetic peptides 13C-labeled at 1 to 4 residues.76,77,79 The higher-than-expect vibrational frequency of the labeled residues most likely results from through H-bonding and/or through space vibrational coupling with the unlabeled residues. (In an α-helix stabilized by i+4 → i H-bonding, three consecutively labeled residues would be H-bonded to unlabeled residues.) The peptides labeled at V36G37G38V39, on the other hand, show weaker spectral features that are shifted further down to 1596-1602 cm−1 (Figure S1). Diminished intensity and frequency of these signals suggest that the C-terminus of the peptides is disordered and the respective peptide units behave like isolated oscillators. Earlier data indicating significant weakening of the relative intensity of the amide I component generated by 13C-labeled α-helical peptide residues upon thermal melting78-81 provide support for this conjecture. The amide II region shows a major band around 1546 cm−1 and a weaker component at lower wavenumbers around 1516 cm−1 for the labeled peptides (Figures 2 and S1). This splitting evidently results from both 13C- and 15N-labeling, but these signals are not diagnostic in terms of secondary structural characterization.
Thus, the peptides in dehydrated state adopt mostly α-helical structure that involves the K16L17V18 sequence. The small fraction of unordered structure involves the V36G37G38V39 segment close to the C-terminus, and possibly a stretch downstream to K16L17V18. This is in partial agreement with NMR data on the APP fragments comprising the Aβ1-42 sequence,41,42 which identified a bend near residues G37G38, and agrees with MD data on the initial α-helical structure of Aβ,56-58 implying that the dry peptide is a biologically meaningful starting material to monitor structural changes upon aggregation in an aqueous medium. The relatively high amide I peak wavenumbers of the peptides (~1660 cm−1) as compared with those of stable α-helices (1647-1657 cm−1)67,84 suggest that the helices formed by the dry peptides are flexible. (Weaker helical H-bonding corresponds to stronger C=O covalent bonding and consequently to higher vibrational frequencies.) This is also in line with the flexible nature of the helix in APP sequence corresponding to Aβ.41,42
3.2. Conformational changes in aqueous buffer
3.2.1. Unlabeled peptides
Transfer into aqueous buffer results in substantial conformational changes in Aβ1-42 and AβpE3-42 peptides and their equimolar combinations. The prominent amide I α-helical feature around 1660 cm−1 is replaced with two major components located near 1630 cm−1 and 1673 cm−1 (Figure 3), which are readily assigned to β-sheet and type I β-turn structures.67,84 The spectra also display residual α-helical features around 1660 cm−1, indicating that during the first two hours of aggregation the peptides are mostly in β-sheet conformation still retaining fractions of α-helix. Distinction between parallel versus antiparallel or in-register versus out-of-register β-sheets cannot be made based solely on the β-sheet wavenumber. On the other hand, the weak, higher frequency (1680-1695 cm−1) component resulting from amide I splitting in antiparallel β-sheets is not resolved well enough apparently because of its diminutive extinction coefficient.84,85 The nature of the β-sheets will be addressed below, based on the data on isotopically labeled peptides. Meanwhile, analysis of spectra of the unlabeled peptides provides the following structural information. First, the β-sheet wavenumbers decrease by 2-5 cm−1 during 2-hour incubation in a D2O-based buffer, owing to amide H/D exchange, but still remain in the 1633-1628 cm−1 range. This strongly suggests that, within this time frame, the peptides form intramolecular rather than intermolecular β-sheets because the latter structures generate lower frequency signals in the 1613-1625 cm−1 range (see Ref. 67 and references therein). Inspection of amide II region of the spectra showed that the amide H/D exchange was close to completion at 2 h incubation in D2O-based buffers (Figure S2), implying no further spectral shifts would be expected upon prolonged exposure to D2O. Second, the peptides adopt a tertiary structure that is not packed too tightly to prevent solvent access to most of the residues. Third, the prominent β-turn components and significant α-helical fractions imply that the peptides fold into a mixed α/β-type structure such as those predicted by MD simulations and CD data for helix-to-fiber intermediates of Aβ.57,58,86 Fourth, systematic spectral differences between Aβ1-42, AβpE3-42 and their combination provide insight into individual structural propensities of the two peptides and mutual structural effects. Finally, data obtained on peptides in buffers of different ionic strengths allow assessment of the role of ionic interactions in the observed structural transitions (see below).
Figure 3.

FTIR spectra of unlabeled Aβ1-42 (black solid line), AβpE3-42 (dotted line), and their 1:1 combination (gray solid line) in D2O-based buffers of 50 mM NaCl, 50 mM Na,K-phosphate, pD 7.2 (a,b,c) and 10 mM Na,K-phosphate (pD 7.2) (d,e,f) for 10 min (a,d), 1 h (b,e), and 2 h (c,f). All spectra are normalized to a total area of 1.0.
