

Abstract
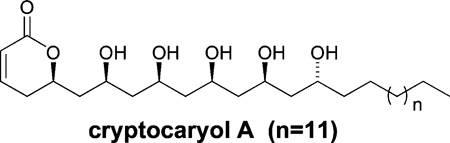
A high-throughput cell-based reporter assay designed to identify small-molecule stabilizers of the tumor suppressor Pdcd4 was used to screen extracts in the NCI Natural Products Repository. Bioassay-guided fractionation of an extract from a Papua New Guinea collection of the tropical tree Cryptocarya sp. provided a series of new 5,6-dihydro-α-pyrone-containing 1,3-polyols (1–8), named cryptocaryols A–H. Their structures were assigned from a combination of NMR, MS, and CD studies in conjunction with NMR database comparisons. Compounds 1–8 were found to rescue Pdcd4 from TPA-induced degradation with EC50 concentrations that ranged from 1.3 to 4.9 µM.
Pdcd4 (programmed cell death 4) is a novel tumor suppressor protein that interacts with the eukaryotic translation initiation factors eIF4A and eIF4G and inhibits the transformation, migration, and invasion of cancer cells in vitro.1–4 Down-regulation of Pdcd4 expression has been associated with the onset of a number of human tumors including colorectal,5 brain,6 ovarian,7 and liver carcinomas.8 In a mouse skin papilloma model, Pdcd4 protein levels were significantly decreased in response to tumor promoters, and the regulation mechanism for Pdcd4 was shown to be phosphorylation-dependent proteosomal degradation.9,10 Given that this tumor suppressor protein has a defined inactivation pathway, stabilization of Pdcd4 levels in cells is an attractive potential target for anticancer therapeutics. Recently, we reported the development of a high-throughput screen designed to identify small-molecule stabilizers of Pdcd411 and the bioassay-guided identification of a series of guanidine alkaloids that were shown to rescue Pdcd4 in a tumor-promoting environment. Herein, we describe a new structural class of Pdcd4-stabilizing compounds obtained from a Papua New Guinea collection of the plant Cryptocarya sp. (Lauraceae, NSC number N098347). The genus Cryptocarya is distributed throughout the tropic, subtropic, and temperate regions of the world, and its members produce an array of secondary metabolites including flavanoids such as cryptochinones A–F,12 pavine and proaporphine alkaloids,13 and a variety of 5,6-dihydro-α-pyrones exemplified by kurzilactone14 and cryptocaryalactone.15 The Cryptocarya-derived α-pyrones display interesting biological activities; for example rugulactone and cryptocaryone were reported to have NF-κB inhibitory activity,16 Z-cryptofolione and cryptomoscatone D2 were shown to be potent inhibitors of the G2 cell cycle checkpoint,17 and 7′,8′-dihydroobolactone inhibited the growth of Trypanosoma brucei brucei with an IC50 of 2.8 µM.18 This paper reports on the isolation, structure elucidation, and Pdcd4-stabilizing activity of cryptocaryols A–H (1–8), a series of new 5, 6-dihydro-α-pyrones that contain repeating 1,3-polyol moieties.
RESULTS AND DISCUSSION
An aliquot of the crude CH2Cl2/MeOH extract of Cryptocarya sp. was subjected to diol flash chromatography eluting sequentially with hexane, EtOAc, and MeOH. The Pdcd4-stabilizing activity was concentrated in the EtOAc eluant, which was further fractionated by C4 flash and Sephadex LH-20 size exclusion chromatography, followed by C2 reversed-phase semipreparative HPLC to yield compounds 1–8.
Cryptocaryol A (1) was isolated as an optically active oil ([α]D + 12, c 0.05, MeOH) with a molecular formula of C30H56O7, as established from HRESIMS measurements. The 1H and 13C NMR spectra for 1 in CD3OD were well resolved (Table 1) and allowed the identification of three partial structures, A–C (Figure 1). Partial structure A contained a conjugated Z-olefin (δH 7.04, ddd, J = 9.8, 6.0, 2.3 Hz, δC 148.6; 5.97, dd, J = 9.8, 1.9 Hz, δC 121.4) coupled to a diastereotopic methylene pair (δHa 2.45, m, δHb 2.36, ddt, J = 18.5, 11.8, 2.6 Hz, δC 31.0). The methylene protons showed HMBC correlations to a carbonyl resonance (δC 167.0) and an oxymethine resonance (δH 4.71 m, δC 76.6), indicative of a 6-substituted-5,6-dihydro-α-pyrone, a common ring system found in Cryptocarya spp. natural products.19 Fragment B had five oxymethine resonances (δH 4.08 m, δC 66.6; δH 3.97 m, δC 69.9; δH 4.00 m, δC 70.2; δH 4.02 m, δC 68.3; δH 3.79 m, δC 69.1) along with six resolved methylenes (δHa 1.94, ddd, J = 14.5, 9.7, 2.3 Hz, δHb 1.67, m, δC 43.9; δH 1.68 m, δC 46.0; δH 1.63 m, δC 45.3; δH 1.59 m, δC 45.9; δH 1.50 m, δC 39.3). COSY and HMBC correlations indicated that each oxymethine was flanked by two methylenes, which helped to establish the presence of a 1,3,5,7,9-polyol moiety. Partial structure C contained a long alkane chain (δH 1.27–1.29 br m, δC 30.5–31.0) and a terminal methyl group (δH 0.89, t, J = 6.9 Hz, δC 14.5). The α-pyrone and 1,3,5,7,9-polyol subunits were joined via COSY and HMBC correlations from H-6 on the pyrone ring to the C-7 diastereotopic methylene and the C-8 oxymethine resonance (Figure 1). Further assignments along the polyol chain were based on the oxymethine 3J-HMBC correlations depicted in Figure 1. Partial structure C was positioned on the terminal portion of the molecule, and its length was deduced to be C14 based on molecular formula considerations. Positive ion HRESIMS/MS fragmentation analysis of 1 showed five sequential losses of 18 amu, corresponding to the elimination of five molecules of H2O from the parent ion, which further supported the planar structure assigned for cryptocaryol A (1).
