

SUMMARY
Network-based molecular modeling of physiological behaviors has proven invaluable in the study of complex diseases such as cancer, but these approaches remain largely untested in contexts involving interacting tissues such as autoimmunity. Here, using Alopecia Areata (AA) as a model, we have adapted regulatory network analysis to specifically isolate physiological behaviors in the skin that contribute to the recruitment of immune cells in autoimmune disease. We use context-specific regulatory networks to deconvolve and identify skin-specific regulatory modules with IKZF1 and DLX4 as master regulators (MRs). These MRs are sufficient to induce AA-like molecular states in vitro in three cultured cell lines, resulting in induced NKG2D-dependent cytotoxicity. This work demonstrates the feasibility of a network-based approach for compartmentalizing and targeting molecular behaviors contributing to interactions between tissues in autoimmune disease.
Graphical Abstract
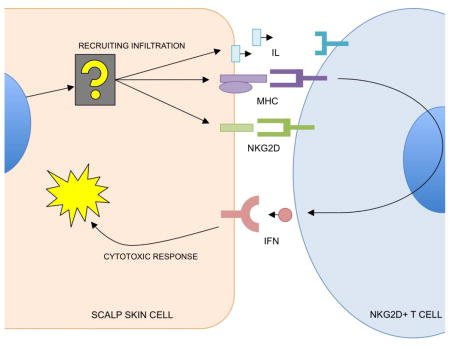
INTRODUCTION
Systems-level analysis using reverse-engineered regulatory networks is an emerging computational discipline that has demonstrated great promise in the study of complex diseases such as cancer and Alzheimer’s disease (Aubry et al., 2015; Chen et al., 2014; Shelanski et al., 2015; Zhang et al., 2013). This approach enables the modeling of complex physiological behaviors as modules of genes (subsets of differentially expressed genes that associate with disease) that are controlled by master regulators (MRs). MRs represent the minimal number of transcription factors (TFs) that are predicted to specifically activate or repress a target module and, by extension, the associated physiological behavior (Carro et al., 2010; Lefebvre et al., 2010). They can be regarded as molecular “switches” that regulate physiological behaviors. The inference of MRs is made possible through the reverse engineering of context-specific regulatory networks using computational algorithms such as ARACNe (Margolin, Nemenman, et al., 2006a).
These MRs are validated biologically and serve as targetable “hubs” governing disease pathology. These approaches have proven highly effective for the study of cell autonomous behaviors in diseases such as cancer, i.e. behaviors of a tumor mass. Physiological behaviors such mesenchymal transformation in glioblastoma (Carro et al., 2010) and oncogenesis in B cell lymphoma or breast cancer (Chen et al., 2014; Mani et al., 2008), as well as onset of Alzheimer’s disease(Zhang et al., 2013) have been functionally linked to a relatively small number of MRs, which in turn become the “bottleneck” that can be used to infer driver mutations in patients(Chen et al., 2014) or become the targets of drug screens for treatment(Shelanski et al., 2015).
However, this type of computational approach is only starting to be implemented to target pathogenic, non-cell autonomous interactions between different tissues such as autoimmune disease. In particular, inferring master regulators cannot be done directly using typical ARACNe-based analysis because of fundamental assumptions made during the generation of a regulatory network: 1) that the samples used are relatively pure or represent the one underlying transcriptional network; and 2) the underlying molecular behavior of a dataset exists at a steady-state such that each sample can be treated as a “snapshot” of regulatory dependency within the overall network(Basso et al., 2005; Califano, Butte, Friend, Ideker, & Schadt, 2012; Margolin, Wang, et al., 2006b). A contaminated sample, particularly by a tissue that exhibits a different context-specific regulatory network, can impair the accuracy of regulatory predictions. Further, when pathogenesis is dependent on the interaction between the two tissues, there will always be an artifact correlation between contaminant gene signatures and the molecular modules that recruit them but are expressed in the other tissue. This makes it difficult to clearly define modules exclusive to one tissue or the other when analyzing gene expression data generated from a mixture of the two tissues.
Alopecia Areata (AA) provides an ideal model for such a study since it is characterized by T cells actively infiltrating the hair follicles and scalp skin that are typically absent in normal skin (Xing et al., 2014). AA typically presents as loss of distinct, random patches of hair that can recover on its own (20–30% of cases). In 5–10% of cases, hair loss can spread to the entire scalp (alopecia totalis), or the entire body (alopecia universalis) (Olsen et al., 1999). Previous research from our lab and others has directly implicated immune genes in AA (Martinez-Mir et al., 2007; Petukhova et al., 2010), many of which are shared with other autoimmune diseases such as Type I diabetes, celiac disease, and rheumatoid arthritis (Betz et al., 2015; Farh et al., 2014; Petukhova et al., 2010). Previous studies have identified infiltration of cytotoxic CD8-positive, NKG2D-positive T cells into the skin of AA mice (Xing et al., 2014), and the pathology of AA involves IFN-gamma-dependent signaling pathways, which are frequently disrupted in association with immune evasion in cancer(Dunn, Bruce, Ikeda, Old, & Schreiber, 2002; Sato et al., 1998).
However, little work has been done to determine if there are intrinsic factors in the “end organ” (the tissue that suffers autoimmune attack) that contribute to the disease, such as scalp skin in AA, making this molecular component an prime target for the analysis. We predict that pathogenic changes in the molecular profile of the scalp skin will contain genes that mediate interactions with the infiltrating T cells. As a corollary, identifying the master regulators will grant regulatory control over the modules that are sufficient to induce immune recruitment. To study this, we leverage context-specific regulatory networks for the regulatory deconvolution of a mixed-signature gene expression profiles of AA patients. The goal of this work was to develop a framework capable of separating mixed AA tissue biopsy gene expression data into skin-specific modules of AA pathology and infiltrate recruitment.
Using this approach, we identify a molecular profile of AA that includes the genetic modules of infiltrate recruitment in the scalp skin by filtering genes that do not accurately map to a skin-specific network. This scalp skin signature allowed the subsequent identification of two master regulators of scalp skin contribution to infiltration: IKZF1 and DLX4. These two genes are expressed in primary scalp biopsies, and are sufficient to induce an AA-like molecular signature and NKG2D-dependent cytotoxicity in independent, wild-type cellular contexts, allowing for direct genetic control induction of immune-mediated cytotoxicity.
