

Abstract
Background
Late–onset Alzheimer's disease (AD) is heritable with 20 genes showing genome wide association in the International Genomics of Alzheimer's Project (IGAP). To identify the biology underlying the disease we extended these genetic data in a pathway analysis.
Methods
The ALIGATOR and GSEA algorithms were used in the IGAP data to identify associated functional pathways and correlated gene expression networks in human brain.
Results
ALIGATOR identified an excess of curated biological pathways showing enrichment of association. Enriched areas of biology included the immune response (p = 3.27×10-12 after multiple testing correction for pathways), regulation of endocytosis (p = 1.31×10-11), cholesterol transport (p = 2.96 × 10-9) and proteasome-ubiquitin activity (p = 1.34×10-6). Correlated gene expression analysis identified four significant network modules, all related to the immune response (corrected p 0.002 – 0.05).
Conclusions
The immune response, regulation of endocytosis, cholesterol transport and protein ubiquitination represent prime targets for AD therapeutics.
Keywords: Alzheimer's disease, dementia, neurodegeneration, immune response, endocytosis, cholesterol metabolism, uniquitination, pathway analysis, ALIGATOR, Weighted gene coexpression network analysis
Background
Alzheimer's disease (AD) affects over 5M Americans: one in eight over the age of 65 and represents >60% of the 6M dementia cases in Europe[1-3]. It is the commonest cause of dementia and imposes a large socioeconomic burden on individuals, their families and society. Prevalence is estimated to treble by 2050: thus understanding the mechanisms underlying this disease and developing treatments for it are essential. This study utilises the largest GWAS sample yet assembled for late-onset AD[4], and is the first to combine GWAS and expression data in a systematic search for the biological pathways underlying the genetic susceptibility to this disorder.
Much of our current understanding of the mechanisms that contribute to AD derives from the genetics of Mendelian forms of the disease: mutations in APP, PSEN1 and PSEN2 cause early onset forms of AD and underpin the amyloid cascade hypothesis[5]. While amyloid deposition is diagnostic of AD, its aetiological contribution to the majority of common late onset AD (LOAD) is unclear and therapeutic strategies addressing the amyloid cascade hypothesis have been unsuccessful[6]. Therefore other therapeutic avenues must be identified and targeted.
LOAD is genetically complex with 56-79% heritability[7]. In the Genetic and Environmental Risk in Alzheimer's Disease (GERAD) dataset[8] approximately 20% of the total trait variance was accounted for by SNPs on the GWAS chip outside the APOE region[9], with the e4 allele of the apolipoprotein E gene[10] accounting for a similar amount[9, 11]. However, a substantial proportion of the genetic variance of late-onset AD is not accounted for by the 20 susceptibility genes currently identified[11]. The remaining genetic variance is likely to be due both to susceptibility genes of small effect which current sample sizes are insufficient to detect, and to rare variants, such as the coding variants in TREM2[12], that are poorly tagged by common variants in GWAS panels. In addition, individual genome-wide signficant genes identified in such studies may themselves not form good therapeutic targets and the areas of biology that they highlight may only give a partial view of the potential therapeutic landscape. In order to gain the maximum useful information about causative pathways that may underpin LOAD and be prime targets for pharmaceutical intervention we performed a robust pathway and integrated gene expression analysis using the largest available GWAS for AD[4].
Methods
Samples and genetic data
The sample comprised 17,008 AD cases and 37,646 control subjects in the primary GWAS analysis, with 8,752 AD cases and 11,312 control subjects in the replication/extension sample and is described in detail elsewhere[4]. Only selected SNPs were genotyped in the replication/extension sample (see Online Methods).
Pathway analyses
We explored whether particular biological pathways were enriched for genetic associations[13-14]in the IGAP data[4]. We used ALIGATOR[13-14], to test whether genes containing signals below the genome-wide significance threshold contribute to a pathway signal. ALIGATOR defines significant genes as having a best single-SNP p-value less than a pre-set threshold. The resulting list of significant genes is compared to replicate gene sets generated by sampling SNPs randomly (thereby correcting for gene size). The method also controls for linkage disequilibrium between genes, and multiple testing of non-independent pathways (see Online Methods). Brown's method [15] was used to test pathway enrichment in the replication data. This method combines multiple SNPs together, explicitly correcting for both LD between SNPs and number of SNPs per gene (see Online Methods). Thus, correction for gene size was applied at both stages of the analysis. We interrogated the externally curated gene ontology (GO), KEGG and MSigDB functional pathway collections (see Online Methods).
Expression correlation analyses
We used the expression data from Gibbs et al.[16] and performed weighted gene correlation network analysis (WGCNA) using the WGCNA package[17], separately on each tissue type to identify clusters of highly correlated genes called ‘modules’. These modules were then tested for enrichment of GWA association signal in ALIGATOR.
Results
The sub-genome-wide significant variation in the IGAP data contains genetic signal, manifest by a significant excess of SNPs at all significance threshold up to p = 0.05 (Supplementary Table 1). This signal is unlikely to be due to uncorrected stratification, since each of the individual Caucasian GWAS samples in the IGAP meta analysis was corrected for ethnic variation using principal components[18].
We first identified a significant excess of biological pathways enriched for association signal in the IGAP data (Table 1, Supplementary Table 2). Using the most significant 18,472 SNPs (p < 8.32 ×10-4) from IGAP[4], covering the top 5% of genes, 177 significantly enriched (p<0.01) curated pathways were identified by ALIGATOR. To ensure that the excess of pathways was not an artifact of LD with genes of strong effect, we performed secondary enrichment analyses removing all genes that lay in the LD region of APOE or any of the genome-wide significant (GWS) genes from the IGAP[4] study. A significant excess of enriched pathways remained (Table 1), showing that the pathways showed significant enrichment independent of the “known” AD genes. Likewise, a significant excess of enriched pathways was observed when the p-value criterion for defining significant SNPs and genes was varied (Supplementary Table 3).
Table 1. Significant excess of enriched pathways remain after removing APOE and the genome-wide significant genes.