Table 1.
NMR Spectroscopic Data (600 MHz, CD3OD) for Cryptocaryols A (1) and C (3)
| 1 | 3 | |||
|---|---|---|---|---|
| no. | δC, type | δH (J in Hz) | δC | δH (J in Hz) |
| 2 | 167.0, C | 166.3 | ||
| 3 | 121.4,CH | 5.97 dd (9.8, 1.9) | 122.9 | 6.06 d (9.7) |
| 4 | 148.6,CH | 7.04 ddd (9.8, 6.0, 2.3) | 147.1 | 7.06 dd (9.7, 5.9) |
| 5a | 31.0,CH2 | 2.45 m | 63.4, CH | 4.03 dd (5.9, 2.7) |
| 5b | 2.36 ddt (18.5, 11.8, 2.6) | |||
| 6 | 76.6,CH | 4.71 m | 79.2 | 4.62 dt (10.2, 2.7) |
| 7a | 43.9,CH2 | 1.94 ddd (14.5, 9.7, 2.3) | 39.4 | 2.09 ddd (14.6, 10.2, 2.5) |
| 7b | 1.67 m | 1.66 m | ||
| 8 | 66.6,CH | 4.08 m | 66.7 | 4.09 m |
| 9 | 46.0,CH2 | 1.68 m | 46.1 | 1.67 |
| 10 | 69.9,CH | 3.97 m | 70.0 | 4.00 m |
| 11 | 45.3,CH2 | 1.63 m | 46.0 | 1.61 m |
| 12 | 70.2,CH | 4.00 m | 70.2 | 3.98 m |
| 13 | 45.9,CH2 | 1.59 m | 45.8 | 1.59 m |
| 14 | 68.3,CH | 4.02 m | 68.3 | 4.02 m |
| 15 | 45.8,CH2 | 1.50 m | 45.3 | 1.51 m |
| 16 | 69.1,CH | 3.79 m | 69.1 | 3.80 m |
| 17 | 39.3,CH2 | 1.43 m | 39.3 | 1.43 m |
| 18 | 26.8,CH2 | 1.32 m | 26.8 | 1.32 m |
| 19–28 | 30.5–31.0,CH2 | 1.27–1.29 br m | 30.5–30.8 | 1.27–1.29 br m |
| 29 | 33.2,CH2 | 1.29 m | 33.2 | 1.27 m |
| 30 | 23.8,CH2 | 1.27 m | 23.8 | 1.30 m |
| 31 | 14.5,CH3 | 0.89 t (6.9) | 14.5 | 0.89 t (6.9) |
Figure 1.
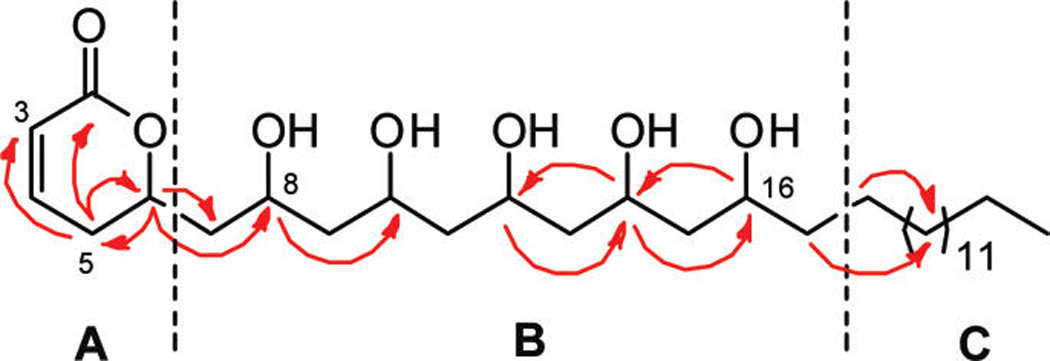
Key HMBC correlations for partial structures A–C in 1.
The stereochemistry of 1 was assigned by a combination of CD spectropolarimetry and Kishi’s universal 13C NMR database analysis, summarized in Figure 2. The ECD spectrum of 1 in MeOH displayed a positive Cotton effect at 256 nm (Δε +0.7); therefore, following the Snatzke rule for α-pyrones,20 the absolute configuration at C-6 was concluded to be R. In an attempt to assign the relative configuration of the polyol chain, compound 1 was reacted with 2,2-dimethoxypropane and pyridinuim p-toluenesulfonate; however the substrate was unstable to the conditions required to form acetonide derivatives. Using an alternative approach, the relative configuration of the polyol system was assigned on the basis of Kishi’s 13C NMR database (Database 2 in CD3OD).21 Kishi’s method is based on the experimental observation that for a 1,3,5-polyol system the C-3 central carbon atom has a characteristic 13C NMR chemical shift that is dependent on the relative configuration at C-1 and C-5 and is largely independent of the steric and stereoelectronic effects of substitutions that are outside of the polyol motif.21 The method has also been shown to be applicable to longer extended polyol systems.21,22 According to the database, the expected chemical shifts for C-3 in a 1,3,5-polyol system are 66.3 ± 0.5 for an anti/anti orientation, 68.6 ± 0.5 for syn/anti or anti/syn, and 70.7 ± 0.5 for a syn/syn arrangement. The relevant oxymethines in 1 were C-10, C-12, and C-14, which resonated at δC 69.9, 70.2, and 68.3, respectively. This revealed that C-10 and C-12 had syn/syn configurations with their neighboring OH groups, while C-14 was either syn/anti or anti/syn (Figure 2). Since the relative configuration at C-12 was established as syn/syn, the configuration at C-14 could be assigned as syn/anti. Thus, the relative configuration of the polyol moiety was revealed to be 8S*, 10S*, 12S*, 14S*, and 16R*. However, due to extensive NMR signal overlap, even in a variety of different NMR solvents, it was not possible to relate the absolute configuration of C-6 (R) with the configuration of C-8 or any of the other chiral carbons in 1.
