

Abstract
The repair of DNA double-strand breaks (DSB) is central to the maintenance of genomic integrity. In tumor cells, the ability to repair DSBs predicts response to radiation and many cytotoxic anti-cancer drugs. DSB repair pathways include homologous recombination and non-homologous end joining (NHEJ). NHEJ is a template-independent mechanism, yet many NHEJ repair products carry limited genetic changes, which suggests that NHEJ includes mechanisms to minimize error. Proteins required for mammalian NHEJ include Ku70/80, the DNA-dependent protein kinase (DNA-PKcs), XLF/Cernunnos and the XRCC4:DNA ligase IV complex. NHEJ also utilizes accessory proteins that include DNA polymerases, nucleases, and other end-processing factors. In yeast, mutations of tyrosyl-DNA phosphodiesterase (TDP1) reduced NHEJ fidelity. TDP1 plays an important role in repair of topoisomerase-mediated DNA damage and 3′-blocking DNA lesions, and mutation of the human TDP1 gene results in an inherited human neuropathy termed SCAN1. We found that human TDP1 stimulated DNA binding by XLF and physically interacted with XLF to form TDP1:XLF:DNA complexes. TDP1:XLF interactions preferentially stimulated TDP1 activity on dsDNA as compared to ssDNA. TDP1 also promoted DNA binding by Ku70/80 and stimulated DNA-PK activity. Because Ku70/80 and XLF are the first factors recruited to the DSB at the onset of NHEJ, our data suggest a role for TDP1 during the early stages of mammalian NHEJ.
Keywords: Non-homologous end joining (NHEJ), TDP1, Ku70/80, XLF, DNA-dependent protein kinase (DNA-PK)
1. Introduction
DNA repair is critical for cell survival following exposure to a wide range of environmental agents, as well as for insuring the integrity of DNA during normal cell division. DNA repair competence is a critical determinant for maintaining genome stability, and failure to perform accurate repair can cause genome alterations that lead to carcinogenesis (1). One of the most cytotoxic forms of DNA damage is the DNA double-strand break (DSB), as unrepaired DSBs can lead to cell death or senescence.
The two mechanisms for repairing DSBs are homologous recombination and non-homologous end joining (NHEJ). Homologous recombination uses a template from an intact chromosome for error-free DSB repair. In contrast, NHEJ does not depend on a template for repair and has the potential to generate mutations. It is now clear that cells have multiple non-homologous repair pathways that have differing likelihoods of generating errors during the repair process (2, 3). Mammalian cells mainly rely on NHEJ for repairing DSBs, especially at points in the cell cycle when a sister chromatid is unavailable as a template for repair (2, 3).
Factors required for classical mammalian NHEJ include the Ku70/80 heterodimer, DNA ligase IV, and proteins that interact with these essential NHEJ factors (2, 3). Binding of Ku70/80 to exposed DNA ends is thought to initiate NHEJ. This is followed by Ku70/80-dependent recruitment of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), XLF and the XRCC4:ligase IV complex (4–9). Numerous interactions between Ku70/80, DNA-PKcs, XLF and the XRCC4:ligase IV complex retain these so-called “core” NHEJ factors at the DSB and stimulates DNA end joining (5–10).
In addition to the core factors that are required for all NHEJ events, DNA end-processing factors play an important role in NHEJ. If the DNA ends are incompatible or damaged, then processing steps to remove, repair or replace damaged or missing nucleotides may be required before ligation can take place. Additional factors implicated in the processing steps of NHEJ include the Artemis nuclease, the Pol X family DNA polymerases μ and λ, polynucleotide kinase/phosphatase (PNKP), aprataxin (APTX) and the APTX-PNKP like factor (APLF) (2, 3).
Tyrosyl-DNA phosphodiesterase (TDP1), a key factor in the repair of DNA damage from topoisomerase I, has the ability to remove covalently bound polypeptides and tyrosyl-phosphates that arise when topoisomerases fail to ligate reaction intermediates (11–14). TDP1 is active on a wide variety of substrates and has been shown to remove 3′ phosphoglycolate, chain terminating nucleosides, alkylated and undamaged nucleosides to leave a 3′ phosphate (12, 15–20). TDP1-deficient cells are, therefore, sensitive to treatment with chain terminating nucleosides, alkylating agents, and calicheamicin, which produces DSBs with 3′ phosphoglycolate (12, 15–20). The importance of TDP1 in DNA repair is highlighted by the observation that mutation of an active-site histidine in human TDP1 (H493R) is associated with the human neurodegenerative disorder spinocerebellar ataxia with axonal neuropathy (SCAN1) (21).
The role of TDP1 in repair of topoisomerase-mediated DNA damage has been well established (14, 18, 22–25), yet a number of studies suggest a broader involvement of TDP1 in DNA repair. In DT-40 cells, TDP1 deficiency resulted in sensitivity to both ionizing radiation and bleomycin (16). SCAN1 cells are reported to be sensitive to calicheamicin, and knockdown of TDP1 in HeLa cells resulted in reproducible, if modest, sensitivity to calicheamicin (18, 26, 27). In human cells, TDP1 is phosphorylated following exposure to ionizing radiation and phosphorylated TDP1 forms nuclear foci that co-localize with λH2AX foci, presumably at DSBs (28). The ability of TDP1 to remove adducts from DNA ends explains its role in NHEJ of damaged ends (14, 16–18, 25, 27), but does not explain why TDP1-deficient yeast exhibited reduced fidelity during NHEJ of ligatable, cohesive ends (29). This latter observation identified TDP1 as part of a mechanism that minimizes error during NHEJ, and possibly as a common participant in NHEJ. We hypothesize that TDP1 participates in NHEJ in mammalian cells. Here, we show that TDP1 interacts with the NHEJ core factors XLF and Ku70/80. TDP1 promoted DNA binding by XLF and Ku70/80, participated in multiprotein:DNA complexes with XLF and Ku70/80, and increased DNA-PK activity. Additionally, XLF stimulated TDP1 enzymatic activity on dsDNA. Our data demonstrate that TDP1 functionally interacts with proteins that are recruited to the DSB during the initial stages of NHEJ and suggest that TDP1 plays a general role during this repair process.
2. Materials and methods
2.1 Plasmids, cloning and mutagenesis
Plasmids pET15b.FLAG.hTDP1, pET15b.hTDP1.H263A and pET15b.hTDP1.H493R were derived from pET15b.hTDP1 by site directed mutagenesis (Quickchange II, Stratagene) using oligonucleotides summarized in supplemental methods. Plasmids pET15b-XLF and pET15b.FLAG.XLF were cloned as follows: the full-length XLF cDNA from pGEX-XLF (a generous gift from Steve Jackson, University of Cambridge, Cambridge, UK) was amplified using 5′ primers Xho.Start.XLF or Xho.FLAG.XLF and 3′ primer XLF.stop.Bam. PCR products were digested with XhoI and BamHI and ligated into pET15b that had been digested with XhoI and BamHI. Plasmid pMAL-XLF was cloned as follows: pGEX-XLF was digested with BamHI and HindIII to release the XLF cDNA, which was gel purified and ligated into pMAL-c2 (NEB) that had been digested with BamHI and HindIII. Plasmid pMAL-XLF-1-224 was derived from pMAL-XLF by site directed mutagenesis (Quickchange II, Stratagene) using oligonucleotides summarized in supplemental methods. Plasmid pClPuroMyc-hTDP1 was a generous gift from Sherif El-Khamisy (University of Sussex, Brighton, UK). All cDNA sequences were confirmed by direct DNA sequence analysis.