| Genes removed (number of genes) | enrichment p<0.05 | enrichment p<0.01 | enrichment p<0.001 | |||
|---|---|---|---|---|---|---|
|
| ||||||
| #path | p | #path | p | #path | p | |
| None | 542 | <0.0002 | 177 | <0.0002 | 40 | <0.0002 |
| APOE+1Mb (77) | 446 | 0.0002 | 131 | 0.0006 | 28 | 0.0008 |
| APOE+1Mb+GWS (98) | 402 | 0.0020 | 116 | 0.0008 | 23 | <0.0002 |
| APOE+1Mb+GWS+1Mb (552) | 336 | 0.0094 | 93 | 0.0066 | 22 | 0.0018 |
Genes containing a SNP with p<8.32×10-4 counted as significant. This corresponds to the top 5% of genes (ranked by most significant SNP) when no genes are removed. 0kb window used to assign SNPs to genes. #path = number of pathways
Many of the 177 pathways with p<0.01 in ALIGATOR are still significantly enriched after removing the APOE region and genes within 1Mb of a genome-wide significant SNP (Table 2, Supplementary Table 4). They remain significantly enriched under a range of p-value criteria for defining significant SNPs, and are also significant under a GSEA analysis [19-20]. This robustness to analysis parameters and methods gives confidence that the enrichments observed by ALIGATOR are genuine. Of the 177 pathways significant at p<0.01 in the ALIGATOR analysis of the IGAP GWAS, 119 are significant (p<0.05) in the replication sample. This is more than expected by chance (see Online Methods), a further confirmation that the pathways highlighted by the ALIGATOR analysis contain genuine signals. Notably, pathway SNPs had significantly lower replication p-values than non-pathway SNPs even after correcting for their p-value in the original IGAP GWAS (2-sided p=0.0237, see Online Methods). Thus, the pathway analyses highlighted which among a set of associated, but not genome-wide significant, SNPs are likely to replicate and therefore be enriched for true signals. To obtain the most strongly enriched pathways in the entire dataset, the p-values from the ALIGATOR analysis were combined with those from the replication study using Fisher's method and corrected for multiple testing of 9,816 pathways using Sidak's formula. Forty-five pathways were significant after multiple testing correction (Sidak p<0.05) in the combined dataset. These pathways are shown in Table 2, grouped into clusters by gene membership, such that pathways with more than 40% of genes in common are gathered in a cluster.
Table 2. Clusters of significant pathways in combined IGAP GWAS and replication data (Sidak-corrected p-value <0.05).
| Cluster | Pathway number | #genes | #sig | p-value | p-value no GWS | Description |
|---|---|---|---|---|---|---|
| 1 | GO: 2455 | 32 | 5 | 3.27E-12 | 5.72E-01 | humoral immune response mediated by circulating immunoglobulin |
| 1 | GO:50776 | 421 | 29 | 3.24E-09 | 1.57E-04 | regulation of immune response |
| 1 | GO: 2684 | 421 | 31 | 3.95E-09 | 2.11E-04 | positive regulation of immune system process |
| 1 | GO:50778 | 271 | 21 | 1.55E-07 | 6.65E-04 | positive regulation of immune response |
| 1 | KEGG 4664 | 78 | 13 | 5.76E-04 | 2.18E-02 | Fc epsilon RI signaling pathway |
| 2 | GO:60627 | 140 | 20 | 1.31E-11 | 2.00E-01 | regulation of vesicle-mediated transport |
| 2 | GO:30100 | 88 | 14 | 6.76E-10 | 1.06E-01 | regulation of endocytosis |
| 2 | GO:45806 | 19 | 6 | 3.91E-07 | 1.77E-02 | negative regulation of endocytosis |
| 2 | GO:48261 | 6 | 3 | 3.89E-06 | 9.82E-01 | negative regulation of receptor-mediated endocytosis |
| 2 | GO:48259 | 30 | 6 | 6.19E-05 | 1.00E+00 | regulation of receptor-mediated endocytosis |
| 3 | GO:30301 | 41 | 8 | 2.96E-09 | 2.51E-01 | cholesterol transport |
| 3 | GO:43691 | 16 | 5 | 3.90E-09 | 2.78E-01 | reverse cholesterol transport |
| 3 | GO:15918 | 42 | 8 | 3.91E-09 | 3.15E-01 | sterol transport |
| 3 | GO:34366 | 8 | 2 | 6.40E-07 | N/A | spherical high-density lipoprotein particle |
| 4 | KEGG 4640 | 81 | 11 | 1.05E-08 | 4.91E-01 | Hematopoietic cell lineage |
| 5 | GO:32434 | 40 | 5 | 1.34E-06 | 1.00E+00 | regulation of proteasomal ubiquitin-dependent protein catabolic process positive regulation of ubiquitin-protein ligase activity involved in mitotic cell |
| 5 | GO:51437 | 70 | 9 | 2.60E-03 | 2.60E-03 | cycle |
| 5 | GO:51439 | 76 | 9 | 3.82E-03 | 3.82E-03 | regulation of ubiquitin-protein ligase activity involved in mitotic cell cycle |
| 5 | REACT 440 | 108 | 11 | 3.89E-03 | 3.89E-03 | REACTOME_CELL_CYCLE_CHECKPOINTS |
| 5 | GO:51443 | 77 | 9 | 9.62E-03 | 9.62E-03 | positive regulation of ubiquitin-protein ligase activity |
| 6 | REACT 539 | 261 | 25 | 2.95E-05 | 6.93E-02 | REACTOME_HEMOSTASIS |
| 7 | GO:30131 | 31 | 7 | 1.20E-03 | 9.13E-01 | clathrin adaptor complex |
| 7 | GO:30119 | 32 | 7 | 1.53E-03 | 9.54E-01 | AP-type membrane coat adaptor complex |
| 7 | GO:44433 | 301 | 31 | 1.01E-02 | 1.00E+00 | cytoplasmic vesicle part |
| 7 | GO:30122 | 9 | 4 | 1.29E-02 | 1.00E+00 | AP-2 adaptor complex |
| 7 | GO:30118 | 39 | 7 | 1.35E-02 | 1.00E+00 | clathrin coat |
| 8 | GO: 6457 | 200 | 12 | 1.60E-03 | 1.00E+00 | protein folding |
To obtain the most strongly enriched pathways in the entire dataset (IGAP GWAS and replication), the p-values from the ALIGATOR analysis (counting the top 5% of genes as significant) were combined with those from the replication study using Fisher's method. The resulting p-values from the combined samples were corrected for multiple testing of 9,816 pathways using Sidak's formula. For each pair of gene sets, an overlap measure K was defined as the number of genes common to both sets divided by the number of genes in the smaller dataset. A gene set was assigned to a cluster if the average K between it and the gene sets already in the cluster was greater than 0.4. If it was not possible to assign a gene set to an existing cluster, a new cluster was started. This procedure was carried out recursively, in descending order of enrichment significance. Clusters containing a significant pathway are listed here, and where more than 5 pathways are significant only the five most significant pathways in each cluster are shown. A complete list of pathways significant at p<0.01 in the ALIGATOR analysis of the IGAP GWAS data is given in Supplementary Table 4. “No GWS” refers to analyses in which genes containing a SNP genome-wide significant (p<5×10-8) in the IGAP GWASdataset (and thus expected to be strongly significant in the replication dataset) are removed from the analysis of the replication data.
This multiple testing correction may be considered conservative since it assumes that the pathways are independent, whereas in fact there is considerable genic overlap between them. Sidak-corrected p-values for the combined IGAP GWAS and replication datasets are therefore given in Supplementary Table 4 for all 177 pathways significant at p<0.01 in the ALIGATOR analysis of the IGAP GWAS. Redundant pathways (i.e. those with high genic overlap with other pathways) were not pruned from our analysis since it is not clear a priori which pathways will give the most significant enrichment and should thus be retained. Pruning a posteriori (i.e. by choosing the most significant pathways) will bias the significance of the combined discovery and replication p-values (making the correction for multiple testing of pathways anticonservative). The pathway clusters given in Table 2 and Supplementary Table 4 are intended to aid interpretation of our results in light of shared gene membership between pathways, by highlighting areas of biology rather than individual pathways.