RESULTS
Initial definition of a pathogenic expression signature in AA reveals the presence of local (scalp skin) and infiltrating (immune) signals
First, we created a molecular signature comparing AA patients to controls to generate a molecular representation of AA. We analyzed a training set of microarray studies of patient biopsies from an initial cohort of 34 unique biopsy samples: 21 AA patients of varying clinical presentations and 13 unaffected controls. We additionally had patient-matched, non-lesional scalp biopsies for 12 of the 21 AA patients. These 34 patients were gathered as the first of two cohorts totaling 96 patients, the remainder of which was saved for validation exercises.
We created an overall gene expression signature by comparing patients of two distinct clinical presentations, patchy AA (AAP) and totalis and universalis (AT/AU) all against unaffected controls. To account for artifacts in the signature associated with secondary effects of infiltration such as hair loss, we then performed hierarchical clustering using this gene signature on a set of patient-matched lesional (no hair, symptomatic) and non-lesional (hair-bearing, asymptomatic) samples. This analysis identified gene clusters that were differentially expressed between these samples, and those that were systemically equivalent across lesional and non-lesional samples. We subsequently removed from the first expression set any genes that fell in clusters correlating with lesional vs nonlesional (no-hair vs hair) states. This primarily removed a significant number (but not all) of the keratin and keratin-associated proteins from the signature.
The resulting gene expression signature, the AAGS (Alopecia Areata Gene Signature), consisted of a total of 136 unique genes (Supplemental Table 1) and provided sufficient information to cluster the entire training cohort into two appropriate superclusters corresponding to the control and disease states (Figure 1A). Clustering these genes by co-expression also revealed two distinct modules of genes, with greater diversity of co-expression in the genes upregulated in the disease state (Figure 1B). As a qualitative measure of the genes differentially expressed between affected and unaffected patients, we analyzed them for functional annotation enrichments. The analysis revealed the presence of HLA genes, immune response elements, and inflammatory and cell death pathway gene expression in the affected patient samples (Figure 1C). The two most significant superclusters of the AAGS were transmembrane signaling peptides (p=2.8×10−11) and secreted cell-cell signaling peptides (p=2.1×10−10). As expected, this list also contains several antigen-presenting elements and immune response elements that are associated with AA and autoimmune disease (Supplemental Table 2, Supplemental Figure S1). These results indicate that there are significant alterations of multiple biological processes in AA-presenting cells. We postulate that some subset of these genes originate from the scalp skin and are required to induce infiltration recruitment.
Figure 1. Gene expression analysis identifies mixed-tissue gene signatures.

(A) Unsupervised hierarchical clustering of a cohort of Alopecia Areata patchy (AAP), totalis/universalis (AT/AU), and unaffected controls (Normal) using the Alopecia Areata gene signature (AAGS). Blue, underexpression; Red, overexpression. (B) Gene co-similarity matrix showing gene clusters. Stronger orange indicates lower dissimilarity in gene expression. Clusters over- and under-expressed in Alopecia Areata are indicated. (C) Graphical representation of genes in the signature and the statistically enriched functional categories associated with them. Blue indicates signaling pathways; yellow, immune/inflammation pathways; orange, HLA; and red indicates cell death pathways. Pathways at p<0.05 FDR corrected were kept for this analysis.
There is also significant evidence for immune-related genes originating from infiltrating immune cells that must be filtered beforehand; or else they could confound the identification of skin-specific molecular programs. Gene markers associated specifically with immune cells were detected as part of the AAGS including CD8a, CXCL9/10, and CCL5/18/20/26 (full list in Supplemental Tables). In primary patient biopsy samples, defining skin-specific molecular behaviors contributing to AA is a difficult task due to the presence of infiltrating T cells and secondary response pathways in AA skin samples.
Leveraging regulatory networks to deconvolve skin and immune signatures in the AAGS into regulatory modules
With clear definitions of the disease signature, we sought to deconvolve the skin molecular program in the AAGS from the molecular program originating in infiltrating immune cells in a systemic, unbiased manner. Rather than using GO pathway enrichment or other annotation-based methods that rely on a priori knowledge and potentially ambiguous annotations, we instead utilize our inferred regulatory networks under the hypothesis that we can filter non-skin (immune infiltrate) gene expression by identifying the genes that cannot be mapped to a skin-specific regulatory network.
A transcriptional regulatory network of the scalp skin was generated using the ARACNe algorithm and associated software suites (see Methods). Specifically, to generate the network we included a cohort of 106 primary scalp skin samples consisting of normal (unaffected) whole skin biopsies and several samples of primary cultured dermal fibroblasts and dermal papilla cells. These sources contain few or no T cell infiltrates. This network represents the regulatory network in un-infiltrated skin-derived tissues, and serves as the cornerstone of the deconvolution, which occurs in two primary steps as detailed in Figure 2.
Figure 2. Identification of IKZF1 and DLX4 as master regulators.

An overall flow of the pipeline used to deconvolve regulators of genes expressed in the end organ (skin) from those expressed in infiltrating tissue (immune cells). (A) Genes (aqua nodes labeled A–F) measured from a complex primary tissue sample are assigned to either end-organ (red, AAGS) or infiltrate (blue) based on whether or not they can be mapped to the skin network. Only genes mapped to the red node are considered for master regulator analysis. Genes mapped to the blue node are pruned away. (B) The resulting pruning of the AAGS provides an end-organ-enriched gene expression signature (aqua nodes) that is mutually exclusive with an Immune Gene Signature (IGS), p=1.77×10−4, that are overexpressed (cluster 1, node sizes proportional to fold change) and suppressed (cluster 2) in Alopecia Areata. (C) To deconvolve the scalp skin regulators, we performed master regulator (MR) analysis on the AAGS and the IGS, yielding candidate regulators of each signature. True MRs in the skin (R2) will only appear when using the AAGS and not in the IGS. Infiltrate regulators (R1) will not be detected using the AAGS. IKZF1 and DLX4 only have significant FDR values when using the AAGS, and are insignificant (FDR=1) when using the IGS, left panel. This analysis establishes IKZF1 and DLX4 as AAGS (aqua squares) master regulators (yellow squares) in the skin, right panel.
For deconvolution of regulatory modules, the genes in the AAGS are directly mapped to the regulatory network (Figure 2A, see methods for details). A gene in the AAGS is only retained if there is a direct regulatory interaction between it and a transcription factor using the regulatory logic of a skin ARACNe network (red, solid edges). Any genes that come uniquely from infiltrating immune cells will not have significant represention in the ARACNe network, and are subsequently removed from the AAGS (black, dotted edges) for skin, and added to an Immune Gene Signature (IGS).