2.2 Protein expression and purification
All XLF and TDP1 proteins were expressed and purified using the following method: BL21(DE3)pLysS Rosetta2™ E. coli cells (EMD Biosciences) were transformed with the relevant plasmid, grown in Luria Broth (LB) at 37°C until the culture reached OD-600 1.0, at which point cells were chilled on ice to reduce culture temperature, transferred to an 18°C shaker incubator, induced with 1 mM IPTG and grown for 18 hours. Cells were harvested by centrifugation (5000xg, 15 min, 4°C) and cell pellets were stored at −80°C. Cells were resuspended in lysis buffer (20 mM Tris pH 7.9, 500 mM NaCl, 5 mM imidazole, 2 mM DTT and 1 mM PMSF) at 5% of original culture volume. Lysozyme was added to 2 mg/ml and cells were lysed (20 min, room temperature (RT)) with gentle mixing. Cell lysate was sonicated (Bransen Digital Sonifier 10%, 8x 20 sec burst), cellular debris was removed by centrifugation (14,000xg, 15 min, 4°C) and the resulting supernatant was bound in batch to 1 ml of Ni-NTA (Qiagen) with gentle end-over-end mixing (1 hour, 4°C), washed in batch with wash buffer (20 mM Tris pH 7.9, 250 mM NaCl, 15 mM Imidazole and 0.5 mM DTT), packed onto a column, washed with 10 column volumes of wash buffer and eluted with 20 mM Tris pH 7.9, 250 mM NaCl and 1 M imidazole. The eluate was diluted to 100 mM NaCl with 20 mM Tris pH 7.9, loaded onto a 1 ml hi-Trap Q column (GE) that had been equilibrated with Q buffer (50 mM Tris pH 7.9, 1 mM EDTA, 2 mM DTT, 5% glycerol) with 50 mM NaCl, washed with 5 ml Q-buffer with 50 mM NaCl and eluted with a 50 mM to 1 M NaCl linear gradient in Q buffer. Peak fractions were pooled and dialyzed (50 mM Tris pH 7.9, 50 mM NaCl, 1 mM EDTA, 2 mM DTT, and 10% glycerol) for 16 hours at 4°C, snap-frozen and stored at −80°C. Protein concentrations were determined by Bradford assay.
2.3 Electrophoretic Mobility Shift Analysis (EMSA)
For examination of DNA binding by TDP1 and NHEJ factors, the 61-nt EMSA.top oligonucleotide was labeled with 32P and annealed to EMSA.bottom to produce a 61-bp duplex with protruding 3′ ends. Labeled DNA (15 fmol) and proteins were incubated for 30 min at 25°C in 10 μl reaction in 50 mM Hepes pH 8.0, 50 mM NaCl, 0.5 mM EDTA pH 8.0, 1 mM DTT and 10% glycerol. For super-shifts, labeled DNA and proteins were combined for 10 min at RT, anti-FLAG antibodies (SIGMA) were added and the reaction was incubated for an additional 20 min at RT. The reactions were resolved on 5% (29:1) acrylamide gels (15 cm gel, 160 V, 220 min or 7.5 cm gel, 100 V, 65 min), after which the gel was dried and 32P was detected by phosphorimager (BioRad PMI).
2.4 Affinity capture “Pull-down” assay
BL21(DE3)pLysS Rosetta2™ E. coli cells were transformed with the relevant plasmid, grown in LB at 37°C to OD-600 1.0, chilled on ice, transferred and 18°C shaker, induced with 1 mM IPTG and grown for 18 hours. Cells were harvested by centrifugation (8000xg, 15 min, 4°C) and resuspended in JL buffer (20 mM Tris-HCl pH 7.4, 200 mM NaCl, 1 mM EDTA, 1 mM DTT) with 1mM PMSF and lysed by sonication as previously described. Cell debris was removed by centrifugation (14,000xg, 30 min, 4°C) and the resulting supernatant was collected as lysate. 100 μl amylose resin (50% slurry) was washed with 500 μl of JL buffer and maltose-binding protein (MBP) fusion proteins were bound to the amylose resin by combining 250 μl cell lysate with the washed resin for 1 hour at 4°C with gentle end-over-end mixing. Resin was collected by centrifugation (4300xg, 1 min, RT), and the supernatant was removed. Resin was washed twice with 1 ml JL buffer and once with binding buffer (50 mM tris-HCl pH 7.4, 100 mM KOAc, 1 mM EDTA, 5% glycerol, 2 mM DTT). 100 μg of his-tagged proteins was added to the resin in binding buffer in a final volume of 300 μl with 1 mg/ml BSA and subject to gentle end-over-end mixing for 1 hour at 4°C. Resin and bound proteins were collected by centrifugation (4300xg, 1 min, RT). Resin pellets were washed three times with 1 ml binding buffer and eluted with 50 μl of elution buffer (column buffer with 10 mM maltose) by gentle agitation at 4°C for 10 min. Resin was collected by centrifugation (4300xg, 1 min, RT) and the supernatant was saved as the eluate. Protein samples were separated on SDS-PAGE, transferred to PVDF membrane (Immobilon P) and individual proteins were detected by Western blot analysis. Antibodies used: Anti-MBP (NEB), 1:10,000. Anti-his6 (GE), 1:1000. Anti-XLF (30) 1:1000.
2.5 Co-immunoprecipitation
HCT 116 cells were transiently transfected with pClPuroMyc-hTDP1 using lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions and cells were harvested 24 hours after transfection. Extracts were prepared and immunoprecipitations (IPs) were carried out previously described (31) (supplemental methods). For detection of proteins in IP, samples were heated to 100°C for 3 min, separated by SDS-PAGE (8%) and Western transferred. Antibodies used in Western blot analysis: Ku80, monoclonal antibody clone 111 (NeoMarkers) 1 μg/ml. Myc-hTDP1, 9E10 myc hybridoma (32) supernatant (undiluted).
2.6 DNA-PK assay
The SignaTECT DNA-PK assay system (Promega) was used according to manufacturer’s instructions with the following modifications. Kinase reactions were conducted with 46 nM purified, recombinant Ku70/80, 230 nM TDP1 protein, 13 nM of purified DNA-PKcs, 1 μg/μl BSA and 3 nM of PstI-linearized pBluescript DNA.