The clusters of multiple pathways were related to the broad categories of immune response, regulation of endocytosis, cholesterol transport, protein ubiquitination and clathrin, with the first three of these being particularly strongly enriched for signal. Since one would expect SNPs showing strong association to be significant upon replication regardless of biology, the analysis was repeated removing genes containing a genome-wide significant SNP in the IGAP GWAS from the analysis of the replication data. From Table 2 it can be seen that the immune-related and ubiquitination pathways are still highly significant. Sidak-corrected p-values for all 177 pathways significant at p<0.01 in the ALIGATOR analysis are shown in Supplementary Table 4. The relationship between the enriched pathways is shown by their shared gene membership (Figure 1). Table 3 lists genes in the clusters identified in Table 2 that are counted as significant (best single-SNP p<8.32×10-4) in the ALIGATOR analysis of the IGAP GWAS and also gene-wide significant (gene-wide p<0.05) in both the IGAP GWAS and the replication data. P-values for all genes counted as significant in the ALIGATOR analysis from the 177 pathways enriched at p<0.01 are given in Supplementary Table 5.
Figure 1. The pathways highlighted by ALIGATOR ontology analyses are related.
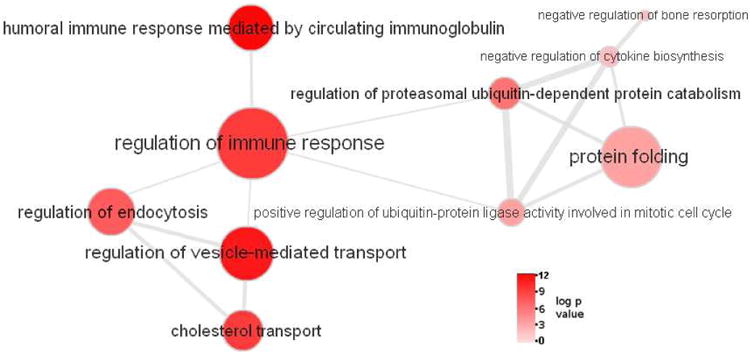
The network was generated in ReVIGO[32] using gene ontology processes identified in ALIGATOR only. Bubble size (and label font size) reflects the frequency of the GO term in the GOA database, bubble colour reflects pathway p-value. Similar GO terms are linked by edges (lines) in the network where line width reflects the degree of similarity between pathways but line length is arbitrary. Strong relationships are revealed between negative regulation of endocytosis and cholesterol transport and many of the pathways are related to the immune response process.
Table 3. Genes in the significant ALIGATOR pathway clusters.
| Entrez ID | Gene Symbol | Best p (IGAP) | Gene-wide p (IGAP) | Best p (REP) | Gene-wide p (REP) |
|---|---|---|---|---|---|
| Cluster 1: Immune response | |||||
| 1191 | CLU | 2.48E-17 | 5.14E-15 | 1.06E-10 | 2.60E-08 |
| 1378 | CR1 | 3.65E-15 | 3.46E-07 | 3.82E-11 | 5.06E-08 |
| 2206 | MS4A2 | 3.28E-10 | 3.68E-09 | 1.81E-04 | 6.54E-06 |
| 3117 | HLA-DQA1 | 3.38E-09 | 1.20E-05 | 5.33E-05 | 8.89E-03 |
| 3123 | HLA-DRB1 | 1.24E-08 | 6.54E-06 | 5.80E-05 | 1.13E-02 |
| 3127 | HLA-DRB5 | 2.87E-07 | 4.78E-05 | 4.56E-04 | 5.23E-03 |
| 1380 | CR2 | 9.35E-07 | 2.99E-02 | 5.76E-05 | 6.41E-03 |
| 3119 | HLA-DQB1 | 2.97E-06 | 3.88E-05 | 3.58E-04 | 6.45E-03 |
| 3635 | INPP5D | 6.62E-06 | 3.33E-03 | 9.93E-06 | 1.02E-04 |
| 102 | ADAM10 | 1.45E-04 | 2.90E-02 | 1.13E-02 | 2.71E-02 |
| Cluster 2: Endocytosis | |||||
| 274 | BIN1 | 3.72E-16 | 4.75E-06 | 3.15E-11 | 5.27E-09 |
| 8301 | PICALM | 1.91E-12 | 1.20E-08 | 2.57E-07 | 2.97E-07 |
| 2206 | MS4A2 | 3.28E-10 | 3.68E-09 | 1.81E-04 | 6.54E-06 |
| 1265 | CNN2 | 1.19E-06 | 3.07E-03 | 2.91E-04 | 2.11E-03 |
| Cluster 3: Cholesterol transport | |||||
| 1191 | CLU | 2.48E-17 | 5.14E-15 | 1.06E-10 | 2.60E-08 |
| 10347 | ABCA7 | 1.70E-09 | 3.00E-07 | 1.43E-06 | 1.02E-06 |
| Cluster 4: Hematopoietic cell lineage | |||||
| 1378 | CR1 | 3.65E-15 | 3.46E-07 | 3.82E-11 | 5.06E-08 |
| 3123 | HLA-DRB1 | 1.24E-08 | 6.54E-06 | 5.80E-05 | 1.13E-02 |
| 3127 | HLA-DRB5 | 2.87E-07 | 4.78E-05 | 4.56E-04 | 5.23E-03 |
| 1380 | CR2 | 9.35E-07 | 2.99E-02 | 5.76E-05 | 6.41E-03 |
| Cluster5: Protein ubiquitination | |||||
| 1191 | CLU | 2.48E-17 | 5.14E-15 | 1.06E-10 | 2.60E-08 |
| 5702 | PSMC3 | 3.70E-06 | 3.04E-05 | 1.55E-02 | 1.15E-02 |
| 5434 | POLR2E | 1.94E-05 | 6.93E-03 | 1.08E-03 | 1.26E-04 |
| 6827 | SUPT4H1 | 1.94E-04 | 2.26E-02 | 2.27E-02 | 2.27E-02 |
| 5706 | PSMC6 | 2.98E-04 | 1.25E-02 | 3.99E-02 | 3.79E-02 |
| 6878 | TAF6 | 4.22E-04 | 1.66E-02 | 6.41E-04 | 6.41E-04 |
| Cluster 6: Hemostasis | |||||
| 1191 | CLU | 2.48E-17 | 5.14E-15 | 1.06E-10 | 2.60E-08 |
| 3635 | INPP5D | 6.62E-06 | 3.33E-03 | 9.93E-06 | 1.02E-04 |
| Cluster 7: Clathrin/AP2 adaptor complex | |||||
| 1191 | CLU | 2.48E-17 | 5.14E-15 | 1.06E-10 | 2.60E-08 |
| 8301 | PICALM | 1.91E-12 | 1.20E-08 | 2.57E-07 | 2.97E-07 |
| 9179 | AP4M1 | 2.16E-04 | 2.13E-03 | 3.74E-04 | 1.57E-04 |
| Cluster 8: Protein folding | |||||
| 1191 | CLU | 2.48E-17 | 5.14E-15 | 1.06E-10 | 2.60E-08 |
| 664618 | HSP90AB4 | 4.62E-04 | 2.30E-03 | 2.19E-02 | 4.48E-02 |
Gene-wide p-values calculated using Brown's method (see Online Methods). Genes shown are those counted as significant (best p<8.32×10-4) in the ALIGATOR analysis of the IGAP GWAS data that are also significant (gene-wide p<0.05) in the replication data. Note that genes in the vicinity of APOE are not included in this table since this region was not genotyped in the replication sample. Such genes were highly significant in the meta analysis (p<1×10-10) and comprise APOC1/2 in cluster 2, APOE and APOC1/2/4 in cluster 4, APOE, PVRL,BCL3 and PVR in cluster 7, APOE in cluster 8.