The IGS was used as a “negative control” signature, adapted from previous work in characterizing cancer immune infiltrates (Bindea et al., 2013). The signatures were defined as a set of genes that are specifically expressed in each immune cell type, including T cells, B cells, mast cells, and macrophages. This step iteratively re-defines the AAGS and IGS by separating those genes whose regulation can be accounted for by an un-infiltrated regulatory network (AAGS) from those that cannot (IGS). By extension, we expected the filtered AAGS to be enriched enough in skin gene expression to generate accurate skin-specific regulons.
As indicated in 2B, 13 infiltrate-specific genes were removed from the AAGS (9.5% of the total signature) when passed through the skin-specific regulatory network. These genes are also listed in Supplemental Table S1. This resulted in two mutually exclusive gene modules (no overlapping genes, p=1.77×10−4), the AAGS and the IGS. A subsequent pathway enrichment analysis further confirmed loss of statistical enrichment of the “T cell activation” and “Immune response” categories (see Supplemental Table S2) while retaining the other clusters including known skin immune response elements (such as the HLA genes). This left a total of 123 genes in the AAGS that we interpret to represent all end-organ programs associated with AA pathology, including end-organ-initiated immune recruitment and immune response (Supplemental Table S1, starred entries).
Note that we have made the distinction between annotations associated with immune cells (e.g. CD8a), and annotations associated with immune response genes (e.g. HLA). The former are removed by the regulatory network as unrepresented in a skin regulatory network. The latter are signature genes that we aim to keep, as they represent the response elements in the skin and are relevant for the pathology of the disease.
Clustering the filtered AAGS revealed two distinct molecular modules that define the transition from unaffected patients (Figure 2B, second panel) to an AA disease state (Figure 2B, third panel). Each node represents a gene in the signature, and its size represents the relative expression in each state (larger means higher expression). We labeled these gene groups: 1) genes whose expression is increased when transitioning into the disease state, and 2) genes whose expression is lost in the transition. This filtered AAGS reflects end-organ-specific gene modules and served as the input to our master regulator analysis.
IKZF1 and DLX4 are master regulators of the skin AAGS and, by extension, infiltrate recruitment
The next step is the most important in identifying end-organ-specific master regulators. We performed master regulator analyses on both the deconvolved AAGS and the IGS independently and in parallel using the scalp skin regulatory network (Figure 2C, first panel, red outline). Using only regulatory interactions represented in skin, we identified the transcriptional regulators that had the highest specificity for the deconvolved AAGS (red arrows) and repeated the analysis for the IGS (black arrows). This step compares the AAGS against the IGS in terms of regulatory logic in the scalp skin, as opposed to direct coverage of gene expression. This analysis assays which transcription factors are the best candidates for the deconvolved AAGS (and not for the IGS) using a molecular regulatory network specific to the skin. We identify skin-specific candidate master regulators by keeping only the candidates that were both significant in AAGS coverage and insignificant for IGS coverage.
Of the significant candidate master regulators specifically for the AAGS, we employed a greedy sort to identify the fewest number of regulators needed to maximize the coverage of the AAGS. We found that two MRs are sufficient to cover >60% of the AAGS: IKZF1 and DLX4. Any additional candidates boosted the coverage by a statistically insignificant margin (<5%). We conclude that the maximum AAGS fidelity (most faithful recreation of the expression signature) and efficiency (fewest necessary regulators) could be achieved through these two genes (IKZF1 p=4.17×10−4, DLX4 p=4.8×10−10 FDR-corrected).
An equivalent master regulator analysis conducted on the IGS modules failed to generate any statistically significant or meaningful MRs when using the scalp-skin regulatory network. Specifically, the best candidates for the AAGS, IKZF1 and DLX4, fall to statistical irrelevance (falling from 1st and 2nd to 159th and 210th, respectively, FDR=1) (Figure 2C, table). Conducting the master regulator analysis on the AAGS without deconvolution fails to generate master regulator candidates at the threshold that is typically expected (both in p-value and signature coverage) due to the presence of contaminating genes in the signature which cannot accurately be mapped to a MR, but nonetheless count against enrichment in the analysis.
These two candidates represent the minimum number of regulators required to recreate the AAGS using regulatory interactions derived from a specific tissue context (scalp skin), distinct from any immune-specific regulatory modules that were deconvolved away using this method. IKZF1 and DLX4 therefore represent a genetic regulatory module in the scalp skin that contributes to AA pathogenesis (Figure 2C, last panel), and may be sufficient to induce infiltration recruitment in an AA-like manner.
The identification of IKZF1 was particularly unusual, as it is a well-established T cell differentiation factor, though it is not without precedent that IKZF1 may have a role in cells outside the immune system (Javierre et al., 2011). However, it is important to note that this analysis does not imply that a master regulator such as IKZF1 has no role in T cells contributing to AA pathogenesis, but rather, that there is significant evidence that IKZF1 additionally functions in the scalp skin to mediate the interactions between the tissues.
Expression of IKZF1 and DLX4 induces an AAGS-like signature in normal huDP and HKs
To validate our master regulator predictions with functional studies, we exogenously overexpressed them in skin-derived cell lines and cultured cells to test for sufficiency in influencing expression of the AAGS. We cloned DLX4 and two isoforms of IKZF1 for exogenous expression in cultured cells. The active IKZF1 isoform served the experimental arm of the study, while the isoform that lacks a DNA binding domain was included as a negative control (IKZF1δ). We expressed these genes in cultured primary human hair follicle dermal papillae (huDP) and human keratinocytes (HK). This experimental system allowed us to directly test two distinct but related hypotheses: 1) IKZF1 and DLX4 can induce AA-like recruitment of immune cells, and 2) they do so through expression in the skin (not the immune infiltrates).
We identified a set of genes that were significantly differentially expressed in the same direction in IKZF1 and DLX4 transfections across both cell types. Unsupervised hierarchical clustering of all samples based on these transcripts reveals clean co-segregation of IKZF1 and DLX4 transfections from IKZF1δ and RFP controls (Figure 3A). Furthermore, we observed that the sub-clustering within these supergroups was not biased based on cell type used (HK did not cluster with HK, and DP did not cluster with DP), supporting that we have identified context-independent effects of master regulator overexpression. Interestingly, we observed that DLX4 transfections resulted in increased levels of IKZF1 transcript and protein, whereas the IKZF1 transfections did not influence DLX4 expression (Figure 3B, 3C).
Figure 3. Exongeous expression of IKZF1 and DLX4 induces a context-independent AA-like gene expression signature.