2.7 TDP1 activity assay with purified enzyme
TDP1 activity assays using a 3′ phosphotyrosine substrate were performed as previously described (19, 24, 29, 33, 34). TDP1 assays using 3′ biotin-adduct substrates were performed as follows: To create the single-stranded 3′ biotin-adduct substrate, TDP1 3′ biotinylated substrate top was 5′ end labeled with 32P. To create the double-stranded TDP1 substrate, 32P-labeled TDP1 3′ ′ biotinylated substrate top was annealed with EMSA.bottom. Purified, recombinant his-tagged TDP1 was combined with 25 fmol of 32P labeled 3′ biotin-adduct substrate in 10 μl of TDP1 reaction buffer (50 mM Tris-Cl, pH 8.5, 100 mM NaCl, 25 μg/ml BSA, 0.5 mM EDTA) with 1% PVA, incubated at 37° C for 15 min and the reaction was stopped by addition of 10 μl of 2x TBE-Urea Sample Buffer (Invitrogen). Samples were boiled for 4 min, chilled on ice then resolved on a 7.5% urea-acrylamide gel at 1800 volt for 2.5 hours. Gel was then dried, exposed overnight and bands were detected using a BioRad Personal Molecular imager phosphorimager. Proteins used for Tdp1 activity assays are shown in Figure S2.
3. Results
3.1 TDP1 and XLF interact to form a TDP1:XLF:DNA complex
Based on our results in yeast, which demonstrate that TDP1 can affect the efficiency and accuracy of NHEJ (29), we sought to determine whether human TDP1 interacts with components of the human NHEJ apparatus. In TDP1-deficient yeast, NHEJ junctions showed an increased frequency of aberrant gap filling by DNA polymerases (29). XLF-deficient cells showed reduced gap filling by DNA polymerases at NHEJ junctions (35). Based on these findings we hypothesized that XLF and TDP1 might collaborate to regulate DNA polymerase action, or general access to DNA 3′ ends, during NHEJ. We looked for evidence of TDP1:XLF physical interactions by using purified maltose-binding protein (MBP)-tagged XLF (MPB-XLF) to pull-down purified his-tagged TDP1 (his6-TDP1) on amylose resin. As shown in Fig. 1A, retention of his6-TDP1 on amylose resin required XLF, which indicates a TDP1:XLF interaction. In similar experiments, we found that MBP-TDP1 retained his6-XLF on amylose resin, and that glutathione-S-transferase-XLF (GST-XLF) was capable of retaining his6-TDP1 on glutathione sepharose, whereas GST alone was not (data not shown). To address the possibility that TDP1:XLF interactions are mediated by DNA we performed pull-down experiments in the presence of ethidium bromide (EtBr). EtBr can interfere with protein:DNA interactions and is frequently used to reduce false positive results mediated by contaminating DNA in protein-protein interaction assays (36–38). Neither EtBr (Fig. 1A) nor DNase treatment (not shown) affected the ability of MBP-XLF to affinity capture his6-TDP1, which indicates that TDP1 and XLF physically interact and that TDP1:XLF interactions are likely not mediated by DNA.
Fig. 1. TDP1 physically interacts with and stimulates DNA binding by XLF.

(A) TDP1 and XLF physically interact. MBP-XLF, or MBP alone, was used to pull down his6-TDP1 on amylose resin as described in materials and methods. Lane 4, EtBr (100 μg/ml) was added to pull down reaction. Lane 6, his6-TDP1 protein positive control for Western blot. Samples were separated by SDS-PAGE, Western transferred and his6-TDP1 was detected using anti-his6 monoclonal antibodies.
(B) Formation of DNA-binding complex when TDP1 and XLF are combined. EMSA was carried out as described in materials and methods using a 5′ 32P-labeled 61-bp duplex oligonucleotide. TDP1 and XLF were used at sub-optimal concentrations such that neither protein bound DNA alone. Bovine Serum Albumin (BSA) was used as indicated.
XLF and TDP1 have been described as exhibiting length-dependent DNA-binding and prefer longer DNAs to shorter substrates (23, 39–41). Physical interaction between XLF and TDP1 may influence the way these proteins bind DNA. To assess the effect of TDP1 on DNA binding by XLF we carried out electrophoretic mobility shift assays (EMSA) using a 5′ 32P-labeled 61-bp duplex DNA probe, which would provide sufficient length for simultaneous binding of multiple proteins. To determine whether TDP1 stimulates DNA binding by XLF, we carried out EMSA using sub-optimal amounts of TDP1 and XLF such that neither protein alone bound DNA (Fig. S1A). When XLF and TDP1 were combined, a stable protein:DNA complex was observed as a distinct band (Fig. 1B). To determine whether this reaction specifically required XLF, we carried out EMSA reactions in which XLF was replaced with BSA. Reactions containing TDP1 and XLF produced a distinct band, while reactions combining TDP1 and BSA did not (Fig. 1B). Similar results were obtained when TDP1 was replaced with BSA (data not shown). These data show that, when combined, XLF and TDP1 form a stable protein:DNA complex.
To identify the protein constituents of the mobility shifted species, we used FLAG-tagged TDP1 in EMSA reactions that included XLF, DNA and anti-FLAG monoclonal antibodies (mAbs). This combination (Fig. 2A) produced a distinct band with lower mobility than the band observed with similar reactions lacking anti-FLAG mAbs. We interpret this “super shift” as a ternary complex of 32P-labeled DNA, FLAG-tagged TDP1, anti-FLAG mAbs, and possibly XLF. Control reactions containing untagged TDP1, XLF and DNA produced a stable protein:DNA complex that was unaffected by treatment with anti-FLAG mAbs (Fig. 2A), which demonstrate the specificity of the anti-FLAG mAbs. Additional controls show that DNA binding by FLAG-TDP1 and untagged TDP1 were stimulated by XLF equally well and that anti-FLAG mAbs harbored no DNA binding activity (Fig. 2A). In similar experiments we combined FLAG-XLF with untagged TDP1 and found that anti-FLAG mAbs super-shifted the protein:DNA complex assembled with FLAG-XLF, but had no affect on similar reactions that contained untagged XLF (Fig. 2B). In both experiments, treatment with anti-FLAG mAbs super-shifted the entire protein:DNA complex, indicating that all of the protein:DNA complexes were TDP1:XLF:DNA with no detectable TDP1:DNA or XLF:DNA sub-populations. We observed that complexes containing FLAG-tagged XLF had modest, but consistent, higher mobility than complexes assembled with untagged XLF. We attribute this to the high negative charge of the FLAG tag (DYDDDDK) and to the fact that XLF is a homodimer that contributes two FLAG tags to each protein:DNA complex.
Fig. 2. Formation of a ternary TDP1:XLF:DNA complex.

(A) Protein:DNA complex formed in the presence of TDP1 and XLF contains TDP1. EMSA was carried out using 100 ng of TDP1 or N-terminally FLAG-tagged TDP1 (FLAG-TDP1), 300 ng of XLF and anti-FLAG monoclonal antibodies (αFLAG) as indicated.