In contrast to ALIGATOR, GSEA uses all genes (rather than using a threshold) and weights these by their significance, so may highlight different biological signals. We therefore performed a secondary analysis of all pathways using GSEA. Pathways significant under GSEA but not ALIGATOR are shown in Supplementary Table 6. Most of these pathways relate to areas of biology already highlighted in the ALIGATOR analysis, the exceptions being synapse, neuronal differentiation and calcium signalling (Supplementary Table 6). Genes contributing to these pathway signals that are significant in both the IGAP GWAS and the replication study are listed in Supplementary Table 7. Notably, these pathways contain large genes. In addition to the differences between ALIGATOR and GSEA described above, the Simes correction for gene size used by GSEA is less stringent for large genes than that used by ALIGATOR, thereby explaining the discrepancy in the results between the methods.
In the ALIGATOR analysis 73.2% of the top 5% of genes mapped to a pathway, leaving a substantial minority of genes unannotated: in addition many annotated genes may possess other functions not currently annotated. Genes with correlated expression patterns display functional similarities and Zhang et al.[21] highlighted modules of co-expressed genes as being important in the aetiology of LOAD. Therefore, in order to overcome the annotation gap and access biologically related signal across the entire genome, we created modules of brain co-expressed genes and tested them for enrichment of association signal in the IGAP GWAS. The dataset we used consisted of gene expression data from four brain regions in a sample of approximately 150 control brains[16], and was independent from that used by Zhang et al.[21]. We used control individuals rather than AD cases so that correlations between expression levels would not be confounded by neuron loss. A weighted gene correlation network analysis (WGCNA)[17] gave 117 modules of co-expressed genes in these data (see Online Methods and Supplementary Table 8): these 117 modules were tested for enrichment of association signal in the IGAP GWAS using ALIGATOR. Four modules were found to be significantly enriched after correcting for multiple testing, and these enrichments were robust to varying p-value criteria and analysis methods (Supplementary Table 9). The four significantly enriched modules, one from each brain region, are all related to the immune response and have overlapping gene membership (Figure 2).
Figure 2. The immune response is enriched in gene co-expression modules from human brain.
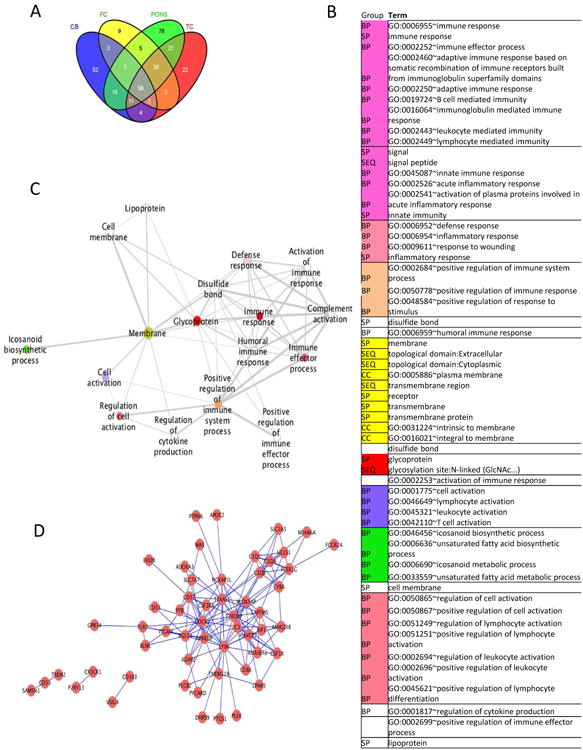
A Venn diagram indicating the number of genes in common across the four modules that were found to be significantly enriched in the IGAP GWAS using ALIGATOR after correcting for multiple testing. Each significant module originates from a different brain region as indicated here (Cb = cerebellum, FC = frontal cortex, TC = temporal cortex). B Network showing the pathways significantly enriched for gene membership among the 151 genes present in at least two of the four most significantly enriched expression modules: the principal biological themes were derived from DAVID[33-34] analysis. Terms from the analysis were filtered at 0.05% FDR, progressively clustered according to average gene similarity at a threshold of 90% and rendered on Cytoscape with the Enrichment Map plugin[35-36]. The diagram shows only the principal (lowest FDR) term for each of the clusters and white nodes indicate a single term that does not cluster with other groups. Coloured nodes indicate a multi-term cluster: the related terms represented by each node are given in C, in increasing significance order. Sources of the functional terms are:
BP = GOTERM_BP_FAT: Gene Ontology biological processes in DAVID's GO Fat Database;
CC = GOTERM_CC_FAT: Cellular Component terms in DAVID's GO Fat Database;
SP = SP_PIR_KEYWORDS: keywords in the Uniprot (Swiss-Prot/Protein Information Resource) database
SEQ = UP_SEQ_FEATURE: Uniprot sequence annotation feature.
The full data are available in Supplementary Table 8
D Network showing the strongest correlations in expression (>0.9 in at least one brain area) between genes present in at least two of the four most significantly enriched expression modules.
The extent to which the overlap in gene membership between these modules is related to the GWAS signal was investigated by examining genes that occurred in multiple modules and testing these for enrichment using ALIGATOR and GSEA (Supplementary Table 10). It can be seen that the set of 151 out of 294 genes that are present in two or more modules consistently showed the most significant enrichment of IGAP signal across a variety of test criteria. Conversely, the set of 143 genes present in only one module showed no significant enrichment for association signal, highlighting the utility of using multiple datasets to produce meaningful co-expression modules. Figure 2 shows the strongest correlations (>0.9 in at least one brain area) between the 151 genes present in two or more modules. It can be seen that the TYROBP gene highlighted by Zhang et al.[21] as an important causal regulator is also a hub gene in this network. Pathways significantly enriched in the 151 genes present in two or more modules are shown in Figure 2, clustered by gene membership. Many of the enriched pathways are immune-related, but some are related to fatty acid metabolism and lipoprotein, further corroborating the results of our analysis of the IGAP GWAS data. A list of the 151 genes is shown in Supplementary Table 11.