Figure 2.
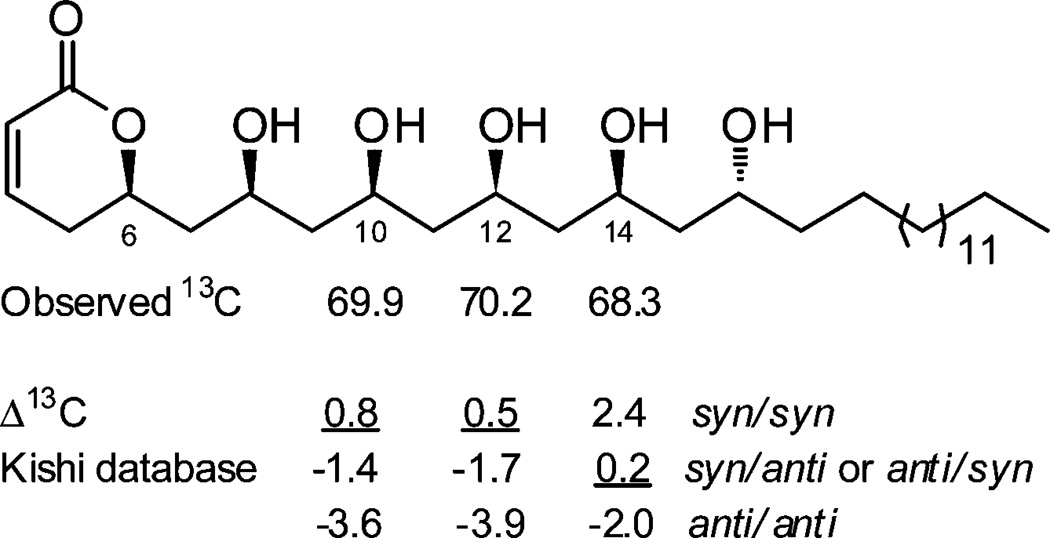
Assignments of configuration by 13C NMR chemical shift analysis for cryptocaryol A (1).
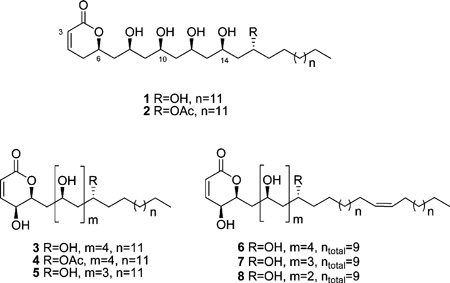
The HRESIMS spectrum of cryptocaryol B (2) showed an addition of 42 amu compared to 1. The 1Hand 13C NMR spectra of 2 were very similar to those of 1 with the exception of a significant downfield shift of one of the oxymethine resonances (δH 5.07 m, δC 72.9), the appearance of an ester carbonyl resonance (δC 173.2), and a singlet methyl (δH 2.03, δC 21.2). These data were consistent with the addition of an acetyl group to one of the alcohol functionalities, and key HMBC correlations placed the acetate group on C-16. Once purified, 2 was found to be unstable in MeOH, and reliable optical rotation and ECD data could not be obtained. However, on the basis of its structural homology with 1 and the close correspondence of its 1H and 13C chemical shift data with those of 1 (see Supporting Information), the configuration of 2 is proposed to be (6R, 8S*, 10S*, 12R*, 14R*, 16R*).