(B) Protein:DNA complex formed in the presence of TDP1 and XLF contains XLF. EMSA was carried out using 100 ng of TDP1, 300 ng of XLF or N-terminally FLAG-tagged XLF (FLAG-XLF) and anti-FLAG monoclonal antibodies (αFLAG) as indicated.
3.2 The C-terminal domain of XLF is required for interaction with TDP1
To identify the determinants of TDP1:XLF interactions we carried out mutational analysis of XLF and used our MBP pull-down assay to see how well XLF mutants interacted with wild-type TDP1. XLF is composed of an N-terminal globular head domain that mediates interactions with XRCC4 (residues 1–141), a coiled-coil domain that mediates homodimerization (residues 142–230) and a C-terminal DNA-binding domain (residues 231–299) (42–46) (Fig. 3A). Removal of the C-terminal DNA-binding domain of XLF produced XLF 1–224, which was unable to bind DNA alone (43), or with TDP1 (Fig. 3B), and did not physically interact with TDP1 (Fig. 3C).
Fig. 3. DNA binding by XLF is required for XLF:TDP1 interactions.

(A) Schematic diagram of XLF.
(B) C-terminally truncated XLF fails to bind DNA in the presence of TDP1. EMSA was carried out as described in materials and methods using 100 ng TDP1 and 300 ng of wild type XLF (WT) or C-terminally truncated XLF mutant XLF 1-224 (ΔC).
(C) C-terminally truncated XLF does not interact with TDP1. MBP-XLF, or XLF 1-224 (ΔC) were used to pull down wild type his6-TDP1. Samples were separated by SDS-PAGE and Western transferred. His6-TDP1 proteins were detected in the pull-down eluates (top) and input (bottom) using anti-his6 monoclonal antibodies. MBP-XLF proteins were detected with anti-MBP polyclonal antibodies (center). Lane 3, his6-TDP1 protein positive control for Western blot.
(D) XLF DNA-binding mutant XLF-K293A fails to bind DNA in the presence of TDP1. EMSA was carried out as described in materials and methods using 100 ng TDP1 and 300 ng of wild type XLF (WT), mutant XLF-K293A (K293A) or BSA.
(E) DNA-binding mutant XLF-K293A interacts with TDP1. MBP-XLF, or XLF-K293A (K293A), were used to pull down wild type his6-TDP1 on amylose resin. Samples were separated by SDS-PAGE and Western transferred. His6-TDP1 proteins were detected in the pull-down eluate (top) and input (bottom) samples using anti-his6 monoclonal antibodies. MBP-XLF and MBP proteins were detected with anti-MBP polyclonal antibodies (center). Lane 7, his6-TDP1 protein positive control for Western blot.
To examine the role of DNA binding by XLF in TDP1:XLF and TDP1:XLF:DNA interactions we used the single amino acid substitution mutant XLF-K293A, which is severely impaired for DNA binding (43). XLF-K293A was unable to bind DNA alone or in the presence of TDP1 (Fig. 3D). TDP1 binding by XLF-K293A was reduced relative to binding by wild type XLF (Fig. 3E). These observations show that DNA binding by XLF plays an important role in formation or stability of the TDP1:XLF:DNA complex and suggest that XLF directly contacts the DNA in this TDP1:XLF:DNA complex. These data also indicate that residues at the extreme C-terminus of XLF, which mediate XLF:Ku70/80 interactions (7), contribute to TDP1:XLF interactions.
3.3 DNA binding by TDP1 is required for stable TDP1:XLF:DNA complexes
We next sought to determine the role of DNA binding by TDP1 in TDP1:XLF:DNA interactions. To do this, we needed TDP1 mutants with reduced DNA-binding activity. TDP1 is composed of a poorly conserved N-terminal domain (residues 1–148) and a well-conserved catalytic domain (residues 149–609) (Fig. 4A) (12, 13, 47). Mutation of TDP1 active site histidine (H) 263 to alanine (A) results in TDP1-H263A, which is catalytically inactive (Fig. S3D) (13). We found that TDP1-H263A has reduced affinity for double-stranded DNA (Fig. S1B) and was unable to form TDP1:XLF:DNA complexes (Fig. 4B). TDP1-H263A retained the ability to physically interact with XLF (Fig. 4C). TDP1 mutation H493R, found in the human SCAN1 disorder, reduced enzyme activity and caused accumulation of the TDP1-H493R:DNA covalent reaction intermediate (24) (Fig. S3E). TDP1-H493R failed to bind DNA alone (Fig. S1B) or in combination with XLF (Fig. 4B). TDP1-H493R had reduced, but detectable, physical interaction with XLF (Fig. 4C). Using anti-his6 antibodies directed against the N-terminal his-tag, we observed a C-terminally truncated form of TDP1-H493R that could interact with XLF (Fig. 4C). These data may suggest that TDP1-H493R may be more susceptible to proteolysis than other TDP1 proteins. These data show that TDP1:XLF interactions are not sufficient for assembly of TDP1 and XLF on DNA and indicate that TDP1 DNA binding, or catalytic activity, plays an important role in formation of TDP1:XLF:DNA complexes. Additionally, these observations illustrate the role of the TDP1 active site in DNA binding by TDP1.
Fig. 4. DNA binding by TDP1 is required for TDP1:XLF:DNA interaction but not for TDP1:XLF physical interaction.

(A) Schematic diagram of TDP1.
(B) Active site, SCAN1 and N-terminal deletion mutants of TDP1 do not bind DNA in the presence of XLF. EMSA was carried out as described in materials and methods using 300 ng of XLF and 100 ng of wild type TDP1 (WT), active site mutant TDP1-H236A (H263A), SCAN1 mutant TDP1-H493R (H493R) or N-terminally truncated TDP1-ΔN (ΔN).
(C) Physical interactions between active site and N-terminal truncation mutants of TDP1 and XLF. MBP-XLF was used to pull down wild type his6-TDP1, N-terminally truncated his6-TDP1-ΔN and active site mutants his6-TDP1-H236A and -H493R. Samples were separated by SDS-PAGE, Western transferred and his-tagged TDP1 proteins were detected in the pull-down eluates using anti-his6 monoclonal antibodies. Lanes 4 and 5, his6-TDP1 and his6-TDP1-ΔN positive controls for Western blot. * indicates C-terminally truncated form of TDP1-H493R that can interact with XLF
It has been suggested that the TDP1 N-terminal domain mediates interactions with DNA repair proteins (28, 48–50). Deletion of the N-terminal domain of TDP1 produced the catalytically active TDP1-ΔN (Fig. S3C) (19), which could physically interact with XLF (Fig. 4C), but had reduced DNA-binding activity (Fig. S1B) and did not form a TDP1:XLF:DNA complex (Fig. 4B). These data demonstrate an unexpected role for the TDP1 N-terminal domain in binding double-stranded DNA.
Like mutants TDP1-H263A and -H493R, TDP1-ΔN was able to physically interact with XLF (Fig. 4C). The observation that TDP1 mutants lacking DNA binding activity retain the ability to physically interact with XLF provides additional evidence that the TDP1:XLF interaction is likely not mediated by DNA. Collectively, these data show that DNA binding by TDP1 is required for TDP1:XLF:DNA complex formation and suggest that TDP1 makes contact with XLF and DNA.