(A) Two-dimensional hierarchical clustering of gene expression measured in human dermal papillae (huDP) and HACAT keratinocytes (HK) transfected with plasmid vectors expressing IKZF1, DLX4, or controls expressing RFP and IKZFδ, an isoform lacking DNA binding domains. The treatment type and cell type for each experiment are indicated at the top of the heatmap. Blue indicates decreased expression and red indicates overexpression. (B) Analysis of IKZF1 and DLX4 mRNA expression in transfected cells in quadruplicate, represented as average ±SEM, normalized to B-actin (C) Western blot confirming IKZF1 and DLX4 proteins. GSEA plots measuring the specificity of AA-like response assayed by differential expression of the AAGS following (D) IKZF1 or (E) DLX4 overexpression. Genes are ranked left to right from most- to least-differentially expressed on the x-axis, barcodes represent the positions of IKZF1 and for DLX4 signature genes. The Enrichment Score (ES) is shown in the plot, and the normalized Enrichment Score (nES) is displayed at the top. The nES is derived from the ES at the “leading edge” of the plot—that is, the first maximal ES peak obtained. The p-value is computed for the nES compared against a randomized null distribution.
We subsequently interrogated the expression data for enrichment of the AAGS genes using gene set enrichment analysis (GSEA). We performed two differential gene expression studies comparing the IKZF1 transfections vs RFP controls and DLX4 transfections vs RFP controls. The results show that the ectopic expression of the MRs is followed by significant enrichment in the induction of the AAGS (IKZF1 p=0.012, DLX4 p=2.08×10−4; Figure 3D, 3E).
IKZF1 and DLX4 expression are sufficient to induce sensitivity to NKG2D-mediated cytotoxicity in normal cultured skin
The experiments of IKZF1 and DLX4 overexpression suggest that these two genes are master regulators capable of mediating the AAGS when applied to HK and huDP. However, the functional relevance of these master regulators to autoimmunity and immune infiltration is whether or not their expression is sufficient to induce a targeted autoimmune response. In order to investigate this ex vivo, we performed experiments measuring the level of cytotoxic cell death in HK and huDP cells when exposed to peripheral blood monocytes (PBMCs).
We again transfected both HK and huDP cells with one of four expression constructs: IKZF1, DLX4, RFP (negative control) or IKZF1δ (negative control). 24 hours post-transfection, these cells were incubated with fresh, purified PBMCs. We additionally cultured human dermal fibroblasts that were patient-matched for the PBMCs used in this experiment. The PBMCs were obtained from a control subject with no history of AA or any other autoimmune disease.
In all comparisons, we observed a statistically significant increase in PBMC-dependent cytotoxicity for the IKZF1 and DLX4 transfections compared to RFP and IKZF1δ controls (Figure 4, center columns, total bar height). The patient-matched PBMCs and RFP-control transfected fibroblasts exhibited no evidence of cytotoxic interactions, as expected in healthy target cells (Figure 4A, center). However, the introduction of IKZF1 and DLX4 were both sufficient to induce an interaction between these previously exclusive cells, resulting in significant increase of total cytotoxicity. In a similar fashion, both huDP (Figure 4B, center) and HK cells (Figure 4C, center) showed a significant increase above background levels in cytotoxic sensitivity to the PBMCs.
Figure 4. Exogenous expression of IKZF1 and DLX4 induces increased NKG2D-dependent PBMC-associated cytotoxicity in three cultured cell types.

The schematic on the left of each row describes the tissues introduced to PBMCs for cytotoxicity assays (in triplicate). Colors indicate host sources (matching colors indicate host-matched tissues). Middle bar graphs present the cytotoxicity values obtained after either 6 hours of incubation (total bar height) or the cytotoxicity observed after 6 hours with the addition of human anti-NKG2D monoclonal antibody (grey bar). The NKG2D-dependent cytotoxicity is the difference between the two (white bar). The right bar graphs report the changes in NKG2D-dependent cytotoxicity normalized to the RFP controls. IKZF1.2B indicates cells transfected with the IKZF1δ vector, and IKZF1.3B indicates the full-length transcript. Y-axis reports cytotoxicity measured as a fraction of maximum cytotoxicity (total cell count). All error bars report ±SEM. ** indicate statistically significant difference from RFP control at FDR<0.05. (A) Dataseries corresponding to WB215J PBMCs and WB215J fibroblasts. (B) WB215J PBMCs against cultured human dermal papillae (huDP). (C) WB215J PBMCs against cultured human keratinocytes (HK).
Since we previously showed that the likely pathogenic immune cell in AA is CD8+, NKG2D+ activated T cells, we also performed all treatments with the addition of an NKG2D-blocking antibody (see Methods) to prevent NKG2D-dependent interactions. In all cases, we observed that blocking NKG2D suppressed the cytotoxicity in both IKZF1 and DLX4 treatments to levels comparable to controls (Figure 4, center panel, grey bars). From the difference between the inhibitor-treated and untreated cells, we can infer the cytotoxicity that is NKG2D-dependent (Figure 4, center panel, white bars), which can be normalized to that observed in controls for a relative fold change analysis. From the NKG2D blockade, we observed a statistically significant increase specifically in IKZF1 and DLX4 transfections across all trials (Figure 4 right panels). There was a large (>50 fold) increase in patient-matched cytotoxicity compared to the control transfection, which again showed no significant cytotoxicity. There was approximately a 2- to 8-fold increase in NKG2D-dependent cytotoxicity compared to both controls, despite a statistically significant but small (<10%) increase in NKG2D-independent cytotoxicity. We conclude from these experiments that IKZF1 and DLX4 are capable of inducing NKG2D-dependent interactions with normal PBMCs that result in toxicity for the transfected cells regardless of the exact tissue type.
Importantly, these experiments establish the cell autonomous function for IKZF1 and DLX4 in the scalp skin as opposed to infiltrating cells, as the exogenous modification was done strictly on normal cultured cells and exposed to healthy PBMCs from a source with no history of autoimmune disease.
MR expression permits reconstruction of a directional skin-specific master regulator module of infiltration recruitment
After establishing that IKZF1 and DLX4 are sufficient to induce the AAGS, we sought to use this data to fully reconstruct the AA master regulator module. ARACNe is capable of detecting direct transcriptional dependencies between a transcription factor (TF) and non-regulatory genes that are potential targets (T) because we can infer that the regulation is TF→T. ARACNe cannot infer directional interactions between TF-TF pairs, and subsequently cannot infer secondary targets of master regulators due to the regulatory equivalence of TFs (Figure 5A, first panel). However, since we have directly perturbed HKs and huDPs with specific master regulators (Figure 5A, asterisks), we can use the gene expression data to infer directionality. If TFB is a target of our master regulator (TFA), then overexpression of TFA will result in the differential expression of TFB and we can infer that TFA→TFB. Subsequently, any marker genes in the signature associated with TFB can be linked to MR as secondary targets TFA →TFB→T (Figure 5A, top panel). If TFB functions upstream of or in parallel with MR then the expression of TFB and T will not be affected by overexpression of TFA (Figure 5A, bottom panel).