We also directly tested the modules described by Zhang et al.[21] for enrichment of association signal in the IGAP GWAS data (Supplementary Table 12). No single module was significantly enriched after correction for multiple testing of modules (“corr p” <0.05), but the most significantly enriched modules are immune-related. Interestingly, the immune/microglia module highlighted by Zhang et al.[21] (#1, yellow) did not show significant enrichment for association signal in the IGAP GWAS under ALIGATOR analysis, although it did show moderate enrichment under GSEA. However, the 108 genes common both to this module and the set of 151 genes present in two or more of the four most significantly enriched modules in our analysis do show enrichment, which becomes progressively more significant as increasingly stringent criteria are used to select significant SNPs and genes (Supplementary Table 13). The genes that are in the Zhang module but not our set of 151 genes show no significant enrichment for association signal under either ALIGATOR or GSEA analysis.
Discussion
This analysis reveals pathways aetiologically related to AD in addition to those identified previously[14, 22]. The current sample[4] is larger than any used before and was imputed on a dense reference panel, giving improved gene coverage, and is therefore likely to be more powerful to detect real associations than any previous study. A larger set of pathways has been analysed than previously and annotations have changed, so gene membership of pathways is not identical to previous studies, though a substantial proportion of genes still fall into the annotation gap and are not currently mapped to any pathway. In the current analysis we also clustered genes that were within 1Mb of each other together in ALIGATOR, to prevent counting a single signal more than once. Secondary analyses were also performed removing genes in the APOE LD region and within 1Mb of the GWS genes. This was done to prevent pathway enrichments being biased by LD between pathway genes and neighbouring genes of strong effect, and to test whether there were significant pathway enrichments independent of “known” AD genes. Such enrichments would increase the interest of novel pathways and genes highlighted by the main analysis Despite these differences, many of the pathways previously identified[14] show enrichment in the IGAP dataset (Supplementary Table 14). These include cholesterol transport, immunity and the synaptic transmission, cholinergic pathway, the latter being the target of the cholinesterase class of drugs widely used in AD.
We used both GWAS and expression data to detect functional pathways associated with AD. ALIGATOR analysis of combined IGAP-GWAS and replication samples highlights four main areas of biology: the immune response, regulation of endocytosis, cholesterol transport and protein ubiquitination. The immune response is particularly significant in the replication sample, even when GWS genes from the IGAP GWAS are excluded. The replication SNPs were not chosen for pathway membership and do not survey the genome randomly, so the lack of significance in some pathway clusters once the GWS genes are removed does not mean that there is no excess signal in these pathways: this may simply not have been measured. However these data indicate that further genes that are involved in the immune response are likely to be implicated in LOAD. Both regulation of endocytosis and cholesterol transport are functions also implicated by the genome-wide significant genes, while the immune response and protein ubiquitination contain fewer genome-wide significant signals[4]. The most significant signals in the GSEA analysis relate to the same biology but add some additional categories related to neurological biology including the synapse and neuronal projection development along with calcium-related signalling, not revealed by ALIGATOR. It is notable that these areas of biology are linked by common gene membership (Figure 1) and their interdependence may also be important in susceptibility to AD.
The additional immune response genes implicated in cluster 1 (Table 3) are plausible AD risk genes: CR2 encodes complement receptor 2 which is present on subsets of B-cells as is the GWS CR1. HLA-DQB1 is in the chromosome 6 HLA locus in common with several GWS loci. INPP5D is genome-wide significant once replication analyses are taken into account[4]. As well as being annotated as having immune system activity, ADAM10 has been proposed as a candidate α-secretase that cleaves APP to prevent the production of β-amyloid[23]. The protein ubiquitination cluster 5 (Table 3) includes two ATPase subunits of the 19S proteasome, PSMC3 and PSMC6, and three proteins involved in transcriptional control, POLR2E, SUPT4H1 and TAF6. CNN2, encoding calponin 2, thought to regulate the actin cytoskeleton[24] appears in the endocytosis cluster, though it can also regulate phagocytosis in macrophages[25]. The additional genes from GSEA include CHRNA2 encoding the neuronal cholinergic receptor, nicotinic, alpha 2 and RAPSN, the receptor-associated protein of the synapse, both of which appear in the synaptic transmission, cholinergic pathway (Supplementary Table 13). CAV1 encodes caveolin 1 which can interact with APOE[26] and is found in caveolae, areas of cholesterol-rich lipid raft involved in endocytosis. CACNA1D encodes the calcium channel, voltage-dependent, L type, alpha 1D subunit, one of a series of alpha subunits that confer channel-specific properties, influences insulin secretion and is a risk gene for type 2 diabetes[27]. Finally, APP itself is highlighted in this analysis: it is annotated in both the synapse and neuronal clusters. Recent findings show that there is at least one rare protective coding variant in APP seen in late onset AD[28] and this signal may reflect this or other relatively rare variants.
Convergent evidence for the importance of the immune response in AD susceptibility was obtained by performing WGCNA on expression data from four brain regions. The four modules that were significantly enriched for association in the IGAP GWAS after multiple testing correction were all related to the immune response, and shared multiple genes in common: INPP5D is GWS[4] and was the only GWS gene found in these modules. The enrichment for association was driven by genes that occurred in two or more of these modules. None of the modules from Zhang et al.[21] was significantly enriched for genetic association after multiple testing correction, though the immune-related modules in their study gave the strongest signal. However, while the microglia module highlighted by Zhang et al.[21] did not show significant enrichment for association, the genes shared in common with our signficant expression modules did, highlighting the utility of using multiple expression datasets in generating biologically-meaningful modules. The TYROBP gene highlighted by Zhang et al. as an important causal regulator is also a hub gene in this network[29].
Regulation of endocytosis, cholesterol transport and ubiquitination were not strongly represented in our WGCNA modules, which may relate to the large size of the modules and the use only of brain gene expression. In addition, co-ordinated gene expression in brain may well reflect differences in distribution of specific cell types or sub-types[30]. The brain expression signatures we used came from non-neurologically compromised brains but it is likely that changes in microglial composition or fate in response to inflammation or infection in these subjects could propagate such co-ordinate changes in gene expression. TREM2 is one of the 151 genes that occur in two or more expression modules (Figure 2) and rare variants in TREM2 are associated with a significant increase in AD susceptibility[12]. TREM2 regulates the phenotype of microglia controlling their downstream activation to an inflammatory or phagocytotic fate, thought to promote or inhibit AD pathogenesis respectively[31]. Thus the expression signature we detect through genome–wide association may also be a marker for changes in microglial phenotypes that act to enhance or inhibit the susceptibility of individuals to AD.