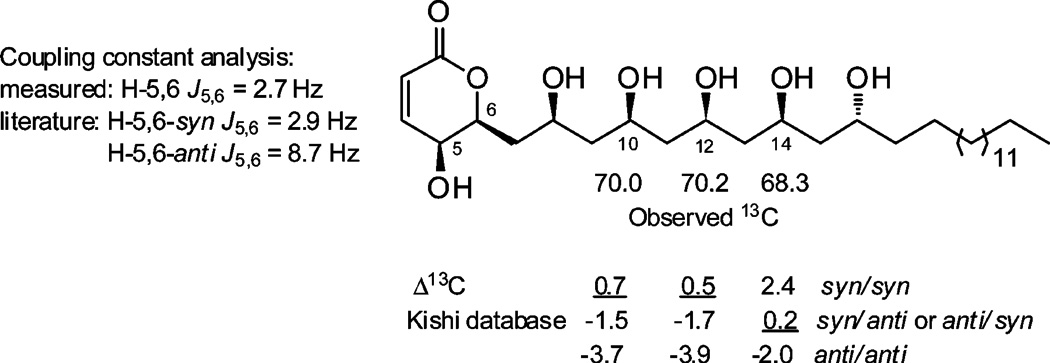
The molecular formula of 3 (C30H56O8 by HRESIMS) had one extra oxygen atom compared to 1, but the same number of double-bond equivalents. Comparison of the 1H and 13C NMR spectra (Table 1) revealed that the major differences between 3 and 1 were the resonances associated with the α-pyrone ring: the methylene pair at C-5 in 1 was replaced with an oxymethine resonance (δH 4.03, dd, J = 5.9, 2.7 Hz; δC 63.4), the oxymethine at C-6 was simplified to a doublet of triplets (δH 4.62, dt, J = 10.2, 2.7 Hz; δC 79.2), and the C-7 resonance was shifted significantly upfield (δC 39.4). Characteristic COSY and HMBC correlations revealed compound 3 had a 5-hydroxy-6-substituted-5,6-dihydro-α-pyrone subunit and a polyol substituent at C-6 that was identical to that of 1. Positive ion HRESIMS/MS fragmentation of 3 showed six sequential losses of H2O (18 amu), which supported the presence of six secondary OH groups. The relative configuration of 3 was established by a combination 1H NMR coupling constant analysis and Kishi’s 13C NMR database comparison, summarized in Figure 3. The H-5/H-6 coupling constant (J = 2.7 Hz) was indicative of a syn relationship for the two protons, comparable to the related 5,6-syn-pyrone sultriecin, where J5,6 was shown to be 2.9 Hz,23,24 in contrast to the 5,6-anti-pyrone, 5-epi-phomalactone with a J5,6 of 8.7 Hz.25 The relative configuration of the polyol system was assigned on the basis of Kishi’s database, with the relevant chemical shifts of C-10 (δC 70.0), C-12 (δC 70.2), and C-14 (δC 68.3) showing good agreement with a syn/syn, syn/syn, and syn/anti configuration, respectively. Since both the specific rotation ([α]D +12, c 0.05, MeOH) and the ECD spectrum of 3 were comparable in sign and magnitude to those of 1, the configuration of 3 was assigned to be (5S, 6S, 8S*, 10S*, 12S*, 14S*, 16R*).
Figure 3.
Relative configuration assignments for cryptocaryol C (3).
Cryptocaryol D (4) had the molecular formula C32H58O9 (by HRESIMS). The 1H and 13C NMR spectra for 4 suggested that it was an acetyl derivative of 3, and HMBC correlations placed the acetate group on C-16. Compound 4 was unstable in solution; however close homology with 3 and good agreement of its 1H and 13C NMR spectroscopic data suggested that the configuration of 4 was (5S, 6S, 8S*, 10S*, 12R*, 14R*, 16R*).
The HRESIMS spectrum of cryptocaryol E (5) indicated a molecular formula of C28H52O7. Comparison of the 1H and 13C NMR spectra for 5 with those of 3 (see Supporting Information) showed that the only significant difference was in the polyol portion of the molecule, where only five oxymethine resonances were present. 2D COSY and HMBC spectra confirmed that compound 5 was the C-16 dehydroxy analogue of 3. A combination of 1H–1H coupling constant analysis and Kishi’s 13C NMR database comparison, in addition to good agreement of the specific rotation ([α]D +12, c 0.05, MeOH) and ECD data with those of 3, established the configuration of 5 as (5S, 6S, 8S*, 10S*, 12S*, 14R*).
HRESIMS data for cryptocaryol F (6) established a molecular formula of C32H58O8. The 1H and 13C NMR spectra of 6 were similar to those of 3 except for an additional disubstituted olefin resonance (2H, δH 5.33, t, J = 5.5 Hz; δC 130.9) that only showed HMBC correlations to the methylene chain portion of the molecule. On the basis of the upfield 13C chemical shift of the allylic methylenes (δC 28.2) the double bond was assigned as Z. The exact position of the double bond within the methylene chain could not be determined by HRESIMS/MS, as the fragmentation pattern showed sequential loss of six H2O units but no diagnostic peaks related to fragmentation of the alkyl chain.
Two additional compounds containing a Z-olefin within the methylene chain were also isolated (7 and 8). Comparison of the HRESIMS and 1H and 13C NMR data recorded for 7 and 8 with those of cryptocaryol F (6) revealed that the only structural differences were in the length of the polyol system and the methylene chain. Cryptocaryol G (7), with a molecular formula of C30H54O7, was the C-16 dehydroxy analogue of 6. Cryptocaryol H (8), with a molecular formula of C28H56O6, lacked OH groups at both C-14 and C-16, and the total number of carbons in its unsaturated alkyl chain was 13 instead of the 15 found in 6. Coupling constant analyses, Kishi database comparisons, optical rotation, and ECD data allowed assignment of the configuration of compounds 6, 7, and 8 as (5S, 6S, 8S*, 10S*, 12S*, 14S*, 16R*), (5S, 6S, 8S*, 10S*, 12S*, 14R*), and (5S, 6S, 8S*, 10S*, 12R*), respectively.
The cryptocaryols were found to rescue Pdcd4 from TPA-induced degradation with EC50's that ranged from 1.3 to 4.9 µM (Table 2). With the exception of compound 6, the 5-hydroxy-6-substituted-5,6-dihydro-α-pyrones 3–5, 7, and 8 were shown to be more potent Pdcd4 stabilizers than the 5-dehydroxy analogues 1 and 2. The C-16 acetate analogues 2 and 4 were essentially equipotent to the corresponding free alcohols 1 and 3. Modifications in the polyol chain length and the addition of an olefin to the long methylene chain of the molecule did not significantly affect the activity of the series. With activity in the low micromolar range, the cryptocaryols represent a new structural class of natural products that can enhance the stability of Pdcd4 in response to tumor-promoting conditions.