In a buffer of near-physiological ionic strength, distinct spectral differences are seen between AβpE3-42 and Aβ1-42 peptides (Figure 3 a,b,c). The AβpE3-42 peptide absorbs stronger in the α-helical region and weaker in the β-sheet region compared to Aβ1-42. The equimolar combination of the two peptides shows even stronger α-helix feature than any of the two peptides, but also has more β-turn and less β-sheet structure. The (Aβ - AβpE) difference spectra presented in Figure S3a show a negative peak in the α-helical region (~1660 cm−1) and a positive one in the β-sheet region (~1620 cm−1), clearly indicating a higher α-helical and lower β-sheet structure in AβpE3-42 as compared to Aβ1-42. The difference between the spectrum of the 1:1 molar combination of AβpE3-42 and Aβ1-42 and the normalized spectral sum of the two peptides, [1:1 – ½(Aβ + AβpE)], displays well-defined positive components near 1672 cm−1 and 1653 cm−1 and a negative component around 1626 cm−1 (Figure S3b). If both peptides retained their individual structures in the 1:1 combination, then these difference spectra would represent flat lines. The observed spectral features indicate significant conformational changes in one or both peptides caused by intermolecular interactions in the combined sample, i.e. increase in the α-helical and turn structures at the expense of β-sheets. The fact that the 1:1 combination spectrum resembles that of AβpE more than Aβ suggests that the pyroglutamylated peptide possesses a stronger capability of propagating its structural features into the mixed peptide sample. Interestingly, the α-helical component in the combined sample is shifted from ~1660 cm−1 down to ~1653 cm−1, as also seen in the raw spectra of Figure 3 a,b,c. Since spectral subtractions involved spectra exposed to D2O for similar time periods, this shift most likely reflects structural differences, such as helix stabilization in the combined peptide sample, rather than amide deuteration effects. The results of curve-fitting reflect these structural differences in a quantitative manner (Figure S4). These data are tabulated in Table 1 and indicate the presence of 10-20% α-helical structure during the first two hours of aggregation. A higher fraction of α-helix in AβpE3-42 agrees with a slower β-sheet conversion of this peptide. In sum, in a near-physiological ionic strength buffer the AβpE3-42 peptide resists conversion of α-helix to β-sheet more than the unmodified Aβ1-42 peptide. In the 1:1 combination, this α-helical propensity is augmented, corroborating the notion that AβpE3-42 is able to retard amyloid fibrillogenesis by opposing α-helix to β-sheet structural transition.65
Table 1.
Secondary structures of Aβ1-42, AβpE3-42, and their 1:1 combinations incubated in 50 mM NaCl + 50 mM Na,K-phosphate buffer (shaded rows) or 10 mM Na,K-phosphate buffer, pD 7.2, as determined by curve-fitting of FTIR amide I bands (see Figure S3). Average percentages for 10 min, 1 h, and 2 h incubation in aqueous media are presented. “Other” refers mostly to irregular structure.
| Aβ1-42 | AβpE3-42 | Aβ1-42 /AβpE3-42 (1:1) | |
|---|---|---|---|
| α-helix | 11.0±2.4 | 18.7±3.2 | 22.9±3.6 |
| 24.8±5.1 | 13.6±1.8 | 16.3±2.1 | |
| β-sheet | 50.4±4.6 | 43.0±4.4 | 34.5±3.9 |
| 38.7±4.8 | 48.9±3.6 | 34.2±3.2 | |
| β-turn | 18.1±3.3 | 20.3±1.7 | 28.4±2.4 |
| 24.0±2.7 | 19.8±1.3 | 27.7±3.6 | |
| other | 20.5±3.9 | 18.0±2.2 | 14.2±1.3 |
| 12.5±3.6 | 17.7±2.6 | 21.8±2.7 |
The structural behavior of the peptides in a low ionic strength buffer is different. Under these conditions, AβpE3-42 displays higher β-sheet structure and reduced fractions of β-turn, α-helix and irregular components compared to Aβ1-42 (Figure 3 d,e,f and Table 1). The (Aβ - AβpE) difference spectra highlight these structural features by a positive component in the 1675-1646 cm−1 region, corresponding to β-turn, α-helix, and irregular structures, and a negative component around 1620-1626 cm−1, corresponding to β-sheet (Figure S3c). These data suggest that a low ionic strength buffer promotes β-sheet formation in the pyroglutamylated peptide faster than in the unmodified peptide, in contrast to the trend in a near-physiological buffer. This result is further supported by the fact that in low ionic strength buffer the β-sheet component of AβpE3-42 is shifted to lower wavenumbers compared to Aβ1-42 (Figure 3 d,e,f), while the opposite is true in the higher ionic strength buffer (Figure 3 a,b,c). Moreover, the spectra of the 1:1 molar combination are similar to those of Aβ1-42 (Figure 3 d,e,f), indicating a dominant structural effect of Aβ1-42 in a low salt buffer. The [1:1 – ½(Aβ + AβpE)] spectra show positive peaks around 1672 cm−1 and 1645 cm−1 and a negative well around 1624 cm−1 (Figure S3d), i.e., the overall fractions of β-turn and unordered structure are increased and the fraction of β-sheet is reduced in the combined peptide sample, a structural feature of Aβ1-42 under these conditions.
