

Abstract
NANOG expression in prostate cancer is highly correlated with cancer stem cell characteristics and resistance to androgen deprivation. However, it is not clear whether NANOG or its pseudogenes contribute to the malignant potential of cancer. We established NANOG- and NANOGP8-knockout DU145 prostate cancer cell lines using the CRISPR/Cas9 system. Knockouts of NANOG and NANOGP8 significantly attenuated malignant potential, including sphere formation, anchorage-independent growth, migration capability, and drug resistance, compared to parental DU145 cells. NANOG and NANOGP8 knockout did not inhibit in vitro cell proliferation, but in vivo tumorigenic potential decreased significantly. These phenotypes were recovered in NANOG- and NANOGP8-rescued cell lines. These results indicate that NANOG and NANOGP8 proteins are expressed in prostate cancer cell lines, and NANOG and NANOGP8 equally contribute to the high malignant potential of prostate cancer.
Keywords: NANOG, NANOGP8, gene knockout, CRISPR/Cas9, prostate cancer
INTRODUCTION
Tumors include a small population of cells with stem cell-like properties, including self-renewal and tumor-initiation capacities [1–5]. These cells are often termed cancer stem-like cells (CSCs) and express pluripotency-related genes [6–9], such as NANOG, OCT3/4, and SOX2, which are essential transcription factors in embryonic stem cells (ESCs) [10, 11]. These transcription factors are involved in various somatic cancers and drive tumor development [12–18]. NANOG is expressed in various cancers, such as ovarian cancer [19], breast cancer [20], colorectal cancer [21], and prostate cancer [22], and it is enriched in CSCs [16]. CD44+CD133+ and CD133+ cells are markers of prostate CSCs in human prostate cancer tissues [23], and these immune cells express higher NANOG mRNA levels compared to their corresponding negative cells. Increased NANOG expression is associated with poor prognosis in various cancers, such as lung cancer [24], oral cancer [25], brain cancer [26], ovarian cancer [19], breast cancer [27], and prostate cancer [22]. Additionally, NANOG expression is negatively correlated with postoperative survival in patients with lung and ovarian cancer [19, 24], and increased NANOG expression in human prostate cancer tissues is correlated with an increased Gleason score, which is an indicator of poor prognosis [22, 28].
NANOG (hereinafter NANOG1 to avoid confusion) is particularly interesting because this gene has at least 10 pseudogenes, but the sequence similarities among these genes confounds analyses of NANOG expression [29]. The NANOGP8 pseudogene has attracted attention because only NANOGP8 encodes the full-length NANOG1 protein with a 2-amino acid substitution, and NANOGP8 is expressed in cancer cells and increases the clonogenicity and tumorigenicity [18, 21, 22, 30] [31–34]. NANOGP8 overexpression in vitro promotes sphere formation and migration in a prostate cancer cell line and drug resistance in a breast cancer cell line [30]. In addition, NANOGP8-overexpressing cells form larger tumors in vivo in immunodeficient mice [30, 33]. However, no antibodies can distinguish NANOG1 and NANOGP8 proteins because of the high similarity between these two proteins. Therefore, the expression of NANOG1 and its pseudogenes has only been analyzed using reverse transcription polymerase chain reaction (RT-PCR) and cDNA sequencing analysis [35]. Most somatic cancer cell lines predominantly express protein-coding NANOGP8 and non-coding NANOGP5 with markedly less NANOG1 expression. In contrast, human ESCs and the NTERA2 cell line, which is derived from a human teratocarcinoma, express large amounts of NANOG1 [35]. Therefore, NANOGP8 is likely a primary contributor of NANOG protein expression in various somatic cancers [35], including prostate cancer. However, the proportion of NANOG protein expression that comes from NANOG1 and NANOG8 in cancer cells is not known. The overexpression of NANOGP8 in prostate cancer cell lines has been shown to increase migration and tumorigenic potential [30], and the overexpression of NANOG1 has been shown to increase migration in an ovarian cancer cell line [19] and increase migration, metastasis, and tumorigenic potential in a breast cancer cell line [27]. However, these previous gain-of-function studies did not include loss-of-function analyses of NANOG1 and NANOGP8 because the sequence similarity makes individual gene knockout without off-target effects difficult. Therefore, a causal role of NANOG1 and NANOGP8 in cancer cells is not clear.
This study established NANOG1- and NANOGP8-knockout, DU145 prostate cancer cell lines using CRISPR/Cas9 system-mediated genetic engineering [36, 37]. In the DU145 prostate cancer cell line with endogenous NANOG1 and NANOGP8 proteins, both NANOG1 and NANOGP8 contributed equally to many properties associated with malignant potential in prostate cancer, including sphere formation, migration, drug resistance, and tumorigenic potential. Our findings suggest that the malignant potential of cancer cells is increased by NANOG protein expression from both NANOGP8 and NANOG1.
RESULTS
Establishment of NANOG1- and NANOGP8-knockout DU145 cell lines and rescue cell lines
Human NANOG1 has at least 10 pseudogenes. NANOG1 and the pseudogene NANOGP8 code for intact NANOG protein. We first generated each gene knockout in DU145 cells (human prostate cancer cell line) using the CRISPR/Cas9 system to evaluate the functions of these two genes [36, 37]. We designed two gRNAs against exon 2 of NANOG1, which codes for the homeodomain, to avoid non-specific effects of the CRISPR/Cas9 system (Figure 1A). Genomic DNA PCR products from each cell were cloned into a plasmid to analyze the targeted NANOG1 genomic region in each transfected cell line. The NANOG1 PCR primers only amplify the NANOG1 genomic region because the forward primer recognizes intron 1 of NANOG1, which is unique among NANOG1 and its pseudogenes (Figure 1A). This primer amplified the targeted NANOG1 genomic region, and amplicon sequence analyses demonstrated that NANOG1−/− #1 and NANOG1−/− #2 harbored 26 bp and 8 bp deletions, respectively, in exon 2 of the NANOG1 gene (Figure 1B). All 16 analyzed sequences from NANOG1−/− #1 and all 8 analyzed sequences from NANOG1−/− #2 exhibited the same deletions. The amplified genomic region in DU145 cells includes a restriction enzyme cleavage site for BfmI, and this cleavage site is lost in NANOG1−/− #1. We performed restriction enzyme analysis of the PCR amplicon to confirm the deletion in NANOG1−/− #1 cells (Figure 1C). BfmI completely digested the amplicon from DU145 cells, but the amplicon from NANOG1−/− #1 cells was not digested. Therefore, we confirmed the deletion of the NANOG1 gene on both alleles in NANOG1−/− #1 cells. This method to confirm the deletion was not available in NANOG1−/− #2 cells because the deleted site did not contain a restriction enzyme site. Therefore, we confirmed the deletion of the NANOG1 gene on both alleles in NANOG1−/− #2 cells using sequencing analysis. The deletions in NANOG1−/− #1 and #2 cells induce frame shifts, which result in premature stop codons. We examined the off-target effects of the gRNAs on NANOG pseudogenes because the gRNA expression constructs that targeted exon 2 of NANOG1 exhibit a high similarity to NANOG pseudogenes. In conclusion, NANOG1−/− #1 cells have a 19 bp and a 5 bp deletion in NANOGP7 and a 124 bp insertion in NANOGP9. However, NANOG1−/− #2 cells harbor no indels in NANOG pseudogenes. NANOG1−/− cell lines exhibited no off-target effects in NANOGP8, which is the only NANOG pseudogene with the ability to produce intact full-length NANOG protein.