Figure 5. The fully reconstructed master regulator module predicts both immune infiltration and severity.

(A) Using the exogenous expression data, it is possible to infer both direct transcriptional MR targets (MR → T), as well as targets regulated by TFs that are targets of the MR (MR → TF → T). Any TF (TFB) that is paired with MRs IKZF1 or DLX4 (TFA) and that exhibits changes in expression upon overexpression of the TFA is regulated by the TFA. Subsequently, any genes (T) in the AAGS that are linked to TFB are secondary targets of TFA (TFB responds). Any TFB that does not respond to transfection of TFA is not regulated by the TFA, so either TFB regulates TFA (TFB stable, left) or both are co-regulated by a third, TFC (TFB stable, right). (B) Using this approach, 78% of AAGS can be mapped to IKZF1 or DLX4 within one indirect TFB. Blue nodes represent AAGS genes that respond to IKZF1 or DLX4 expression, size of nodes scaled to the fold change experimentally observed (only nodes having at least 25% change are shown) (C) Using these targets, we generate single numeric scores of IKZF1 and DLX4 transcriptional activity and used them to create classifiers for AA severity. AA samples are then imposed over the search space to assess accuracy (top chart). The table provides quantitation and statistics for separation of presentations across territories in the search space (NC:unaffected, AAP:patchy AA, AT/AU: totalis/universalis). Centroid representations can be used to show how populations transition into disease states by moving across the trained boundaries (bottom chart; AAP-N: nonlesional, AAP-L: lesional).
Using this logic, we reconstructed the regulatory module to measure the full extent of the coverage obtained by overexpressing IKZF1 and DLX4 in these cellular contexts. We mapped any downstream targets of TFs that both (1) respond to IKZF1/DLX4 expression in the experiments and (2) are predicted to have mutual information with the expressed master regulator by ARACNe to the regulatory module. 78% of the responding AAGS are within 2° of downstream separation from the master regulators IKZF1 and DLX4 based on these criteria (Figure 5B, full module listed in Supplementary Table S4).
IKZF1 and DLX4 can be used to predict both immune infiltration and disease severity in an independent cohort
As validation of this module, we returned to our original AA array cohort and performed a machine-learning analysis. We attempted to classify an validation AA set into control and affected samples using only the inferred IKZF1 and DLX4 activity. Using the earlier training set from Figure 1, we arrayed the samples into a search space of two dimensions: the consensus activity of IKZF1 (x-axis) and the consensus activity of DLX4 (y-axis) (see Methods). From the training set, we generated a topographical map of the consensus activity space to define ranges of IZKF1 and DLX4 activity associated with control samples, patchy AA, and AT/AU samples (Figure 5C, black lines). The region in 5C closest to the origin of the plot represents the lowest combined IKZF1 and DLX4 activity; its upper bound (the lower black line) is the support vector machine (SVM) margin that maximizes the difference between control and all AA patients. The next upper bound (the upper black line) represents the SVM margin that maximizes the separation of AT/AU patients from AAP.
Using these measures of MR activity, we turned to the validation set and tested for the predictive power of these parameters in separating patients and controls. We observed a strong ability to separate samples into disease and control states, in addition to clinical severity (Figure 5C, top, p<1×10−5). A centroid map of each patient subgroup more clearly reveals how the transition of patient groups from Normal to AAP and AT/AU is reflected by relative IKZF1 and DLX4 activity (Figure 5C, bottom). For comparison, we also included a centroid for the AAP non-lesional sample biopsies, which were not included in the training set.
Deconvolution applied to independent autoimmune diseases identifies known genes
For comparison, and to provide proof-of-concept for the generalizability of our approach, we downloaded publicly available gene expression datasets for atopic dermatitis (AD) (Suárez-Fariñas et al., 2011) and psoriasis (Ps) (Yao et al., 2008). We generated gene expression signatures for each disease by comparing lesional biopsies to unaffected biopsies, similar to our AA analysis (Figure 6A, Supplemental Figure S2, Supplemental Table S3). A direct comparison of the genes within the AA, AD, and Ps signatures revealed statistically significant evidence that the AAGS is distinct from both AD and Ps (p=0.003 and 3.93×10−13, respectively). By contrast, comparison of the AD and Ps signatures to each other revealed statistically significant evidence for shared molecular signatures and, by extension, possible shared molecular pathology (p=0.0173).
Figure 6. Deconvolved regulatory modules can be generated for Alopecia Areata, psoriasis, and atopic dermatitis using the same naïve framework.

(A) Disease-associated gene expression signatures for psoriasis (Ps) and atopic dermatitis (AD) can be clearly defined by differential expression. Comparison of these signatures to the Alopecia Areata (AA) gene signature reveals that the AA signature is statistically distinct from both Ps and AD signatures (Fisher’s Exact Test), whereas there is statistical evidence for some sharing between the Ps and AD signatures. (B) Translating these signatures into regulatory modules reveals entirely different master regulators governing AD and Ps compared to AA. Yellow nodes = AA gene signature; blue nodes = AD gene signature; aqua nodes = Ps gene signature; orange = AA MR; dark blue = AD MR; cyan = Ps MR. The list of top five AD and Ps master regulators are provided, ranked by coverage of the corresponding signature. Also provided are the p-values of each MR without deconvolution (IGS p-value). * indicates published regulators. † indicates an MR common to AD and Ps.
These two signatures were applied to our pipeline. Figure 6B reports the top five master regulators identified after the analysis, ranked by their total coverage of the appropriate disease signatures (Ps or AD). Also provided are the ranks of the master regulators using the corresponding deconvolved IGS. The results indicate that the key regulatory hubs associated with AA (specifically IKZF1 and DLX4) are unique to AA. Each disease was assigned its own unique list of master regulators, but there additionally was overlap of two candidate MRs in AD and Ps: SMAD2 and HLTF. SMAD2 and TGFBR1 are transcription factors with published evidence of involvement in psoriasis, and our pipeline was able to identify them with no a priori evidence, using a basic definition of a Ps gene expression signature (Doi, Shibata, Kiyokane, & Otsuki, 2003; Gambichler, Terras, & Skrygan, 2013). These results demonstrate the effectiveness of modeling complex genetic behaviors as regulatory modules to differentiate mechanisms of pathology.