3.2.2. Isotopically labeled peptides
Analysis of proteins or peptides by isotope-edited FTIR spectroscopy provides site-specific structural information as well as such details as parallel versus antiparallel or in-register versus out-of-register β-sheets.80 For example, in aligned β-strands, through H-bonding or through space 13C=O-13C=O vibrational coupling between peptide units in adjacent strands results in lower frequency (~1591-1594 cm−1) amide I mode whereas in the absence of in-register alignment 13C-12C couplings generate higher frequency (~1601-1604 cm−1) components of enhanced intensity.47,82,83
The amide I FTIR spectra of the peptides 13C,15N-labeled either at K16L17V18 or V36G37G38V39 display a major peak around 1628-1637 cm−1 and a well-defined component around 1670-1673 cm−1 (Figures 4, 5) that are assigned to β-sheet and β-turn structures, respectively, as in the case of unlabeled peptides. Comparison of these spectra with those of the unlabeled peptides (Figure 3) identifies an additional component near 1598-1604 cm−1 that is evidently generated by the isotope-labeled residues. Spectral features in the 1650-1660 cm−1 region indicate significant fractions of α-helix. The results of curve-fitting, shown in Figure S5, identify β-sheet, β-turn, and α-helical components in Aβ1-42 and AβpE3-42 peptides and their combinations. In certain cases, an additional component at 1642-1647 cm−1 was present, which most likely is generated by irregular (unordered) structure. The secondary structural contents of the isotope-labeled peptide samples agree with those of the unlabeled peptides (Table 1) when the component generated by the isotope-labeled segment around 1600 cm−1 is assigned to β-sheet structure (see below).
Figure 4.

FTIR spectra of Aβ1-42 (black solid line), AβpE3-42 (dotted line), and their 1:1 combination (gray solid line) labeled either at K16L17V18 (a, b, c) or V36G37G38V39 (d, e, f) in D2O-based buffers of 50 mM NaCl, 50 mM Na,K-phosphate, pD 7.2, for 10 min (a,d), 1 h (b,e), and 2 h (c,f). Inset text in each panel intends to facilitate identification of spectra. For example, “AβpEVGGV 1 h” means AβpE3-42 peptide labeled at V36G37G38V39 incubated in buffer for 1 h. All spectra are normalized to a total area of 1.0.
Figure 5.

FTIR spectra of Aβ1-42 (black solid line), AβpE3-42 (dotted line), and their 1:1 combination (gray solid line) labeled either at K16L17V18 (a, b, c) or V36G37G38V39 (d, e, f) in D2O-based buffers of 10 mM Na,K-phosphate, pD 7.2, for 10 min (a,d), 1 h (b,e), and 2 h (c,f). Inset text is explained under Figure 4. All spectra are normalized to a total area of 1.0.
In the buffer of near-physiological ionic strength, the β-sheet wavenumber of Aβ1-42 is 4-5 cm−1 lower than that of AβpE3-42 at similar times of exposure to the buffer, suggesting faster β-sheet formation in the unmodified peptide. With peptides labeled at K16L17V18 (Figure 4 a,b,c), the pyroglutamylated peptide has more α-helical structure whereas the unmodified peptide features more β-turn structure. In case of peptides labeled at V36G37G38V39, again the β-sheet component of the unmodified peptide is shifted toward lower frequencies, and AβpE3-42 has more β-turn structure than Aβ1-42 (Figure 4 d,e,f). The consensus is that, under these conditions, Aβ1-42 undergoes a more efficient transition to β-sheet structure whereas AβpE3-42 tends to retain more α-helix or turn structures. The spectra of 1:1 combinations of the two peptides labeled at K16L17V18 resemble those of AβpE3-42, suggesting the pyroglutamylated peptide may have a stronger potency to dictate it structural features within the mixed sample, in agreement with data on unlabeled peptides (Figure 3 a,b,c).
In the low ionic strength buffer, the β-sheet peak of the unmodified peptide occurs at similar or higher wavenumbers compared to AβpE3-42, i.e., in this case AβpE3-42 undergoes β-sheet transition more readily (Figure 5). Also, Aβ1-42 contains higher fractions of β-turn than AβpE3-42. These structural features are in agreement with those obtained on the unlabeled peptides (Figure 3 d,e,f) and indicate that low ionic conditions facilitate β-sheet formation in AβpE3-42 more than in Aβ1-42. Given the notoriously unstable, polymorph-prone nature of the prefibrillar structural intermediates of Aβ peptide,87-89 identification of distinct structural propensities in multiple experiments on peptides from different sources certainly adds credibility to the observed structural trends. It should be noted here that despite clear differences between β-sheet propensities, the relative contents of β-turn and α-helical structures vary within certain ranges, indicating low energy barrier between these structures and possibly related to the inherent structural polymorphism of Aβ peptides.
3.3. Structural models
The important question is what structural information is obtained from the analysis of the signals generated by the isotope-labeled amino acid residues. The signal of the labeled residues is located at 1602 ±1 cm−1 for Aβ1-42, AβpE3-42, and their 1:1 combination. Considering the main β-sheet wavenumbers of 1628-1637 cm−1 and using a harmonic oscillator model, where the vibrational frequency of a diatomic molecule with masses m1 and m2 is proportional to (1/m1 + 1/m2)1/2, one can expect 13C=O vibrational wavenumbers around 1592-1600 cm−1. On the other hand, the diagnostic signal of 13C=O-13C=O couplings in β-sheets is in the range of 1591-1594 cm−1 and that of 13C-12C couplings is within 1601-1604 cm−1 (see above). All this information leads to the following structural constraints for the early oligomers. First, the β-sheet wavenumbers of both unlabeled and labeled segments strongly suggest formation of intramolecular β-sheets, because intermolecular β-sheets absorb at lower frequencies (see section 3.2.1). Second, both K16L17V18 and V36G37G38V39 segments are in β-sheet conformation. Third, all, or at least most, isotopically labeled amino acid residues are involved in 13C=O-12C=O rather than 13C=O-13C=O vibrational couplings. Fourth, considering the moderate size of the peptides, the large fraction of β-sheet (up to 70%), and the presence of considerable fraction of α-helix, intramolecular parallel β-sheet formation is hardly possible hence can be ruled out with high confidence. The structure of the monomer of Aβ can be roughly presented as a core β-hairpin composed of two strands and a turn or loop that may be stabilized by a salt bridge between Asp23 and Lys28 (see Ref. 90), and a short α-helix (Figure 6). This model is consistent with the aforementioned four constraints. The N-terminus appears to be the most likely segment to harbor the transient α-helix, which has been identified in this work and in earlier studies.54-58
Figure 6.