Table 2.
Pdcd4-Stabilizing Activity of Compounds 1–8
EC50 values represent the minimum concentration required for 50% recovery of the Pdcd4-luciferase signal from TPA-induced degradation.
Positive control.
EXPERIMENTAL SECTION
General Experimental Procedures
Optical rotations were recorded on a Perkin-Elmer 241 polarimeter using a 1 dm cell in the solvent indicated. Ultraviolet–visible spectra were run as methanol solutions on a Varian Cary 50-Bio UV–vis scanning spectrophotometer. ECD spectra were recorded on a Jasco J-720 spectropolarimeter using a 1 cm cell in the solvent indicated. NMR spectra were recorded on a Bruker Avance DRX-600 spectrometer operating at 600 MHz for 1H nuclei and 150 MHz for 13C nuclei, equipped with a 3 mm CPTCI probe. Residual solvent signals were used as reference: CD3OD δH 3.30; δC 49.05. High-resolution mass spectra were recorded on an Agilent Q-TOF 6520 mass spectrometer. Low-resolution mass spectra were recorded on an Agilent Series 1100 LC-MS. Normal-phase flash chromatography was carried out on Applied Separations SPE 2 g diol cartridges. Reversed-phase flash chromatography was carried out on Bakerbond Wide-Pore C4 40 µm packing. Size-exclusion flash chromatography was preformed on Amersham Biosciences Sephadex LH-20 resin. Semipreparative reversed-phase HPLC was run on a Varian ProStar 215 HPLC system using a Chromanetics Lichosorb C2 column (310 µm; 250 × 10 mm).
Plant Collection, Extraction, and Isolation
The Cryptocarya sp. sample was collected in the East Sepik Province, Papua New Guinea, in July 1995. It was identified by Professor Doel Soejarto, Director of the Pharmacognosy Field Station of the University of Illinois, Chicago, and a voucher specimen is maintained at The Field Museum, Chicago, IL (NSC number N098455). The dried and ground plant material (307 g) was repeatedly extracted with CH2Cl2 –MeOH (1:1) and 100% MeOH, according to the methodology described by McCloud,26 to give the organic solvent crude extract (10.57 g). A portion of the crude organic extract (250 mg) was subjected to diol flash chromatography eluting sequentially with hexanes, EtOAc, and MeOH. The Pdcd4-stabilizing activity was concentrated in the EtOAc fraction, which was further subjected to C4 flash chromatography eluting with MeOH–H2O (9:1), Sephadex LH-20 size exclusion chromatography eluting with hexanes–CH2Cl2–MeOH (2:5:1), followed by semipreparative C2 reversed-phase HPLC eluting with an isocratic mixture of MeCN–H2O (1:1) to yield 1 (0.5 mg, 0.20% extract weight), 2 (1.1 mg, 0.44% extract weight), 3 (1.5 mg, 0.60% extract weight), 4 (0.9 mg, 0.36% extract weight), 5 (0.3 mg, 0.12% extract weight), 6 (1.0 mg, 0.40% extract weight), 7 (2.0 mg, 0.80% extract weight), and 8 (1.8 mg, 0.72% extract weight).
Cryptocaryol A (1): clear oil; [α]D +12 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 208 (4.04) nm; ECD (MeOH) λmax (Δε) 202 (+2.7), 208 (+2.3), 256 (+0.7) nm; 1H NMR (CD3OD, 600 MHz) and 13C NMR (CD3OD, 150 MHz) data, see Table 1; HRESIMS m/z [M + H]+ 529.4105 (calcd for C30H57O7, 529.4099).
Cryptocaryol B (2): clear oil; UV (MeOH) λmax (log ε) 208 (4.01) nm; 1H NMR (CD3OD, 600 MHz) δ 7.04 (1H, ddd, J = 9.7, 6.0, 2.4 Hz, H-4), 5.97 (1H, dd, J = 9.7, 1.8 Hz, H-3), 5.07 (1H, m, H-16), 4.70 (1H, m, H-6), 4.08 (1H, m, H-8), 3.96 (2H, m, H-10/12), 3.78 (1H, m, H-14), 2.45 (1H, dt, J = 18.7, 4.9 Hz, H-5a), 2.36 (1H, ddt, J = 18.7, 11.6, 2.4 Hz, H-5B), 2.03 (3H, s, OCOCH3), 1.94 (1H, ddd, J = 14.3, 9.8, 2.3 Hz, H-7a), 1.72 (1H, ddd, J = 14.6, 12.9, 3.0 Hz, H-15a), 1.65 (2H, m, H-7b/11a), 1.62 (4H, m, H-9/13), 1.59 (2H, m, H-17), 1.57 (2H, m, H-11b/15b), 1.34–1.28 (26H, br m, CH2), 0.89 (3H, t, J = 7.0 Hz, H-31); 13C NMR (CD3OD, 150 MHz) δ 173.2 (OCOCH3), 167.0 (C-2), 148.6 (C-4), 121.4 (C-3), 76.6 (C-6), 72.9 (C-16), 69.93 (C-12), 69.87 (C-10), 67.5 (C-14), 66.6 (C-8), 45.9 (C-13), 45.8 (C-9), 45.3 (C-11), 43.9 (C-7), 43.3 (C-15), 36.0 (C-17), 33.1 (C-29), 31.0–30.5 (CH2), 26.4 (C-18), 23.8 (C-30), 21.2 (OCOCH3) 14.5 (C-31); HRESIMS m/z [M + H]+ 571.4181 (calcd for C32H59O8, 571.4204).