Figure 1. Generation of NANOG1- and NANOGP8-knockout DU145 cell lines using the CRISPR/Cas9 system.
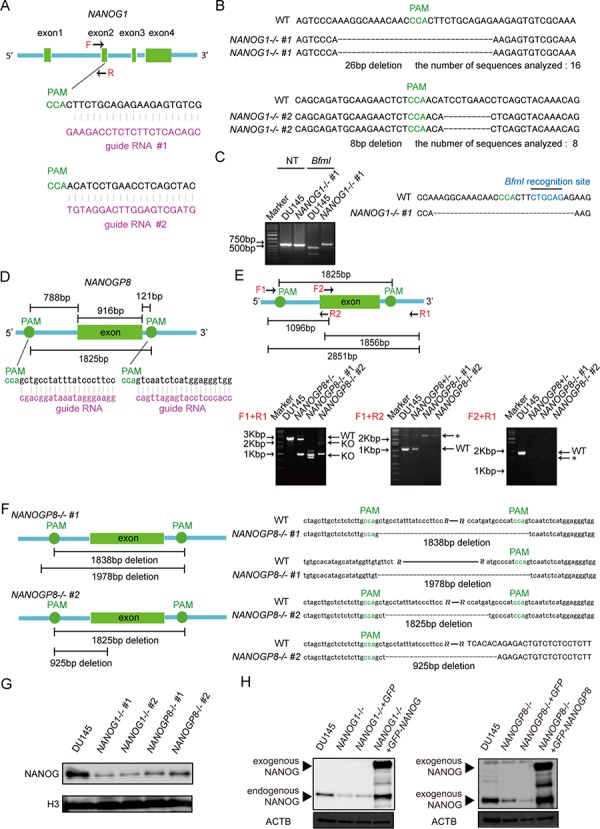
A. Schematic representation of the NANOG1-targeting gRNA sequences. Arrows indicate primer positions. PAM, protospacer adjacent motif. B. Two NANOG1−/− cell lines were established from DU145 cells. The deleted sequences in the NANOG1−/− #1 and NANOG1−/− #2 cell lines are presented. The number of sequences analyzed for each cell line is indicated below the deleted sequences. C. Confirmation of the genotype of NANOG1-/- cells. The NANOG1 genomic regions in the indicated cells were amplified using the NANOG1-specific primers indicated in Figure 1A. Amplicons digested by BfmI were separated in the depicted agarose gel. NT, no treatment. D. Schematic representation of the NANOGP8-targeting gRNA sequences. E. Upper panel: Schematic representation of the NANOGP8 genomic region, targeted PAM positions, and primer positions. Arrows indicate primer positions. Lower panel: Genotyping of NANOGP8-/- cells. The NANOGP8 genomic region was analyzed by PCR. Amplicons were separated in agarose gels. Using the F1 + R1 primer set, the 2851 bp wild type region (WT) was amplified in DU145 cells, whereas shorter amplicons (KO) were detected in NANOGP8-knockout cell lines. Neither the F1 + R2 primer set nor the F2 + R1 primer set amplified the NANOGP8 genomic region in NANOGP8−/− cells. Asterisks indicate non-specific bands. F. Establishment of two NANOGP8−/− cell lines from DU145 cells. NANOGP8-/- #1 cells have deletions of 1838 bp and 1978 bp, and NANOGP8-/- #2 cells have deletions of 1825 bp and 925 bp. G. NANOG protein levels in the indicated cell lines. NANOG protein expression was analyzed by Western blot analysis. H3 was used as the loading control. H. Establishment of NANOG1- and NANOGP8-rescued cell lines. NANOG expression in these rescued cell lines was analyzed by Western blot. Exogenous NANOG indicates GFP-NANOG and GFP-NANOGP8 proteins in each rescued cell lines. ACTB was used as the loading control.
To delete the NANOGP8 gene, we designed two gRNAs outside of NANOGP8 (Figure 1D). Because most NANOG pseudogenes, including NANOGP8, are intronless genes with highly conserved sequences, it is difficult to specifically amplify the exon of NANOGP8. It is also challenging to quickly examine the genotypes of NANOGP8-targeted clones using the single-gRNA approach employed for NANOG1 (Figure 1A and 1D). We designed three primer sets to screen for NANOGP8 gene deletion. Primer set F1 + R1 amplified a 2851-bp region of the NANOGP8 gene in DU145 cells, and the amplicon was apparently shorter in the NANOGP8 gene knockout cell line (Figure 1E). Primer sets F1 + R2 and F2 + R1 could not amplify the NANOGP8 genomic region in the NANOGP8 gene knockout cell line (Figure 1E). These primers identified two NANOGP8−/− cell lines. Sequence analysis of the PCR products revealed that NANOGP8−/− #1 had a 1838-bp and a 1978-bp deletion, and NANOGP8−/− #2 had a 1825-bp and a 925-bp deletion (Figure 1F), which indicated that we generated two independent NANOGP8-knockout DU145 cell lines. Unfortunately, we were unsuccessful in our attempt to establish NANOG1- and NANOGP8-double knockout cell lines (Supplementary Figure 1).
We analyzed NANOG protein expression in each cell line using Western blot to examine whether NANOG1 and NANOGP8 contribute to the production of NANOG protein in DU145 cells. NANOG protein expression decreased significantly in the NANOG1- and NANOGP8-knockout cell lines (Figure 1G). NANOG1 is the primary contributor of NANOG expression in ESCs, but NANOG protein is primarily derived from NANOGP8 in DU145 cells, as shown by PCR-based analyses [35]. Therefore, we designed three “multi-NANOG” primer sets with high similarity to NANOG pseudogenes, with the exception of NANOGP3 and NANOGP6, which are too different from the other NANOG pseudogenes to be amplified by a common primer set (Figure 2A and 2B). Sequence analyses indicated that primer 1 predominantly amplified NANOGP8, primer 2 amplified NANOGP8, NANOGP1 and NANOG1, and primer 3 mainly amplified NANOGP4 and NANOGP5. The upper amplicon of primer 2 was derived from NANOGP1 and NANOG1 cDNA, which are derived from each pre-mRNA that included intron 3 (Figure 2A). Therefore, we conclude that each primer exhibited a PCR bias (Figure 2A and 2B), and RT-PCR and sequence analyses of cloned cDNA are not appropriate for examining the proportion of NANOG expression from each gene. DU145 cells actually express all 7 NANOG genes, including NANOG1 and NANOGP8. We established a NANOG1-rescued cell line and a NANOGP8-rescued cell line from NANOG1−/− #1 cells and NANOGP8−/− #1 cells, respectively, to avoid non-specific effects of the CRISPR/Cas9 system. NANOG protein expression was recovered in each rescued cell line, which suggests that exogenous NANOG1 protein promotes NANOGP8 expression, and vice versa (Figure 1H). Taken together, our results indicate that DU145 cells express both NANOGP8 and NANOG1.
Figure 2. Unequal amplification of transcripts corresponding to NANOG1 and its pseudogenes by PCR.
