

Abstract
Failure of embryonic neural tube closure results in the second most common class of birth defects known as neural tube defects (NTDs). While NTDs are likely the result of complex multigenic dysfunction, it is not known whether polymorphisms in epigenetic regulators may be risk factors for NTDs. Here we characterized Baf155msp3, a unique ENU-induced allele in mice. Homozygous Baf155mps3 embryos exhibit highly penetrant exencephaly, allowing us to investigate the roles of an assembled, but malfunctional BAF chromatin remodeling complex in vivo at the time of neural tube closure. Evidence of defects in proliferation and apoptosis were found within the neural tube. RNA-Seq analysis revealed that surprisingly few genes showed altered expression in Baf155 mutant neural tissue, given the broad epigenetic role of the BAF complex, but included genes involved in neural development and cell survival. Moreover, gene expression changes between individual mutants were variable even though the NTD was consistently observed. This suggests that inconsistent gene regulation contributes to failed neural tube closure. These results shed light on the role of the BAF complex in the process of neural tube closure and highlight the importance of studying missense alleles to understand epigenetic regulation during critical phases of development.
Keywords: neural tube defect, epigenetic regulation, Baf155msp3, apoptosis, proliferation
Introduction
Neural tube defects (NTDs) result when the embryonic neural tube, which gives rise to the adult brain and spinal cord, fails to close completely. NTDs are the second most common birth defect in humans, occurring in 1 in 1000 live births worldwide (Copp, 2005; Detrait et al., 2005). NTDs include caudal neural tube closure defects such as spina bifida and cranial NTDs such as exencephaly. These devastating birth defects occur due to disruption in the intricate process of neural tube closure, which requires the coordination of many molecular and cellular functions. The complexity of this process is illustrated by the fact that over 200 mutations in the mouse have been identified that result in NTDs (Harris and Juriloff, 1999; Harris and Juriloff, 2010). These affected genes play roles in multiple processes, including cell cycle regulation, neurogenesis, cell viability, developmental signaling, and epigenetic regulation of transcription (Copp and Greene, 2010). Despite the body of knowledge from studies in animal models, very few gene mutations have been identified in human NTDs using a single gene approach. Moreover, based on twin studies and epidemiological studies, NTDs in humans are likely the result of multiple genetic alterations and therefore many genes would be expected to contribute to this complex disease etiology (Leck, 1974; Elwood et al., 1992). It is possible that studying global gene modifiers might contribute to our understanding of the causation of NTDs. Epigenetic mediators represent a good candidate for global gene expression modifiers, and their role in the NTD disease etiology has been poorly characterized.
DNA exists in the nucleus as chromatin, a highly ordered structure, which consists of organized units called nucleosomes, DNA wrapped around a histone protein octomer (Luger et al., 1997). Epigenetics refers to heritable changes in gene transcription that are independent of DNA sequence, most commonly thought of at the level of chromatin. There are two main types of epigenetic processes—covalent modifications that are directly added to the chromatin (DNA methylation and histone modifications) and movement of nucleosomes along the DNA (chromatin remodeling). Chromatin remodeling requires an ATPase to hydrolyze ATP for energy to disrupt histone-DNA contacts and move chromatin proteins along the DNA. Overall, this process alters chromatin structure and changes DNA accessibility to transcriptional regulatory factors (Clapier and Cairns, 2009). The dynamic process of neural tube closure requires rapid changes in gene expression, which might be achieved through chromatin remodeling. NTDs can arise as a consequence of mutations in genes that mediate epigenetic regulation (Harris and Juriloff, 2010). Of the >200 mouse NTD mutants, ∼5% involve disruptions in genes encoding epigenetic regulators, which are involved in all facets of epigenetic regulation. This includes DNA methylation [Dnmt3l (Hata et al., 2002), Dnmt3b (Okano et al., 1999)], histone methylation or acetylation (Kat2a [Gcn5] (Bu et al., 2007), Cbp (Tanaka et al., 2000), p300 (Yao et al., 1998), Hdac4 (Vega et al., 2004), Sirt1 (Cheng et al., 2003)), and chromatin remodeling (Smarcc1 (Kim et al., 2001), Smarca4 (Bultman et al., 2000), Cited2 (Dunwoodie et al., 1998; Bamforth et al., 2001), Cerc2 (Banting, 2004)). Smarcc1 and Smarca4 encode BAF155 and BRG1, respectively, core components of an ATP-dependent chromatin remodeling complex.
The mammalian BRG1/BRM associated factor ATP-dependent chromatin remodeling complex (BAF complex) is estimated to contain 15 protein subunits encoded by 26 genes (Ronan et al., 2013) and is part of the Swi/Snf family of chromatin remodelers originally described in yeast (Nasmyth, 1987). In many organisms, including mice and humans, investigation of the BAF chromatin remodeling complex in different cell types indicates significant heterogeneity in subunit association. For example, the BAF complex is composed of different protein isoforms in embryonic stem (ES) cells, developing cardiomyocytes, and neural progenitor cells, suggesting there are tissue and cell-type specific roles for the complex during development (Ho and Crabtree, 2010). However, the core components of the complex, ATPase BRG1 or BRM, along with BAF155, BAF170, and BAF47/INI5, have been isolated from all cell types studied to date and can remodel nucleosomes in vitro at the same efficiency as the fully intact BAF chromatin remodeling complex (Phelan et al., 1999). This core set of BAF proteins is particularly important in vivo, as complete loss of BRG1, BAF155, or BAF47 all result in peri-implantation embryonic death and heterozygous loss of expression can lead to variable penetrance cranial NTDs (Bultman et al., 2000; Kim et al., 2001; Mandel and Gozes, 2007; Ho et al., 2009).
The core component BAF155 (encoded by the Smarcc1 gene) also plays an important role in maintenance of the BAF complex. BAF155 protects BAF complex proteins from degradation and maintains their nuclear localization (Chen and Archer, 2005; Sohn et al., 2007). BAF155 and BRG1 show a near-perfect overlap of association on the ES cell genome (Ho et al., 2009), indicating assembled complexes on chromatin. While it is known that BAF155 is important during early development (Kim et al., 2001; Sun et al., 2007) it has been difficult to study the function of the protein during this time due to the early lethality of Baf155 null mouse embryos. Twenty percent of BAF155 heterozygous embryos exhibit exencephaly with increased cranial proliferation observed 4 days after the time of neural tube closure (Kim et al., 2001). However, the role of BAF155 has not been determined during the time of neural tube closure.
Here, we characterize a missense mutation allele of the Smarcc1 gene (called Smarcc1msp3 or Baf155msp3). Mice homozygous for the Baf155msp3 allele show 81% incidence of exencephaly. The BAF155msp3 protein is expressed and the BAF core complex can assemble in vivo, providing a unique means to study the function of the BAF chromatin remodeling complex during embryogenesis. The molecular basis of the NTD is not due to aberrant BAF complex localization within the cell, disruption of neural tube patterning, or premature differentiation. However, proliferation and apoptosis are misregulated in the mutants, suggesting a potential mechanism underlying the NTD. Gene expression analysis of cranial tissue at the time of neural tube closure shows that genes involved in cell survival and neuronal development are misregulated and that mutant tissue shows significantly more variability in gene expression than wild-type (WT). Overall, these studies lend new insight into the functions of the BAF chromatin remodeling complex during the critical stage of neural tube closure.
Materials and Methods
Mouse Strains
Baf155msp3 (Smarcc1msp3) was originally identified on a mixed genetic background (BALB/cJ; C57BL/6J) in an ENU screen (Buac et al., 2008). The mutation in Smarcc1 was identified as described in the results. Mice have been maintained on a mixed C57BL/6J:129S1/SvlmJ background as a 3rd generation cross and heterozygous carriers were mated to produce the embryos and results presented here. Further crossing into C57BL/6J results in more penetrant developmental delay. For timed pregnancies, noon of the day of an observed vaginal plug was designated E0.5. At dissection, the embryonic phenotype was recorded and a portion of the yolk sac used for genotyping.
