

Abstract
Purpose
To evaluate dabrafenib, a selective BRAF inhibitor, combined with trametinib, a selective MEK inhibitor, in patients with BRAF V600–mutant metastatic colorectal cancer (mCRC).
Patients and Methods
A total of 43 patients with BRAF V600–mutant mCRC were treated with dabrafenib (150 mg twice daily) plus trametinib (2 mg daily), 17 of whom were enrolled onto a pharmacodynamic cohort undergoing mandatory biopsies before and during treatment. Archival tissues were analyzed for microsatellite instability, PTEN status, and 487-gene sequencing. Patient-derived xenografts were established from core biopsy samples.
Results
Of 43 patients, five (12%) achieved a partial response or better, including one (2%) complete response, with duration of response > 36 months; 24 patients (56%) achieved stable disease as best confirmed response. Ten patients (23%) remained in the study > 6 months. All nine evaluable during-treatment biopsies had reduced levels of phosphorylated ERK relative to pretreatment biopsies (average decrease ± standard deviation, 47% ± 24%). Mutational analysis revealed that the patient achieving a complete response and two of three evaluable patients achieving a partial response had PIK3CA mutations. Neither PTEN loss nor microsatellite instability correlated with efficacy. Responses to dabrafenib plus trametinib were comparable in patient-derived xenograft–bearing mice and the biopsied lesions from each corresponding patient.
Conclusion
The combination of dabrafenib plus trametinib has activity in a subset of patients with BRAF V600–mutant mCRC. Mitogen-activated protein kinase signaling was inhibited in all patients evaluated, but to a lesser degree than observed in BRAF-mutant melanoma with dabrafenib alone. PIK3CA mutations were identified in responding patients and thus do not preclude response to this regimen. Additional studies targeting the mitogen-activated protein kinase pathway in this disease are warranted.
INTRODUCTION
Missense mutation of the v-raf murine sarcoma viral oncogene homolog B (BRAF) gene is present in 5% to 10% of metastatic colorectal cancers (mCRCs).1,2 BRAF encodes a protein kinase in the mitogen-activated protein kinase (MAPK) pathway that may be rendered constitutively active by substitution at valine 600 (V600), most commonly to glutamic acid (V600E). Patients with mCRC whose tumors harbor BRAF V600 mutations generally respond poorly to standard therapies,3–8 with a median progression-free survival (PFS) of 2.5 months after second-line chemotherapy8 and median overall survival (OS) < 1 year, versus > 2 years for patients with BRAF wild-type mCRC.9 New therapeutic approaches for patients with BRAF-mutated mCRC are critically needed.
Patients with BRAF-mutated mCRC are more likely to be female and older, with right-sided primary tumors and an unusual pattern of metastatic spread, including frequent peritoneal and distant lymph node involvement.2,9 BRAF mutation defines a unique molecular subtype of CRC, commonly originating from serrated adenomas with low rates of chromosomal instability and high rates of hypermethylation and microsatellite instability (MSI).10–12 MSI is a favorable prognostic marker in early-stage CRC, but may not improve outcomes in the context of BRAF V600E mCRC.9,11,13
BRAF inhibition with vemurafenib or dabrafenib has resulted in significantly prolonged PFS and OS in patients with BRAF-mutated metastatic melanoma.14–19 Trametinib, targeting mitogen-activated extracellular signal–related kinase kinase (MEK), downstream of BRAF in the MAPK pathway, has similarly been associated with improved PFS and OS compared with conventional chemotherapy in BRAF-mutated metastatic melanoma.20,21 These pivotal studies led to the approval of vemurafenib, dabrafenib, and trametinib by the US Food and Drug Administration, revolutionizing treatment of BRAF V600–mutated melanoma.
In contrast to BRAF-mutant melanoma, mCRC with the same BRAF V600 mutation has shown a marked lack of sensitivity to BRAF or MEK inhibitor monotherapy in early clinical trials. Whereas approximately 50% of patients with metastatic melanoma responded to vemurafenib, the response rate was only 5% among 19 assessable patients with BRAF-mutated mCRC.16,17,22 One of nine assessable patients with BRAF-mutated CRC experienced partial response (PR) after dabrafenib monotherapy.18 The phase I/II trial of trametinib alone showed no responses in a small number of patients with BRAF-mutant mCRC.23
This was predicted by preclinical studies, which suggested that BRAF or MEK inhibitors alone do not produce sustained MAPK pathway inhibition in BRAF-mutant CRC, likely because of feedback signals that reactivate MAPK signaling.24–26 This is hypothesized to be a major factor underlying the lack of clinical response to these agents in BRAF-mutant CRC. However, laboratory studies have also suggested that combined inhibition of BRAF and MEK can lead to improved suppression of MAPK signaling and increased efficacy.24,25 Combined inhibition of BRAF and MEK with dabrafenib plus trametinib has been studied extensively in patients with unresectable or metastatic BRAF V600–mutated melanoma and was recently granted accelerated US Food and Drug Administration approval after demonstrating an acceptable safety profile with a statistically significant improvement in PFS and response rates compared with dabrafenib alone.27
We report here on a 43-patient expansion cohort of the dabrafenib plus trametinib combination study,27,28 comprising patients with BRAF V600–mutated mCRC, to test the hypothesis that the MAPK pathway is a valid therapeutic target in this population. Conducting clinical trials in the patient population with BRAF-mutated mCRC presents several challenges, including the relative rarity of BRAF mutations, the failure to routinely test for the mutation, and the aggressive nature of the disease. Thus, a strong emphasis was placed on correlative science to guide subsequent investigations aimed at personalizing therapy to improve clinical outcomes for patients with BRAF-mutated mCRC.