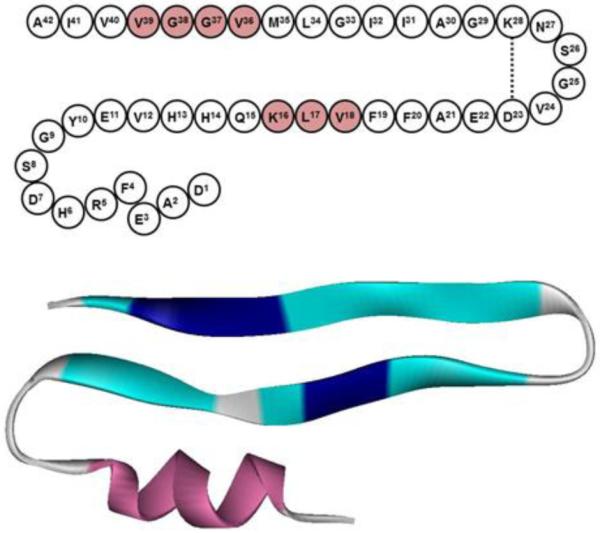
(a) The amino acid sequence of Aβ1-42 arranged in a structure involving a β-hairpin stabilized by Asp23-Lys28 ionic bridge, and an N-terminal stretch that may assume α-helical conformation. The isotopically labeled amino acids are shaded. (b) Ribbon model for Aβ monomer, composed of a β-hairpin and an N-terminal α-helix. The segments K16L17V18 and V36G37G38V39 are both involved in the β-hairpin and are marked by dark blue color.
How do the monomers aggregate into early oligomers? The absence of intermolecular H-bonding and the absence of 13C=O-13C=O vibrational couplings help to explore this question. Figure 7 presents a number of possibilities for oligomer formation via interactions between the core β-hairpin structures. Parallel in-register stacking of the β-sheets with the aggregation axis perpendicular to the hairpin plane (Figure 7a), reminiscent to the structure of fibrils, would generate strong through-space 13C=O-13C=O couplings. Antiparallel stacking would also involve such couplings between the K16L17V18 segments (Figure 7 b). Since 13C=O-13C=O coupling has not been seen, these two models are invalid. The next two models involve aggregation through non-H-bonding (i.e., ionic or hydrophobic) interactions between the N-terminal strand of one peptide and the C-terminal strand of the other, either in parallel or antiparallel arrangements (Figure 7 c,d). The two strands from neighboring hairpins that are involved in intermolecular interactions can be arranged either laterally, like in regular β-sheets, or stacked like in steric zippers,91 possibly involving side chain interdigitation. Stacking can proceed alternately, when each hairpin has one strand above and the other below the neighboring strands (Figure 7 c), or the whole hairpins themselves can go up and down consecutively (Figure 7 d). Other geometries are possible, such as a staircase architecture where each hairpin goes up by one step. These structures are plausible because they involve 13C=O-12C=O and not 13C=O-13C=O couplings, in agreement with the data. Next we consider formation of dimers by interactions between the N-terminal strands or between C-terminal strands, followed by propagation of aggregation by interactions between C-terminal strands or N-terminal strands, parallel (Figure 7 e) or antiparallel (Figure 7 f). These structures predict 13C=O-13C=O couplings, which is not seen. Thus, peptide oligomers most likely form through interaction schemes shown in Figure 7 c,d.
Figure 7.

Schematic models for Aβ oligomerization through interactions between the core β-hairpin structures. Two arrows in each molecule represent two β-strands, connected by a loop or turn. The monomers are colored gray and pink, and the isotopically labeled segments are indicated by darker color. In all cases, the structures are stabilized by intramolecular H-bonding and intermolecular non-H-bonding contacts, i.e. ionic and/or hydrophobic interactions. The plane of the picture is parallel to the hairpin plane. In a) and b), the aggregation axis is perpendicular, and in c) to f) it is coplanar to the picture plane.