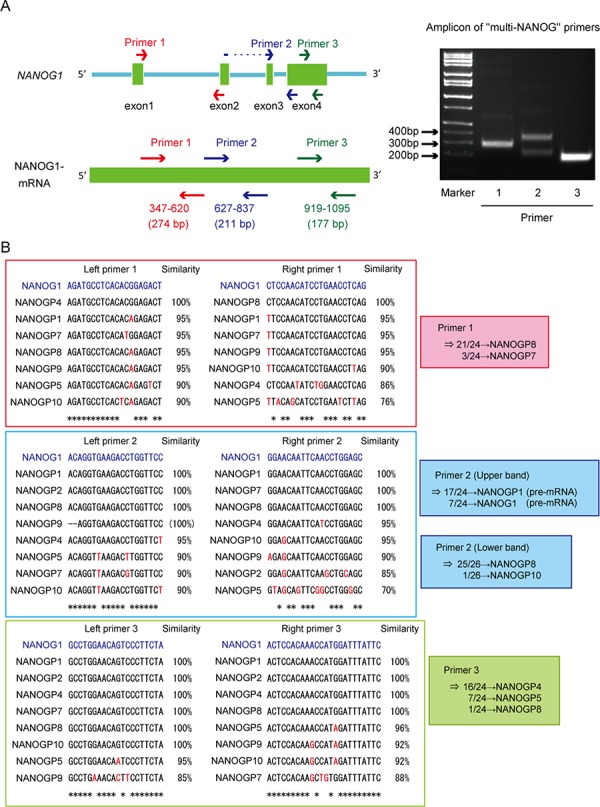
A. Left panel: Schematic representation of the NANOG1 gene (top) and NANOG1 mRNA (bottom). Arrows indicate “multi-NANOG” primer positions. DU145 cDNA was amplified using each “multi-NANOG” primer set. Right panel: Amplicons were separated in the depicted agarose gel. B. Left panels: Multiple sequence alignments of NANOG1 and its pseudogenes. Primer sequences are presented in blue at the top of each panel, and base differences between NANOG1 and its pseudogenes are indicated in red. PCR products generated using each “multi-NANOG” primer set were cloned into plasmids, and their sequences were analyzed. Right panel: Results of sequence analyses.
NANOG1 and NANOGP8 knockout decreases the clonogenic potential of DU145 cells
Malignant tumor cells exhibit well-known aggressive and anchorage-independent cell growth, and a high clonogenic potential. We first examined NANOG1−/− and NANOGP8−/− cell proliferation in a monolayer to examine whether NANOG1 and NANOGP8 were involved in these well-known tumor cell properties. However, each knockout cell line exhibited a similar growth rate as DU145 cells (Figure 3A). NANOGP8-expressing prostate cancer cells exhibit greater clonogenic potential [30]. We utilized a colony formation assay to examine the role of NANOG1 and NANOGP8 in self-renewal. The colony-forming capacity of NANOG1−/− and NANOGP8−/− cells was decreased compared to parental DU145 cells (Figure 3B). The sphere-forming capacity of prostate cancer cells is highly associated with tumorigenic potential [34]. Therefore, we examined the sphere-forming capacity of NANOG1−/− and NANOGP8−/− cells and evaluated the number of spheres that formed after 2 weeks. Five independent experiments demonstrated that the sphere-forming capacity of NANOG1−/− and NANOGP8−/− cells decreased to approximately 50% compared to DU145 cells (Figure 3C). We also evaluated the sphere-forming capacity of NANOG1- and NANOGP8-rescued cell lines and demonstrated that the number of formed spheres in NANOG1- and NANOGP8-rescued cell lines increased significantly compared to the non-rescued cell lines (Figure 3D). The size of the formed spheres in NANOG1−/− and NANOGP8−/− cell lines was generally smaller than the parental DU145 cells, and this phenotype was also recovered in NANOG1- and NANOGP8-rescued cell lines (Figure 3E). We also utilized the soft agar colony formation assay to examine clonogenic potential under anchorage-independent conditions. The clonogenic potential under anchorage-independent conditions was lower in the NANOG1−/− and NANOGP8−/− cell lines compared to DU145 cells. This phenotype was also recovered in NANOG1- and NANOGP8-rescued cell lines (Figure 3F). Therefore, we concluded that NANOG1 and NANOGP8 increase the clonogenic potential of DU145 cells.
Figure 3. Knockout of NANOG1 and NANOGP8 decreases clonogenic potential.
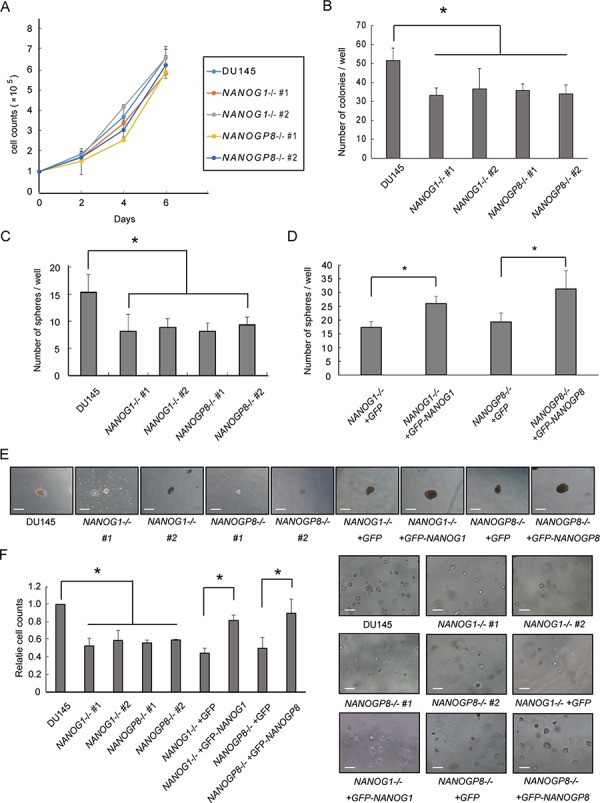
A. Adherent cell growth was assessed by cell counting at the indicated times. Error bars indicate means ± SE (n = 3). B. Anchorage-dependent colony formation assay. A total of 100 cells were seeded in 6-well plates, and colonies consisting of at least 50 cells were counted after 14 days. The number of colonies for each cell line is compared with the number of colonies for parental DU145 cells. Error bars indicate means ± SE (n = 4). *p < 0.05. C–D. Sphere-forming assay. Each cell line was plated at 2000 cells/well in low-attachment 6-well plates and cultured in serum-free epithelial basal medium (Cambrex) supplemented with B27, insulin, EGF, and basic fibroblast growth factor for 2 weeks. Sphere-forming capacity was assessed based on the number of spheres observed at 14 days. Error bars indicate means ± SE (n = 5). *p < 0.05. C) The numbers of spheres for the NANOG1- and NANOGP8-knockout cell lines were compared with the number of spheres for DU145 cells. D) The numbers of spheres for the NANOG1- and NANOGP8-rescued cell lines were compared with the numbers of spheres for the GFP-expressing knockout cell lines. E. Representative microscopic images of sphere formation for the indicated cell lines. Scale bars represent 300 μm. F. Clonogenic potential under anchorage-independent growth conditions was examined using the soft agar colony formation assay. The microscopic appearance of each cell line in soft agar is depicted on the right side of the figure. Each relative cell count at 8 days was calculated as a ratio compared with DU145 cells. Error bars indicate means ± SE (n = 3). *p < 0.05. Scale bars represent 300 μm.