PATIENTS AND METHODS
Patient Selection
Patients with histologically confirmed mCRC with either a BRAF V600E or V600K mutation were eligible to enroll. Eligibility criteria also included measurable disease according to RECIST (version 1.1) and Eastern Cooperative Oncology Group performance status of 0 or 1. Exclusion criteria included history of central serous retinopathy, retinal vein occlusion, and serious cardiac comorbidities. The study was approved by the institutional review board at each site. All patients provided written informed consent before any study procedures.
Study Design
This open-label phase I/II study assessed the safety, pharmacokinetics, pharmacodynamics, and clinical activity of combination therapy with dabrafenib plus trametinib.27,28 Here we report results for a portion of part B: the mCRC cohort of patients with BRAF V600–mutated tumors, including a pharmacodynamic expansion cohort (Data Supplement).
Pharmacodynamic and Exploratory Biomarker Assessments
Patients in the pharmacodynamic cohort consented to mandatory tumor biopsies before treatment (within 21 days before starting study treatment) and on day 15 of study treatment (± 7 days). Formalin-fixed paraffin-embedded (FFPE) and flash-frozen core biopsy samples were collected. Pathway inhibition was assessed in the paired tumor tissue for change in expression levels of phosphorylated ERK (P-ERK) and AKT (P-AKT) by immunohistochemistry (IHC).29 The frozen paired tumor tissue was used for reverse-phase protein array (RPPA) analysis (George Mason University, Manassas, VA).30
Targeted next-generation sequencing (NGS) was performed for a 487-candidate cancer gene panel (Data Supplement). PTEN and epidermal growth factor receptor (EGFR) IHC were conducted at Ventana Medical Systems (Tucson, AZ) using rabbit monoclonal antibody D4.3 or 5B7, respectively. MSI status was evaluated using Promega MSI Analysis System (version 1.2 [product No. MD1641]; Madison, WI). DNA was extracted using Qiagen QIAamp DNA FFPE Tissue Kit (product No. 56404; Valencia, CA) Details on IHC, RPPA, and NGS are provided in the Data Supplement.
Patient-Derived Xenografts
A companion protocol with a separate consent form permitted collection of additional tissue at the time of tumor biopsies in the pharmacodynamic cohort for patient-derived xenograft (PDX) development at the Jackson Laboratory (Sacramento, CA). Mice bearing subcutaneous PDXs (seven to 10 per group) were treated with vehicle (0.1% Tween-20 or 0.5% hydroxypropyl methylcellulose and 0.2% Tween-80), dabrafenib (30 mg/kg per day), and trametinib (1 or 0.6 mg/kg per day) by oral gavage for 21 days at University of California San Francisco (San Francisco, CA) or MD Anderson Cancer Center (Houston, TX; details provided in Data Supplement).
Statistical Methods
The objectives of enrolling the part B colorectal cohort were to collect safety, clinical activity, and pharmacodynamic response in paired biopsies among patients with BRAF-mutant mCRC. PFS was summarized with Kaplan-Meier methodology using medians and 95% CIs (estimated using Brookmeyer-Crowley method).
RESULTS
Patient Population
Between January 3, 2011, and April 25, 2013, 43 patients with BRAF V600–mutated mCRC were enrolled at eight centers: 26 patients in the original efficacy cohort, and 17 patients in the pharmacodynamic expansion cohort with mandatory tumor biopsies before treatment and on day 15 of treatment. All patients initiated treatment with dabrafenib 150 mg twice daily and trametinib 2 mg daily. Baseline characteristics of patients with mCRC are listed in Table 1.
Table 1.
Baseline Patient Demographic and Clinical Characteristics (N = 43)
| Characteristic | No. (%) |
|---|---|
| Age, years | |
| Mean | 55 |
| SD | 13 |
| Female sex | 34 (79) |
| ECOG performance status | |
| 0 | 24 (56) |
| 1 | 19 (44) |
| BRAF V600E mutation | 43 (100) |
| No. of disease sites at screening | |
| < 3 | 22 (51) |
| ≥ 3 | 21 (49) |
| No. of lines of prior systemic anticancer therapy* | |
| 0 | 1 (2) |
| 1 | 6 (14) |
| 2 | 14 (33) |
| ≥ 3 | 22 (51) |
| Prior EGFR inhibitor | 20 (47) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; EGFR, epidermal growth factor receptor; SD, standard deviation.
Prior chemotherapy, immunotherapy, hormonal, biologic, or small-molecule targeted therapy regimens.
Safety
Adverse events (AEs) were consistent with those reported in patients with metastatic melanoma treated with dabrafenib 150 mg twice daily plus trametinib 2 mg daily27 (Table 2). The most frequent AEs were nausea, pyrexia, and fatigue. Pyrexia was the most common reason for dose interruption, occurring in 13 patients (30%), and dose reduction, occurring in 12 patients (28%). One patient (2%) discontinued treatment because of pyrexia. Left ventricular ejection fraction (LVEF) reduction occurred in eight patients (19%), including two grade 3 events. LVEF reduction led to dose reduction in five patients (12%) and treatment discontinuation in one patient (2%). Grade 1 ocular events (eg, photophobia, blurred vision, and visual impairment) occurred in three patients (7%). No grade 4 pyrexia, LVEF reduction, or ocular events were reported. No grade 5 AEs were reported. Overall, 17 AEs (40%) led to dose reduction, 25 AEs (58%) led to dose interruption, and four patients (9%) discontinued treatment because of an AE.