3.4 XLF stimulates TDP1 activity on double-strand DNA, but not on single-strand DNA
We used the ability of TDP1 to remove a 3′ biotin adduct from DNA (19) to compare the effect of XLF on TDP1 activity on the single-stranded (ss) and double-stranded (ds) DNA substrates diagramed in Figure 5. To generate the ssDNA substrate (ss61-bio) a 3′ biotinylated 61-mer was 5′ end labeled with 32P. To generate the dsDNA substrate (ds61-bio) the labeled ss61-bio was annealed to a complementary 61-mer to produce 3′ extensions. In the assay, varying amounts of purified recombinant TDP1 (Fig. S2) were combined with the appropriate DNA substrate in the presence or absence of purified recombinant 60 nM XLF, reactions were terminated by the addition of urea-containing sample buffer, reaction products were boiled then separated by denaturing gel electrophoresis and the labeled strand was detected by phosphorimager. As shown in Figure 5, XLF had no effect on the ability of TDP1 to remove the 3′ biotin adduct from the ssDNA substrate. In the absence of XLF, TDP1 was more active on the ssDNA than the dsDNA substrate. In contrast, TDP1 was significantly more active on dsDNA than on ssDNA when XLF was present in the reaction (Fig. 5B and C). These data indicate that interactions between XLF and TDP1 facilitate the ability of TDP1 to act on dsDNA.
Fig. 5. XLF stimulates TDP1 activity on dsDNA, but not on ssDNA.

(A) Removal of 3′ biotin adduct from single-strand DNA template ss61-bio by TDP1. (Top) Schematic diagram of single-strand DNA substrate ss61-bio and TDP1 product ss61-P.
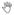 indicates 32P. - indicates no TDP1. (Bottom) 32P-labeled ss61-bio was combined with TDP1 (4.7, 9.4, 18.75, and 37.5 nM) in with 0 or 60 nM XLF.
indicates 32P. - indicates no TDP1. (Bottom) 32P-labeled ss61-bio was combined with TDP1 (4.7, 9.4, 18.75, and 37.5 nM) in with 0 or 60 nM XLF.
(B) Removal of 3′ biotin adduct from double-strand DNA template ds61-bio by TDP1. (Top) Schematic diagram of double-strand DNA substrate ds61-bio and TDP1 product ds61-P. (Bottom) 32P-labeled ds61-bio (ds61-bio) was combined with TDP1 (4.7, 9.4, 18.75, and 37.5 nM) in with 0 or 60 nM XLF. (C) Quantitation of TDP1 activity in 3 independent experiments shows that XLF has no effect on TDP1 activity on ssDNA, but increases TDP1 activity on dsDNA. Error bars represent standard deviation.
3.5 TDP1 interacts with Ku70/80 and stimulates DNA binding
The Ku70/80 heterodimer interacts with several proteins that are required for NHEJ and is thought to play an important role in assembly of the NHEJ apparatus (2, 3). To determine whether TDP1 interacts with Ku70/80 we expressed myc-tagged human TDP1 in HCT 116 cells and used anti-myc antibodies to immunoprecipitate (IP) TDP1 and TDP1-interacting proteins. Western blot analysis of the IP revealed the presence of Ku80 in IPs of cells transfected with the myc-TDP1 expression vector, pClPuroMyc-hTDP1, but not in IPs of untransfected cells (Fig. 6A). These data show that TDP1 interacts with Ku70/80 in human cell extracts.
Fig. 6. TDP1 functionally interacts with Ku70/80.

(A) Ku70/80 co-immunoprecipitates with myc-tagged TDP1. Extracts were prepared from pClPuroMyc-hTDP1 and untransfected cells. Myc-TDP1 was immunoprecipitated from lysates using anti-myc mAbs, immune complexes were separated by SDS-PAGE and Western transferred. Myc-TDP1 was detected using anti-myc mAbs (α-myc). Ku80 was detected using anti-Ku80 mAbs (α-Ku80).
(B) TDP1 stimulates DNA binding by Ku70/80. EMSA was carried out as described in materials and methods using 100 ng of Ku70/80 and 300 ng of wild type TDP1 (WT) or N-terminally truncated mutant TDP1-ΔN (ΔN).
(C) TDP1 binds to Ku70/80-bound DNA. EMSA optimized for Ku70/80 binding was carried out essentially as described in materials and methods with 1% polyvinyl alcohol to stabilize Ku70/80:DNA complexes. 100 ng of Ku70/80, 300 ng of FLAG-TDP1, anti-Ku80 mAbs (α80) and anti-FLAG mAbs (αF) as indicated.
(D) TDP1 stimulates DNA-PK activity. DNA-PK assays were carried out using 0.23 μM of wild type TDP1 (WT) or N-terminally truncated mutant his6-TDP1-ΔN (ΔN). Average of 2 experiments done in triplicate and normalized to reactions without TDP1.
If the interaction between TDP1 and Ku70/80 affects DNA binding by Ku70/80 the interaction could have a significant impact on NHEJ as a whole. To determine whether TDP1 stimulates DNA binding by Ku70/80 we carried out EMSA under sub-optimal binding conditions that did not produce stable DNA binding by Ku70/80 or TDP1 and found that TDP1 stimulated DNA binding by Ku70/80 (Fig. 6B). Under these conditions, TDP1-ΔN had only a subtle effect on DNA binding by Ku70/80, which indicated a role for the TDP1 N-terminal domain in TDP1 functional interactions with Ku70/80 (Fig. 6B).
To determine the effect of TDP1 on Ku70/80 bound to DNA, we optimized EMSA conditions for DNA binding by Ku70/80. Addition of 1% polyvinyl alcohol to EMSA reactions containing Ku70/80 improved DNA binding by Ku70/80 and produced two well-defined species that we interpret as Ku70/80:DNA complexes. Under these conditions, addition of TDP1 resulted in a new protein:DNA complex with a unique mobility (Fig. 6C). Addition of anti-Ku80 mAbs super-shifted all of the protein:DNA complex (Fig. 6C), which demonstrated that all of the detectable protein:DNA species contained Ku70/80. Anti-FLAG antibodies super-shifted a fraction of the protein:DNA complexes (Fig. 6C). The fraction of protein:DNA complexes that were not super-shifted had a banding pattern identical to that produced by Ku70/80 binding in the absence of TDP1. Given the importance of the TDP1 N-terminal domain in TDP1:Ku70/80 interactions (Fig. 6B), it is likely that recognition of the N-terminal FLAG tag by anti-FLAG antibodies super-shifts some FLAG-TDP1:Ku70/80 complexes, and disrupts or prevents the formation of others.