DISCUSSION
Systemic generation and analysis of gene regulatory networks and gene expression data capitalizing on genome-wide profiling has proven promising in the study of complex diseases. Integrative projects to interrogate functional interactions have recently been leveraged in genome-wide expression signature deconvolution (Bindea et al., 2013; Newman et al., 2015) and cross-tissue interactions(MacNeil et al., 2015) in diabetes and atherosclerosis (Hecker, Goertsches, Engelmann, Thiesen, & Guthke, 2009; Keller et al., 2008). These studies have been invaluable in identifying infiltrating gene signatures, which provides insight into the types of pathogenic immune infiltrates associated with disease; and they have helped identify driver genes from eQTLs and other genomic association tests, similar to the systematic algorithms being developed in cancer and Alzheimer’s disease research (Chen et al., 2014; Zhang et al., 2013) by providing significant genome-level coverage of regulatory activity and tissue-level gene panels of interacting tissues.
However, particularly in contexts such as AA, little has been done to characterize the modular regulation of discrete pathogenic molecular behaviors within a gene expression profile, and how they translate to physiological interactions between tissues of the disease. Modeling physiological traits as genetic programs controlled by “master regulators” provides a uniquely powerful perspective in the study of complex disease. The approach canalizes large gene expression signatures into a relatively few number of selected master regulators that subsequently become the targets of manipulation via gene therapies or drugs and small molecules.
Here, we extend the application of regulatory networks to interrogate the complex molecular state of a mixed sample of end organ (scalp skin) and infiltrating (immune infiltrates) tissue in AA by comparing regulatory networks of different skin contexts (infiltrated and normal). We establish that in addition to their typical use for identifying the key regulatory hubs governing molecular phenotype switches, these networks can be used to isolate and compartmentalize molecular behaviors that originate from different tissues based on whether or not they are accurately represented in an independent context-specific network. This allows for more precise identification of tissue-specific molecular programs from a mixed sample that contribute to an integrated, interactive physiological behavior such as immune infiltration. Using this pipeline, we were able to reconstruct the master regulators mediating infiltration from the skin not only in the context of AA, but our analysis of psoriasis and atopic dermatitis provides additional candidates for the genetic regulation of immune infiltration in skin autoimmune diseases in general, and demonstrates the general applicability of the approach.
Aside from the direct implications in Alopecia Areata pathology, this work provides the proof-of-principle for two key, generalizable notions: (1) a complex interaction between two tissues can be modeled as a quantifiable, molecular gene expression modules; and (2) these modules and their regulators can be extracted from expression data, compartmentalized a tissue, and co-opted to induce the associated interaction in normal cell types. This was evidenced by our ability to recapitulate the AAGS upon ectopic expression of master regulators IKZF1 and DLX4, and to subsequently induce enhanced cytotoxicity in non-AA cell lines using normal (non-AA) PBMCs solely via the manipulation of IKZF1 and DLX4 expression within the target end organ itself (no genetic manipulation of the PBMCs).
Specifically, our analysis identified master regulators that are sufficient to induce interactions with immune cells when expressed solely in scalp skin. Even in a patient-matched context with samples from a healthy, AA-unaffected patient, IKZF1 and DLX4 expression were sufficient to induce aberrant NKG2D-depedent interactions between dermal fibroblasts and PBMCs resulting in cytotoxicity. These interactions were not present in control transfections and they were repeated in two other (non-patient-matched) cell types, indicating that the expression of IKZF1 or DLX4 is sufficient to induce interactions with normal immune cells regardless of the specific target tissue or host matching. The identification of IKZF1 and DLX4 would have been impossible without our network-based deconvolution, as the significant presence of infiltrating signature in the original AAGS would have prevented any accurate identification of candidate MRs. Instead, network-based deconvolution identified master regulators that are capable of inducing specific molecular interactions in any of several molecular contexts that are completely independent of AA itself.
The identification of IKZF1 was unexpected, since IKZF1 is widely studied in the context of T cell differentiation (Kleinmann, Geimer Le Lay, Sellars, Kastner, & Chan, 2008). However, its identification came solely from using a deconvolved AA signature, and not the immune gene signature, using regulatory logic derived from skin. Had we relied on public databases, previous literature, or GO annotations to filter our gene expression data, we would have disregarded and removed IKZF1 entirely due to extensive annotation as a T cell differentiation factor. Instead, by turning to regulatory networks, we were able to identify the possibility that local expression of IKZF1 could have a pathogenic relevance independent of its established role directly in immune cells.
While IKZF1 is well characterized in the context of immune cells, a role for IKZF1 outside of immune cells is not without precedent in the literature. The losses of IKZF1 and DLX4 loci are also associated with oncogenesis in colorectal, lung, and breast cancers, and low-grade squamous intraepithelial lesions (Javierre et al., 2011; Sakane et al., 2015; Tomida et al., 2007). These studies obtain their genomic information directly from tumor masses, indicating that somatic losses of these two loci can contribute to cancer pathophysiology as end organ genomic alterations. Our studies into IKZF1 and DLX4 as master regulators inducing immune infiltration support these results and raise the possibility that the loss of these loci may contribute to immune evasion in cancer. Further, these observations, and the identification of IKZF1 and DLX4 as master regulators of immune infiltration recruitment, provide support that there is a function for IKZF1 outside of its role as a T cell-specific differentiation factor, and raises support for the hypothesis that autoimmunity in AA and tumor immune-evasion exist at opposite extremes of normal immune interactions. The loss of the master regulators of immune infiltration is associated with cancer, and their overexpression is associated with the onset of autoimmune disease in AA.
We have shown that systems biology and network analysis can be used to model the molecular mechanisms mediating interactions between two distinct tissues, identify the key regulators, and use them to re-create the interactive trait in other contexts. While the output for the validation of these master regulators was ultimately induction of cell death, the function of these master regulators in the context of autoimmune disease is to induce a molecular profile that ultimately signals to and recruits immune infiltrates. Up to this point, applications of systems biology have mainly been to identify “breakpoints” in cell-autonomous molecular behaviors of cancers. The controlled induction of cross-tissue interactions, particularly those involving the immune system, invites potentially significant avenues for modeling complex genetic traits with regulatory networks that has previously not been feasible, and provides a proof-of-concept framework that can be used to actively compartmentalize molecular behaviors for study even in complex diseases involving interactions between different tissues.
EXPERIMENTAL METHODS
This section contains a description of the less common or unique methods implemented in this study. The remaining methods are detailed in Extended Experimental Methods.