Table 2.
Adverse Events
| Adverse Event | No. (%) |
|
|---|---|---|
| Grade 3 | Any Grade | |
| Any | 25 (58) | 42 (98) |
| Nausea | 2 (5) | 27 (63) |
| Pyrexia | 5 (12) | 27 (63) |
| Fatigue | 3 (7) | 23 (53) |
| Chills | 1 (2) | 21 (49) |
| Vomiting | 3 (7) | 20 (47) |
| Diarrhea | 1 (2) | 15 (35) |
| Headache | 0 | 15 (35) |
| Anemia | 7 (16) | 12 (28) |
| Constipation | 0 | 11 (26) |
| Decreased appetite | 2 (5) | 11 (26) |
| Peripheral edema | 0 | 11 (26) |
NOTE. Listed are all adverse events reported in ≥ 25% of patients, irrespective of whether causal relationship was likely. Six grade 4 events independent of attribution were reported: dyspnea, thrombocytopenia, hypotension, large intestine obstruction, pulmonary embolism, and sepsis syndrome.
Efficacy
Of 43 patients enrolled, five (12%) achieved a PR or better (Fig 1A), including one patient (2%) with a complete response (CR), with duration of response > 36 months (ongoing; last data cut, January 15, 2015). This patient achieved a CR by week 32 of study treatment, with complete resolution of a large tumor mass invading through the abdominal wall (Fig 1B). Among the 43 patients, only the patient achieving the CR had not received prior systemic therapy. Two of four patients achieving a PR and the patient achieving a CR had confirmed responses. A total of 24 patients (56%) achieved stable disease as their best confirmed response. Overall, 16 patients (37%) experienced a reduction in target lesion size by RECIST of ≥ 10%. Median PFS was 3.5 months (95% CI, 3.4 to 4.0 months), and median duration of study treatment was 3.6 months (range, 0.3 to 36.8 months). Ten patients (23%) remained in study treatment > 6 months (Fig 1C).
Fig 1.

(A) Waterfall plot of maximum percent reduction in target lesion size by RECIST. Horizontal lines at + 20% and − 30% denote boundaries of stable disease. (B) Computed tomography images and photographs from patient achieving complete response. (C) Time receiving study treatment plot.
Pharmacodynamic Analysis of Paired Biopsy Specimens
Of 17 patients enrolled onto the pharmacodynamic cohort, paired pretreatment and day-15 treatment biopsies with evaluable tumor content were available for nine patients. Biopsies from the other eight patients were not evaluable, either because the pre- or during-treatment samples lacked tumor cells or because during-treatment samples were not collected. All nine during-treatment biopsies showed reduced levels of P-ERK relative to pretreatment biopsies (paired t test P < .001; Figs 2A and 2B). However, the mean decrease (± standard deviation) in P-ERK in these patients was 47% ± 24% (median, 37%), which was significantly less than the mean decrease of 75% ± 21% (median, 84%) in P-ERK observed among patients with BRAF-mutated melanoma treated with dabrafenib alone (Fig 2C).31 No consistent changes in the levels of P-AKT after treatment were observed (Fig 2D). Within this limited patient sample, no clear correlations between changes in pharmacodynamic markers after treatment and response could be determined.
Fig 2.

Pharmacodynamic biomarkers from nine evaluable paired pre- and during-treatment (day 15, 2 to 4 hours after dabrafenib [D] plus trametinib [T] dosing) tumor biopsies. (A) Representative images of phosphorylated ERK (P-ERK) immunohistochemistry staining in pre- (left) and during-treatment (right) biopsies. (B) H scores for P-ERK and (C) percent change in P-ERK H score in patients with BRAF V600–mutant colorectal cancer (CRC) treated with dabrafenib (150 mg twice daily) and trametinib (2 mg daily) as compared with patients with BRAF-mutant melanoma treated with dabrafenib only (70 to 200 mg twice daily; P < .001 by paired t test).18 (D) Phosphorylated AKT H scores before and during treatment. (E) Change in abundance of specific proteins or phosphoproteins in during-treatment biopsies relative to paired pretreatment biopsies was analyzed by reverse-phase protein array. Targets showing greatest average increase (yellow) or decrease (blue) after treatment are shown. Specific targets of interest are labeled, with mitogen-activated protein kinase pathway targets shown in green and mammalian target of rapamycin pathway targets shown in purple. PDGFRβ, platelet-derived growth factor receptor–β.
Analysis of paired biopsies by RPPA showed modulation of MAPK targets, including decreased levels of phosphorylated MAPK signaling components after treatment and increased levels of BIM, a key proapoptotic protein known to be induced on MAPK inhibition (Fig 2E).32 Decreases in mammalian target of rapamycin (mTOR) pathway targets were also observed, consistent with previous studies suggesting that mTOR activity is predominantly regulated by MAPK signaling in BRAF-mutant cancers.33 We also observed a marked decrease in the proliferation marker Ki67. Increases in the levels of platelet-derived growth factor receptor–β and STAT3 phosphorylation on tyrosine 705 were also seen, both of which have been implicated in resistance to BRAF inhibition.34,35 Our analysis did not detect a clear increase in phosphorylation at any of the five EGFR phosphorylation sites analyzed after therapy with dabrafenib plus trametinib (Data Supplement).