3.6 TDP1 stimulates phosphorylation by DNA-PK
In vertebrates, Ku70/80 is the DNA binding subunit of DNA-PK. The DNA-PK catalytic subunit (DNA-PKcs) is a core NHEJ factor and DNA-PKcs autophosphorylation helps to regulate DSB repair pathway choice (51, 52). Assembly of the DNA-PK holoenzyme at exposed DNA termini can block DNA end processing. Activation of the kinase and subsequent trans autophosphorylation causes dramatic structural changes that lead to DNA-PK inactivation and dissociation of DNA-PKcs from Ku70/80 and DNA, which permits access to DNA ends and may regulate the transition to subsequent steps in the NHEJ reaction (53–57). Factors that affect DNA binding by Ku70/80 may affect DNA-PK activity and progression through the NHEJ reaction. We found that wild type TDP1 stimulated DNA-PK activity by approximately 2.5-fold, while the TDP1 N-terminal deletion mutant, TDP1-ΔN, which did not stimulate DNA binding by Ku70/80 did not (Fig. 6D). These data show that TDP1 is capable of regulating DNA-PK activity via the TDP1 N-terminal domain. We suggest that TDP1 helps to coordinate events at DNA ends during the NHEJ reaction. The 3′ nucleosidase activity of TDP1 allows it to act as a negative regulator of some end processing events by generating 3′ phosphates that prevent the action of DNA polymerases and likely prevent the action of DNA ligases. TDP1 can also contribute to regulation of DNA end accessibility through control of DNA-PK activity.
4. Discussion
The NHEJ core factors constitute an apparatus capable of joining DNA ends with little to no dependence on sequence homology. As a result, NHEJ is mechanistically prone to error and may rejoin inappropriate partners (e.g. translocations) or alter the DNA sequence at the break point to cause mutation. Despite this error-prone mechanism, many NHEJ events are accurately repaired (29, 58–60). In addition to the core NHEJ factors, a number of end-processing proteins participate in NHEJ to make non-ligatable DNA ends suitable substrates for ligase IV. These accessory factors influence the frequency and patterns of error at NHEJ junctions.
Unlike accessory factors PNKP, APTX and APLF, which are recruited through interactions with XRCC4 (2), TDP1 does not appear to bind to XRCC4 (data not shown). Instead, TDP1 contacts the NHEJ apparatus through interactions with XLF and Ku70/80. TDP1 alters the DNA-binding properties of XLF and Ku70/80. Most significantly, we found that XLF stimulates TDP1 activity on dsDNA, but not on ssDNA.
TDP1 has been shown to preferentially bind ssDNA or blunt dsDNA over dsDNAs with 5′ or 3′ extensions (40, 61, 62). In keeping with this preference, TDP1 alone preferentially removed a 3′ biotin adduct from ssDNA as compared to a dsDNA substrate with a biotin adduct on a 3′ extension. Addition of XLF to these reactions had no effect on TDP1 activity on the ssDNA substrate, which was expected as XLF is a dsDNA binding protein (41). XLF and TDP1 stimulate DNA binding by one another and form a TDP1:XLF:DNA complex, which is mediated by TDP1:XLF physical interactions. We found that XLF increased TDP1 activity on dsDNA, and that when XLF was present dsDNA was the preferred TDP1 substrate. These data show that the interaction between TDP1 and XLF has functional consequences that support a role for TDP1 in DSB repair.
Previously, TDP1 was identified as a contaminant in highly purified Ku70/80 from human placenta (17) and we found that TDP1 immunoprecipitates from HCT 116 lysates contained Ku70/80. Together, these data demonstrate a stable TDP1:Ku70/80 interaction. We went on to determine that TDP1 stimulates DNA binding by Ku70/80. Finally, TDP1 stimulates the protein kinase activity of DNA-PK. Ku70/80 is the DNA binding subunit of DNA-PK and the stimulatory effect of TDP1 on DNA-PK may be, in part, due to its interaction with Ku70/80. While we have not ruled out the possibility that TDP1:Ku70/80 interactions may be mediated by DNA, the stimulatory effect of TDP1 on DNA-PK demonstrates the functional relevance of TDP1:Ku70/80 interactions.
Defects in NHEJ core factors can prevent immunoglobulin gene rearrangement and can cause immunodeficiency (2). However, this is not true for all NHEJ factors. XLF is required for NHEJ, yet XLF−/− mice are not immunodeficient because the fundamental role of XLF in NHEJ is masked by functional redundancy with ATM (63, 64). Similarly, functional redundancy of DNA-PKcs with ATM permits V(D)J signal joint formation in DNA-PKcs-deficient cells (65–67). These observations argue that immunodeficiency does not always accompany defects in NHEJ, and that the lack of immunodeficiency in TDP1−/− mice (68, 69) does not rule out participation of TDP1 in some NHEJ reactions.
Several observations implicate TDP1 in DSB repair. TDP1 is a substrate for ATM and DNA-PK and TDP1 phosphorylation by these DSB repair kinases is triggered by topoisomerase I (TopI) associated DSBs, as indicated by co-localization with λH2AX foci (28, 49). Additional reports indicate that TDP1 deficiency leads to altered DSB repair (16, 26). TDP1 interacts with poly(ADP-ribose) polymerase 1 (PARP1) (50), which has been implicated in NHEJ through interactions with DNA-PK (70–72). Our previous findings show that, in yeast, TDP1-deficiency has a significant impact on NHEJ (29), and we show here that human TDP1 interacts with human NHEJ factors. In contrast, TDP1+/+ and TDP1−/− murine astrocytes showed little difference λH2AX focus formation in response to H2O2, camptothecin or ionizing radiation (69). It has been reported that mouse astrocytes are radioresistant with reduced levels of ATM, and lack 53BP1 focus formation following exposure to ionizing radiation (73). 53BP1 plays an important role in preventing DNA end resection and promoting NHEJ (74) and the unusual behavior of 53BP1 in murine astrocytes suggests that DSB repair may be unconventional in this cell type.
The majority of DSBs produced by environmental exposure to DNA damaging agents have chemically altered ends that require processing before ligation can occur. Because removing chemically damaged bases is critically important to DSB repair, cells have multiple end-processing factors. One mechanism for processing of damaged 3′ termini is TDP1. Removal of a damaged 3′ nucleoside by TDP1 leaves a 3′ phosphate, which can be removed by PNKP (40, 75–77). An important NHEJ end-processing factors is the Artemis nuclease, which can act alone as a 5′ exonuclease and as part of the DNA-PKcs:Artemis complex as a 5′- or 3′-endonuclease (2, 78). Additionally, APLF possess activities that can trim single-stranded 3′ extensions at a DSB (79, 80). Finally, Ku70/80 has been shown to possess dRp/AP lyase activity (81). While these enzymes are capable of removing damaged nucleotides that might block ligation, some can also act on undamaged DNA and may facilitate joining of non-complementary ends.