NANOG1 and NANOGP8 knockout decreases DU145 cell migration capability
NANOG controls cell migration in cancer [19] [38]. Therefore, wound-healing assays were performed to examine the effect of NANOG1 and NANOGP8 on prostate cancer cell migration. Migration was decreased in NANOG1−/− and NANOGP8−/− cells by 40–60%, and this phenotype was recovered in the NANOG1- and NANOGP8-rescued cell lines. The rescued cell lines exhibited similar migration to parental DU145 cells in this study (Figure 4A–4C). Decreased E-cadherin expression in cancer cells leads to epithelial-mesenchymal transition (EMT) [39]. The transcription factor Snail represses E-cadherin expression [40] and accelerated EMT promotes cell migration. SNAIL, which encodes SNAIL, is recently reported to be a transcriptional target of NANOG in reprograming cells [41]. We also found that NANOG1−/− and NANOP8−/− DU145 cells exhibited increased E-cadherin expression and decreased Snail expression and that the increased E-cadherin and decreased Snail expression were abolished in NANOG1- and NANOGP8-rescued cells (Figure 4D). These results indicate that NANOG1 and NANOGP8 are involved in the promotion of migration capacity in DU145 cells.
Figure 4. Knockout of NANOG1 and NANOGP8 decreases migratory potential.
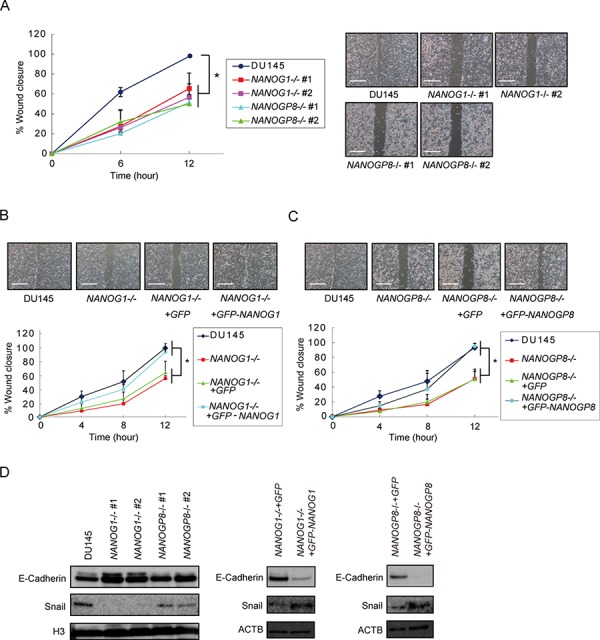
A–C. Wound healing assay in the indicated cell lines. The width of wound closure in DU145 cells after 12 hours was set to 100%. Error bars indicate means ± SE (n = 3). *p < 0.05. Scale bars represent 500 μm. D. E-cadherin and Snail expression levels in the indicated cell lines. E-cadherin and Snail expression were analyzed by Western blot. H3 and ACTB were used as loading controls.
NANOG1 and NANOGP8 knockout increases docetaxel sensitivity in DU145 cells
NANOG1 and NANOGP8 overexpression increases drug resistance in cancer cells [30], and NANOG knockdown increases drug sensitivity in cancer cells [42]. Therefore, we evaluated the viability of NANOG1−/− and NANOGP8−/− cells using an MTS assay 48 hours after docetaxel administration. NANOG1- and NANOP8-knockout cells showed increased sensitivity to docetaxel (Figure 5A), and docetaxel sensitivity was similar between the NANOG1- and NANOGP8-rescued cell lines and parental cells (Figure 5B and 5C). We also evaluated the number of residual cells from each cell line 72 hours after 10 nM docetaxel treatment in vitro (Figure 5D). These results indicate that NANOG1 and NANOGP8 decrease drug sensitivity in DU145 cells.
Figure 5. Knockout of NANOG1 and NANOGP8 increases sensitivity to docetaxel.
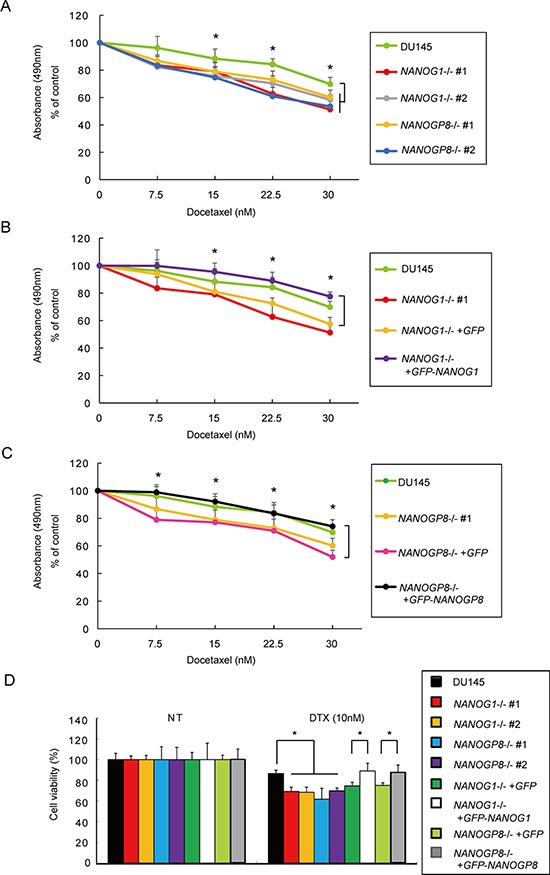
A–C. Cell viability was assessed by an MTS assay after docetaxel administration. Each cell line was seeded in a 48-well plate at 2.5 × 104 cells/well; 24 hours later, docetaxel was added at concentrations of 7.5, 15, 22.5, or 30 nM. Cell viability was evaluated after 48 hours of docetaxel treatment. Error bars indicate means ± SE (n = 9), and data are presented as percentages relative to the indicated cell lines. *p < 0.05. D. Each cell line was seeded in a 6-well plate at 5 × 104 cells/well; 24 hours later, docetaxel was added at a concentration of 10 nM. Residual cell numbers were counted 72 hours after the administration of 10 nM docetaxel (DTX). NT, no treatment. Error bars indicate means ± SE (n = 3), and the data are presented as percentages relative to the indicated cell lines. *p < 0.05.
NANOG1 and NANOGP8 knockout decreases the in vivo tumorigenicity of DU145 cells
Tumorigenic potential is promoted by NANOGP8, but not NANOG1, overexpression in cancer cells [30]. We subcutaneously injected 2 × 106 NANOG1-knockout cells, NANOGP8-knockout cells, or parental cells into non-obese diabetic/severe combined immunodeficient (NOD-SCID) mice to determine whether NANOG depletion influences tumor development in vivo. NANOG1- and NANOGP8-knockout cells exhibited significantly decreased tumorigenic potential compared to parental cells (Figure 6A and 6B). We next examined whether this phenotype was recovered by exogenous NANOG1 and NANOGP8 expression. NANOG1- and NANOGP8-rescued cells showed increased tumorigenic potential compared to NANOG1−/−+GFP and NANOGP8−/−+GFP cells, respectively (Figure 6A and 6B). These results demonstrated that NANOGP8 and NANOG1 increase the tumorigenic potential of DU145 cells in vivo.
Figure 6. Tumorigenic potential was reduced in NANOG1- and NANOGP8-knockout DU145 cells.