Molecular Analyses on Archived Tissues
Mutational analysis was performed on FFPE primary tumor samples available from 15 patients (Fig 3A). As expected, the majority of tumors harbored mutations in the Wnt/β-catenin and p53 pathways.10 No clear correlation between the presence of these alterations and clinical response was evident.
Fig 3.
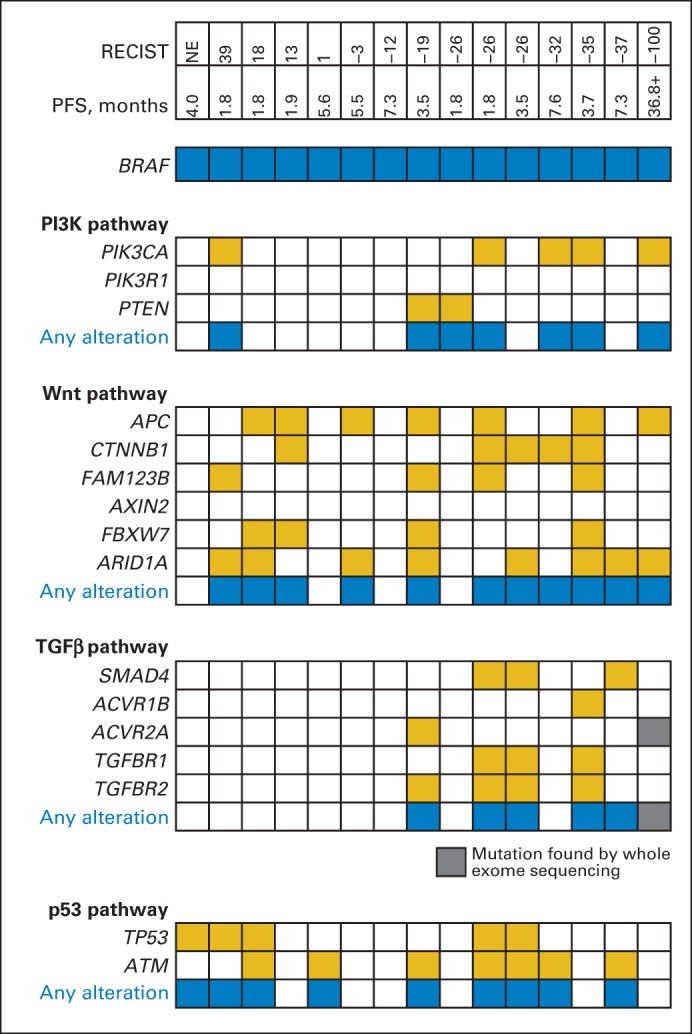
487-gene mutational analysis of archival tumor specimens available from 15 patients. RECIST value is maximum percent reduction in measurement of target lesions from baseline. Whole-exome sequencing was performed on tumor from patient achieving complete response. PDGFRβ, platelet-derived growth factor receptor–β; PFS, progression-free survival; PI3K, phosphatidylinositol 3-kinase.
Interestingly, PIK3CA mutations at known hotspots in exons 9 and 20 were identified in five of 15 evaluable patients, including in three of five patients with a PR or CR. All but one patient with a PIK3CA mutation achieved a reduction in target lesion size by RECIST. A separate analysis for PTEN loss by IHC was performed in 20 patients. PTEN loss was identified in four of the 19 patients with interpretable PTEN status, all of whom achieved a reduction in target lesion size by RECIST. There was no difference in PFS by PTEN status (Appendix Fig A1A, online only). In addition, all patients with tumors harboring transforming growth factor beta (TGF-β) pathway alterations (six of 15) had a reduction in target lesion size by RECIST, although again, definitive correlations cannot be established because of the limited sample size.
MSI analysis using a polymerase chain reaction–based assay was performed on tumor specimens from 29 patients, with 24 specimens yielding interpretable results; eight (33%) of these 24 tumors were microsatellite unstable. No statistically significant difference in PFS was observed between the MSI and MSS subpopulations (Appendix Fig A1B, online only). Total EGFR, evaluable in archival tumor specimens from 22 patients, did not correlate with PFS (median EGFR membrane H score, 85).
PDXs
We undertook an exploratory effort to generate PDX models from pretreatment biopsies obtained from five patients enrolled onto this study, because these may represent valuable tools for future correlative studies. PDX models were successfully generated from four of five patients and were successfully expanded for drug testing (Fig 4A). Response in PDX tumors after 21 days of treatment with dabrafenib plus trametinib mirrored the response in the patients' biopsied lesions from which they were derived, ranging from partial regression to progressive disease (Fig 4B).
Fig 4.