Cryptocaryol C (3): clear oil; [α]D +16 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 208 (3.97) nm; ECD (MeOH) λmax (Δε) 217 (+0.7), 270 (+0.09) nm; 1H NMR (CD3OD, 600 MHz) and 13C NMR (CD3OD, 150 MHz) data, see Table 1; HRESIMSm/z [M + H]+ 545.4063 (calcd for C30H57O8, 545.4048).
Cryptocaryol D (4): clear oil; UV (MeOH) λmax (log ε) 208 (4.07) nm; 1H NMR (CD3OD, 600 MHz) δ 7.06 (1H, dd, J = 9.7, 5.9 Hz, H-4), 6.06 (1H, d, J = 9.7 Hz, H-3), 5.07 (1H, m, H-16), 4.62 (1H, dt, J = 10.2, 2.7 Hz, H-6), 4.09 (1H, m, H-8), 4.03 (1H, dd, J = 5.9, 2.7 Hz, H-5), 4.00 (1H, m, H-10), 3.97 (1H, m, H-12), 3.78 (1H, m, H-14), 2.09 (1H, ddd, J = 14.7, 10.3, 2.3 Hz, H-7a), 2.03 (3H, s, OCOCH3), 1.72 (1H, m, H-15a), 1.66 (3H, m, H-7b/9), 1.60 (2H, m, H-13), 1.58 (1H, m, H-15b), 1.56 (2H, m, H-17), 1.34–1.28 (26H, br m, CH2), 0.89 (3H, t, J = 7.0 Hz, H-31); 13C NMR (CD3OD, 150 MHz) δ 173.2 (OCOCH3), 166.3 (C-2), 147.1 (C-4), 122.9 (C-3), 79.2 (C-6), 72.9 (C-16), 70.0 (C-10/12), 67.5 (C-14), 66.7 (C-8), 63.4 (C-5), 46.1 (C-9), 45.8 (C-13), 45.3 (C-11), 43.3 (C-15), 39.4 (C-7), 36.0 (C-17), 33.1 (C-29), 30.8–30.5 (CH2), 26.4 (C-18), 23.8 (C-30), 21.2 (OCOCH3), 14.5 (C-31); HRESIMS m/z [M + H]+ 587.4169 (calcd for C32H59O9, 578.4154).
Cryptocaryol E (5): clear oil; [α]D +12 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 208 (3.98) nm; ECD (MeOH) λmax (Δε) 211 (+1.5), 266 (+0.2) nm; 1H NMR (CD3OD, 600 MHz) δ 7.06 (1H, dd, J = 9.7, 5.9 Hz, H-4), 6.07 (1H, d, J = 9.7 Hz, H-3), 4.62 (1H, dt, J = 10.2, 2.7 Hz, H-6), 4.09 (1H, m, H-8), 4.04 (1H, dd, J = 5.9, 2.7 Hz, H-5), 4.03 (1H, m, H-12), 4.01 (1H, m, H-10), 3.80 (1H, m, H-14), 2.09 (1H, ddd, J = 14.7, 10.2, 2.4 Hz, H-7a), 1.67 (3H, m, H-7b/9), 1.62 (2H, m, H-11), 1.51 (2H, m, H-13), 1.43 (2H, m, H-15), 1.43–1.33 (26H, br m, CH2), 0.89 (3H, t, J = 7.0 Hz, H-29); 13C NMR (CD3OD, 150 MHz) δ 166.3 (C-2), 147.1 (C-4), 122.9 (C-3), 79.2 (C-6), 70.1 (C-10), 69.1 (C-14), 68.3 (C-12), 66.7 (C-8), 63.4 (C-5), 46.1 (C-9), 46.0 (C-11), 45.8 (C-13), 39.4 (C-7), 39.3 (C-15), 33.1 (C-27), 30.9–30.5 (CH2), 26.8 (C-16), 23.8 (C-28), 14.5 (C-29); HRESIMS m/z [M + H]+ 501.3819 (calcd for C28H53O7, 501.3786).
Cryptocaryol F (6): clear oil; [α]D +8 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 208 (4.01) nm; ECD (MeOH) λmax (Δε) 217 (+0.7), 270 (+0.08) nm; 1H NMR (CD3OD, 600 MHz) δ 7.05 (1H, dd, J = 9.7, 5.9 Hz, H-4), 6.06 (1H, d, J = 9.7 Hz, H-3), 5.33 (2H, t, J = 5.5 Hz, CH2CH=CHCH2), 4.62 (1H, dt, J = 10.2, 2.6 Hz, H-6), 4.09 (1H, m, H-8), 4.04 (1H, dd, J = 5.9, 2.6 Hz, H-5), 4.02 (1H, m, H-14), 4.00 (1H, m, H-10), 3.98 (1H, m, H-12), 3.80 (1H, m, H-16), 2.10 (1H, ddd, J = 14.6, 10.3, 2.5 Hz, H-7a), 2.02 (4H, m, CH2CH=CHCH2), 1.70 (1H, m, H-9a), 1.68 (1H, m, H-7b), 1.66 (2H, m, H-13), 1.62 (2H, m, H-11), 1.59 (1H, m, H-b), 1.51 (2H, m, H-15), 1.43 (2H, m, H-17), 1.34–1.27 (22H, br m, CH2), 0.89 (3H, t, J = 7.0 Hz, H-33); 13C NMR (CD3OD, 150 MHz) δ 166.3 (C-2), 147.1 (C-4), 130.9 (CH2CH=CHCH2), 122.9 (C-3), 79.2 (C-6), 70.2 (C-12), 70.0 (C-10), 69.1 (C-16), 68.3 (C-14), 66.7 (C-8), 63.4 (C-5), 46.1 (C-13), 46.0 (C-11), 45.8 (C-15), 45.3 (C-9), 39.4 (C-7), 39.3 (C-17), 33.1 (C-31), 30.9–30.4 (CH2), 28.2 (CH2CH=CHCH2), 26.8 (C-18), 23.8 (C-32), 14.5 (C-33); HRESIMS m/z [M + H]+ 571.4203 (calcd for C32H59O8, 571.4204).