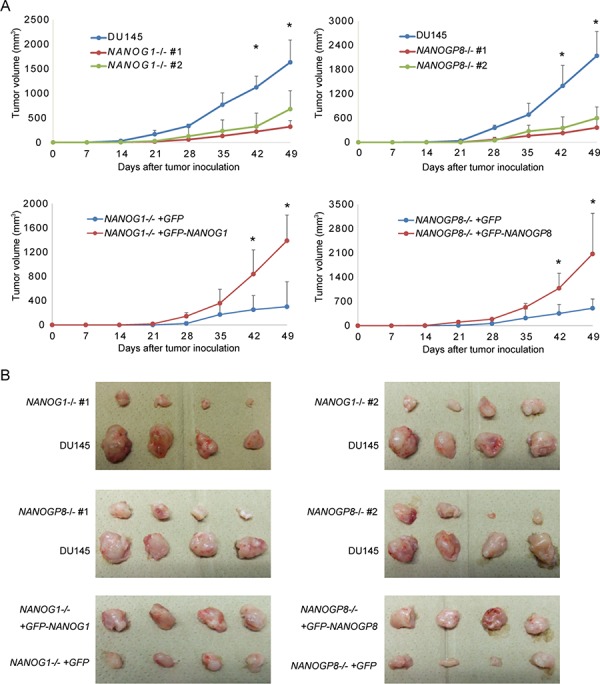
A. Tumor development in vivo. DU145 and its derivatives (2 × 106 cells) were implanted subcutaneously in NOD-SCID mice. Tumor appearance and development were observed for 7 weeks. Tumor volume was calculated using the following formula: tumor volume (mm3) = length × (width)2 /2. The data for the indicated time points are presented as means ± SE (n = 4). *p < 0.05. B. Tumor images in the indicated cell lines 49 days after inoculation.
DISCUSSION
Elucidating the link between transcription factors and cancer properties is of interest because improves our understanding of how cancer cells promote malignancy through the regulation of gene expression pattern. NANOG1 is a critical transcription factor that enables the pluripotent properties of ESCs [43] [44]. Recent studies suggested a role for NANOG1 in the progression of malignancy using knockdown systems [16] [19] [42]. NANOG1 knockdown using RNAi likely exerts an off-target effect on NANOGP8 mRNA, and vice versa, because of the high similarity between NANOG1 and NANOGP8 mRNA. Moreover, knockdown using RNAi cannot completely turn off target genes, and the possibility that cells with residual gene function influence other cells cannot be excluded, especially in tumor development. Therefore, knockdown experiments are not suitable for functional analyses of NANOG1 and NANOGP8 genes. Jeter et al. reported that the overexpression of NANOGP8, but not NANOG1, in prostate cancer cell lines increases migration capacity and tumorigenic potential [30]. However, Lu et al. reported that NANOG1 overexpression in breast cancer cells increases migration capacity, metastasis, and tumorigenic potential [27], and Siu et al. reported that NANOG1 overexpression increased the migration capacity of an ovarian cancer cell line [19]. Therefore, the effects of NANOG1 and NANOGP8 on malignant potential remain controversial. This study used the CRISPR/Cas9 system to establish NANOG1- and NANOGP8-knockout prostate cancer cell lines and examine the function of NANOG1 and NANOGP8 genes in prostate cancer cells. We found that DU145 cells express NANOGP8 and NANOG1, in contrast to an earlier report [35]. As mentioned above (Figure 2), we demonstrated that RT-PCR and cloning analyses are not suitable for the quantification of the expression of genes with many pseudogenes with highly similar mRNA sequences, such as NANOG1, because primers do not evenly amplify each cDNA despite perfectly matched primer-target sequences. Our results revealed an equivalent proportion of NANOG1 and NANOGP8 protein expression and an equivalent function of NANOG1 and NANOGP8 genes in the regulation of the malignant potential of DU145 prostate cancer cells.
Notably, one double-strand break (DSB) caused by one gRNA in NANOG1-knockout prostate cancer cells led to the same length deletion in both alleles in the establishment of knockout cell lines using the CRISPR/Cas9 system, and two DSBs caused by two gRNAs in NANOGP8-knockout cells led to different length deletions on each allele. In general, a DSB in mammalian cells can be repaired by one of two general repair pathways, non-homologous end-joining (NHEJ) and homologous recombination (HR) [45], and a DSB at a target locus induced by CRISPR/Cas9 can be repaired through either NHEJ or homology-directed repair (HDR) [46]. DSBs caused by CRISPR/Cas9 are repaired through NHEJ in the absence of a homologous repair donor, which results in indels [46]. Both alleles should have different indels because NHEJ of the DSBs in both alleles are independently repaired. Therefore, we speculate that CRISPR/Cas9-mediated DSBs at the same sites in both alleles may be repaired by HR after the repair of one allele by the NHEJ pathway, which results in NANOG1-knockout cells with the same deletions on both alleles. The DNA deletion resulting from the two CRISPR/Cas9-mediated DSBs at separate target sites in NANOGP8-knockout cells was longer. Therefore, it may be more difficult to repair these DSBs through the HR pathway, which results in different deletions on each allele.
NANOG1- and NANOGP8-rescued cell lines exhibited increased endogenous NANOG protein expression compared to the parental knockout cell lines (Figure 1H). The increased endogenous NANOG protein in the NANOG1-rescued, NANOG1-knockout cell line was likely derived from NANOGP8, and vice versa, because only NANOG1 and the NANOG pseudogene NANOGP8 encode full-length NANOG protein. The NANOG1 protein binds to its own gene promoter and promotes its own expression in ESCs [44]. Therefore, we hypothesize that NANOG1 and NANOGP8 reciprocally promote each other's expression in prostate cancer cells. The reciprocal promotion between NANOG1 and NANOGP8 may be important for the maintenance of malignant potential. Notably, we could not detect substantial NANOG-GFP expression in either NANOG1- or NANOGP8-rescued cell lines using fluorescence microscopy, which suggests that excessive NANOG protein expression is lethal to somatic cancer cells, as speculated previously [30].
In our study, knockout of NANOG1 and NANOGP8 did not alter cell growth, but knockout of each gene significantly decreased malignant potential, including colony formation (Figure 3B, 3C, and 3F), migration (Figure 4A), drug resistance (Figure 5A), and tumorigenicity (Figure 6A). These results suggest that NANOG activates cancer cell properties but not cell growth. These data are consistent with the function of NANOG1 in ESCs, in which NANOG1 forms a core module that regulates genes related to cell properties, and cell growth-related genes are regulated by a MYC module [47].
Our results demonstrated that NANOG1 and NANOGP8 activate migration capacity via E-cadherin expression. NANOG1 overexpression in ovarian cancer cells has been shown to enhance migration capacity, which is accompanied by decreased E-cadherin, caveolin-1, FOXJ1, and FOXO1 expression [19]. In addition, TALEN-mediated NANOG1 deletion in HeLa cells decreases migration capacity, which is accompanied by increased E-cadherin expression and decreased N-cadherin and vimentin expression [48]. Furthermore, E-cadherin promotes epithelial cell-cell adhesions, and decreased E-cadherin expression is important for EMT [39].