ARACNe
To generate a context-specific transcriptional interaction network for scalp skin, we employed the ARACNe algorithm(Margolin, Nemenman, et al., 2006a) on a set on 128 microarray experiments independent of the analytic cohorts in this study. These experiments represent platform-matched (Affymetrix U133 2Plus) data acquired on whole skin samples from a mixture of normal whole skin biopsies, AA patient biopsies, microdissected dermal papillae, and separated dermis and epidermis samples. These samples collectively provide the heterogeneity required for accurate detection of transcriptional dependencies in the scalp skin. The experiments were pooled and postprocessed as described above, and a standard ARACNe analysis was performed. The ARACNe software suite is available from the Califano lab website, http://wiki.c2b2.columbia.edu/califanolab/index.php/Software.
Master Regulator Analysis
Master regulators for a specific gene expression signature were defined as transcription factors whose direct ARACNe-predicted targets (regulon) are statistically enriched in the gene expression signature. Each TF’s regulon was tested for enrichment of the AAGS using Fisher’s Exact Test, FDR=0.05. This analysis allows for the ranking and determination of the minimum number of TFs required to specifically cover a gene expression module associated with a physiological trait.. http://wiki.c2b2.columbia.edu/califanolab/index.php/Software/MARINA
Master regulator activity classifiers
The ARACNe-predicted targets of IKZF1 and DLX4 were integrated with the exogenous gene expression studies to identify all genes in the AAGS that could be mapped as targets of IKZF1 and DLX4. This was done by intersecting the ARACNe regulons of IKZF1 and DLX4 with the AAGS. The intersection of these two sets was then screened in the expression studies for any genes that responded with at least 25% fold change. This set of genes was used to construct a consensus “meta-activity” for the IKZF1 and DLX4 loci. The rank-normalized change of each gene across the AA patient cohort was integrated into an average as a consensus measure of the relative activity of the parent MR.
These values were subsequently used to define a two-dimensional search space, (X ×Y) where X=IKZF1 meta-activity and Y=DLX4 meta-activity, to classify each of the patients in the AA training set. The meta-activity vectors were rank transformed such that the minimum values were bound to the origin of the search space (0,0), and such that activity measures were positive. This transformation has no influence on the results other than projecting the search space into a more intuitive grid for display purposes, in which both axes are bound between [0,n] where n is positive.
Classification in this space was done using a modified nonlinear, soft-margin support vector machine algorithm. The algorithm is formalized:
The algorithm defines a vector set (A × B), that exists within the search space (X ×Y )such that every given pair (a, b) maximizes the likelihood ratio f (a, b). This function is defined such that Sj is the next order of disease severity to Si, and QI and QIII are the quadrants I and III of the grid created by the hyperplanes (a × R)and (R ×b). Samples in the training set are mapped to each grid with known molecular subtypes and the likelihood ratio is computed for the segregation of subtypes defined by S. The severity ranking used for S was Normal<Mild<Severe. Each coordinate set in (A × B) therefore defines the points to a nonlinear plane that maximizes the separation between samples of different molecular classes in the IKZF1/DLX4 meta-activity space.
Cytotoxicity assay
PBMC-dependent cytotoxicity was measured using the CyoTox 96 Non-Radioactive Cytotoxicity Assay available through Promega. For the processing of samples and solutions, we followed manufacturer protocols. The optimization for PBMC:target was done as below, but using variable concentrations (1:1, 5:1, 10:1) (Supplemental Figure S3).
Cytotoxicity experiments were set up in 96-well format, with each treatment done in triplicate. Transfections were done 36 hours prior to the experiment. The day of the experiment, HK and huDP cells were trypsinized and diluted with DMEM into working stocks. The target concentration per well was 80k cells in 50ul DMEM, combined with 800k PBMCs. The NKG2D inhibitor was the Human NKG2D mAb (clone 149810) from R&D Biosystems (Cat. MAB139), used at a final concentration of 20ug/ml. Each transfection was allocated in triplicate according to manufacturer instructions.
Supplementary Material
Acknowledgments
We would like to thank Julian Mackay-Wiggan and the Alopecia Areata Registry sites for patient samples. JCC and JEC were supported by the Kirschstein NRSA postdoctoral training grant in Medical Genetics, T32GM082271 (to AMC). Funding for the project was provided by the Alopecia Areata Initiative, R01AR056101 (to AMC), R21AR061881 (to AMC and RC), and the Locks of Love Foundation. AJ was supported by the Dermatology Foundation Physician Scientist Career Development Award and by the Louis V. Gerstner Jr Scholars Program.
Footnotes
AUTHOR CONTRIBUTIONS
The overall framework of this study was conceived by AMC in collaboration with JCC. AMC and JCC wrote the manuscript. JCC designed and implemented all computational analysis and experimental work, and analyzed the data. RC provided his guidance and expertise for the project. AJ acquired sample biopsies and processed them for microarray analysis with JEC.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aubry S, Shin W, Crary JF, Lefort R, Qureshi YH, Lefebvre C, et al. Assembly and interrogation of Alzheimer’s disease genetic networks reveal novel regulators of progression. PloS One. 2015;10(3):e0120352. doi: 10.1371/journal.pone.0120352. http://doi.org/10.1371/journal.pone.0120352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse engineering of regulatory networks in human B cells. Nature Genetics. 2005;37(4):382–390. doi: 10.1038/ng1532. http://doi.org/10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- Betz RC, Petukhova L, Ripke S, Huang H, Menelaou A, Redler S, et al. Genome-wide meta-analysis in alopecia areata resolves HLA associations and reveals two new susceptibility loci. Nature Communications. 2015;6:5966. doi: 10.1038/ncomms6966. http://doi.org/10.1038/ncomms6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–795. doi: 10.1016/j.immuni.2013.10.003. http://doi.org/10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Califano A, Butte AJ, Friend S, Ideker T, Schadt E. Leveraging models of cell regulation and GWAS data in integrative network-based association studies. Nature Genetics. 2012;44(8):841–847. doi: 10.1038/ng.2355. http://doi.org/10.1038/ng.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463(7279):318–325. doi: 10.1038/nature08712. http://doi.org/10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Alvarez MJ, Talos F, Dhruv H, Rieckhof GE, Iyer A, et al. Identification of Causal Genetic Drivers of Human Disease through Systems-Level Analysis of Regulatory Networks. Cell. 2014;159(2):402–414. doi: 10.1016/j.cell.2014.09.021. http://doi.org/10.1016/j.cell.2014.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi H, Shibata MA, Kiyokane K, Otsuki Y. Downregulation of TGFbeta isoforms and their receptors contributes to keratinocyte hyperproliferation in psoriasis vulgaris. Journal of Dermatological Science. 2003;33(1):7–16. doi: 10.1016/s0923-1811(03)00107-5. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nature Immunology. 2002;3(11):991–998. doi: 10.1038/ni1102-991. http://doi.org/10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2014 doi: 10.1038/nature13835. http://doi.org/10.1038/nature13835. [DOI] [PMC free article] [PubMed]