We previously proposed a model in which TDP1 acts at undamaged 3′ ends to generate 3′ phosphates, which prevents error-inducing DNA synthesis during NHEJ (29). While TDP1 is not active against DNA ends with 3′ phosphates, it binds tightly to 3′ phosphate ends and may further protect the exposed 3′ terminus (23). Our current data indicates that TDP1 participation in NHEJ is likely mediated by interactions with Ku70/80 and XLF. Because Ku70/80 and XLF are thought to be among the first NHEJ factors to bind to the DSB (2, 5), we suggest that TDP1 is recruited to the break during the early stages of NHEJ. This timing may allow TDP1 to act at exposed 3′ ends prior to assembly of DNA-PK, which has been shown to block TDP1 access to the 3′ nucleoside (18). We suggest that the function of TDP1 in NHEJ is production of the 3′ phosphate, which we have shown can influence the events that take place at DNA ends (29).
Supplementary Material
Highlights.
End-processing factor TDP1 physically interacts with the NHEJ factor XLF to form a TDP1:XLF:DNA complex
XLF stimulates TDP1 enzymatic activity on dsDNA
TDP1 also interacts with Ku70/80 and stimulates DNA binding by Ku70/80
TDP1 stimulates the protein kinase activity of DNA-PK
Acknowledgments
We thank Kayla Kohlmeier and Anjelica Peacock for help with TDP1:XLF and TDP1:XRCC4 interactions, respectively. We gratefully acknowledge funding from the UIC Cancer Center and NIH awards CA52814 (to JLN) and CA87651 (to LAH and JLN).
Footnotes
Conflict of interest statement
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Branzei D, Foiani M. The DNA damage response during DNA replication. Curr Opin Cell Biol. 2005;17:568–75. doi: 10.1016/j.ceb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiruvella KK, Liang Z, Wilson TE. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol. 2013:5. doi: 10.1101/cshperspect.a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mari PO, Florea BI, Persengiev SP, Verkaik NS, Bruggenwirth HT, Modesti M, Giglia-Mari G, Bezstarosti K, Demmers JA, Luider TM, Houtsmuller AB, van Gent DC. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc Natl Acad Sci U S A. 2006;103:18597–602. doi: 10.1073/pnas.0609061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yano K, Morotomi-Yano K, Wang SY, Uematsu N, Lee KJ, Asaithamby A, Weterings E, Chen DJ. Ku recruits XLF to DNA double-strand breaks. EMBO Rep. 2008;9:91–6. doi: 10.1038/sj.embor.7401137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yano K, Chen DJ. Live cell imaging of XLF and XRCC4 reveals a novel view of protein assembly in the non-homologous end-joining pathway. Cell Cycle. 2008;7:1321–5. doi: 10.4161/cc.7.10.5898. [DOI] [PubMed] [Google Scholar]
- 7.Yano K, Morotomi-Yano K, Lee KJ, Chen DJ. Functional significance of the interaction with Ku in DNA double-strand break recognition of XLF. FEBS Lett. 2011;585:841–6. doi: 10.1016/j.febslet.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costantini S, Woodbine L, Andreoli L, Jeggo PA, Vindigni A. Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA Repair (Amst) 2007;6:712–22. doi: 10.1016/j.dnarep.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 9.Leber R, Wise TW, Mizuta R, Meek K. The XRCC4 gene product is a target for and interacts with the DNA-dependent protein kinase. J Biol Chem. 1998;273:1794–801. doi: 10.1074/jbc.273.3.1794. [DOI] [PubMed] [Google Scholar]
- 10.Hsu HL, Yannone SM, Chen DJ. Defining interactions between DNA-PK and ligase IV/XRCC4. DNA Repair (Amst) 2002;1:225–35. doi: 10.1016/s1568-7864(01)00018-0. [DOI] [PubMed] [Google Scholar]
- 11.Pouliot JJ, Yao KC, Robertson CA, Nash HA. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science. 1999;286:552–5. doi: 10.1126/science.286.5439.552. [DOI] [PubMed] [Google Scholar]
- 12.Pommier Y, Huang SY, Gao R, Das BB, Murai J, Marchand C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2) DNA Repair (Amst) 2014;19:114–29. doi: 10.1016/j.dnarep.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Interthal H, Pouliot JJ, Champoux JJ. The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily. Proc Natl Acad Sci U S A. 2001;98:12009–14. doi: 10.1073/pnas.211429198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nitiss KC, Malik M, He X, White SW, Nitiss JL. Tyrosyl-DNA phosphodiesterase (Tdp1) participates in the repair of Top2-mediated DNA damage. Proc Natl Acad Sci U S A. 2006;103:8953–8. doi: 10.1073/pnas.0603455103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alagoz M, Wells OS, El-Khamisy SF. TDP1 deficiency sensitizes human cells to base damage via distinct topoisomerase I and PARP mechanisms with potential applications for cancer therapy. Nucleic Acids Res. 2014;42:3089–103. doi: 10.1093/nar/gkt1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murai J, Huang SY, Das BB, Dexheimer TS, Takeda S, Pommier Y. Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells. J Biol Chem. 2012;287:12848–57. doi: 10.1074/jbc.M111.333963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inamdar KV, Pouliot JJ, Zhou T, Lees-Miller SP, Rasouli-Nia A, Povirk LF. Conversion of phosphoglycolate to phosphate termini on 3′ overhangs of DNA double strand breaks by the human tyrosyl-DNA phosphodiesterase hTdp1. The Journal of biological chemistry. 2002;277:27162–8. doi: 10.1074/jbc.M204688200. [DOI] [PubMed] [Google Scholar]
- 18.Zhou T, Akopiants K, Mohapatra S, Lin PS, Valerie K, Ramsden DA, Lees-Miller SP, Povirk LF. Tyrosyl-DNA phosphodiesterase and the repair of 3′-phosphoglycolate-terminated DNA double-strand breaks. DNA Repair (Amst) 2009;8:901–11. doi: 10.1016/j.dnarep.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Interthal H, Chen HJ, Champoux JJ. Human Tdp1 cleaves a broad spectrum of substrates, including phosphoamide linkages. J Biol Chem. 2005;280:36518–28. doi: 10.1074/jbc.M508898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang SY, Murai J, Dalla Rosa I, Dexheimer TS, Naumova A, Gmeiner WH, Pommier Y. TDP1 repairs nuclear and mitochondrial DNA damage induced by chain-terminating anticancer and antiviral nucleoside analogs. Nucleic Acids Res. 2013;41:7793–803. doi: 10.1093/nar/gkt483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, Stockton DW, Lupski JR. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet. 2002;32:267–72. doi: 10.1038/ng987. [DOI] [PubMed] [Google Scholar]