Our data indicate that NANOG1 and NANOGP8 are relevant to drug resistance. Previous studies demonstrated that NANOG1 and NANOGP8 overexpression in MCF7 cells upregulates several detoxification genes, including ABCG2, Bcl-2, ALDH1A1, and CD133 [30], and disruption of NANOG1 decreases the expression of MDR1 [48]. MDR1 encodes a P-glycoprotein that pumps various foreign substances out of cells. Various studies showed that NANOG1 depletion decreases tumorigenicity [16, 48], and NANOG1 overexpression increases tumorigenicity [27]. Lu et al. used an inducible NANOG1 transgenic mouse model and reported that ectopic NANOG1 expression upregulates the PDGFRa gene [27], which encodes an alpha-type platelet-derived growth factor receptor that drives tumorigenesis and metastasis in various cancers [49]. Emerging evidence also suggests that NANOGP8 overexpression increases tumorigenicity [30, 33]. Our results indicate that NANOG1 and NANOGP8 are involved in the tumorigenic potential of prostate cancer cells, which is consistent with these previous studies.
In our study, the phenotypes (e.g., sphere formation capacity, migration capability, drug resistance, and tumorigenic potential) of NANOG1- and NANOGP8-rescued cell lines only recovered to the levels of the parental DU145 cell line despite an excess expression of NANOG protein in the rescued cell lines. We hypothesize that this effect resulted from a loss of the reciprocal promotion between NANOG1 and NANOGP8 in the NANOG1- and NANOGP8-rescued cell lines.
We attempted to establish a NANOG1- and NANOGP8-double knockout cell line to examine whether an NANOG1 and NANOGP8 double knockout exerted a positive effect on the loss of malignant potential. However, we were unsuccessful in our attempt. We isolated more than 300 colonies that were candidates for the double knockout, but we did not find cells with deletions of both NANOG1 and NANOGP8 on both alleles. We identified 2 NANOG1-knockout cells out of 46 candidate colonies and 2 NANOGP8-knockout cells out of 24 candidate colonies, which suggests that the knockout of both NANOG1 and NANOGP8 genes in DU145 cells is lethal (Supplementary Figure 1).
In summary, we established NANOG1- and NANOGP8-knockout prostate cancer cell lines using the CRISPR/Cas9 system. Our results indicate that NANOG1 and NANOGP8 are expressed equally and that both genes activate many properties that are associated with the malignant potential in prostate cancer cells, including sphere formation capacity, migration capability, drug resistance, and tumorigenic potential.
MATERIALS AND METHODS
Cell culture
The androgen-independent human prostate cell line DU145 was purchased from the American Type Culture Collection (Rockville, MD). DU145 and its derivatives were cultured in RPMI-1640 medium (Nacalai Tesque) supplemented with 10% FBS (Biowest), 100 U/ml penicillin, and 100 μg/ml streptomycin (Penicillin-streptomycin mixed solution; Nacalai Tesque). These cells were incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2. Growth curves were generated for each cell line as follows. Each cell line was seeded in a 6-well plate at 1 × 105 cells/well and incubated for up to 6 days. Cell growth was assessed by cell counting every other day. Cells were trypsinized before reaching 70% confluency and transferred to 10-cm dishes. All experiments were conducted using passage-matched parental cells.
Establishment of DU145NANOG1−/−, DU145NANOGP8−/−, and NANOG-rescue cell lines
The targeted gRNA expression oligos were introduced into the pX330 vector [46] (Addgene). The sequences of these oligos are shown in Supplementary Table 1. A mixture of 1 μg of pX330 plasmid DNA containing each target gRNA sequence and 0.5 μg of pPGKpuro (Addgene) was transfected into suspended DU145 (1 × 105 cells) [36]. NEON (Invitrogen) electroporation was used to transfect the plasmids, and transfected cells were cultured in medium containing 1.0 μg/ml puromycin for 2 days for selection. Surviving cells were trypsinized and diluted in medium for colony formation. Single colonies were selected, and each colony was passaged and genotyped. DNA was isolated using a DNeasy Blood & Tissue Kit (Qiagen). The genomic region surrounding the CRISPR/Cas9 target site for each gene was PCR amplified, and PCR products were purified using a QIAquick Gel Extraction Kit (QIAGEN) according to the manufacturer's protocol. The amplicons were cloned into the pCR-BluntII-TOPO vector (Invitrogen). Each colony was selected, and the amplicon sequences were analyzed using a 3100 Genetic Analyzer (ABI). Supplementary Table 1 shows the primer sequences.
NANOG-rescued DU145 cell lines and control cell lines were generated by transducing 30 μg of plasmid (CAG-GFP-NANOG1-IRES-Puro, CAG-GFP-NANOGP8-IRES-Puro, and -GFP-IRES-Puro) into suspended NANOG1−/− cells and NANOGP8−/− cells (2 × 107 cells). Single colonies were selected after a 0.5-μg/ml puromycin selection. NANOG protein expression levels in each cell line were examined using Western blotting.
In vivo tumorigenicity experiments
DU145 and its derivatives (2 × 106 cells) were implanted subcutaneously in NOD-SCID mice. These NOD-SCID mice, aged 5 weeks, were purchased from CLEA Japan and maintained in a temperature-controlled, pathogen-free room. All animals were handled according to approved protocols and the guidelines of the Animal Committee of Osaka University (Osaka, Japan). Mouse appearance and tumor development were observed for 7 weeks. Tumor volume was calculated according to the following formula: tumor volume (mm3) = length × (width)2/2 [50].
Soft agar colony formation assay
A CytoSelect 96-well Cell Transformation Assay (Soft Agar Colony Formation) Kit (Cell Biolabs) was used to evaluate anchorage-independent growth according to the manufacturer's instructions. Briefly, DU145 and its derivatives (1.5 × 103 cells) were mixed with an agar solution, seeded into wells, and culture medium was added to each well. Cells were incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2 for 8 days. The colonies were lysed after agar solubilization, and CyQuant GR Dye was used to quantify anchorage-independent growth using a 485/520 nm filter set.
Sphere culture
Spheres of DU145 and its derivatives formed as previously described [51]. Briefly, each cell line (2 × 103 cells) was plated on low-attachment 6-well dishes (Corning). Cells were cultured in a serum-free epithelial basal medium (Cambrex) supplemented with B27, 4 μg/ml insulin (Sigma-Aldrich), 20 ng/ml EGF, and 20 ng/ml basic fibroblast growth factor (bFGF; Invitrogen) for 2 weeks. Sphere forming capacity was assessed based on the number of colonies.
MTS assay
Docetaxel was purchased from Aventis Pharmaceuticals (Sanofi-Aventis). Each cell line was seeded in a 48-well plate at 2.5 × 104 cells/well, and docetaxel was added at concentrations of 7.5, 15, 22.5, or 30 nM 24 hours later. Cell viability was evaluated after 48 hours of docetaxel treatment using a CellTiter 96 Aqueous One Solution Cell Proliferation Assay Kit (Promega) as previously described [52].
Migration assay
Migration ability was measured using a Culture-Insert in a μ-Dish 35 mm (Ibidi) as previously described [53]. Briefly, each cell line was suspended at a concentration of 5 × 105 cells/μl, and 3.5 × 104 cells were placed in each well. The wells were removed gently after a 24-hour incubation. The width of the scratch was measured at the beginning and every 4–6 hours during cell migration, and the wound closure rate was quantified as previously described [54].