As the main motivation for genetic analysis of complex traits is to understand the biology of disease and inform the search for treatments, interpretation of genetic signals in a biologically meaningful way is essential. Pathway analyses that integrate multiple dense sources of data provide a means of starting to do this. Identifying strong susceptibility targets also highlights potential drug targets. While expression analyses alone can provide important clues about aetiology of disease, integrating them with genetic data which identify causative factors underlying susceptibility to disease ensures that the gene expression signatures revealed are related to disease aetiology rather than secondary effects, making the pathways highlighted by the analysis primary targets for therapy. This study implicates regulation of endocytosis and protein ubiquitination, in addition to cholesterol metabolism, as potential therapeutic targets in AD. It strongly reinforces the critical role of the immune system in conferring AD susceptibility: gaining a detailed mechanistic understanding of the events within the immune system that predispose to AD and investigating how to address these mechanisms should now be a priority for AD research.
Online Methods
IGAP meta-analysis data
The main dataset was reported by the International Genomics of Alzheimer's Project (IGAP) consortium[4] and consists in total of 17,008 cases and 37,646 controls. The full details of the samples and methods for conduct of the GWA studies are provided in the respective manuscripts[4, 8, 22, 37-39]. This sample of AD cases and controls comprises 4 data sets taken from genome-wide association studies performed by GERAD, EADI, CHARGE and ADGC[40].
Each of these datasets was imputed with Impute2 software using 1000 genomes data (release Dec2010) as a reference panel. In total 11,863,202 SNPs were included in the SNPs allelic association result file. To make our analysis as conservative as possible, we only included autosomal SNPs which passed stringent quality control criteria, i.e. we included only SNPs with minor allele frequencies (MAF) ≥0.01 and INFO score greater than or equal to 0.8 in each individual study, resulting in 7,055,881 with SNPs which are present in at least 40% of the AD cases and 40% of the controls in the analysis. We corrected all individual SNPs p-values for genomic control (GC) λ=1.087. These SNPs are well imputed on a large proportion of the sample, which increases confidence in the accuracy of the association analysis upon which the pathway and gene-wide analyses are based.
Replication data
11,632 SNPs with p-values <10-3 in the IGAP meta-analysis were successfully genotyped in a replication sample comprising 8,492 cases and 11,392 controls (see primary IGAP manuscript[40] for more details).
Assignment of SNPs to genes
SNPs were assigned to genes if they were located within the genomic sequence lying between the start of the first and the end of the last exon of any transcript corresponding to that gene, as defined by NCBI. The chromosome and location for all currently known human SNPs was taken from the dbSNP132 database, as was their assignment to genes (using build 37.1). In total, we retained 2,804,431 (39.7% of the total) SNPs which annotated 28,636 unique genes with 1-16,514 SNPs per gene. Pathway analyses were also performed using 10kb and 60kb windows around genes to assign SNPs to genes.
Assignment of genes to functional gene sets
Genes were assigned to a series of functional gene sets defined by five independent sources: 1) Gene ontology (GO) [41] (http://www.geneontology.org/; downloaded 11/6/2011), 2) Kyoto Encyclopedia of Genes and Genomes (KEGG) [42] (http://www.genome.jp/kegg/; downloaded 27/6/2011), 3) the “canonical pathways” collection from the Molecular Signatures Database v3.0 (MsigDB) (http://www.broadinstitute.org/gsea/msigdb/index.jsp) accessed on 2/2/2011. We restricted our analysis to a total of 9,816 functional gene sets containing between 3 and 500 genes: 8,888 in GO, 234 in KEGG and 694 in MsigDB.
Statistical analysis
Gene-wide significance
Gene-wide significance was calculated by combining single-SNP p-values while controlling for LD and different number of marker's per gene using Brown's method[15] adopted for set-based analysis of genetic data[43].
ALIGATOR analysis
ALIGATOR was then used to test the list of gene-wide significance measures for enrichment within functional gene sets as previously described in Holmans et al.[13]. Unlike methods designed for gene-expression data (where there is typically only one measurement per gene), ALIGATOR corrects for variable numbers of SNPs per gene. ALIGATOR takes a list of significant SNPs and coverts this into a list of the genes in which these SNPs lie. Each gene is counted once regardless of how many significant SNPs it contains, thus eliminating the influence of linkage disequilibrium (LD) between SNPs within genes Replicate gene lists of the same length as the original are generated by randomly sampling SNPs (thus correcting for variable gene size). The lists are used to obtain p- values for enrichment for each gene set, to correct these for testing multiple non-independent gene sets, and to test whether the number of significantly enriched gene sets is higher than expected.
To minimise the possibility of multiple significant genes in a pathway that are close together reflecting the same association signal due to LD, we conservatively grouped significant genes that were <1Mb apart and located in the same functional gene set into one signal. To remove the possibility of a small gene set being deemed significantly enriched based on just one signal, we only classed gene sets as being enriched if they carried at least two signals
To assess the potential of any bias caused by LD with strong association signals that had been previously identified in these samples, we also performed ALIGATOR analysis after excluding three sets of genes: a) all genes within 1Mb of APOE (77 genes), b) all genes within 1 Mb of APOE and the 21 genes containing a SNP reaching genome-wide significance (p<5×10-8) in the IGAP meta-analysis (98 genes) c) all genes within 1Mb of APOE or any of the 21 genome-wide significant genes (552 genes). The same p-value criteria for defining significant SNPs are used for all these analyses.
Definition of significant SNPs and genes for ALIGATOR
ALIGATOR requires that a set of significant SNPs are chosen to define the list of significant genes used to determine pathway enrichment [13]. As the genetic signal extends beyond the genome-wide significant genes (Supplementary Table 1), the p-value cut off used to select these SNPs should be fairly lax. For our primary analysis, we selected SNPs such that 5% of the genes would be deemed significant. When no gene window was used to assign SNPs to genes, this required 18,472 SNPs, with a p-value criterion for inclusion of 8.32×10-4. When a 10kb window was used to assign SNPs to genes, the SNP p-value criterion required to cover the top 5% of genes is reduced to 5.39×10-4, (14,385 SNPs) and using a 60kb window reduces the criterion still further, to 1.66×10-4 (7,807 SNPs). Supplementary Table 2 shows the number of significantly enriched pathways using each of these windows. It can be seen that using a 0kb window gives a more significant excess of enriched pathways than the 10kb or 60kb windows. Thus, the 0kb window was used for all analyses presented in this paper. To ensure that the results of the ALIGATOR analyses are not dependent on the choice of p-value criterion for defining significant genes, secondary analyses were performed using a range of p-value criteria.
Gene-set-enrichment (GSEA) analysis
As a further validation of the ALIGATOR results, and to show that the results of our analyses are not driven by the choice of p-value cut-off for defining significant genes, gene-set enrichment analysis (GSEA) was performed on the gene sets nominally-significant (p<0.05) in the ALIGATOR analyses using the method described in[44]. Rather than defining a list of significant genes, GSEA ranks all genes in order of a gene-wide association statistic, and tests whether the genes in a particular gene set have higher rank overall than would be expected by chance, weighted by the values of their gene-wide association statistic (thus giving more weight to significant genes). Following Wang and colleagues, in order to allow for varying numbers of SNPs per gene, the gene-wide statistic used was the Simes-corrected single-SNP p-value[45]. Since the GSEA method is known to be sensitive to very strongly associated genes, the analysis was performed removing all genes within 1Mb of APOE, and also the 21 genome-wide significant genes.