Cryptocaryol G (7): clear oil; [α]D +7 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 208 (4.11) nm; ECD (MeOH) λmax (Δε) 217 (+1.7), 270 (+0.2) nm; 1H NMR (CD3OD, 600 MHz) δ 7.06 (1H, dd, J = 9.7, 5.9 Hz, H-4), 6.06 (1H, d, J = 9.7 Hz, H-3), 5.34 (2H, t, J = 5.5 Hz, CH2CH=CHCH2), 4.63 (1H, dt, J = 10.2, 2.6 Hz, H-6), 4.10 (1H, m, H-8), 4.04 (1H, dd, J = 5.9, 2.6 Hz, H-5), 4.02 (1H, m, H-12), 4.00 (1H, m, H-10), 3.80 (1H, m, H-14), 2.10 (1H, ddd, J = 14.6, 10.3, 2.6 Hz, H-7a), 2.02 (4H, m, CH2CH=CHCH2), 1.67 (3H, H-7a/9), 1.63 (2H, m, H-11), 1.52 (2H, m, H-13), 1.43 (2H, m, H-15), 1.32–1.27 (22H, br m, CH2), 0.89 (3H, t, J = 7.0 Hz, H-31); 13C NMR (CD3OD, 150 MHz) δ 166.3 (C-2), 147.1 (C-4), 130.9 (CH2CH=CHCH2), 122.9 (C-3), 79.2 (C-6), 70.1 (C-10), 69.1 (C-14), 68.3 (C-12), 66.7 (C-8), 63.4 (C-5), 46.1 (C-9), 46.0 (C-11), 45.8 (C-13), 39.4 (C-7), 39.3 (C-15), 33.1 (C-29), 28.2 (CH2CH=CHCH2), 30.9–30.4 (CH2), 26.8 (C-16), 23.8 (C-30), 14.5 (C-31); HRESIMS m/z [M + H]+ 527.3947 (calcd for C30H55O7, 527.3942).
Cryptocaryol H (8): clear oil; [α]D +7 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 208 (3.92) nm; ECD (MeOH) λmax (Δε) 217 (+0.5), 270 (+0.09) nm; 1H NMR (CD3OD, 600 MHz) δ 7.06 (1H, dd, J = 9.7, 5.6 Hz, H-4), 6.07 (1H, d, J = 9.7 Hz, H-3), 5.34 (2H, t, J = 5.5 Hz, CH2CH=CHCH2), 4.63 (1H, dt, J = 10.2, 2.6 Hz, H-6), 4.09 (1H, m, H-8), 4.05 (1H, m, H-10), 4.04 (1H, dd, J = 5.6, 2.6 Hz, H-5), 3.81 (1H, m, H-12), 2.10 (1H, ddd, J = 14.7, 10.2, 2.3 Hz, H-7a), 2.02 (4H, m, CH2CH=CHCH2), 1.72 (1H, m, H-9a), 1.66 (1H, ddd, J = 14.7, 10.2, 2.7 Hz, H-7b), 1.60 (1H, m, H-9b), 1.53 (2H, m, H-11), 1.44 (2H, m, H-13), 1.32–1.27 (22H, br m, CH2), 0.89 (3H, t, J = 7.0 Hz, H-29); 13C NMR (CD3OD, 150 MHz) δ 166.3 (C-2), 147.1 (C-4), 130.9 (CH2CH=CHCH2), 122.9 (C-3), 79.2 (C-6), 69.1 (C-12), 68.1 (C-10), 66.7 (C-8), 63.4 (C-5), 46.7 (C-9), 45.7 (C-11), 39.4 (C-7), 39.3 (C-17), 33.1 (C-27), 28.2 (CH2CH=CHCH2), 30.9–30.4 (CH2), 26.8 (C-14), 23.8 (C-28), 14.5 (C-29); HRESIMS m/z [M + H]+ 483.3663 (calcd for C28H51O6, 483.3682).
Pdcd4 Assay
Stabilization of Pdcd4 was assessed as previously described.11 In brief, HEK293 cells expressing a fusion protein comprised of a fragment of Pdcd4 containing the regulatory region (amino acids 39–91) and luciferase were plated (2000 cells/well, 40 µL/well) in 384-well opaque white plates and allowed to attach overnight (18 h). TPA (final concentration 10 nM) was added followed (within 15 min) by test samples or controls. Following an 8 h incubation, luciferase activity was measured 10–15 min after the addition of Steadylite Plus (Perkin-Elmer) reagent. Controls were DMSO only (no TPA), TPA only, and TPA + rapamycin (100 nM final). The activities of compounds were calculated using the following formula: Activitytarget (%) = (RLUcompound+TPA − RLUTPA)/(RLUDMSO − RLUTPA) × 100.