Patient-derived xenograft (PDX) –bearing mice (seven to 10 per group) treated with vehicle or dabrafenib (D) plus trametinib (T) daily for 21 days. (A) Plotted is change in PDX tumor volume after treatment, relative to initial tumor volume (all P < .001 by unpaired t test). (B) Computed tomography images showing lesion in patient who was biopsied to generate PDX pretreatment and at maximal response (weeks 32, 8, 3, and 8, respectively). First patient had spinal intramuscular mass biopsied, which regressed by 23% after 8 weeks of treatment and was not measurable by week 16 (overall best response in patient, confirmed partial response). Second patient had liver lesion biopsied, which regressed by 41% after 8 weeks of treatment (overall best response in patient, stable disease). Third patient had superficial paraumbilical nodule biopsied. This patient was not evaluable (ended study treatment because of toxicity), but imaging at week 3 showed early response in biopsied lesion. Fourth patient had posterior vaginal mass resected pretreatment, with anterior rectal recurrence by first restaging (overall best response in patient, progressive disease). If positron emission tomography was performed, maximum standardized uptake value (SUV) is noted for biopsied lesion.
DISCUSSION
Our study suggests that dual MAPK pathway blockade with the BRAF inhibitor dabrafenib and the MEK inhibitor trametinib can lead to meaningful clinical benefit in a subset of patients with BRAF-mutant mCRC. This heavily pretreated population of patients with a poor prognosis mutational subtype of mCRC achieved several PRs and a durable CR ongoing for > 3 years. In addition, 56% of patients achieved stable disease as their best confirmed response, and 23% of patients remained in the study > 6 months. We believe this study represents an important therapeutic step forward for patients with BRAF-mutant mCRC. However, the median PFS for all patients was only 3.5 months. Although this is greater than the median PFS of 2.5 months observed with standard chemotherapy,8 it is substantially less than the median PFS of 9.4 months observed with dabrafenib plus trametinib in patients with BRAF-mutant melanoma.27
Our data suggest that suboptimal MAPK pathway inhibition by dabrafenib plus trametinib in BRAF-mutant mCRC may be a major factor underlying the more limited efficacy observed in these patients. Indeed, our pharmacodynamic analyses of paired pretreatment and during-treatment biopsies showed that although the combination of inhibitors suppressed MAPK signaling, the degree of inhibition was significantly less than what has been achieved in BRAF- mutant melanoma with dabrafenib alone. This finding is critical, because studies have suggested that robust MAPK pathway suppression is required for response in BRAF-mutant cancers.14 The importance of MAPK in suppression in BRAF-mutant mCRC is also supported by a recent study demonstrating that the first mechanisms of acquired resistance identified in patients experiencing initial clinical benefit from BRAF inhibitor combinations (including dabrafenib and trametinib) all involve components of the MAPK pathway and lead to MAPK reactivation.36 Although it is possible that other signaling pathways play an important role in this disease, these data suggest that effective suppression of MAPK signaling is paramount in BRAF-mutant CRC and that therapeutic strategies capable of achieving improved MAPK suppression are needed.
It is likely that we achieved suboptimal MAPK pathway inhibition despite dual MAPK pathway blockade with dabrafenib and trametinib. This suggests that feedback reactivation of MAPK signaling may be limiting the effectiveness of the regimen. Recent preclinical studies have suggested that EGFR may drive resistance to BRAF inhibitors in many (but perhaps not all) BRAF-mutant CRCs, likely by leading to feedback activation of RAS, which can reactivate the MAPK pathway and other important signaling pathways.25,37 Although some studies have proposed that the mechanism involves increases in EGFR phosphorylation on MAPK inhibition, other studies have observed increased signaling downstream of EGFR with no increase in EGFR phosphorylation. We did not observe a clear increase in EGFR phosphorylation in paired during-treatment biopsies by RPPA, which is more consistent with the latter model. Several clinical trials are evaluating combinations of EGFR antibodies and BRAF inhibitors.38–42 Initial results of these studies suggest that combination of an EGFR antibody and a BRAF inhibitor together with a MEK inhibitor, phosphatidylinositol 3-kinase (PI3K) inhibitor, or irinotecan may be more effective than two-drug strategies.40–42 Given its comparable tolerability profile relative to BRAF inhibitors alone, the combination of dabrafenib plus trametinib represents a promising backbone for therapeutic combinations that provide some degree of MAPK pathway suppression, regardless of whether MAPK activity is driven by EGFR-dependent or -independent resistant signals. The combination of dabrafenib, trametinib, and the EGFR antibody panitumumab is being evaluated in an ongoing clinical trial.40
In an effort to identify the subset of patients with BRAF-mutant CRC most likely to derive benefit from the combination of dabrafenib plus trametinib, we performed several exploratory biomarker analyses. Although small numbers of patients limited the power of these analyses, we found that mutations in the p53 and Wnt/β-catenin pathways and MSI status did not clearly predict response to or outcome with this therapy. The potential association of PI3K pathway alterations and improved response is surprising, given that PI3K pathway activation has previously been proposed as a mechanism of resistance to BRAF and MAPK pathway inhibition based on differential in vitro sensitivity in a panel of BRAF-mutated CRC cell lines.43 Two of our four PDX models had activating PIK3CA mutations and demonstrated regression with the combination, supporting the utility of in vivo models for biomarker discovery in BRAF-mutant CRC.44 Additional biomarker studies will be required to better define the subpopulation of patients with BRAF-mutant mCRC most likely to respond to this therapeutic strategy, including investigation of a potential association between response and TGF-β pathway alterations.