Western blot analysis
Anti-human NANOG (D73G4), anti-human ACTB (13E5), anti-human H3 (3H1), and anti-human Snail (C15D3) antibodies were purchased from Cell Signaling Technology. The anti-human E-cadherin (CD324) antibody was purchased from BD Biosciences. Cell lysates were separated using SDS-PAGE, and separated proteins were transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 3% skim milk and incubated overnight at 4°C with the primary antibodies. Signals were detected using Chemi-Lumi One or Chemi-Lumi One Super (Nacalai Tesque) and an ImageQuant LAS 4000 mini system (GE Healthcare).
Statistical analysis
The results are reported as the mean ± SD. A two-tailed unpaired Student's t-test was used to determine the statistical significance of differences between two groups, and Tukey's test was used to determine the statistical significance of differences between more than three groups. Probability values of p < 0.05 were considered statistically significant. Statistical analyses were conducted using JMP9 (SAS Institute).
SUPPLEMENTARY FIGURE AND TABLE
Acknowledgments
We thank M. Okado for technical assistance and members of the GTS laboratory for discussion and support.
Footnotes
GRANT SUPPORT
This study was supported by MEXT KAKENHI Grant No. 26670156.
CONFLICTS OF INTEREST
The authors declare that no competing interests exist.
REFERENCES
- 1.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res. 2003;63:5821–8. [PubMed] [Google Scholar]
- 3.Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, Reilly JG, Chandra D, Zhou J, Claypool K, Coghlan L, Tang DG. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006;25:1696–708. doi: 10.1038/sj.onc.1209327. [DOI] [PubMed] [Google Scholar]
- 4.O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 5.Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, Butler PD, Yang GP, Joshua B, Kaplan MJ, Longaker MT, Weissman IL. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466:133–7. doi: 10.1038/nature09161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module Map of Stem Cell Genes Guides Creation of Epithelial Cancer Stem Cells. Cell Stem Cell. 2008;2:333–44. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Clem B, Zuba-Surma EK, El-Naggar S, Telang S, Jenson AB, Wang Y, Shao H, Ratajczak MZ, Chesney J, Dean DC. Mouse Fibroblasts Lacking RB1 Function Form Spheres and Undergo Reprogramming to a Cancer Stem Cell Phenotype. Cell Stem Cell. 2009;4:336–47. doi: 10.1016/j.stem.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, Bernard L, Viale G, Pelicci PG, Di Fiore PP. Biological and Molecular Heterogeneity of Breast Cancers Correlates with Their Cancer Stem Cell Content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 10.Chambers I, Tomlinson SR. The transcriptional foundation of pluripotency. Development. 2009;136:2311–22. doi: 10.1242/dev.024398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silva J, Nichols J, Theunissen TW, Guo G, van Oosten AL, Barrandon O, Wray J, Yamanaka S, Chambers I, Smith A. Nanog Is the Gateway to the Pluripotent Ground State. Cell. 2009;138:722–37. doi: 10.1016/j.cell.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suvà ML, Riggi N, Bernstein BE. Epigenetic Reprogramming in Cancer. Science. 2013;339:1567–70. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic Expression of Oct-4 Blocks Progenitor-Cell Differentiation and Causes Dysplasia in Epithelial Tissues. Cell. 2005;121:465–77. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 14.Piestun D, Kochupurakkal BS, Jacob-Hirsch J, Zeligson S, Koudritsky M, Domany E, Amariglio N, Rechavi G, Givol D. Nanog transforms NIH3T3 cells and targets cell-type restricted genes. Biochem Biophys Res Commun. 2006;343:279–85. doi: 10.1016/j.bbrc.2006.02.152. [DOI] [PubMed] [Google Scholar]
- 15.Gangemi RMR, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P, Ravetti GL, Zona GL, Daga A, Corte G. SOX2 Silencing in Glioblastoma Tumor-Initiating Cells Causes Stop of Proliferation and Loss of Tumorigenicity. Stem Cells. 2009;27:40–8. doi: 10.1634/stemcells.2008-0493. [DOI] [PubMed] [Google Scholar]
- 16.Jeter CR, Badeaux M, Choy G, Chandra D, Patrawala L, Liu C, Calhoun-Davis T, Zaehres H, Daley GQ, Tang DG. Functional Evidence that the Self-Renewal Gene NANOG Regulates Human Tumor Development. Stem Cells. 2009;27:993–1005. doi: 10.1002/stem.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Po A, Ferretti E, Miele E, Smaele ED, Paganelli A, Canettieri G, Coni S, Marcotullio LD, Biffoni M, Massimi L, Rocco CD, Screpanti I, et al. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO, J. 2010;29:2646–58. doi: 10.1038/emboj.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zbinden M, Duquet A, Lorente-Trigos A, Ngwabyt S-N, Borges I, Altaba AR. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010;29:2659–74. doi: 10.1038/emboj.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siu MKY, Wong ESY, Kong DSH, Chan HY, Jiang L, Wong OGW, Lam EW-F, Chan KKL, Ngan HYS, Le X-F, Cheung AN. Stem cell transcription factor NANOG controls cell migration and invasion via dysregulation of E-cadherin and FoxJ1 and contributes to adverse clinical outcome in ovarian cancers. Oncogene. 2013;32:3500–9. doi: 10.1038/onc.2012.363. [DOI] [PubMed] [Google Scholar]
- 20.Alldridge L, Metodieva G, Greenwood C, Al-Janabi K, Thwaites L, Sauven P, Metodiev M. Proteome Profiling of Breast Tumors by Gel Electrophoresis and Nanoscale Electrospray Ionization Mass Spectrometry. J Proteome Res. 2008;7:1458–69. doi: 10.1021/pr7007829. [DOI] [PubMed] [Google Scholar]
- 21.Ishiguro T, Sato A, Ohata H, Sakai H, Nakagama H, Okamoto K. Differential expression of nanog1 and nanogp8 in colon cancer cells. Biochem Biophys Res Commun. 2012;418:199–204. doi: 10.1016/j.bbrc.2011.10.123. [DOI] [PubMed] [Google Scholar]
- 22.Miyazawa K, Tanaka T, Nakai D, Morita N, Suzuki K. Immunohistochemical expression of four different stem cell markers in prostate cancer: High expression of NANOG in conjunction with hypoxia-inducible factor-1?, expression is involved in prostate epithelial malignancy. Oncol Lett. 2014;8:985–92. doi: 10.3892/ol.2014.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective Identification of Tumorigenic Prostate Cancer Stem Cells. Cancer Res. 2005;65:10946–51. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 24.Du Y, Ma C, Wang Z, Liu Z, Liu H, Wang T. Nanog, a novel prognostic marker for lung cancer. Surg Oncol. 2013;22:224–9. doi: 10.1016/j.suronc.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 25.Chiou S-H, Yu C-C, Huang C-Y, Lin S-C, Liu C-J, Tsai T-H, Chou S-H, Chien C-S, Ku H-H, Lo J-F. Positive Correlations of Oct-4 and Nanog in Oral Cancer Stem-Like Cells and High-Grade Oral Squamous Cell Carcinoma. Clin Cancer Res. 2008;14:4085–95. doi: 10.1158/1078-0432.CCR-07-4404. [DOI] [PubMed] [Google Scholar]