- Gambichler T, Terras S, Skrygan M. TGFβ/Smad signalling in psoriatic epidermis models exposed to salt water soaks and narrowband ultraviolet B radiation. Cytokine. 2013;64(1):35–38. doi: 10.1016/j.cyto.2013.06.311. http://doi.org/10.1016/j.cyto.2013.06.311. [DOI] [PubMed] [Google Scholar]
- Hecker M, Goertsches RH, Engelmann R, Thiesen HJ, Guthke R. Integrative modeling of transcriptional regulation in response to antirheumatic therapy. BMC Bioinformatics. 2009;10(1):262. doi: 10.1186/1471-2105-10-262. http://doi.org/10.1186/1471-2105-10-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javierre BM, Rodriguez-Ubreva J, Al-Shahrour F, Corominas M, Graña O, Ciudad L, et al. Long-range epigenetic silencing associates with deregulation of Ikaros targets in colorectal cancer cells. Molecular Cancer Research : MCR. 2011;9(8):1139–1151. doi: 10.1158/1541-7786.MCR-10-0515. http://doi.org/10.1158/1541-7786.MCR-10-0515. [DOI] [PubMed] [Google Scholar]
- Keller MP, Choi Y, Wang P, Davis DB, Rabaglia ME, Oler AT, et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Research. 2008;18(5):706–716. doi: 10.1101/gr.074914.107. http://doi.org/10.1101/gr.074914.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinmann E, Geimer Le Lay AS, Sellars M, Kastner P, Chan S. Ikaros represses the transcriptional response to Notch signaling in T-cell development. Molecular and Cellular Biology. 2008;28(24):7465–7475. doi: 10.1128/MCB.00715-08. http://doi.org/10.1128/MCB.00715-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre C, Rajbhandari P, Alvarez MJ, Bandaru P, Lim WK, Sato M, et al. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Molecular Systems Biology. 2010;6:377. doi: 10.1038/msb.2010.31. http://doi.org/10.1038/msb.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNeil LT, Pons C, Arda HE, Giese GE, Myers CL, Walhout AJM. Transcription Factor Activity Mapping of a Tissue-Specific in vivo Gene Regulatory Network. Cell Systems. 2015;1(2):152–162. doi: 10.1016/j.cels.2015.08.003. http://doi.org/10.1016/j.cels.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani KM, Lefebvre C, Wang K, Lim WK, Basso K, Dalla-Favera R, Califano A. A systems biology approach to prediction of oncogenes and molecular perturbation targets in B-cell lymphomas. Molecular Systems Biology. 2008;4(1):169. doi: 10.1038/msb.2008.2. http://doi.org/10.1038/msb.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla-Favera R, Califano A. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006a;7 Suppl 1(Suppl 1):S7. doi: 10.1186/1471-2105-7-S1-S7. http://doi.org/10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin AA, Wang K, Lim WK, Kustagi M, Nemenman I, Califano A. Reverse engineering cellular networks. Nature Protocols. 2006b;1(2):662–671. doi: 10.1038/nprot.2006.106. http://doi.org/10.1038/nprot.2006.106. [DOI] [PubMed] [Google Scholar]
- Martinez-Mir A, Zlotogorski A, Gordon D, Petukhova L, Mo J, Gilliam TC, et al. Genomewide scan for linkage reveals evidence of several susceptibility loci for alopecia areata. American Journal of Human Genetics. 2007;80(2):316–328. doi: 10.1086/511442. http://doi.org/10.1086/511442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nature Methods. 2015;12(5):453–457. doi: 10.1038/nmeth.3337. http://doi.org/10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen E, Hordinsky M, McDonald-Hull S, Price V, Roberts J, Shapiro J, Stenn K. Alopecia areata investigational assessment guidelines. National Alopecia Areata Foundation. Journal of the American Academy of Dermatology. 1999 Feb; doi: 10.1016/s0190-9622(99)70195-7. [DOI] [PubMed] [Google Scholar]
- Petukhova L, Duvic M, Hordinsky M, Norris D, Price V, Shimomura Y, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466(7302):113–117. doi: 10.1038/nature09114. http://doi.org/10.1038/nature09114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakane J, Taniyama K, Miyamoto K, Saito A, Kuraoka K, Nishimura T, et al. Aberrant DNA methylation of DLX4 and SIM1 is a predictive marker for disease progression of uterine cervical low-grade squamous intraepithelial lesion. Diagnostic Cytopathology. 2015 doi: 10.1002/dc.23256. n/a n/a. http://doi.org/10.1002/dc.23256. [DOI] [PubMed]
- Sato M, Goto S, Kaneko R, Ito M, Sato S, Takeuchi S. Impaired production of Th1 cytokines and increased frequency of Th2 subsets in PBMC from advanced cancer patients. Anticancer Research. 1998;18(5D):3951–3955. [PubMed] [Google Scholar]
- Shelanski M, Shin W, Aubry S, Sims P, Alvarez MJ, Califano A. A systems approach to drug discovery in Alzheimer’s disease. Neurotherapeutics : the Journal of the American Society for Experimental NeuroTherapeutics. 2015;12(1):126–131. doi: 10.1007/s13311-014-0335-5. http://doi.org/10.1007/s13311-014-0335-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez-Fariñas M, Tintle SJ, Shemer A, Chiricozzi A, Nograles K, Cardinale I, et al. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. The Journal of Allergy and Clinical Immunology. 2011;127(4):954–64. e1–4. doi: 10.1016/j.jaci.2010.12.1124. http://doi.org/10.1016/j.jaci.2010.12.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomida S, Yanagisawa K, Koshikawa K, Yatabe Y, Mitsudomi T, Osada H, Takahashi T. Identification of a metastasis signature and the DLX4 homeobox protein as a regulator of metastasis by combined transcriptome approach. Oncogene. 2007;26(31):4600–4608. doi: 10.1038/sj.onc.1210242. http://doi.org/10.1038/sj.onc.1210242. [DOI] [PubMed] [Google Scholar]
- Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nature Medicine. 2014;20(9):1043–1049. doi: 10.1038/nm.3645. http://doi.org/10.1038/nm.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Richman L, Morehouse C, de los Reyes M, Higgs BW, Boutrin A, et al. Type I interferon: potential therapeutic target for psoriasis? PloS One. 2008;3(7):e2737. doi: 10.1371/journal.pone.0002737. http://doi.org/10.1371/journal.pone.0002737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153(3):707–720. doi: 10.1016/j.cell.2013.03.030. http://doi.org/10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.