Genotyping
DNA samples were genotyped using a custom TaqMan assay (Applied Biosystems) with Taqman probes designed across the site of the msp3 mutation specific for both the WT allele (Vic-CTC-CTG-TTG-TAA-CTG-C) and the ENU induced mutant allele (Fam-CTC-CTG-TTT-TAA-CTG-C). The following primers were used, forward primer: TTT-GCA-GAT-GAG-CAG-GAT-GAA-GAA and reverse primer: TCT-CAT-TTC-AGG-CCT-AAA-TAA-ACT-TTT-ACC-T. PCR reactions were carried out in 2× Taqman Universal Fast PCR Master Mix (Applied Biosystems, 4366072), 10 μM each genic primer, and 100 nM of each allele-specific probe. Cycling conditions were 95°C for 10 min, 40 cycles of 95°C for 3 s, and 60°C for 30 min. Relative quantitation of the two alleles was determined in an endpoint assay for genotyping.
Analysis of Mutant Phenotype
Embryos were fixed in 4% paraformaldehyde (PFA) in PBS and processed for whole mount or cryosection (8 μm) in situ hybridization. Whole-mount and section RNA in situ hybridizations were performed as described (Holmes and Niswander, 2001) with digoxygenin-labeled antisense riboprobes. Sections were imaged on a Nikon Eclipse 80i microscope. For immunohistochemistry, embryos were dissected and fixed in 4% PFA in PBS, washed 3 times with PBS, cryopreserved in 30% sucrose, embedded in OCT, and sectioned (8 μM). For cell cycle analysis, BrdU (50 mg/kg) in PBS was injected intraperitoneally into pregnant dams 30 min prior to euthanasia and embryo processing. For apoptosis, the Apop-tag In Situ Apoptosis Detection Kit (Millipore, S7160) was used following the protocol to indirectly detect apoptotic cells by the TUNEL method. For immunostaining, tissue sections were treated with PBS/3% goat serum for 1 h at room temperature. Primary antibody was added and incubated at 4°C overnight. For detection, Alexa-conjugated secondary antibodies (Molecular Probes) were incubated for 1 h at room temperature. Hoechst was added along with the secondary antibodies as a nuclear stain. Stained sections were imaged with Zeiss LSM510 META confocal microscope.
Antibodies Used
Antibodies against the following proteins were used: TUJ1 (Covance, MMS425P, 1:500), phospho-histone H3 (Cell Signaling, 9701, 1:200), BrdU (Novus Biologicals, NB500-169, 1:200), p75NTR (Promega, G3231, 1:100), BRG1 (Santa Cruz, SC-17796 1:200), BAF155 (Santa Cruz, SC-10756, 1:200), BAF170 (Santa Cruz, SC17838, 1:200), and BAF60a (BD Transduction Laboratories, 611728, 1:500). Antibodies from the Developmental Studies Hybridoma Bank were used at a 1:10 dilution including PAX3 and PAX6.
Immunoprecipitation Assay and Western Blots
Embryonic day E11.5 embryos were placed in lysis buffer (50 mM Tris-HCl at pH7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, including 1 tablet protease inhibitor (Roche Complete) per 10 mls lysate) and incubated on ice for 10 min. Tissue debris was pelleted at 12,500 rpm for 10 min at 4°C, and the supernatant was incubated with primary antibodies overnight at 4°C. The lysates were incubated with Protein A or G Sepharose beads for 2 h, followed by washing of the immunoprecipitates three times with lysis buffer and elution of bound proteins in SDS–PAGE sampling buffer for 10 min at 100°C. Western blots were performed as described previously (Chen et al., 2005) using primary antibodies as listed. Secondary antibodies used were goat anti-rabbit (172–1019, Bio-Rad), and goat anti-mouse (172- 1011, Bio-Rad).
RNA-Seq Library Preparation and Analysis
Three somite matched E9.5 embryos were collected of both Baf155+/+ and Baf155Mssp3/Mssp3 alleles. Cranial tissue above the pharyngeal arches and otic vesicles was harvested and stored at −80°C until all samples were collected. RNA was isolated using the RNEasy mini kit from Qiagen (74106). On average, 1400 ng of total RNA was obtained per sample. RNA concentration and A260/A280 ratio was analyzed using the Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific). Libraries were made using the Illumina TruSeq RNA Sample Preparation Kit V2 (RS-122-2001). Libraries were analyzed on the 2100 Bioanalyzer (Agilent Technologies) for proper size and integrity. For high-throughput RNA-Sequencing analysis, the UC Denver Genomics Core used an Illumina High Seq 2000. Mapping and bioinformatics analysis was done in collaboration with the Bioinformatics Shared Resources. The raw data from the RNA set results are available through GEO (accession GSE53459). The sequenced libraries generated 804,339,023 unfiltered reads of ∼100 bps in length. Each replicate represents at least 96.9 million reads, a density sufficient for qualitative analysis of gene expression (Phelan et al., 1999). The ∼100 bp reads were aligned to the MM9 mouse genome using Gsnap, (Wu and Nacu, 2010) allowing a mismatch of 4% and an indel cost of 2.0. The mapped transcripts were visualized in the UCSC genome browser to bioinformatically verify differential expression in transcript counts and/or transcript length, as well as splicing of introns.
Data Analysis and Interpretation
For measurements of differential gene expression, edgeR software (Robinson et al., 2010) was used. Raw read counts were established for each RefSeq gene. In cases where more than one transcript was annotated, the transcript with the highest overall counts was used as proxy for the gene in question, yielding a data table with a single count entry for each gene per sample. The data table was trimmed to remove all genes where zero counts were reported in one or more of the 6 samples, with the exception of genes where either all the mutant samples, or all the WT samples showed zero counts, and the opposing side of the paradigm showed counts in each sample. This resulted in a gene data table with 15,734 entries. The data set was used for differential expression analyses using edgeR. Comparisons of all three mutant samples versus all three WT samples were performed, as well as separate comparisons of each individual mutant sample against all three WT samples; resulting gene lists were used for Ingenuity Pathway Analysis to reveal biological features. Overall sample relationship was visualized using the multidimensional scaling feature of edgeR. Raw gene count numbers were normalized using DESeq (Anders and Huber, 2010), and normalized counts were used to calculate the coefficient of variation for each gene in the normal samples, as well as determine for each gene the relationship of the counts in each sample relative to the mean of the normalized counts derived from the three WT samples for the pertinent gene. To obtain distance from mean, the ratio of normalized counts divided by mean of the normal underwent logarithmic transformation. All genes were ranked according to their coefficient of variation, and distance from mean was plotted for each gene for normal as well as for mutant samples.
Results
Baf155msp3 Mutants Exhibit Cranial Neural Tube Defects
The Msp3 phenotype was originally discovered in a mouse ENU mutagenesis screen (Buac et al., 2008); however, the specific gene mutation was not identified. As outlined below, we identified the ENU-induced mutation in the Smarcc1 gene, encoding BAF155, a component of the Brg1/Brama-associated factor (BAF) ATP-dependent chromatin remodeling complex (BAF complex). For the remainder of the paper we refer to the mutant gene as Baf155msp3 and the protein as BAF155msp3. Baf155msp3/+ heterozygotes are indistinguishable from WT littermates and they are viable and fertile. Homozygous Baf155msp3/msp3 mutant embryos display cranial NTDs and developmental delay (combined phenotypes at 92% penetrance, exencephaly at 81% frequency and developmental delay with and without exencephaly at 30% frequency). on a mixed C57BL/6J; 129SvlmJ background (Fig. 1 and Table 1). The embryonic phenotypes range in severity and include exencephaly and developmental delay, defined as being at least 2 somites less than the least somite number of the non-mutant littermates [Fig. 1(A–C)]. For example, Figure 1(A) shows embryos from a single litter at embryonic day 9.5 (E9.5), just after the time of neural tube closure. Mutant embryo 1 (MUT 1) is exencephalic from the forebrain through the hindbrain but not delayed (25 somites), MUT 2 shows midbrain and hindbrain exencephaly and developmental delay (21 somites), MUT 3 is severely delayed (14–15 somites), and hence cranial neural closure cannot be scored. Other observed defects are an enlarged pericardium (∼12% of mutants) and, at later stages, variable epidermal edema. Most homozygous mutants can survive until E15.5 but no homozygotes are found at the time of weaning (Table 1). The cause of death in the Baf155msp3/msp3 mutants is currently unclear and was not explored here.
Figure 1.