Supplementary Material
ACKNOWLEDGMENT
We thank D. Newman (NCI) and T. McCloud (SAIC-Frederick) for the plant extract, and M. Dyba and S. Tarasov (Biophysics Resource, SBL, NCI-Frederick) for assistance with the HRLCMS studies. This research was supported in part by the Intramural Research Program of NIH, National Cancer Institute, Center for Cancer Research. This project was also funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
ASSOCIATED CONTENT
Supporting Information. 1H NMR, 13C NMR, LRESIMS, and HRESIMS spectra for compounds 1–8 are available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Yang HS, Jansen AP, Komar AA, Zheng X, Merrick WC, Costes S, Lockett SJ, Sonenberg N, Colburn NH. Mol. Cell. Biol. 2003;23:26–37. doi: 10.1128/MCB.23.1.26-37.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang HS, Jansen AP, Nair R, Shibahara K, Verma AK, Cmarik JL, Colburn NH. Oncogene. 2001;20:669–676. doi: 10.1038/sj.onc.1204137. [DOI] [PubMed] [Google Scholar]
- 3.Leupold JH, Yang HS, Colburn NH, Asangani I, Post S, Allgayer H. Oncogene. 2007;26:4550–4562. doi: 10.1038/sj.onc.1210234. [DOI] [PubMed] [Google Scholar]
- 4.Nieves-Alicea R, Colburn NH, Simeone AM, Tari AM. Breast Cancer Res. Treat. 2009;114:203–209. doi: 10.1007/s10549-008-9993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S. Oncogene. 2008;27:2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Liu W, Chao T, Zhang Y, Yan X, Gong Y, Qiang B, Yuan J, Sun M, Peng X. Cancer Lett. 2008;272:197–205. doi: 10.1016/j.canlet.2008.06.034. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Wei Z, Gao F, Zhang X, Zhou C, Zhu F, Wang Q, Gao Q, Ma C, Sun W, Kong B, Zhang L. Anticancer Res. 2008;28:2991–2996. [PubMed] [Google Scholar]
- 8.Zhang H, Ozaki I, Mizuta T, Hamajima H, Yasutake T, Eguchi Y, Ideguchi H, Yamamoto K, Matsuhashi S. Oncogene. 2006;25:6101–6112. doi: 10.1038/sj.onc.1209634. [DOI] [PubMed] [Google Scholar]
- 9.Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. Science. 2006;314:467–471. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- 10.Schmid T, Jansen AP, Baker AR, Hegamyer G, Hagan JP, Colburn NH. Cancer Res. 2008;68:1254–1260. doi: 10.1158/0008-5472.CAN-07-1719. [DOI] [PubMed] [Google Scholar]
- 11.Blees JS, Schmid T, Thomas CL, Baker AR, Benson L, Evans JR, Goncharova EI, Colburn NH, McMahon JB, Henrich CJ. J. Biomol. Screen. 2010;15:21–29. doi: 10.1177/1087057109351028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chou T-H, Chen J-J, Lee S-J, Chiang MY, Yang C-W, Chen I-S. J. Nat. Prod. 2010;73:1470–1475. doi: 10.1021/np100014j. [DOI] [PubMed] [Google Scholar]
- 13.Wu T-S, Lin F-W. J. Nat. Prod. 2001;64:1404–1407. doi: 10.1021/np010258i. [DOI] [PubMed] [Google Scholar]
- 14.Fu X, Sévenet T, Hamid A, Hadi A, Remy F, Païs M. Phytochemistry. 1993;33:1272–1274. [Google Scholar]
- 15.Drewes SE, Horn MM, Shaw RS. Phytochemistry. 1995;40:321–323. [Google Scholar]
- 16.Meragleman TL, Scudiero DA, Davis RE, Staudt LM, McCloud TG, Cardellina JH, II, Shoemaker RH. J. Nat. Prod. 2009;72:336–339. doi: 10.1021/np800350x. [DOI] [PubMed] [Google Scholar]
- 17.Sturgeon CM, Cinel B, Díaz-Marrero A, McHardy LM, Ngo M, Andersen RJ, Roberge M. Cancer Chemother. Pharmacol. 2008;61:407–413. doi: 10.1007/s00280-007-0483-y. [DOI] [PubMed] [Google Scholar]
- 18.Davis RA, Demirkiran O, Sykes ML, Avery VM, Suraweera L, Fechner GA, Quinn RJ. Bioorg. Med. Chem. Lett. 2010;20:4057–4059. doi: 10.1016/j.bmcl.2010.05.091. [DOI] [PubMed] [Google Scholar]
- 19.Davies-Coleman MT, Rivett DEA. Prog. Chem. Org. Nat. Prod. 1989;55:1–35. [Google Scholar]
- 20.Snatzke G. Angew. Chem. Int. Ed. Engl. 1968;7:14–25. [Google Scholar]
- 21.Kobayashi Y, Tan C-H, Kishi Y. Helv. Chim. Acta. 2000;83:2562–2571. [Google Scholar]
- 22.Phuwapraisirisan P, Matsunaga S, Fusetani N. Org. Lett. 2005;7:2233–2236. doi: 10.1021/ol050648m. [DOI] [PubMed] [Google Scholar]
- 23.Ohkuma H, Naruse N, Nishiyama Y, Tsuno T, Hoshino Y, Sawada Y, Konishi M, Oki T. J. Antibiot. 1992;45:1239–1249. doi: 10.7164/antibiotics.45.1239. [DOI] [PubMed] [Google Scholar]
- 24.Burke CP, Swingle MR, Honkanen RE, Boger DL. J. Org. Chem. 2010;75:7505–7513. doi: 10.1021/jo1010203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Z-C, Jiang X-B, Wang Z-M, Zhou W-S. J. Chem. Soc. Perkin Trans. 1997;1:317–321. [Google Scholar]
- 26.McCloud TG. Molecules. 2010;15:4526–4563. doi: 10.3390/molecules15074526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.