Although PDXs have been increasingly used in preclinical studies, to our knowledge, this is the first study in patients with mCRC to report prospective PDX testing from during-study biopsies and correlate the findings with clinical response. Encouragingly, the responsiveness of these PDX models to dabrafenib plus trametinib seemed to recapitulate the responsiveness of the individual patient tumor lesions from which they were derived. PDX models may thus be valuable representative models with which to study individual responsiveness or resistance in tumors, overcoming an existing barrier to performing detailed correlative analyses, which are typically limited by the finite amount of tissue obtained through standard biopsies. The routine generation of PDX models in future clinical trials may help to accelerate efforts to develop more effective therapies for BRAF-mutant mCRC.
Overall, we believe our study provides proof of concept that the MAPK pathway is a valid therapeutic target in BRAF-mutant mCRC and that effective targeting of this pathway has the potential to produce meaningful clinical responses. Even though combined BRAF and MEK inhibition led to a decrease in MAPK pathway activity in all patients, the degree of MAPK inhibition achieved remains suboptimal. Additional studies evaluating therapeutic strategies designed to more effectively target the MAPK signaling pathway in BRAF-mutant CRC are in progress.
Supplementary Material
Acknowledgment
We thank Emanuel Petricoin and Isela Gallagher for contributing to the reverse-phase protein array analyses; Byron Hann, Julia Malato, Neal Goodwin, Van Morris, Ji Wu, and Feng Tian for contributing to the patient-derived xenograft experiments; and Amy J. Markowitz for editorial assistance.
Glossary Terms
- BRAF:
an isoform of RAF. See Raf.
- MAPK (mitogen-activated protein kinase):
a family of enzymes that form an integrated network influencing cellular functions such as differentiation, proliferation, and cell death. These cytoplasmic proteins modulate the activities of other intracellular proteins by adding phosphate groups to their serine/threonine amino acids.
- MEK (MAPK-ERK kinase):
a protein kinase, MEK is activated by c-Raf through phosphorylation of specific serine residues. Activation of ERK by activated MEK may lead to translocation of ERK to the nucleus, resulting in activation of specific transcription factors.
- microsatellite instability (MSI):
an alteration in the length of the microsatellites from cell to cell.
Appendix
Fig A1.

Kaplan-Meier curves for progression-free survival (PFS) by (A) PTEN expression (loss or present) and (B) microsatellite stability (MSS) or instability (MSI). HR, hazard ratio.
Footnotes
Supported by GlaxoSmithKline and in part by a Damon Runyon Clinical Investigator Award and Awards No. P50CA127003 and K08CA166510 from the National Cancer Institute (NCI), National Institute of Health (NIH; R.B.C.); by Award No. K08CA175143 from the NCI, NIH (C.E.A.); by NIH Clinical and Translational Science Award No. UL1 RR024148 and NIH Cancer Center Support Grant No. CA016672 to MD Anderson Cancer Center (G.F.); and by Awards No. R01CA172670 and R01CA184843 from the NCI (S.K.).
Terms in blue are defined in the glossary, found at the end of this article and online at www.jco.org.
Presented in part at the 49th Annual Meeting of the American Society of Clinical Oncology (ASCO), Chicago, IL, May 31-June 4, 2013, and 50th ASCO Annual Meeting, Chicago, IL, May 30-June 3, 2014.
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
Clinical trial information: NCT01072175.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: Ryan B. Corcoran, Chloe E. Atreya, Gerald S. Falchook, David P. Ryan, Johanna C. Bendell, Omid Hamid, Wells A. Messersmith, Adil Daud, Razelle Kurzrock, Mariaelena Pierobon, Shonda Little, Keith Orford, Monica Motwani, Kiran Patel, Alan P. Venook, Scott Kopetz
Collection and assembly of data: Ryan B. Corcoran, Chloe E. Atreya, Gerald S. Falchook, Eunice L. Kwak, David P. Ryan, Johanna C. Bendell, Omid Hamid, Wells A. Messersmith, Adil Daud, Razelle Kurzrock, Mariaelena Pierobon, Peng Sun, Elizabeth Cunningham, Shonda Little, Keith Orford, Monica Motwani, Kiran Patel, Alan P. Venook, Scott Kopetz
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600–Mutant Colorectal Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Ryan B. Corcoran
Honoraria: GlaxoSmithKline
Consulting or Advisory Role: Avidity Nanomedicines, Taiho Pharmaceutical, Merrimack, Genentech
Chloe E. Atreya
Consulting or Advisory Role: Bayer Diagnostics
Research Funding: GlaxoSmithKline, Novartis
Gerald S. Falchook
Employment: Sarah Cannon Research Institute, HealthONE
Research Funding: GlaxoSmithKline, Millennium Pharmaceuticals, EMD Serono, Celgene, MedImmune, Genmab, Vegenics, Novartis, AstraZeneca, Incyte, ARMO BioSciences, Kolltan Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Handbook of Targeted Cancer Therapy
Travel, Accommodations, Expenses: Millennium Pharmaceuticals, Sarah Cannon Research Institute, EMD Serono
Eunice L. Kwak
Travel, Accommodations, Expenses: Amgen
David P. Ryan
Honoraria: UpToDate
Consulting or Advisory Role: MedImmune