Baf155msp3/msp3 embryos show neural tube defects. (A–C) Lateral views of embryos at the indicated stages. (A) Within a single litter, three individual homozygous Baf155msp3 mutant (MUT1–3) embryos show a range of phenotypes including failure of neural tube closure and developmental delay as compared to wild-type (WT). (B) E12.5 Baf155msp3/msp3 mutant shows exencephaly without other apparent defects. (C) E15.5 mutant with exencephaly and coloboma. Red scale bar=1 mm, blue scale bars=2 cm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Table 1. Phenotype and Genotype of Embryos Resulting from Cross Between Heterozygous Baf155msp3/+ Mice.
| +/+ | +/− | −/− | Phenotype of Baf155msp3/msp3 Embryos | |||
|---|---|---|---|---|---|---|
|
| ||||||
| Exencephaly [with dev delay] | dev Delay [unable to score exencephaly] | Normal Morphology | ||||
| E9.5 | 50 | 114 | 49 | 41 [9] | 5 | 3 |
| E10.5 | 38 | 63 | 37 | 30 [8] | 3 | 4 |
| E11.5 | 6 | 18 | 9 | 8 [4] | – | 1 |
| E12.5 | 12 | 15 | 6 | 4 [1] | 2 | – |
| E13.5 | 6 | 12 | 8 | 6 [0] | 2 | – |
| E14.5 | 2 | 4 | 3 | 2 [0] | 1 | – |
| E15.5 | – | 2 | 2 | 2 [0] | – | – |
| E16.5 | 2 | 4 | – | – | – | – |
| Adult | 18 | 33 | – | – | – | – |
Mapping the ENU-Induced Mutation to Baf155
Baf155msp3 was originally identified on a mixed genetic background (BALB/cJ; C57BL/6J) in an ENU mutagenesis screen and localized to mouse chromosome 9 using traditional linkage mapping (Buac et al., 2008). During subsequent outcrossing to C57BL/6J, the critical interval was narrowed to a 2 Mb region (rs13480408-D9Mit37; NCBI Build36 chr9: 108,655,684-110,607,406). Resequencing of exons in the interval was completed using an exon-based hybrid selection strategy (Illumina Genome Analyzer, Broad Institute, Mouse Mutant Resequencing Initiative). Filtering of coding or splicing homozygous variants not present in dbSNP revealed a single variant in Smarcc1 that was not present in the parental BALB/cJ strain. This C to A variant was confirmed with Sanger sequencing and a TaqMan SNP genotyping assay. Subsequently, mice were crossed with 129SvlmJ to generate a mixed C57BL/6J;129SvlmJ background and RNA-Seq (Illumina Hi Seq 2000) was performed on WT and mutant cranial tissue (results discussed below), and the resulting data was analyzed for variants. Variants were filtered similar to the first analysis overlapping the linkage disequilibrium region between 103 and 110 Mb on chromosome 9 (defined by the variant calling program). Nineteen nonsynonymous SNPs were identified, but only the Smarcc1 C to A variant was novel and not present in dbSNP. These two independent genomic analyses identify the causative ENU-induced mutation in Smarcc1, encoding BAF155.
This C to A substitution is predicted to cause a threonine to lysine substitution at amino acid 416 of BAF155 [NP_033237; Fig. 2(A)]. The BAF155 protein is comprised of one Chromo domain, one SWIRM domain, and one SANT domain. The affected amino acid is not within a conserved domain but is located N-terminal to the SWIRM domain in the BAF155 protein. The missense mutation site and surrounding sequences are conserved between H. sapien, M. musculus, R. norvegicus, and D. rerio, but not in S. cerevisiae [Fig. 2(A)], suggesting this residue may be functionally important.
Figure 2.
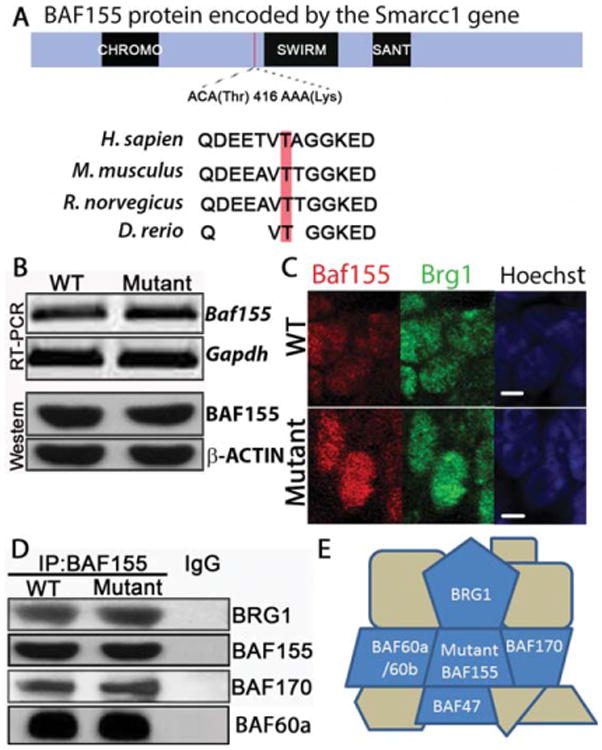
BAF155msp3 associates with other core BAF complex proteins. (A) Schematic of the protein domain structure of BAF155. The msp3 ENU-induced mutation causes a C to A change at Chr9 bp 110022668 (Build 36) in the Smarcc1 gene and leads to a threonine to lysine conversion of amino acid 416 in the BAF155 protein. This missense mutation alters an evolutionarily conserved amino acid in a conserved region, but not in a known structural domain. (B) RT-PCR (top) and Western blot (bottom) analysis of Baf155 expression from E11.5 cranial tissue lysate. Gapdh and β-Actin serve as internal standards. (C) Confocal microscope images of neuroepithelial sections of E9.5 WT and mutant cranial neural tubes. Panels represent wild-type and mutant nuclei stained with antibodies against BAF155 (red) and BRG1 (green). Hoechst stains nuclei (blue). White scale bars = 2 μm. (D) Immunoprecipitation of protein extracts from E11.5 mutant and wild-type embryos using anti-BAF155 antibody followed by western blotting with anti-BRG1, anti-BAF155, anti-BAF170 and anti-BAF60a. (E) Schematic of the BAF complex proteins associated with the mutant BAF155 protein. BAF155 T416K associates with BRG1 BAF60a and BAF170 (IP), and BAF47 and BAF60a/b (yeast two-hybrid, supplemental data). BRG1, BAF170, BAF47, and BAF155 are considered core complex proteins.
Mutant Baf155 RNA and Protein are Present and the Protein is Localized to the Nucleus
Missense mutations within protein-coding genes may affect mRNA stability, as well as protein stability or function. To understand the molecular consequences of the C to A missense mutation in the Baf155 gene, we analyzed mRNA and protein expression from E11.5 mutant and WT embryos. RT-PCR analysis showed the presence of Baf155 mRNA in Baf155msp3/msp3 embryos indicating that the transcript is not subject to nonsense-mediated decay [Fig. 2(B)]. Furthermore, Western blot analysis confirmed the presence of full length BAF155 in homozygous mutant embryos at similar levels to WT [Fig. 2(B)]. Together, these data suggest that BAF155 mRNA and protein stability are not compromised in the Baf155 mutant.
The BAF complex is normally localized to the nucleus. Mutations in the BAF155 nuclear localization signal (NLS) result in incorrect cytoplasmic localization of BAF155, BAF47, and the ATPase BRG1, indicating that BAF155 maintains the subcellular localization of the BAF complex in the nucleus (Sohn et al., 2007). To test the possibility that subcellular localization of BAF155 and BRG1 may be compromised in the Baf155msp3 mutants, we examined their localization in cryosections from WT and mutant embryos. In mutants, BRG1 and BAF155 are present in the nucleus at high levels while they are not detected in the cytoplasm, comparable to WT [Fig. 2(C)]. This suggests that subcellular mislocalization of BAF155 or other BAF proteins does not account for the functional deficit seen in Baf155msp3/msp3 mutants.