The data suggest that in a buffer of near-physiological ionic strength Aβ1-42 undergoes α-helix to β-sheet transition more readily than AβpE3-42, while the opposite is true under low ionic strength conditions (Figures 3-5, Table 1). The presence of a residual α-helix in the transient structure of the peptide is likely to hinder β-sheet formation and aggregation (see Figure 6). The protofibrils and fibrils do not contain any α-helix, i.e. this structure disappears along the way of fibrillogenesis. It is logical to assume that at low ionic strength the helix in Aβ1-42 is more stable than in AβpE3-4, while at higher ionic strength it becomes less stable. These effects are likely associated with the difference in charge of the N-termini of Aβ1-42 and AβpE3-42. According to the model presented in Figure 6, the N-terminal helix would be stabilized by a set of side chain ionic contacts, such as Asp1-Lys16, Glu3-Lys16, Asp1-Arg5, Arg5-Glu11. These ionic interactions would be strengthened under low ionic strength conditions, thus stabilizing the helical structure and retarding β-sheet formation. Helix-stabilizing ionic interactions involving Asp1 and Glu3 would not occur in AβpE3-42 where Asp1 is removed and Glu3 is cyclized. These considerations provide a plausible explanation for a slower α-helix to β-sheet transition for Aβ1-42 compared to AβpE3-42 at low ionic strength and an opposite trend at higher ionic strength. This conclusion is in accord with earlier findings suggesting that the N-terminus of Aβ peptide controls the transition of the oligomers into protofibrils.52 Further exploration of the mechanism of distinct structural propensities of the unmodified and pyroglutamylated Aβ peptides and the location of the transient α-helix will require additional research, involving peptides isotopically labeled within the N-terminus.
4. Conclusions
Aβ peptides originate from APP, including its α-helical transmembrane domain, and undergo complex, evidently multistep conformational changes leading to the formation of extracellular amyloid plaques, the hallmark of AD. These transitions include the initial α-helix to β-sheet conversion, dimerization, formation of early oligomers that are the most toxic species, further aggregation into protofibrils and final fibrillogenesis. While the mature fibrils have been very well structurally characterized, structural studies on the oligomers have proved more challenging mainly because of their unstable, dynamic nature, especially for time-consuming methods such as NMR. Here we have employed isotope-edited FTIR to capture the initial structural changes in Aβ1-42 and the pyroglutamylated peptide AβpE3-42, i.e. a transition from mostly α-helical structure to mostly β-sheet structure. Data on unlabeled peptides and those 13C,15N-labeled at two distinct segments have allowed identification of the most plausible modes of oligomer formation. Distinct differences have been detected between α-helix and β-sheet propensities of Aβ1-42 and AβpE3-42 and transitions from the former to the latter in two buffers of different ionic strengths. These data imply involvement of ionic interactions in α-helix to β-sheet transitions, and the observed ionic strength dependence of the structural transitions are likely associated with the loss of anionic charges of Asp1 and Glu3 side chains in AβpE3-42. Structural models have been generated for the peptide monomers and oligomers (Figures 6, 7). Oligomers form by lateral or partially stacked non-H-bonding (i.e. ionic and/or nonpolar staggering) interactions between the N-terminal strand of one peptide monomer with the C-terminal strand of the other in parallel or antiparallel manner. These early oligomers, still retaining considerable fractions of α-helix, apparently contribute to Aβ cytotoxicity and their structural characterization is important to understand the mechanism of their toxic impact. While the present work identifies important structural constraints for the oligomers, further studies, involving isotopic labeling at additional segments, including the N-terminus, plus studies by higher resolution methods such as NMR, will lead to a better understanding of the structural basis of cytotoxicity of the unmodified and pyroglutamylated Aβ peptides.
Supplementary Material
5. Acknowledgements
This work was partially supported by NIH grant 1R03AI097591 and SEED grant from the College of Sciences of the University of Central Florida. Financial assistance from Dr. Bo Chen (Department of Physics, University of Central Florida) in purchasing the isotopically labeled peptides is gratefully acknowledged.
List of abbreviations
- Aβ
amyloid β peptide
- AβpE
pyroglutamylated Aβ peptide
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- CD
circular dichroism
- ER
endoplasmic reticulum
- FTIR