Patents, Royalties, Other Intellectual Property: McGraw Hill: chapter royalties
Johanna C. Bendell
No relationship to disclose
Omid Hamid
Consulting or Advisory Role: GlaxoSmithKline
Speakers' Bureau: GlaxoSmithKline, Genentech
Research Funding: Genentech, GlaxoSmithKline
Wells A. Messersmith
Consulting or Advisory Role: OncoMed, Immunomedics
Research Funding: GlaxoSmithKline, Pfizer, Roche/Genentech, Millennium Pharmaceuticals, OncoMed, Immunomedics
Adil Daud
Stock or Other Ownership: OncoSec
Consulting or Advisory Role: Novartis, Genentech, Merck
Research Funding: Merck, Bristol-Myers Squibb, GlaxoSmithKline, Novartis, Amgen, OncoSec
Razelle Kurzrock
Stock or Other Ownership: RScueRx
Honoraria: Lynx Group, Cedars-Sinai, Jubilant Biosys, National Comprehensive Cancer Network, Janssen, Dedham Group, Frankel Group, Translational Science–Segal Cancer Center, Clearview Healthcare Partners, American Society of Clinical Oncology, Cancer Therapy and Research Center–External Advisory Board San Antonio, University of Arizona, Merck, American Association for Cancer Research, Cancer Treament Centers of America, SAIC National Cancer Institute Coordinating Center for Clinical Trials Investigational Drug Steering Committee, Usha Mahajani Foundation, Sequenom, Pfizer, Genetech
Consulting or Advisory Role: Sequenom
Research Funding: EMD Serono, Centocor Ortho Biotech, EMD Serono, GlaxoSmithKline, Exelixis, XBiotech, GlaxoSmithKline, Novartis, Merck, Roche/Genentech, EMD Serono, Genetech
Patents, Royalties, Other Intellectual Property: Patent, Patent, Patent, Patent
Travel, Accommodations, Expenses: WIN, Sequenom, American Association for Cancer Research, Caris Life Sciences
Mariaelena Pierobon
Stock or Other Ownership: Theranostics Health
Consulting or Advisory Role: Perthera
Research Funding: GlaxoSmithKline
Patents, Royalties, Other Intellectual Property: Theranostics Health, George Mason University, Istituto Superiore di Santa (Italy)
Peng Sun
Employment: GlaxoSmithKline, Novartis
Stock or Other Ownership: GlaxoSmithKline
Elizabeth Cunningham
Employment: GlaxoSmithKline, Merck
Stock or Other Ownership: GlaxoSmithKline, Merck
Shonda Little
Employment: GlaxoSmithKline, Johnson and Johnson
Stock or Other Ownership: GlaxoSmithKline, Johnson and Johnson
Keith Orford
Employment: GlaxoSmithKline
Stock or Other Ownership: GlaxoSmithKline
Monica Motwani
Employment: GlaxoSmithKline, Abbvie
Stock or Other Ownership: GlaxoSmithKline
Yuchen Bai
Employment: GlaxoSmithKline, Johnson and Johnson
Kiran Patel
Employment: GlaxoSmithKline
Stock or Other Ownership: GlaxoSmithKline
Alan P. Venook
Consulting or Advisory Role: Gilead Sciences
Research Funding: Bayer (Inst), Onyx Pharmaceuticals (Inst), Genentech/Roche (Inst), Novartis (Inst)
Patents, Royalties, Other Intellectual Property: Royalties from Now UptoDate for authoring and maintaining two chapters
Travel, Accommodations, Expenses: Halozyme, Merck Serono, Genentech/Roche, Bristol-Myers Squibb
Scott Kopetz
Consulting or Advisory Role: Amgen, Roche, GlaxoSmithKline, Janssen, Bristol-Myers Squibb, Agendia, Merrimack, Sysmex, Bayer, Taiho Pharmaceutical, sanofi-aventis, Array BioPharma
Research Funding: Roche, Amgen, GlaxoSmithKline, sanofi-aventis, Sysmex, Biocartis, Guardant Health, Agendia
REFERENCES
- 1.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 2.Tie J, Gibbs P, Lipton L, et al. Optimizing targeted therapeutic development: Analysis of a colorectal cancer patient population with the BRAF(V600E) mutation. Int J Cancer. 2011;128:2075–2084. doi: 10.1002/ijc.25555. [DOI] [PubMed] [Google Scholar]
- 3.Samowitz WS, Sweeney C, Herrick J, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res. 2005;65:6063–6069. doi: 10.1158/0008-5472.CAN-05-0404. [DOI] [PubMed] [Google Scholar]
- 4.Yokota T, Ura T, Shibata N, et al. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br J Cancer. 2011;104:856–862. doi: 10.1038/bjc.2011.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Price TJ, Hardingham JE, Lee CK, et al. Impact of KRAS and BRAF gene mutation status on outcomes from the phase III AGITG MAX trial of capecitabine alone or in combination with bevacizumab and mitomycin in advanced colorectal cancer. J Clin Oncol. 2011;29:2675–2682. doi: 10.1200/JCO.2010.34.5520. [DOI] [PubMed] [Google Scholar]
- 6.Bokemeyer C, Van Cutsem E, Rougier P, et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: Pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48:1466–1475. doi: 10.1016/j.ejca.2012.02.057. [DOI] [PubMed] [Google Scholar]
- 7.Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 8.Morris V, Overman MJ, Jiang ZQ, et al. Progression-free survival remains poor over sequential lines of systemic therapy in patients with BRAF-mutated colorectal cancer. Clin Colorectal Cancer. 2014;13:164–171. doi: 10.1016/j.clcc.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117:4623–4632. doi: 10.1002/cncr.26086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst. 2013;105:1151–1156. doi: 10.1093/jnci/djt173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dienstmann R, Guinney J, Delorenzi M, et al. Colorectal Cancer Subtyping Consortium (CRCSC) identification of a consensus of molecular subtypes. J Clin Oncol. 2014;32(suppl 15s):215s. abstr 3511. [Google Scholar]