BAF155msp3 Mutant Protein Can Still Associate with the Core Remodeling Complex
BAF155, along with ATPase BRG1, BAF170, and BAF47, are considered core components in the BAF remodeling complex because they are sufficient to remodel nucleosomes in vitro at the same efficiency as the fully intact BAF remodeling complex (Phelan et al., 1999). BAF155 interacts directly with two core complex proteins (BRG1 and BAF47) as well as with BAF60a (Sohn et al., 2007) in the region that encompasses the BAF155msp3 missense mutation, T416K. To test the possibility that the interaction between BAF155 and the BAF core complex is disrupted by the T416K mutation we performed coimmunoprecipitation assays using E11.5 mutant and WT embryos. These studies revealed that BRG1, BAF170, and BAF60a still associate with BAF155msp3 in vivo and in a similar ratio as compared to the WT, despite the amino acid change [Fig. 2(D)]. Additionally, yeast two-hybrid analysis indicated that BAF155msp3 can still interact directly with BAF47 and BAF60a in vitro (Supporting Information Fig. 1). These experiments indicate that the mutant BAF155msp3 protein still interacts with the core components of the BAF remodeling complex and suggest the mutant protein could interact with other BAF complex proteins in vivo as well [summarized in Fig. 2(E)]. These protein interaction data, as well as the longer survival of Baf155msp3/msp3 embryos relative to Baf155 null embryos (Kim et al., 2001; Sun et al., 2007) suggest that the BAF155msp3 mutant protein is present and can interact molecularly in the remodeling complex, but its function may be partially compromised. Thus, it appears that Baf155msp3 is a missense allele and the mouse may exhibit a hypomorphic phenotype.
Cell Proliferation is Decreased and Cell Death is Increased in Baf155msp3/msp3 Neural Progenitor Cells
NTDs can result when the spatiotemporal control of neural tube closure is disrupted (Copp and Greene, 2010) due to any of several different mechanisms including: reduced neural progenitor cell proliferation, changes in cell fate, decreased cell survival, or defects in patterning. To investigate if the NTD might arise because of improper patterning, we examined a battery of molecular markers that reflect the dorsal–ventral or anterior–posterior axis patterning of E9.5 embryos, just after closure of the neural tube. Antibody staining and RNA in situ hybridization showed no change in the expression pattern of Shh, PAX6, PAX3, Fgf8, and FoxG1 in mutant neural tubes when compared to WT somite matched embryos [Fig. 3(A–F)]. Neural crest cells are specified during neural tube closure and abnormal expression of neural crest genes could also reflect altered patterning (Trainor, 2005). Therefore, we examined markers characteristic of early neural crest specification including p75NTR, Tfap2a, and Sox10 [Fig. 3(G–I)] and again found no change in the expression pattern as compared to WT. These results suggest that neural patterning is not disrupted in the Baf155msp3 mutant.
Figure 3.

BAF155 function is not necessary for early neural patterning. (A–C) Transverse sections of E9.5 embryos followed by immunofluorescence for dorsal/ventral markers Sonic Hedgehog (Shh) (A), NKX2.2 (B), and PAX3 (C). (D, E) Lateral and (F) frontal views of whole mount RNA in situ hybridization on E10.0 WT and Baf155msp3/msp3 mutant embryos with probes to anterior/posterior markers Fgf8 (D) and FoxG1 (E and F). (G) Confocal images of transverse sections of E10.5 WT and mutant embryos incubated with anti-p75NTR antibody to visualize the enteric neural crest cells. Hoechst stains nuclei. (H) Transverse sections of E10.5 rostral spinal cords hybridized with TFAP2α probe marking early migrating neural crest cells. (I) Lateral view of E10.5 WT and mutant embryos hybridized with Sox10 probe to mark early neural crest cells. White scale bars = 100 μm, Red scale bars = 1 mm.
The final step in neural tube closure is the apposition of the two neural folds, which grow and bend closer together partially by increasing the number of neural progenitor cells, and finally, when the two folds meet, tissue fusion. The tight control of cell proliferation is necessary for the neural folds to meet properly at the fusion point. At E9.5, the vast majority of the neural ectoderm is comprised of proliferating neural progenitor cells. Too much or too little cell proliferation can abrogate neural tube closure. Previous studies noted an increase in cell proliferation in the telencephalic striatum region of exencephalic heterozygous Baf155null embryos at E13.5 (Kim et al., 2001). Thus, we asked if the proliferation rate might be altered in Baf155msp3/msp3 mutant embryos at the time of neural tube closure. The mitotic rate of neuroepithelial cells in somite-matched E9.5 Baf155+/+ and mutant Baf155msp3/msp3 embryos was compared using phospho-Histone 3 (pH 3). In the hindbrain, there was a statistically significant decrease in the mitotic index (pH 3+ cells/1000 neuroepithelial cells) of Baf155msp3/msp3 mutants as compared to WT (20 ± 9 vs. 71 ± 10) [Fig. 4(A,D)]. We also quantified the number of cells in S phase by performing bromodeoxyuridine (BrdU) incorporation experiments [Fig. 4(A,E)]. There was no apparent difference in number of cells in S phase in mutant as compared to WT neural tubes at E9.5. These studies suggest that cells are cycling in the Baf155msp3 mutant, but there may be a mitosis defect in the neuroepithelium.
Figure 4.
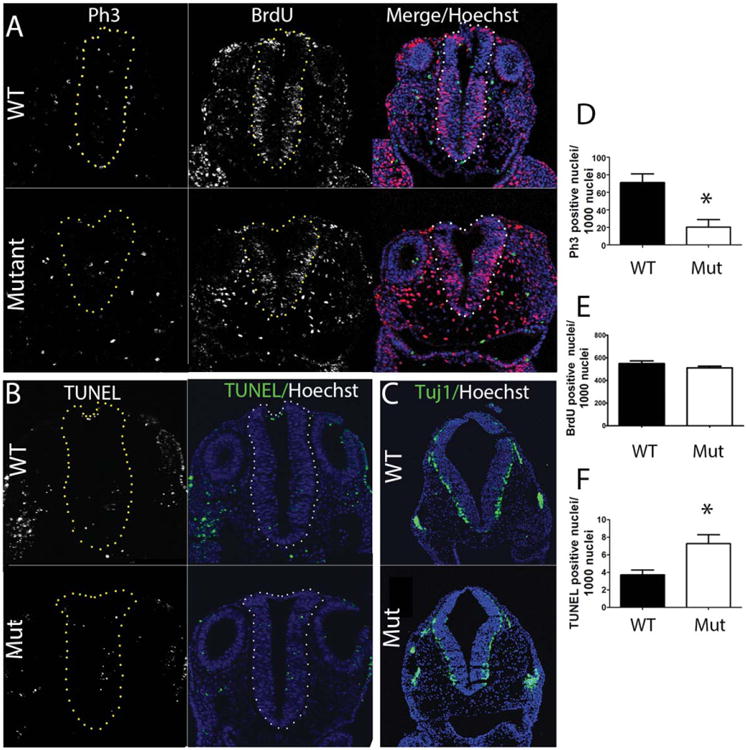
BAF155 function is necessary to maintain proliferation and cell survival. (A) Transverse sections of E9.5 hindbrains of WT and Baf155msp3 mutant embryos followed by immunofluorescence for proliferation marker p-H3 (green), S phase marker BrdU (half hour BrdU incorporation) (red) and merged image with Hoechst stained nuclei. (B) Transverse sections of E9.5 hindbrains in WT and Mutant embryos to visualize apoptotic cells by TUNEL staining. (C) Confocal images of transverse sections through rostral spinal cords. Anti-Tuj1 antibody was used to visualize neural differentiation in neuroepithelial cells. (D) Quantification of antiphospho-H3 positive cells in WT and Mutant hindbrain neural tube. (E) Quantification of anti-BrdU positive cells in WT and Mutant hindbrain neural tube. (F) Quantification of apoptosis by TUNEL staining in WT and Mutant hindbrain neural tube. Ratio of p-H3, BrdU, or TUNEL positive cells were calculated by dividing the number of positive stained cells by the total number of neuroepithelial cells, indicated by Hoechst staining, then multiplying by 1000. Error bars indicate SEM of at least 3 sections from 3 biological replicates. (*) p < 0.01, Student's t-test. The neural tube is outlined by yellow or white dots.