Fourier transform infrared
- HFIP
hexafluoroisopropanol
- MD
molecular dynamics
References
- 1.Thal DR, Walter J, Saido TC, Fändrich M. Acta Neuropathol. 2015;129:167–182. doi: 10.1007/s00401-014-1375-y. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J, Allsop D. Trends Pharmacol. Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 3.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. J. Neurosci. 1993;13:1676–87. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dickson DW. Neurobiol. Aging. 1997;18:S21–S26. doi: 10.1016/s0197-4580(97)00065-1. [DOI] [PubMed] [Google Scholar]
- 5.Kirkitadze MD, Bitan G, Teplow DB. J. Neurosci. Res. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- 6.Klein WL, Stine WB, Teplow DB. Neurobiol. Aging. 2004;25:569–580. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 7.Resende R, Ferreiro E, Pereira C, Resende de Oliveira C. Neuroscience. 2008;155:725–737. doi: 10.1016/j.neuroscience.2008.06.036. [DOI] [PubMed] [Google Scholar]
- 8.Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, Teplow DB, Shea JE, Ruotolo BT, Robinson CV, Bowers MT. Nat. Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masters CL, Selkoe DJ. Cold Spring Harb. Perspect. Med. 2012;2:a006262. doi: 10.1101/cshperspect.a006262. doi: 10.1101/cshperspect.a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benilova I, Karran E, De Strooper B. Nat. Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 11.Tay WM, Huang D, Rosenberry TL, Paravastu AK. J. Mol Biol. 2013;425:2494–2508. doi: 10.1016/j.jmb.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sebollela A, Mustata GM, Luo K, Velasco PT, Viola KL, Cline EN, Shekhawat GS, Wilcox KC, Dravid VP, Klein WL. ACS Chem. Neurosci. 2014;5:1238–1245. doi: 10.1021/cn500156r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerasoli E, Ryadnov MG, Austen BM. Front. Chem. 2015;3:17. doi: 10.3389/fchem.2015.00017. doi: 10.3389/fchem.2015.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lal R, Lin H, Quist AP. Biochim. Biophys. Acta. 2007;1768:1966–1975. doi: 10.1016/j.bbamem.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arispe N, Diaz JC, Simakova O. Biochim. Biophys. Acta. 2007;1768:1952–1965. doi: 10.1016/j.bbamem.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 16.Gillman AL, Jang H, Lee J, Ramachandran S, Kagan BL, Nussinov R, Teran Arce F. J. Phys. Chem. B. 2014;118:7335–7344. doi: 10.1021/jp5040954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferreiro E, Oliveira CR, Pereira CM. Neurobiol. Dis. 2008;30:331–342. doi: 10.1016/j.nbd.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Ferreiro E, Baldeiras I, Ferreira IL, Costa RO, Rego AC, Pereira CF, Oliveira CR. Int. J. Cell Biol. 2012:735206. doi: 10.1155/2012/735206. doi: 10.1155/2012/735206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demuro A, Parker I. J. Neurosci. 2013;33:3824–3833. doi: 10.1523/JNEUROSCI.4367-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reddy PH, Tripathi R, Troung Q, Tirumala K, Reddy TP, Anekonda V, Shirendeb UP, Calkins MJ, Reddy AP, Mao P, Manczak M. Biochim. Biophys. Acta. 2012;1822:639–649. doi: 10.1016/j.bbadis.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pagani L, Eckert A. Int. J. Alzheimers Dis. 2011:925050. doi: 10.4061/2011/925050. doi: 10.4061/2011/925050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Correia SC, Santos RX, Santos MS, Casadesus G, Lamanna JC, Perry G, Smith MA, Moreira PI. Curr. Alzheimer Res. 2013;10:406–419. doi: 10.2174/1567205011310040006. [DOI] [PubMed] [Google Scholar]
- 23.Chi TY, Wang LH, Ji XF, Shen L, Zou LB. J. Asian Nat. Prod. Res. 2013;15:1013–1022. doi: 10.1080/10286020.2013.821982. [DOI] [PubMed] [Google Scholar]
- 24.Lei H, Zhao CY, Liu DM, Zhang Y, Li L, Wang XL, Peng Y. J. Asian Nat. Prod. Res. 2014;16:854–864. doi: 10.1080/10286020.2014.939586. [DOI] [PubMed] [Google Scholar]
- 25.Malinow R. Curr. Opin. Neurobiol. 2012;22:559–563. doi: 10.1016/j.conb.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim T, Vidal GS, Djurisic M, William CM, Michael E, Birnbaum ME, Garcia KC, Hyman BT, Carla J, Shatz CJ. Science. 2013;341:1399–1404. doi: 10.1126/science.1242077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernandez MA, Klutkowski JA, Freret T, Wolfe MS. J. Biol. Chem. 2014;289:31043–31052. doi: 10.1074/jbc.M114.581165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bayer TA, Wirths O. Acta Neuropathol. 2014;127:787–801. doi: 10.1007/s00401-014-1287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, Tayler K, Wiltgen B, Hatami A, Rönicke R, Reymann K, Hutter-Paier B, Alexandru A, Jagla W, Graubner S, Glabe CG, Demuth HU, Bloom GS. Nature. 2012;485:651–655. doi: 10.1038/nature11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S. Neuron. 1995;14:457–466. doi: 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- 31.Wu G, Miller RA, Connolly B, Marcus J, Renger J, Savage MJ. Neurodegener. Dis. 2014;14:53–66. doi: 10.1159/000353634. [DOI] [PubMed] [Google Scholar]