- 26.Elsir T, Edqvist P-H, Carlson J, Ribom D, Bergqvist M, Ekman S, Popova SN, Alafuzoff I, Ponten F, Nistér M, Smits A. A study of embryonic stem cell-related proteins in human astrocytomas: Identification of Nanog as a predictor of survival. Int J Cancer. 2014;134:1123–31. doi: 10.1002/ijc.28441. [DOI] [PubMed] [Google Scholar]
- 27.Lu X, Mazur SJ, Lin T, Appella E, Xu Y. The pluripotency factor nanog promotes breast cancer tumorigenesis and metastasis. Oncogene. 2014;33:2655–64. doi: 10.1038/onc.2013.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CMA, Hubaud A, Stadler B, Choi M, Bar M, Tewari M, Liu A, et al. HIF Induces Human Embryonic Stem Cell Markers in Cancer Cells. Cancer Res. 2011;71:4640–52. doi: 10.1158/0008-5472.CAN-10-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Booth HAF, Holland PWH. Eleven daughters of NANOG. Genomics. 2004;84:229–38. doi: 10.1016/j.ygeno.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 30.Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, Repass J, Zaehres H, Shen JJ, Tang DG. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene. 2011;30:3833–45. doi: 10.1038/onc.2011.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ambady S, Malcuit C, Kashpur O, Kole D, Holmes WF, Hedblom E, Page RL, Dominko T. Expression of NANOG and NANOGP8 in a variety of undifferentiated and differentiated human cells. Int J Dev Biol. 2010;54:1743–54. doi: 10.1387/ijdb.103192sa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J, Wang X, Li M, Han J, Chen B, Wang B, Dai J. NANOGP8 is a retrogene expressed in cancers. FEBS, J. 2006;273:1723–30. doi: 10.1111/j.1742-4658.2006.05186.x. [DOI] [PubMed] [Google Scholar]
- 33.Uchino K, Hirano G, Hirahashi M, Isobe T, Shirakawa T, Kusaba H, Baba E, Tsuneyoshi M, Akashi K. Human Nanog pseudogene8 promotes the proliferation of gastrointestinal cancer cells. Exp Cell Res. 2012;318:1799–807. doi: 10.1016/j.yexcr.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 34.Zhang J, Wang X, Chen B, Xiao Z, Li W, Lu Y, Dai J. The human pluripotency gene NANOG/NANOGP8 is expressed in gastric cancer and associated with tumor development. Oncol Lett. 2010;1:457–63. doi: 10.3892/ol_00000081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palla AR, Piazzolla D, Abad M, Li H, Dominguez O, Schonthaler HB, Wagner EF, Serrano M. Reprogramming activity of NANOGP8, a NANOG family member widely expressed in cancer. Oncogene. 2014;33:2513–9. doi: 10.1038/onc.2013.196. [DOI] [PubMed] [Google Scholar]
- 36.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell. 2013;153:910–8. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–9. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borrull A, Ghislin S, Deshayes F, Lauriol J, Alcaide-Loridan C, Middendorp S. Nanog and Oct4 overexpression increases motility and transmigration of melanoma cells. J Cancer Res Clin Oncol. 2012;138:1145–54. doi: 10.1007/s00432-012-1186-2. [DOI] [PubMed] [Google Scholar]
- 39.Hao L, Ha JR, Kuzel P, Garcia E, Persad S. Cadherin switch from E- to N-cadherin in melanoma progression is regulated by the PI3K/PTEN pathway through Twist and Snail. Br J Dermatol. 2012;166:1184–97. doi: 10.1111/j.1365-2133.2012.10824.x. [DOI] [PubMed] [Google Scholar]
- 40.Tran DD, Corsa CAS, Biswas H, Aft RL, Longmore GD. Temporal and Spatial Cooperation of Snail1 and Twist1 during Epithelial-Mesenchymal Transition Predicts for Human Breast Cancer Recurrence. Mol Cancer Res. 2011;9:1644–57. doi: 10.1158/1541-7786.MCR-11-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gingold JA, Fidalgo M, Guallar D, Lau Z, Sun Z, Zhou H, Faiola F, Huang X, Lee D-F, Waghray A, Schaniel C, Felsenfeld DP, et al. A Genome-wide RNAi Screen Identifies Opposing Functions of Snai1 and Snai2 on the Nanog Dependency in Reprogramming. Mol Cell. 2014;56:140–52. doi: 10.1016/j.molcel.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Du Y, Shi L, Wang T, Liu Z, Wang Z. Nanog siRNA plus Cisplatin may enhance the sensitivity of chemotherapy in esophageal cancer. J Cancer Res Clin Oncol. 2012;138:1759–67. doi: 10.1007/s00432-012-1253-8. [DOI] [PubMed] [Google Scholar]
- 43.Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M, Yamanaka S. The Homeoprotein Nanog Is Required for Maintenance of Pluripotency in Mouse Epiblast and ES Cells. Cell. 2003;113:631–42. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- 44.Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, et al. Core Transcriptional Regulatory Circuitry in Human Embryonic Stem Cells. Cell. 2005;122:947–56. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pardo B, Gómez-González B, Aguilera A. DNA Repair in Mammalian Cells. Cell Mol Life Sci. 2009;66:1039–56. doi: 10.1007/s00018-009-8740-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, Cantor AB, Orkin SH. A Myc Network Accounts for Similarities between Embryonic Stem and Cancer Cell Transcription Programs. Cell. 2010;143:313–24. doi: 10.1016/j.cell.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding Y, Qing Yu A, Lin Li C, Fang J, Zeng Y, Sheng Li D. TALEN-mediated Nanog disruption results in less invasiveness, more chemosensitivity and reversal of EMT in Hela cells. Oncotarget. 2014;5:8393–401. doi: 10.18632/oncotarget.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hatano K, Yamaguchi S, Nimura K, Murakami K, Nagahara A, Fujita K, Uemura M, Nakai Y, Tsuchiya M, Nakayama M, Nonomura N, Kaneda Y. Residual Prostate Cancer Cells after Docetaxel Therapy Increase the Tumorigenic Potential via Constitutive Signaling of CXCR4, ERK1/2 and c-Myc. Mol Cancer Res. 2013;11:1088–100. doi: 10.1158/1541-7786.MCR-13-0029-T. [DOI] [PubMed] [Google Scholar]
- 51.Borrull A, Ghislin S, Deshayes F, Lauriol J, Alcaide-Loridan C, Middendorp S. Nanog and Oct4 overexpression increases motility and transmigration of melanoma cells. J Cancer Res Clin Oncol. 2012;138:1145–54. doi: 10.1007/s00432-012-1186-2. [DOI] [PubMed] [Google Scholar]
- 52.Matsushima-Miyagi T, Hatano K, Nomura M, Li-Wen L, Nishikawa T, Saga K, Shimbo T, Kaneda Y. TRAIL and Noxa Are Selectively Upregulated in Prostate Cancer Cells Downstream of the RIG-I/MAVS Signaling Pathway by Nonreplicating Sendai Virus Particles. Clin Cancer Res. 2012;18:6271–83. doi: 10.1158/1078-0432.CCR-12-1595. [DOI] [PubMed] [Google Scholar]
- 53.Angelucci C, Maulucci G, Lama G, Proietti G, Colabianchi A, Papi M, Maiorana A, De Spirito M, Micera A, Balzamino OB, Di Leone A, Masetti R, et al. Epithelial-Stromal Interactions in Human Breast Cancer: Effects on Adhesion, Plasma Membrane Fluidity and Migration Speed and Directness. PLoS ONE. 2012;7:e50804. doi: 10.1371/journal.pone.0050804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.