A decrease in cell survival within the neural tissue may result in too few cells to allow proper apposition and fusion of the neural folds, resulting in a NTD. To determine if neural progenitor cell survival was affected in mutant embryos we used a TUNEL assay to visualize apoptotic cells in somite-matched E9.5 WT and mutant neural epithelium. This showed a statistically significant increase in TUNEL positive cells per 1000 nuclei in the hindbrain at E9.5 in the Baf155msp3/msp3 mutant as compared to WT (7.2 ± 1.0 vs. 3.7 ± 0.5) [Fig. 4(B,F)]. Together, these studies suggest that BAF155 function is necessary to maintain the normal rates of proliferation and cell survival in the neuroepithelium.
As neural precursors begin to differentiate starting around E10.5, they exit the cell cycle and leave the ventricular zone to reside at the periphery of the neural tube. If differentiation does not occur at the correct time, this can contribute to an excess or a deficiency in cell number for effective neural fold juxtaposition and ultimate fusion to form the neural tube. To determine whether or not neural differentiation was compromised in the homozygous mutants we evaluated Tuj1 expression, a known neural differentiation marker, in the spinal cord and hindbrain of E10.5 somite matched Baf155+/+ and Baf155msp3/msp3 embryos. Mutant and WT embryos exhibit the same characteristic expression pattern of Tuj1 [Fig. 4(C)] and therefore neural differentiation does not appear to be affected in the homozygous mutant.
Gene Expression Levels are More Variable in Baf155msp3 Mutant Cranial Tissue
Averaged Sample Analysis
BAF155, as a component of a chromatin remodeling complex, is thought to be involved in the regulation of gene expression. Baf155 knockdown in ES cells results in misregulation of hundreds of genes (Ho et al., 2009). The Baf155msp3 allele presents a unique case for gene expression analysis wherein the BAF complex appears to be intact, in contrast to disruption of the BAF complex when BAF155 function is lost (Chen and Archer, 2005; Sohn et al., 2007). To examine gene expression in the Baf155msp3 mutant at the time of neural tube closure, we performed RNA-Seq (Geo accession GSE53459) and compared gene expression levels between homozygous WT and homozygous mutant embryos. RNA was isolated from somite matched (21–23 somites) and phenotype matched cranial tissue of homozygous WT and homozygous mutant embryos (3 of each genotype) at E9.5 just after the time of neural tube closure. At E9.5, the vast majority of the embryonic neural tube is comprised of proliferating neural progenitor cells, constituting a relatively homogeneous cell population. After extensive analysis of the RNA-Seq data comparing all 3 mutants to all 3 WT replicates, 78 genes were identified as being upregulated on average in the mutant samples while 22 genes were downregulated. The full list of differentially expressed genes in all 3 mutant replicates collectively, as well as p-values, FDR, and fold change, is shown in Supporting Information Table 1. To facilitate an understanding of global gene expression differences, ingenuity pathway analysis (IPA) was used to assign functional categories to the up-and down-regulated genes in the mutants. The most significant molecular and cellular functions of these genes were: (1) cellular movement (1.46E-04 > p > 4.12E-02), (2) cell death and survival (2.69E-04 > p > 4.12E-02), and (3) cell growth and proliferation (2.69E-04 > p > 3.41E-02). Many neuronal genes were significantly misregulated as well.
Individual Sample Analysis
We used multidimensional scaling (MDS) analysis to conduct an unsupervised examination of the relationship between the global gene expression in each individual mutant and the WT cranial tissue samples. As seen in Figure 5(A), a relationship between the genotypic subtype and gene expression was clearly observed. The WT replicates were tightly grouped in the MDS plot and showed little dimensional variance. In contrast, the mutant replicates did not group together and showed increased variance in both dimensions 1 and 2. Overall, there was a trend towards higher variance of gene expression in the mutant samples as compared to the WT samples, leading to a more dispersed MDS plot where the mutants do not cluster with the WT controls and each mutant does not cluster with the other mutants.
Figure 5.
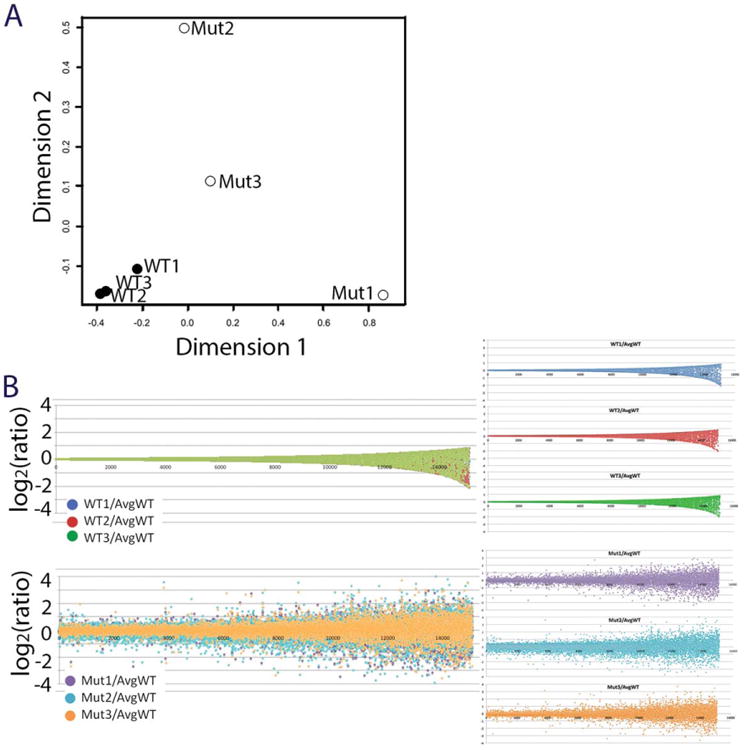
Variable gene expression in Baf155msp3/msp3 mutant cranial tissue. (A) A multidimensional scaling (MDS) plot of differentially expressed genes from cranial tissue of three somite matched WT and three somite matched Baf155msp3/msp3 mutants. (B) Genes were ranked in ascending fashion according to their coefficient of variation observed in wild-type samples, and for each gene, the distance from mean was plotted as the log2 of the ratio of the expression level of a gene in an individual sample normalized to the mean of the expression level of the respective gene in the wild-type samples. Top: All three wild-type samples; bottom: all three mutant samples; right: individual samples, from top: wild-type 1 (blue), wild-type 2 (red), wild-type 3(green), mutant 1 (purple), mutant 2 (turquoise), mutant 3 (orange).
On the basis of the lack of clustering between the individual mutants when global gene expression was compared, the averaged gene expression analysis did not illustrate the nuances of gene expression in the Baf155msp3 mutant samples. Therefore, in a more exploratory analysis, we asked if the observed transcriptional changes were uniformly distributed in the three biological replicates between the homozygous mutant group and as compared to the homozygous WT groups. For each sample, we established for each gene a “distance from mean” parameter by normalizing the expression level of any given gene to the arithmetic mean of the expression of the respective gene in the WT samples; this ratio underwent logarithmic transformation to obtain the distance value. All genes were ranked by their coefficient of variation in the WT samples, and the distance value was plotted as shown in Figure 5(B) (WT in top panel, mutants in bottom panel). The average WT plot showed a relatively tight spacing of all data points in a “trumpet” shape. This coincides well with the low degree of variation we identified in the differential expression test. The same plot for the mutant samples showed a larger spread of gene expression ratios, and mutant replicate 2 (Mut2 in turquoise) seems to have the greatest difference in the ratio of gene expression as compared to both remaining mutants and WT controls. Again, this correlates well with the differential expression tests.
To identify whether there are groups of genes that may contribute to this gene expression variability more than others, we generated three separate mutant differential gene expression lists by directly comparing each mutant gene count to the average of the WT gene counts. This analysis shows collections of genes from each individual mutant as opposed to the genes misregulated in all of the mutants. Over 2149 genes were differentially expressed in Mutant replicate 2 (Mut2) as opposed to 188 genes in Mutant replicate 1 (Mut1) or 215 genes in Mutant replicate 3 (Mut3) (based on DESeq testing, and counting genes with padj<0.05). When we directly compared these three differentially expressed gene lists, we identified a subset of 27 genes that was significantly misregulated in all of the mutants (Supporting Information Table 2). Interestingly, many of the genes on this list do not overlap with Supporting Information Table 1, comparing all mutant samples to all WT samples, because there is often one gene outlier that is not significantly misregulated in each individual. Pathway analysis using IPA suggests that no canonical pathways are shared between these 27 genes, but 6 of these genes are predicted to be within a nucleic acid metabolism network. Overall, these results suggest that gene expression in the mutants is more varied than in the WT samples, and there is little overlap in the functionality of the most variable genes between individuals in the Baf155msp3 mutant group.