- 13.Goldstein J, Tran B, Ensor J, et al. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H) Ann Oncol. 2014;25:1032–1038. doi: 10.1093/annonc/mdu100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: A phase 1 dose-escalation trial. Lancet. 2012;379:1893–1901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 20.Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 2012;13:782–789. doi: 10.1016/S1470-2045(12)70269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 22.Kopetz S, Desai J, Chan E, et al. Phase II pilot study of vermurafenib in patients metastatic, BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33:4032–4038. doi: 10.1200/JCO.2015.63.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Infante JR, Fecher LA, Falchook GS, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012;13:773–781. doi: 10.1016/S1470-2045(12)70270-X. [DOI] [PubMed] [Google Scholar]
- 24.Corcoran RB, Dias-Santagata D, Bergethon K, et al. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal. 2010;3:ra84. doi: 10.1126/scisignal.2001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corcoran RB, Ebi H, Turke AB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montero-Conde C, Ruiz-Llorente S, Dominguez JM, et al. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013;3:520–533. doi: 10.1158/2159-8290.CD-12-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson DB, Flaherty KT, Weber JS, et al. Combined BRAF (dabrafenib) and MEK inhibition (trametinib) in patients with BRAF V600–mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014;32:3697–3704. doi: 10.1200/JCO.2014.57.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCarty KS, Jr, Szabo E, Flowers JL, et al. Use of a monoclonal anti-estrogen receptor antibody in the immunohistochemical evaluation of human tumors. Cancer Res. 1986;46(suppl):4244s–4248s. [PubMed] [Google Scholar]
- 30.Wulfkuhle JD, Berg D, Wolff C, et al. Molecular analysis of HER2 signaling in human breast cancer by functional protein pathway activation mapping. Clin Cancer Res. 2012;18:6426–6435. doi: 10.1158/1078-0432.CCR-12-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Falchook GS, Long GV, Kurzrock R, et al. Dose selection, pharmacokinetics, and pharmacodynamics of BRAF inhibitor dabrafenib (GSK2118436) Clin Cancer Res. 2014;20:4449–4458. doi: 10.1158/1078-0432.CCR-14-0887. [DOI] [PubMed] [Google Scholar]
- 32.Faber AC, Corcoran RB, Ebi H, et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011;1:352–365. doi: 10.1158/2159-8290.CD-11-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corcoran RB, Rothenberg SM, Hata AN, et al. TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF-mutant melanoma. Sci Transl Med. 2013;5:196ra198. doi: 10.1126/scitranslmed.3005753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Celtikci B, Carson R, Johnston PG, et al. The role of JAK1/2-STAT3 as acute resistance mechanism to MEK inhibition in BRAF-mutant colorectal cancer cell lines. J Clin Oncol. 2014;32(suppl 15s):236s. abstr 3594. [Google Scholar]
- 36.Ahronian LG, Sennott EM, Van Allen EM, et al. Clinical acquired resistance to RAF inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discov. 2015;5:358–367. doi: 10.1158/2159-8290.CD-14-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 38.Yaeger R, Cercek A, O'Reilly EM, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21:1313–1320. doi: 10.1158/1078-0432.CCR-14-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tabernero J, Chan E, Baselga J, et al. VE-BASKET, a Simon 2-stage adaptive design, phase II, histology-independent study in nonmelanoma solid tumors harboring BRAF V600 mutations (V600m): Activity of vemurafenib (VEM) with or without cetuximab (CTX) in colorectal cancer (CRC) J Clin Oncol. 2014;32(suppl 15s):217s. abstr 3518. [Google Scholar]
- 40.Atreya C, Van Cutsem E, Bendell J, et al. Updated efficacy of the MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E mutated (BRAFm) metastatic colorectal cancer (mCRC) J Clin Oncol. 2015;(suppl 15s):33. abstr 103. [Google Scholar]
- 41.Schellens J, van Geel R, Bendell J, et al. Final biomarker analysis of the phase I study of the selective BRAF V600 inhibitor encorafenib (LGX818) combined with cetuximab with or without the α-specific PI3K inhibitor alpelisib (BYL719) in patients with advanced BRAF-mutant colorectal cancer. Presented at the 106th Annual Meeting of the American Association for Cancer Research; April 18-22, 2015; Philadelphia, PA. [Google Scholar]
- 42.Hong D, Morris V, El Osta B, et al. Phase Ib study of vemurafenib in combination with irinotecan and cetuximab in patients with BRAF-mutated metastatic colorectal cancer and advanced cancers. J Clin Oncol. 2015;(suppl 15s):33. abstr 3511. [Google Scholar]
- 43.Mao M, Tian F, Mariadason JM, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res. 2013;19:657–667. doi: 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kopetz S, Lemos R, Powis G. The promise of patient-derived xenografts: The best laid plans of mice and men. Clin Cancer Res. 2012;18:5160–5162. doi: 10.1158/1078-0432.CCR-12-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.