Clustering of significantly-enriched gene sets
To aid functional interpretation, gene sets significantly enriched in the ALIGATOR analysis were assigned to clusters according to the genes they contain. This was done as follows: For each pair of gene sets, an overlap measure K was defined as the number of genes in common to both sets divided by the number of genes in the smaller dataset. A gene set was assigned to a cluster if the average K between it and the gene sets already in the cluster was greater than 0.4. If it was not possible to assign a gene set to an existing cluster, a new cluster was started. This procedure was carried out recursively, in descending order of enrichment significance.
Pathway analysis of replication data
Pathway-wide evidence of association in these data was assessed by aggregating the p-values of all SNPs in the pathway using the method of Brown. This is a generalisation of Fisher's method for combining p-values in situations where the p-values are not independent, and was adapted to genetic association data by Moskvina et al. (2011)[43]. LD between SNPs was estimated using the December 2010 release of the 1000 Genomes data (the same release that was used to impute the data for the IGAP meta-analysis).
The pathways of interest contain several genes with very significant associations in the IGAP meta analysis that are also strongly associated in the replication study. Since the presence of such genes can give rise to a significant Brown p-value even in the absence of signal from the remainder of the pathway, the analysis of the replication data was repeated removing all SNPs from genes containing a genome-wide significant (p<5×10-8) SNP in the IGAP meta-analysis.
Effect of varying p-value criterion on pathway analysis
A significant excess of enriched pathways is still observed (Supplementary Table 3) when the p-value criterion for defining significant SNPs (and, thus, genes) is varied. Again, the significantly enriched pathways from Table 3 also show significant enrichment over a range of p-value criteria (Supplementary Table 4), thus giving extra confidence that the enrichments are genuine, and not an artefact of how the significant genes are defined. This is confirmed by observing that many of the significantly enriched pathways from the ALIGATOR analysis also have significant p-values in the GSEA analysis (Table 3 and Supplementary Table 4).
Replication of pathway analysis results
Further confirmation of pathway significance was gained from a replication sample in which a subset of SNPs from the main IGAP study (excluding the APOE region) were studied: these showed a significant enrichment of association signal in the pathways identified by the pathway analysis of the IGAP meta-analysis data. Pathway-wide Brown p-values derived from the replication data (see online methods) are given in Supplementary Table 4 for all 177 pathways enriched at p<0.01 in the original ALIGATOR analysis. Of these pathways, 119 have a Brown p<0.05 when all SNPs in the pathway are included, and 97 have a Brown p<0.05 after removal of SNPs from genes with a genome-wide significant SNP in the IGAP meta-analysis. These are considerably higher than expected by chance, and indicate the presence of genuine AD risk variants in these pathways, even outside the “known” AD risk genes.
Genes containing a significant SNP (p<8.32×10-4) in the IGAP meta-analysis that are also nominally significant (gene-wide p<0.05) in the replication data are shown in Table 4 for the pathway clusters listed in Table 2. Gene-wide p-values for all genes containing a significant SNP (p<8.32×10-4) in the IGAP meta-analysis that lie in any of the 177 pathways enriched at p<0.01 in the ALIGATOR analysis are shown in Supplementary Table 5.
As a final test of whether SNPs that lie in pathways of interest are enriched for association signal in the replication data, a regression analysis of replication p-value on pathway membership was performed. Specifically, the 5297 replication SNPs that lay within gene boundaries were sorted in order of their IGAP meta-analysis p-value. The list was then pruned by removing SNPs within 100kb of a more significant SNP from the IGAP meta-analysis. This left 730 SNPs. The pruning procedure was carried out to prevent the regression analyses being biased by clusters of neighbouring SNPs with similar p-values. Of these pruned and filtered SNPs 163 were in the 177 pathways enriched at p<0.01 in the ALIGATOR analysis and were found to have significantly lower p-values than the genic SNPs not in the pathways (p=5.57×10-5). However, the ALIGATOR analysis preferentially selects pathways enriched for significant SNPs in the IGAP meta analysis and the IGAP meta analysis p-value is a highly significant predictor of replication p-value (p<2×10-16). While this shows that pathway SNPs are selected from genes that are likely to be true positives, in order to demonstrate an advantage of pathway membership (in terms of replication p-value) over and above that conferred by being significant in the IGAP meta-analysis, a linear regression was carried out of –log(replication p) on –log(IGAP meta p) and pathway membership simultaneously. Pathway SNPs had significantly lower replication p-values than non-pathway SNPs even after correcting for IGAP meta p-value (2-sided p=0.0237). This provides further evidence of the utility of pathway analysis in highlighting true positive signals.
Description of Gibbs expression data
The brain expression data are described in Gibbs et al.[16] and the GEO database reference for the dataset is GSE15745.
Description of WGCNA and derivation of co-expressed modules
Present/absent calls were made on the dataset by detection p-value. Any single probeset from a sample was designated absent with a p-value >0.1. If more than half the probesets in the dataset were absent, they were flagged for removal. In addition, a sample was removed if the number of missing probesets were above 2 standard deviations from the mean of the dataset. After removing probesets with over 50% absence, the remaining data was normalised. The influence of age and post-mortem interval (PMI) on the dataset was accounted for by performing regression according to these values and taking the residuals.
All arrays were separated by tissue type. Each tissue sample set was further assessed for outliers by hierarchical clustering. Any branch of arrays at the top of the dendrogram that contained less than 10% of the total number of arrays was removed so that the further analysis would not be charactersing these small sub-groups but focus on more global patterns of gene expression. This pruning was continued until each of the principal branches on the dendrogram contained over 10% of all arrays. The final sample numbers are given in Supplementary Table 15.
Weighted gene correlation network analysis was performed in the R environment using the WGCNA package[17] and performed separately on each tissue type. The dataset was collapsed so that multiple probes were reduced to single gene values based on gene annotation of the probesets obtained from Biomart. For duplicate probesets, the largest mean value for the sample was selected.
Soft-thresholding powers were selected by plotting a range of candidate powers against connectivity measures and observing the values where connectivity began to decrease. These all occurred between power values of 6 and 8 for the 4 tissue types. The modules were then created with this soft-threshold power (using a minimum module size of 20), and the component gene names of the modules extracted. The modules were then tested for enrichment for association signal in the IGAP GWAS as pathways in ALIGATOR using the same thresholds for defining significant SNPs (p<8.32×10-4) as previously.
Supplementary Material
Research in Context.