Discussion
NTDs in humans have a complex etiology and it is likely that multiple genes contribute to the incidence of this common birth defect, but the specific genetic causes of NTDs are still largely unknown. Moreover, it is unknown whether polymorphisms in epigenetic regulators may be risk factors for NTDs in humans. Studies in model organisms like the mouse can provide insight into the genetics underlying NTDs. Indeed, mutations and deletions in a number of epigenetic regulators can cause NTDs in mice. In this study, we focused on the BAF chromatin remodeling complex, as mutations in two members of the BAF complex cause NTDs in mice, yet the functions of the BAF complex at the time of neural tube closure have not been studied. Here we characterized the first missense allele of the Baf155 gene, Baf155msp3, which exhibits a cranial NTD. We found that the mutant protein can still interact with the core BAF complex proteins BRG1, BAF170, BAF60a, and BAF47 and properly localize to the nucleus. This unique allele allowed an investigation of the cause of the NTD in the presence of an assembled core BAF complex. In Baf155msp3 mutant embryos, we discovered a reduction in neural progenitor cell proliferation and an increase in cell death, revealing a possible mechanism underlying the NTD. We analyzed transcriptional control in the Baf155 mutant using RNA-Seq and discovered misregulation of genes involved in apoptosis and proliferation. We also found increased variability in gene expression in the mutant as compared to WT, the meaning of which will be speculated on below.
The BAF complex can act as both a repressor and activator of genes, and has been found to refine the gene transcriptional network in ES cells to maintain pluripotency (Zhang et al., 2007; Ho et al., 2009; Schaniel et al., 2009). Using RNA-Seq, we analyzed transcriptional control in the cranial tissue of the mutant embryo as compared to WT. Most of the differentially expressed genes in the Baf155msp3 mutant embryo are upregulated, including neural development genes discussed below, suggesting that BAF155 and the BAF complex largely play a repressive role during neurulation. Consistent with the expectation that the BAF complex provides more global, rather than gene-specific, transcriptional regulation, we did not find strong single gene candidates to explain the underlying mechanism of the NTD. Instead, we observed a broader dysregulation in expression of genes and gene families that are associated with NTDs when disrupted. While the fold change in gene expression was not particularly striking across all the mutants, it is not unreasonable to suggest that small gene expression changes in combination may result in an NTD. As previously noted, neural tube closure is a highly dynamic process that requires precise coordination of complex cellular functions to proceed to completion. Moreover, many of the misregulated genes in Supporting Information Table 1 are associated with NTDs: mutations in Abl (Koleske et al., 1998), Frem2 (Jadeja et al., 2005; Timmer, 2005), as well as other genes discussed below result in NTDs.
Baf155 Regulation of Cell Survival During Neural Development
Proper control of proliferation and cell survival are critical for neural tube closure. In the Baf155msp3 mouse mutants we identified both a decrease in cell proliferation and an increase in apoptosis. RNA-Seq revealed several misregulated genes that contribute to cell survival, including the downregulated genes Abl1 and ApoB, and the upregulated genes Adora3, Fam72a, SST, and Map3k12. Mouse embryos mutant for Apolipoprotein B (ApoB) exhibit exencephaly (30% incidence) and excessive cell death in the hindbrain (Huang et al., 1995). Knock-out of Abl1 kinase in combination with Abl related gene (ARG) results in NTDs and disruption of the actin cytoskeleton of the neuroepithelium (Koleske et al., 1998). Although the mechanism underlying the increased apoptosis in Baf155msp3 mutants is unclear, altered expression of these genes may contribute to the increase of cell death in the neural epithelium of the Baf155 mutant.
BAF155 can be phosphorylated by and interact with CyclinE (Shanahan et al., 1999) in the Rb pathway as well as Akt (Foster et al., 2006) in the PI3K/AKT pathway. However, we did not detect misregulation of Rb or PI3K pathway genes in the mutant, suggesting these signaling pathways are not disrupted and BAF155 may play other transcriptional roles in controlling proliferation. While many genes known to control proliferation were misregulated in the mutant cranial tissue, no specific cyclins or cell cycle genes were differentially expressed that could account for the observed reduction in proliferation. However, there is decreased expression of the centrosomal gene Poc1a. Inhibition of centrosomal genes can lead to cell cycle defects and cell death (Doxsey et al., 2005). For example, inhibition of the centrosome protein Nde1 was shown to delay cell cycle reentry in cell lines, and to reduce mitosis in zebrafish (Kim et al., 2011). Together these data suggest that Baf155 may directly or indirectly regulate cell proliferation and cell death via transcriptional control.
Regulation of Neural Development Genes by Baf155
BAF subunits are heterogeneously expressed and assembled in various cell types and these different complexes can have distinct functions. BAF subunit mutations have been reported in many human neurologic diseases including Autism, Schizophrenia, Coffin-Siris syndrome, sporadic mental retardation (Ronan et al., 2013); however, these neurological diseases are not accompanied by NTDs according to the Online Mendelian inheritance of Man database (OMIM). Different protein assemblies of the BAF complex exist in the developing central nervous system, including the neural progenitor BAF complex (npBAF) and neuronal BAF complex (nBAF) (Lessard et al., 2007). These have specific functions in the regulation of proliferation and differentiation of mammalian neural stem cells. Later loss of BRG1 function in neurons (Brg1fl/fl;Nestin-Cre) and transcriptional analysis of E12.5 mutant brains revealed an upregulation of genes within the Notch and Shh pathways. While it is believed that BAF155 is present in BAF complexes in all cell types, we did not see gene expression changes in the Baf155msp3 mutant at E9.5 that might reflect alterations in Notch or Shh signaling. This difference in gene control between the Brg1fl/fl;Nestin-Cre and the Baf155msp3 mice could reflect temporal differences in BAF complex function during brain development. Many of the upregulated genes in Baf155msp3 mutants at the time of neural tube closure contribute to neural development, including Spon1, Doc3, Topb2, Map3k12, Cdk18, Sema3c, Sst, Igta4, Epha3, Fzd2, Fzd3, Ablim3, and Plcg2. While no differentiation defect was observed in the Baf155msp3 mutants at and shortly after the time of neural tube closure, the modest misregulation of many neural development genes may contribute to the NTD phenotype. Particularly interesting are the genes associated with NTDs, namely the Wnt receptors Fzd2 and Fzd3. The Wnt/Fzd (PCP) pathway controls aspects of neural development, including neural tube closure, in mice and humans (Juriloff and Harris, 2012). Although the Fzd2−/− mouse does not show NTD, in a double heterozygous cross with another PCP component, Fzd+/− :VanglLp/+, fully penetrant cranial NTDs are observed (Yu et al., 2010). Also, Fzd3−/−; Fzd6−/− mutants present with the most severe NTD, craniorachischisis (Wang, 2006). This supports the idea that NTDs can arise due to aberrant expression of multiple risk alleles.
Potential Roles of Baf155 in the Baf155msp3 Mutant
Hemizygous Brg1 or Baf155 mice show defects in neural tube closure, suggesting a dosage-sensitive role for BAF complexes in neural development (Bultman et al., 2000; Kim et al., 2001). In the Baf155msp3/msp3 mutant, the core components of the complex are intact and the RNA and protein levels are not significantly changed, yet the neural tube defect is highly penetrant. Our studies suggest an alternative mechanism for epigenetic control of neural tube closure, in addition to dosage of the BAF proteins. One possibility is that the BAF155msp mutation disrupts the interaction of BAF155 with transcription factors or chromatin maintenance proteins. For example, it has been suggested that intrinsic disorder within the BAF protein structures may explain diverse roles of chromatin remodeling proteins (Sandhu, 2009), and the point mutation within our allele occurs in an intrinsically disordered region based on Phyre analysis (data not shown). However, we observed no difference between the mutant and WT protein for association to BAF60, which directly interacts within the region of the Baf155 T416K mutation. It remains possible that another protein may interact within this disordered region and disruption of this interaction may lead to NTDs.