- 32.Kuo YM, Emmerling MR, Woods AS, Cotter RJ, Roher AE. Biochem. Biophys. Res. Commun. 1997;237:188–191. doi: 10.1006/bbrc.1997.7083. [DOI] [PubMed] [Google Scholar]
- 33.Perez-Garmendia R, Gevorkian G. Curr. Neuropharmacol. 2013;11:491–498. doi: 10.2174/1570159X11311050004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russo C, Violani E, Salis S, Venezia V, Dolcini V, Damonte G, Benatti U, D'Arrigo C, Patrone E, Carlo P, Schettini G. J. Neurochem. 2002;82:1480–1489. doi: 10.1046/j.1471-4159.2002.01107.x. [DOI] [PubMed] [Google Scholar]
- 35.He W, Barrow CJ. Biochemistry. 1999;38:10871–10877. doi: 10.1021/bi990563r. [DOI] [PubMed] [Google Scholar]
- 36.Schlenzig D, Manhart S, Cinar Y, Kleinschmidt M, Hause G, Willbold D, Funke SA, Schilling S, Demuth HU. Biochemistry. 2009;48:7072–7078. doi: 10.1021/bi900818a. [DOI] [PubMed] [Google Scholar]
- 37.Schlenzig D, Rönicke R, Cynis H, Ludwig HH, Scheel E, Reymann K, Saido T, Hause G, Schilling S, Demuth HU. J. Neurochem. 2012;121:774–784. doi: 10.1111/j.1471-4159.2012.07707.x. [DOI] [PubMed] [Google Scholar]
- 38.D'Arrigo C, Tabaton M, Perico A. Biopolymers. 2009;91:861–873. doi: 10.1002/bip.21271. [DOI] [PubMed] [Google Scholar]
- 39.Jawhar S, Wirths O, Bayer TA. J. Biol. Chem. 2011;286:38825–38832. doi: 10.1074/jbc.R111.288308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mandler M, Walker L, Santic R, Hanson P, Upadhaya AR, Colloby SJ, Morris CM, Thal DR, Thomas AJ, Schneeberger A, Attems J. Acta Neuropathol. 2014;128:67–79. doi: 10.1007/s00401-014-1296-9. [DOI] [PubMed] [Google Scholar]
- 41.Nadezhdin KD, Bocharova OV, Bocharov EV, Arseniev AS. Acta Naturae. 2011;3:69–76. [PMC free article] [PubMed] [Google Scholar]
- 42.Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, Sanders CR. Science. 2012;336:1168–1171. doi: 10.1126/science.1219988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko RA. Proc. Natl. Acad. Sci. USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paravastu AK, Leapman RD, Yau WM, Tycko R. Proc. Natl. Acad. Sci. USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Döbeli H, Schubert D, Riek R. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim YS, Liu L, Axelsen PH, Hochstrasser RM. Proc. Natl. Acad. Sci. USA. 2008;105:7720–7725. doi: 10.1073/pnas.0802993105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paul C, Axelsen PH. J. Am. Chem. Soc. 2005;127:5754–5755. doi: 10.1021/ja042569w. [DOI] [PubMed] [Google Scholar]
- 48.Balbach JJ, Petkova AT, Oyler NA, Antzutkin ON, Gordon DJ, Meredith SC, Tycko R. Biophys. J. 2002;83:1205–1216. doi: 10.1016/S0006-3495(02)75244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE, Smith SO. Nat. Struct. Mol. Biol. 2010;17:561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chimon S, Shaibat MA, Jones CR, Calero DC, Aizezi B, Ishii Y. Nat. Struct. Mol. Biol. 2007;14:1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
- 51.Yu L, Edalji R, Harlan JE, Holzman TF, Lopez AP, Labkovsky B, Hillen H, Barghorn S, Ebert U, Richardson PL, Miesbauer L, Solomon L, Bartley D, Walter K, Johnson RW, Hajduk PJ, Olejniczak ET. Biochemistry. 2009;48:1870–1877. doi: 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]
- 52.Haupt C, Leppert J, Rönicke R, Meinhardt J, Yadav JK, Ramachandran R, Ohlenschläger O, Reymann KG, Görlach M, Fändrich M. Angew. Chem. Int. Ed. Engl. 2012;51:1576–1579. doi: 10.1002/anie.201105638. [DOI] [PubMed] [Google Scholar]
- 53.Scheidt HA, Morgado I, Rothemund S, Huster D, Fändrich M. Angew. Chem. Int. Ed. Engl. 2011;50:2837–2840. doi: 10.1002/anie.201007265. [DOI] [PubMed] [Google Scholar]
- 54.Kirkitadze MD, Condron MM, Teplow DB. J. Mol. Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 55.Ono K, Condron MM, Teplow DB. Proc. Natl. Acad. Sci. USA. 2009;106:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Urbanc B, Cruz L, Ding F, Sammond D, Khare S, Buldyrev SV, Stanley HE, Dokholyan NV. Biophys. J. 2004;87:2310–2321. doi: 10.1529/biophysj.104.040980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu Y, Shen J, Luo X, Zhu W, Chen K, Ma J, Jiang H. Proc. Natl. Acad. Sci. USA. 2005;102:5403–5407. doi: 10.1073/pnas.0501218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vandersteen A, Masman MF, De Baets G, Jonckheere W, van der Werf K, Marrink SJ, Rozenski J, Benilova I, De Strooper B, Subramaniam V, Schymkowitz J, Rousseau F, Broersen K. J. Biol. Chem. 2012;287:36732–36743. doi: 10.1074/jbc.M112.394635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tomaselli S, Esposito V, Vangone P, van Nuland NA, Bonvin AM, Guerrini R, Tancredi T, Temussi PA, Picone D. ChemBioChem. 2006;7:257–267. doi: 10.1002/cbic.200500223. [DOI] [PubMed] [Google Scholar]