As the main motivation for genetic analysis of complex traits is to understand the biology of disease and inform the search for treatments, interpretation of genetic signals in a biologically meaningful way is essential. Pathway analyses that integrate multiple dense sources of data provide a means of starting to do this. Identifying strong susceptibility targets also highlights potential drug targets. While expression analyses alone can provide important clues about aetiology of disease, integrating them with genetic data which identify causative factors underlying susceptibility to disease ensures that the gene expression signatures revealed are related to disease aetiology rather than secondary effects, making the pathways highlighted by the analysis primary targets for therapy. This study implicates regulation of endocytosis and protein ubiquitination, in addition to cholesterol metabolism, as potential therapeutic targets in AD. It strongly reinforces the critical role of the immune system in conferring AD susceptibility: gaining a detailed mechanistic understanding of the events within the immune system that predispose to AD and investigating how to address these mechanisms should now be a priority for AD research.
Acknowledgments
This work was made possible by the generous participation of the control subjects, the patients, and their families. GERAD was supported by the Wellcome Trust, Medical Research Council (MRC), Alzheimer's Research UK (ARUK) and the Welsh Assembly Government. ADGC and CHARGE were supported by the National Institutes of Health, National Institute on Aging (NIH-NIA) supported this work. CHARGE was also supported by Erasmus Medical Center and Erasmus University. IGAP was funded by the French National Fondation on Alzheimer's disease and related disorders, the Centre National de Génotypage and the Institut Pasteur de Lille, Inserm, FRC (fondation pour la recherche sur le cerveau) and Rotary. This work has been developed and supported by the LABEX (laboratory of excellence program investment for the future) DISTALZ grant (Development of Innovative Strategies for a Transdisciplinary approach to ALZheimer's disease). The French National Fondation on Alzheimer's disease and related disorders and the Alzheimer's Association provided support for I-GAP meetings, communication and some funds to each consortium to support analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hebert LE, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Archives of neurology. 2003;60(8):1119–22. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 2.Plassman BL, et al. Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology. 2007;29(1-2):125–32. doi: 10.1159/000109998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson RS, et al. Sources of variability in estimates of the prevalence of Alzheimer's disease in the United States. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2011;7(1):74–9. doi: 10.1016/j.jalz.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lambert JC, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nature genetics. 2013 doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardy J. The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem. 2009;110(4):1129–34. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- 6.Giacobini E, Gold G. Alzheimer disease therapy-moving from amyloid-beta to tau. Nat Rev Neurol. 2013 doi: 10.1038/nrneurol.2013.223. [DOI] [PubMed] [Google Scholar]
- 7.Gatz M, et al. Role of genes and environments for explaining Alzheimer disease. Archives of general psychiatry. 2006;63(2):168–74. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- 8.Harold D, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nature genetics. 2009;41(10):1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee SH, et al. Estimation and partitioning of polygenic variation captured by common SNPs for Alzheimer's disease, multiple sclerosis and endometriosis. Hum Mol Genet. 2013;22(4):832–41. doi: 10.1093/hmg/dds491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261(5123):921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 11.So HC, et al. Evaluating the heritability explained by known susceptibility variants: a survey of ten complex diseases. Genetic epidemiology. 2011;35(5):310–7. doi: 10.1002/gepi.20579. [DOI] [PubMed] [Google Scholar]
- 12.Guerreiro R, et al. TREM2 Variants in Alzheimer's Disease. The New England journal of medicine. 2012 doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holmans P, et al. Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. American journal of human genetics. 2009;85(1):13–24. doi: 10.1016/j.ajhg.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones L, et al. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer's disease. PloS one. 2010;5(11):e13950. doi: 10.1371/journal.pone.0013950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown MB. A method for combining non-independent, one-sided tests of significance. Biometrics. 1975;31:978–992. [Google Scholar]
- 16.Gibbs JR, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6(5):e1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nature genetics. 2006;38(8):904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 19.Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature genetics. 2003;34(3):267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 20.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang B, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013;153(3):707–20. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lambert JC, et al. Implication of the immune system in Alzheimer's disease: evidence from genome-wide pathway analysis. Journal of Alzheimer's disease : JAD. 2010;20(4):1107–18. doi: 10.3233/JAD-2010-100018. [DOI] [PubMed] [Google Scholar]
- 23.O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu KC, Jin JP. Calponin in non-muscle cells. Cell Biochem Biophys. 2008;52(3):139–48. doi: 10.1007/s12013-008-9031-6. [DOI] [PubMed] [Google Scholar]
- 25.Huang QQ, et al. Role of H2-calponin in regulating macrophage motility and phagocytosis. J Biol Chem. 2008;283(38):25887–99. doi: 10.1074/jbc.M801163200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yue L, et al. Apolipoprotein E enhances endothelial-NO production by modulating caveolin 1 interaction with endothelial NO synthase. Hypertension. 2012;60(4):1040–6. doi: 10.1161/HYPERTENSIONAHA.112.196667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reinbothe TM, et al. The human L-type calcium channel Cav1.3 regulates insulin release and polymorphisms in CACNA1D associate with type 2 diabetes. Diabetologia. 2013;56(2):340–9. doi: 10.1007/s00125-012-2758-z. [DOI] [PubMed] [Google Scholar]
- 28.Jonsson T, et al. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature. 2012;488(7409):96–9. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 29.Langfelder P, Mischel PS, Horvath S. When is hub gene selection better than standard meta-analysis? PloS one. 2013;8(4):e61505. doi: 10.1371/journal.pone.0061505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuhn A, et al. Population-specific expression analysis (PSEA) reveals molecular changes in diseased brain. Nat Methods. 2011;8(11):945–7. doi: 10.1038/nmeth.1710. [DOI] [PubMed] [Google Scholar]
- 31.Forabosco P, et al. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol Aging. 2013;34(12):2699–714. doi: 10.1016/j.neurobiolaging.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Supek F, et al. REVIGO summarizes and visualizes long lists of gene ontology terms. PloS one. 2011;6(7):e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 34.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merico D, et al. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PloS one. 2010;5(11):e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hollingworth P, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nature genetics. 2011;43(5):429–35. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seshadri S, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303(18):1832–40. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naj AC, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nature genetics. 2011;43(5):436–41. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lambert JCea. Extended meta-analysis of 74,538 individuals identifies 11 new susceptibility loci for Alzheimer's disease. 2013 doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harris MA, et al. The Gene Ontology (GO) database and informatics resource. Nucleic acids research. 2004;32(Database issue):D258–61. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanehisa M, et al. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic acids research. 2012;40(Database issue):D109–14. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moskvina V, et al. Evaluation of an approximation method for assessment of overall significance of multiple-dependent tests in a genomewide association study. Genetic epidemiology. 2011;35(8):861–6. doi: 10.1002/gepi.20636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang K, Li M, Bucan M. Pathway-based approaches for analysis of genomewide association studies. American journal of human genetics. 2007;81(6):1278–83. doi: 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simes RJ. An improved Bonferroni-type procedure for multiple tests of significance. Biometrika. 1986;73:751–754. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.