Another possibility is that interaction with specific genomic targets that regulate the developmental program of neural tube closure may be disrupted. In ES cells, BAF155 is bound to promoter or genic regions of ∼5,630 distinct genes (Ho et al., 2009). Moreover, esBAF facilitates Stat3 genomic binding, and can act either synergistically or antagonistically with the Polycomb Repressive Complex Group (PcG) in ES cells (Ho et al., 2011). In HeLa cells, BAF proteins associate with up to 77% of genic DNA (Euskirchen et al., 2011). Given the potentially large set of genomic targets, there are surprisingly few genes misregulated overall in Baf155msp mutant cranial tissue at E9.5, especially considering that at least some of these misexpressed genes are likely indirect targets. There is also very little overlap in the data-sets of differentially expressed genes between our point mutant and knock-out/knock-down of Brg1 or other BAF members in ES cells (Ho et al., 2009), neural progenitors or neurons (Lessard et al., 2007). This lack of overlap likely reflects the differences between mutant alleles: knock-down of one component presumably results in incorrect ratios of complex proteins, whereas our point mutant does not apparently change the levels of complex proteins and the complex is largely intact. Thus, the point mutation may be expected to reveal more specific targets relative to neural tube closure.
Gene Expression is More Variable in Baf155msp3 Cranial Tissue
The genes highlighted above show strong statistical significance in terms of changes in gene expression in the averaged mutant samples versus averaged WT samples. However, the RNA-Seq analysis also raised another interesting finding in that there is an increase in the degree of variability of gene expression between individual Baf155msp3/msp3 embryos. This appears to be unique to the Baf155msp3 embryos, and perhaps other epigenetic regulators, because in gene expression studies of other NTD mutants, high gene expression variability was not observed (Pyrgaki et al., 2011). On the basis of EdgeR and IPA analysis, there is little overlap in the most variable genes between individual mutants, and there does not appear to be a group of genes that mediates this variability. However, Topoisomerase 2β(Top2b) and Sirtuin7(Sirt7) (a histone deacetylase) are differently expressed in all the mutant embryos and stand out as globally acting proteins that may contribute to further differences in gene expression between the individual mutants. Top2b plays a critical role in axonal development in mice (Yang, 2000) and it has been suggested to be involved in DNA repair (Yamane et al., 1997). Sirt7 can preferentially deacetylate H3K18, interact with BRG1/BRM, and is suggested to regulate RNA Polymerase I transcription in many cell types (Barber et al., 2012; Tsai et al., 2012). It is possible that global alterations in histone acetylation could contribute to a greater variability in gene expression. Variable gene expression might also be due to differential occupancy of the BAF complex on the genome, or disruption of other proteins that interact with the BAF complex to modulate its function. Despite the lack of overlap of gene expression changes between Baf155msp3 and other BAF complex gene knockouts and the varying degree of gene expression changes between Baf155msp3 mutants, our mouse model shows a consistent NTD phenotype. Neural tube closure is a complex, multigenic process and our data indicate that inconsistent regulation of genes by a chromatin remodeling complex with reduced function can lead to developmental defects during this highly dynamic phase of embryonic development.
Supplementary Material
Acknowledgments
The authors thank members of the Niswander lab for helpful discussions throughout this project. They also thank Dr. Jim Huntley (UC Boulder) for assistance in RNA-Seq library preparation. Antibodies obtained from the Developmental Studies Hybridoma Bank were developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242. Thank you to Jennifer Moran and the Mouse Mutant Resequencing Initiative for help with exon-based hybrid selection. Lee Niswander is an Investigator of the Howard Hughes Medical Institute.
Contract grant sponsor: Department of Pediatrics, a Cancer Center Support; contract grant number: P30CA046934.
Contract grant sponsor: the Bioinformatics Shared Resource, and the CCTSI.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamforth SD, Bragança J, Eloranta JJ, Murdoch JN, Marques FI, Kranc KR, Farza H, Henderson DJ, Hurst HC, Bhattacharya S. Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nat Genet. 2001;29:469–474. doi: 10.1038/ng768. [DOI] [PubMed] [Google Scholar]
- Banting GS. CECR2, a protein involved in neurulation, forms a novel chromatin remodeling complex with SNF2L. Hum Mol Genet. 2004;14:513–524. doi: 10.1093/hmg/ddi048. [DOI] [PubMed] [Google Scholar]
- Barber MF, Michishita-Kioi E, Xi Y, Tasselli L, Kioi M, Moqtaderi Z, Tennen RI, Paredes S, Young NL, Chen K, Struhl K, Garcia BA, Gozani O, Li W, Chua KF. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature. 2012;487:114–118. doi: 10.1038/nature11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu P, Evrard YA, Lozano G, Dent SYR. Loss of Gcn5 acetyltransferase activity leads to neural tube closure defects and exencephaly in mouse embryos. Mol Cell Biol. 2007;27:3405–3416. doi: 10.1128/MCB.00066-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buac K, Watkins-Chow DE, Loftus SK, Larson DM, Incao A, Gibney G, Pavan WJ. A Sox10 expression screen identifies an amino acid essential for Erbb3 function. PLoS Genet. 2008;4:e1000177. doi: 10.1371/journal.pgen.1000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree G. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell. 2000;6:1287–1295. doi: 10.1016/s1097-2765(00)00127-1. [DOI] [PubMed] [Google Scholar]
- Chen J, Archer TK. Regulating SWI/SNF subunit levels via protein-protein interactions and proteasomal degradation: BAF155 and BAF170 limit expression of BAF57. Mol Cell Biol. 2005;25:9016–9027. doi: 10.1128/MCB.25.20.9016-9027.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2005;38:228–233. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- Copp A. Neurulation in the cranial region–normal and abnormal. J Anat. 2005;207:623–635. doi: 10.1111/j.1469-7580.2005.00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, Greene NDE. Genetics and development of neural tube defects. J Pathol. 2010;220:217–230. doi: 10.1002/path.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detrait ER, George TM, Etchevers HC, Gilbert JR, Vekemans M, Speer MC. Human neural tube defects: Developmental biology, epidemiology, and genetics. Neurotoxicol Teratol. 2005;27:515–524. doi: 10.1016/j.ntt.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxsey S, Zimmerman W, Mikule K. Centrosome control of the cell cycle. Trends Cell Biol. 2005;15:303–311. doi: 10.1016/j.tcb.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Dunwoodie SL, Rodriguez TA, Beddington RS. Msg1 and Mrg1, founding members of a gene family, show distinct patterns of gene expression during mouse embryogenesis. 1998;72:27–40. doi: 10.1016/s0925-4773(98)00011-2. [DOI] [PubMed] [Google Scholar]
- Elwood J, Little J, Elwood J. Epidemiology and control of neural tube defects. Vol. 20 Oxford University Press; New York: 1992. [Google Scholar]
- Euskirchen GM, Auerbach RK, Davidov E, Gianoulis TA, Zhong G, Rozowsky J, Bhardwaj N, Gerstein MB, Snyder M. Diverse roles and interactions of the SWI/SNF chromatin remodeling complex revealed using global approaches. PLoS Genet. 2011;7:e1002008. doi: 10.1371/journal.pgen.1002008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KSJ, McCrary WJ, Ross JS, Wright CF. Members of the hSWI/SNF chromatin remodeling complex associate with and are phosphorylated by protein kinase B/Akt. Oncogene. 2006;25:4605–4612. doi: 10.1038/sj.onc.1209496. [DOI] [PubMed] [Google Scholar]
- Harris M, Juriloff D. Mini-review: Toward understanding mechanisms of genetic neural tube defects in mice. Teratology. 1999;60:292–305. doi: 10.1002/(SICI)1096-9926(199911)60:5<292::AID-TERA10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Harris MJ, Juriloff DM. An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res Part A: Clin Mol Teratol. 2010;88:653–669. doi: 10.1002/bdra.20676. [DOI] [PubMed] [Google Scholar]