- 60.Abedini A, Raleigh DP. Protein Eng. Des. Sel. 2009;22:453–459. doi: 10.1093/protein/gzp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cerf E, Sarroukh R, Tamamizu-Kato S, Breydo L, Derclaye S, Dufrêne YF, Narayanaswami V, Goormaghtigh E, Ruysschaert JM, Raussens V. Biochem. J. 2009;421:415–423. doi: 10.1042/BJ20090379. [DOI] [PubMed] [Google Scholar]
- 62.Sun N, Hartmann R, Lecher J, Stoldt M, Funke SA, Gremer L, Ludwig HH, Demuth HU, Kleinschmidt M, Willbold D. J. Pept. Sci. 2012;18:691–695. doi: 10.1002/psc.2456. [DOI] [PubMed] [Google Scholar]
- 63.Tekirian TL, Yang AY, Glabe C, Geddes JW. J. Neurochem. 1999;73:1584–1589. doi: 10.1046/j.1471-4159.1999.0731584.x. [DOI] [PubMed] [Google Scholar]
- 64.Sanders HM, Lust R, Teller JK. Peptides. 2009;30:849–854. doi: 10.1016/j.peptides.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matos JO, Goldblatt G, Jeon J, Chen B, Tatulian SA. J. Phys. Chem. B. 2014;118:5637–5643. doi: 10.1021/jp412743s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stine WB, Jr., Dahlgren KN, Krafft GA, LaDu MJ. J. Biol. Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 67.Tatulian SA. Methods Mol. Biol. 2013;974:177–218. doi: 10.1007/978-1-62703-275-9_9. [DOI] [PubMed] [Google Scholar]
- 68.Schilling S, Lauber T, Schaupp M, Manhart S, Scheel E, Böhm G, Demuth HU. Biochemistry. 2006;45:12393–12399. doi: 10.1021/bi0612667. [DOI] [PubMed] [Google Scholar]
- 69.Bouter Y, Dietrich K, Wittnam JL, Rezaei-Ghaleh N, Pillot T, Papot-Couturier S, Lefebvre T, Sprenger F, Wirths O, Zweckstetter M, Bayer TA. Acta Neuropathol. 2013;126:189–205. doi: 10.1007/s00401-013-1129-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Salvadores N, Shahnawaz M, Scarpini E, Tagliavini F, Soto C. Cell Rep. 2014;7:261–268. doi: 10.1016/j.celrep.2014.02.031. [DOI] [PubMed] [Google Scholar]
- 71.Sreerama N, Woody RW. In: Circular Dichroism: Principles and Applications. Berova N, Nakanishi K, Woody RW, editors. John Wiley & Sons; Hoboken, N.J.: 2000. pp. 601–620. [Google Scholar]
- 72.Kelly SM, Price NC. Curr. Protein Pept. Sci. 2000;1:349–384. doi: 10.2174/1389203003381315. [DOI] [PubMed] [Google Scholar]
- 73.Bakshi K, Liyanage MR, Volkin DB, Middaugh CR. Methods Mol. Biol. 2014;1088:247–253. doi: 10.1007/978-1-62703-673-3_17. [DOI] [PubMed] [Google Scholar]
- 74.Ludlam CF, Arkin IT, Liu XM, Rothman MS, Rath P, Aimoto S, Smith SO, Engelman DM, Rothschild KJ. Biophys. J. 1996;70:1728–1736. doi: 10.1016/S0006-3495(96)79735-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tatulian SA, Jones LR, Reddy LG, Stokes DL, Tamm LK. Biochemistry. 1995;34:4448–4456. doi: 10.1021/bi00013a038. [DOI] [PubMed] [Google Scholar]
- 76.Tatulian SA, Tamm LK. Biochemistry. 2000;39:496–507. doi: 10.1021/bi991594p. [DOI] [PubMed] [Google Scholar]
- 77.Venyaminov SY, Hedstrom JF, Prendergast FG. Proteins. 2001;45:81–89. doi: 10.1002/prot.1126. [DOI] [PubMed] [Google Scholar]
- 78.Huang R, Kubelka J, Barber-Armstrong W, Silva RAGD, Decatur SM, Keiderling TA. J. Am. Chem. Soc. 2004;126:2346–2354. doi: 10.1021/ja037998t. [DOI] [PubMed] [Google Scholar]
- 79.Barber-Armstrong W, Donaldson T, Wijesooriya H, Silva RAGD, Decatur SM. J. Am. Chem. Soc. 2004;126:2339–2345. doi: 10.1021/ja037863n. [DOI] [PubMed] [Google Scholar]
- 80.Decatur SM. Acc. Chem. Res. 2006;39:169–175. doi: 10.1021/ar050135f. [DOI] [PubMed] [Google Scholar]
- 81.Decatur SM. Biopolymers. 2000;54:180–185. doi: 10.1002/1097-0282(200009)54:3<180::AID-BIP40>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 82.Petty SA, Decatur SM. J. Am. Chem. Soc. 2005;127:13488–13489. doi: 10.1021/ja054663y. [DOI] [PubMed] [Google Scholar]
- 83.Petty SA, Decatur SM. Proc. Natl. Acad. Sci. USA. 2005;102:14272–14277. doi: 10.1073/pnas.0502804102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krimm S, Bandekar J. Adv. Protein Chem. 1986;38:181–364. doi: 10.1016/s0065-3233(08)60528-8. [DOI] [PubMed] [Google Scholar]
- 85.Venyaminov SY, Kalnin NN. Biopolymers. 1990;30:1259–1271. doi: 10.1002/bip.360301310. [DOI] [PubMed] [Google Scholar]
- 86.Liu D, Xu Y, Feng Y, Liu H, Shen X, Chen K, Ma J, Jiang H. Biochemistry. 2006;45:10963–10972. doi: 10.1021/bi060955f. [DOI] [PubMed] [Google Scholar]
- 87.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 88.Jeong JS, Ansaloni A, Mezzenga R, Lashuel HA, Dietler G. J. Mol. Biol. 2013;425:1765–1781. doi: 10.1016/j.jmb.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 89.Tycko R. Neuron. 2015;86:632–645. doi: 10.1016/j.neuron.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tycko R. Annu. Rev. Phys. Chem. 201;62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nelson R, Sawaya MR, Balbirnie M, Madsen AØ, Riekel C, Grothe R, Eisenberg D. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.