- Hata K, Okano M, Lei H, Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002;129:1983–1993. doi: 10.1242/dev.129.8.1983. [DOI] [PubMed] [Google Scholar]
- Ho L, Crabtree G. Chromatin remodelling during development. Nature. 2010;463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Jothi R, Ronan J, Cui K, Zhao K, Crabtree G. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc Natl Acad Sci U S A. 2009;106:5187–5191. doi: 10.1073/pnas.0812888106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Miller EL, Ronan JL, Ho WQ, Jothi R, Crabtree GR. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat Cell Biol. 2011;13:903–913. doi: 10.1038/ncb2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes G, Niswander L. Expression ofslit-2 andslit-3 during chick development. Dev Dyn. 2001;222:301–307. doi: 10.1002/dvdy.1182. [DOI] [PubMed] [Google Scholar]
- Huang LS, Voyiaziakis E, Markenson DF, Sokol KA, Hayek T, Breslow JL. apo B gene knockout in mice results in embryonic lethality in homozygotes and neural tube defects, male infertility, and reduced HDL cholesterol ester and apo A-I transport rates in heterozygotes. J Clin Invest. 1995;96:2152–2161. doi: 10.1172/JCI118269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Bulger M, Pazin MJ, Kobayashi R, Kadonaga JT. ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell. 1997;90:145–155. doi: 10.1016/s0092-8674(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Jadeja S, Smyth I, Pitera JE, Taylor MS, van Haelst M, Bentley E, McGregor L, Hopkins J, Chalepakis G, Philip N, Perez Aytes A, Watt FM, Darling SM, Jackson I, Woolf AS, Scambler PJ. Identification of a new gene mutated in Fraser syndrome and mouse myelencephalic blebs. Nat Genet. 2005;37:520–525. doi: 10.1038/ng1549. [DOI] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ. A consideration of the evidence that genetic defects in planar cell polarity contribute to the etiology of human neural tube defects. Birth Defects Res Part A: Clin Mol Teratol. 2012;94:824–840. doi: 10.1002/bdra.23079. [DOI] [PubMed] [Google Scholar]
- Kim JK, Huh SO, Choi H, Lee KS, Shin D, Lee C, Nam JS, Kim H, Chung H, Lee HW, Park SD, Seong RH. Srg3, a mouse homolog of yeast SWI3, is essential for early embryogenesis and involved in brain development. Mol Cell Biol. 2001;21:7787–7795. doi: 10.1128/MCB.21.22.7787-7795.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Zaghloul NA, Bubenshchikova E, Oh EC, Rankin S, Katsanis N, Obara T, Tsiokas L. Nature cell biology 2011 KimNde1-mediated inhi-2. Nat Cell Biol. 2011;13:351–360. doi: 10.1038/ncb2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleske AJ, Gifford AM, Scott ML, Nee M, Bronson RT, Miczek KA, Baltimore D. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron. 1998;21:1259–1272. doi: 10.1016/s0896-6273(00)80646-7. [DOI] [PubMed] [Google Scholar]
- Leck I. Causation of neural tube defects: Clues from epidemiology. Br Med Bull. 1974;30:158–163. doi: 10.1093/oxfordjournals.bmb.a071187. [DOI] [PubMed] [Google Scholar]
- Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron. 2007;55:201–215. doi: 10.1016/j.neuron.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mäader A, Richmond R, Sargent D, Richmond T. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Mandel S, Gozes I. Activity-dependent neuroprotective protein constitutes a novel element in the SWI/SNF chromatin remodeling complex. J Biol Chem. 2007;282:34448–34456. doi: 10.1074/jbc.M704756200. [DOI] [PubMed] [Google Scholar]
- Nasmyth K. The determination of mother cell-specific mating type switching in yeast by a specific regulator of HO transcription. EMBO J. 1987;6:243–248. doi: 10.1002/j.1460-2075.1987.tb04745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Phelan ML, Sif S, Narlikar GJ, Kingston RE. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell. 1999;3:247–253. doi: 10.1016/s1097-2765(00)80315-9. [DOI] [PubMed] [Google Scholar]
- Pyrgaki C, Liu A, Niswander L. Grainyhead-like 2 regulates neural tube closure and adhesion molecule expression during neural fold fusion. Dev Biol. 2011;353:38–49. doi: 10.1016/j.ydbio.2011.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronan JL, Wu W, Crabtree GR. From neural development to cognition: Unexpected roles for chromatin. Nat Rev Genet. 2013;14:347–359. doi: 10.1038/nrg3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu KS. Intrinsic disorder explains diverse nuclear roles of chromatin remodeling proteins. J Mol Recognit. 2009;22:1–8. doi: 10.1002/jmr.915. [DOI] [PubMed] [Google Scholar]
- Schaniel C, Ang YS, Ratnakumar K, Cormier C, James T, Bernstein E, Lemischka IR, Paddison PJ. Smarcc1/Baf155 couples self-renewal gene repression with changes in chromatin structure in mouse embryonic stem cells. Stem Cells. 2009;27:2979–2991. doi: 10.1002/stem.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanahan F, Seghezzi W, Parry D, Mahony D, Lees E. Cyclin E associates with BAF155 and BRG1, components of the mammalian SWI-SNF complex, and alters the ability of BRG1 to induce growth arrest. 1999;19:1460–1469. doi: 10.1128/mcb.19.2.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn D, Lee K, Lee C, Oh J, Chung H, Jeon S, Seong R. SRG3 interacts directly with the major components of the SWI/SNF chromatin remodeling complex and protects them from proteasomal degradation. 2007;282:10614–10624. doi: 10.1074/jbc.M610563200. [DOI] [PubMed] [Google Scholar]
- Sun F, Tang F, Yan AY, Fang HY, Sheng HZ. Expression of SRG3, a chromatin-remodelling factor, in the mouse oocyte and early preimplantation embryos. Zygote. 2007;15:129–138. doi: 10.1017/S096719940600400X. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Naruse I, Hongo T, Xu M, Nakahata T, Maekawa T, Ishii S. Extensive brain hemorrhage and embryonic lethality in a mouse null mutant of CREB-binding protein. Mech Dev. 2000;95:133–145. doi: 10.1016/s0925-4773(00)00360-9. [DOI] [PubMed] [Google Scholar]
- Timmer JR. Tissue morphogenesis and vascular stability require the Frem2 protein, product of the mouse myelencephalic blebs gene. Proc Natl Acad U S A. 2005;102:11746–11750. doi: 10.1073/pnas.0505404102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trainor PA. Specification of neural crest cell formation and migration in mouse embryos. Semi Cell Develop Biol. 2005;16:683–693. doi: 10.1016/j.semcdb.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Tsai YC, Greco TM, Boonmee A, Miteva Y, Cristea IM. Functional Proteomics establishes the interaction of SIRT7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Mol Cell Proteomics. 2012;11:60–76. doi: 10.1074/mcp.A111.015156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, McAnally J, Pomajzl C, Shelton JM, Richardson JA, Karsenty G, Olson EN. Histone Deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–566. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, Crabtree GR. Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. 1996;10:2117–2130. doi: 10.1101/gad.10.17.2117. [DOI] [PubMed] [Google Scholar]
- Wang Y. The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J Neurosci. 2006;26:2147–2156. doi: 10.1523/JNEUROSCI.4698-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane K, Kawabata M, Tsuruo T. A DNA-topoisomerase-II-binding protein with eight repeating regions similar to DNA-repair enzymes and to a cell-cycle regulator. Eur J Biochem. 1997;25:794–799. doi: 10.1111/j.1432-1033.1997.00794.x. [DOI] [PubMed] [Google Scholar]
- Yang X. DNA topoisomerase II and neural development. Science. 2000;287:131–134. doi: 10.1126/science.287.5450.131. [DOI] [PubMed] [Google Scholar]
- Yao TP, Oh SP, Fuchs M, Zhou ND, Ch'ng LE, Newsome D, Bronson RT, Li E, Livingston DM, Eckner R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998;93:361–372. doi: 10.1016/s0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]
- Yu H, Smallwood PM, Wang Y, Vidaltamayo R, Reed R, Nathans J. Frizzled 1 and frizzled 2 genes function in palate, ventricular septum and neural tube closure: general implications for tissue fusion processes. Development. 2010;137:3707–3717. doi: 10.1242/dev.052001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Chambers KJ, Faller DV, Wang S. Reprogramming of the SWI/SNF complex for co-activation or co-repression in prohibitin-mediated estrogen receptor regulation. Oncogene. 2007;26:7153–7157. doi: 10.1038/sj.onc.1210509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.