- 22.Das BB, Dexheimer TS, Maddali K, Pommier Y. Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19790–5. doi: 10.1073/pnas.1009814107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dexheimer TS, Stephen AG, Fivash MJ, Fisher RJ, Pommier Y. The DNA binding and 3′-end preferential activity of human tyrosyl-DNA phosphodiesterase. Nucleic Acids Res. 2010;38:2444–52. doi: 10.1093/nar/gkp1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Interthal H, Chen HJ, Kehl-Fie TE, Zotzmann J, Leppard JB, Champoux JJ. SCAN1 mutant Tdp1 accumulates the enzyme--DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 2005;24:2224–33. doi: 10.1038/sj.emboj.7600694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barthelmes HU, Habermeyer M, Christensen MO, Mielke C, Interthal H, Pouliot JJ, Boege F, Marko D. TDP1 overexpression in human cells counteracts DNA damage mediated by topoisomerases I and II. J Biol Chem. 2004;279:55618–25. doi: 10.1074/jbc.M405042200. [DOI] [PubMed] [Google Scholar]
- 26.Zhou T, Lee JW, Tatavarthi H, Lupski JR, Valerie K, Povirk LF. Deficiency in 3′-phosphoglycolate processing in human cells with a hereditary mutation in tyrosyl-DNA phosphodiesterase (TDP1) Nucleic Acids Res. 2005;33:289–97. doi: 10.1093/nar/gki170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Povirk LF. Processing of Damaged DNA Ends for Double-Strand Break Repair in Mammalian Cells. ISRN Molecular Biology. 2012;2012:1–16. doi: 10.5402/2012/345805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das BB, Antony S, Gupta S, Dexheimer TS, Redon CE, Garfield S, Shiloh Y, Pommier Y. Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA-PK. EMBO J. 2009;28:3667–80. doi: 10.1038/emboj.2009.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bahmed K, Nitiss KC, Nitiss JL. Yeast Tdp1 regulates the fidelity of nonhomologous end joining. Proc Natl Acad Sci U S A. 2010;107:4057–62. doi: 10.1073/pnas.0909917107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanakahi L. Effect of the inositol polyphosphate InsP(6) on DNA-PK-dependent phosphorylation. Mol Cancer Res. 2011;9:1366–76. doi: 10.1158/1541-7786.MCR-11-0230. [DOI] [PubMed] [Google Scholar]
- 31.Jayaram S, Gilson T, Ehrlich ES, Yu XF, Ketner G, Hanakahi L. E1B 55k-independent dissociation of the DNA ligase IV/XRCC4 complex by E4 34k during adenovirus infection. Virology. 2008;382:163–70. doi: 10.1016/j.virol.2008.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–6. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y. Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst) 2003;2:1087–100. doi: 10.1016/s1568-7864(03)00116-2. [DOI] [PubMed] [Google Scholar]
- 34.Antony S, Marchand C, Stephen AG, Thibaut L, Agama KK, Fisher RJ, Pommier Y. Novel high-throughput electrochemiluminescent assay for identification of human tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors and characterization of furamidine (NSC 305831) as an inhibitor of Tdp1. Nucleic Acids Res. 2007;35:4474–84. doi: 10.1093/nar/gkm463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akopiants K, Zhou RZ, Mohapatra S, Valerie K, Lees-Miller SP, Lee KJ, Chen DJ, Revy P, de Villartay JP, Povirk LF. Requirement for XLF/Cernunnos in alignment-based gap filling by DNA polymerases lambda and mu for nonhomologous end joining in human whole-cell extracts. Nucleic Acids Res. 2009;37:4055–62. doi: 10.1093/nar/gkp283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lai JS, Herr W. Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc Natl Acad Sci U S A. 1992;89:6958–62. doi: 10.1073/pnas.89.15.6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lechner MS, Laimins LA. Inhibition of p53 DNA binding by human papillomavirus E6 proteins. J Virol. 1994;68:4262–73. doi: 10.1128/jvi.68.7.4262-4273.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen TN, Goodrich JA. Protein-protein interaction assays: eliminating false positive interactions. Nat Methods. 2006;3:135–9. doi: 10.1038/nmeth0206-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Interthal H, Champoux JJ. Effects of DNA and protein size on substrate cleavage by human tyrosyl-DNA phosphodiesterase 1. Biochem J. 2011;436:559–66. doi: 10.1042/BJ20101841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Debethune L, Kohlhagen G, Grandas A, Pommier Y. Processing of nucleopeptides mimicking the topoisomerase I-DNA covalent complex by tyrosyl-DNA phosphodiesterase. Nucleic Acids Res. 2002;30:1198–204. doi: 10.1093/nar/30.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu H, Pannicke U, Schwarz K, Lieber MR. Length-dependent binding of human XLF to DNA and stimulation of XRCC4.DNA ligase IV activity. J Biol Chem. 2007;282:11155–62. doi: 10.1074/jbc.M609904200. [DOI] [PubMed] [Google Scholar]
- 42.Malivert L, Ropars V, Nunez M, Drevet P, Miron S, Faure G, Guerois R, Mornon JP, Revy P, Charbonnier JB, Callebaut I, de Villartay JP. Delineation of the Xrcc4-interacting region in the globular head domain of cernunnos/XLF. J Biol Chem. 2010;285:26475–83. doi: 10.1074/jbc.M110.138156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andres SN, Vergnes A, Ristic D, Wyman C, Modesti M, Junop M. A human XRCC4-XLF complex bridges DNA. Nucleic Acids Res. 2012;40:1868–78. doi: 10.1093/nar/gks022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roy S, Andres SN, Vergnes A, Neal JA, Xu Y, Yu Y, Lees-Miller SP, Junop M, Modesti M, Meek K. XRCC4’s interaction with XLF is required for coding (but not signal) end joining. Nucleic Acids Res. 2012;40:1684–94. doi: 10.1093/nar/gkr1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hammel M, Rey M, Yu Y, Mani RS, Classen S, Liu M, Pique ME, Fang S, Mahaney BL, Weinfeld M, Schriemer DC, Lees-Miller SP, Tainer JA. XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. J Biol Chem. 2011;286:32638–50. doi: 10.1074/jbc.M111.272641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Y, Chirgadze DY, Bolanos-Garcia VM, Sibanda BL, Davies OR, Ahnesorg P, Jackson SP, Blundell TL. Crystal structure of human XLF/Cernunnos reveals unexpected differences from XRCC4 with implications for NHEJ. EMBO J. 2008;27:290–300. doi: 10.1038/sj.emboj.7601942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davies DR, Interthal H, Champoux JJ, Hol WG. The crystal structure of human tyrosyl-DNA phosphodiesterase, Tdp1. Structure. 2002;10:237–48. doi: 10.1016/s0969-2126(02)00707-4. [DOI] [PubMed] [Google Scholar]
- 48.Hudson JJ, Chiang SC, Wells OS, Rookyard C, El-Khamisy SF. SUMO modification of the neuroprotective protein TDP1 facilitates chromosomal single-strand break repair. Nat Commun. 2012;3:733. doi: 10.1038/ncomms1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chiang SC, Carroll J, El-Khamisy SF. TDP1 serine 81 promotes interaction with DNA ligase IIIalpha and facilitates cell survival following DNA damage. Cell Cycle. 2010;9:588–595. doi: 10.4161/cc.9.3.10598. [DOI] [PubMed] [Google Scholar]
- 50.Das BB, Huang SY, Murai J, Rehman I, Ame JC, Sengupta S, Das SK, Majumdar P, Zhang H, Biard D, Majumder HK, Schreiber V, Pommier Y. PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res. 2014;42:4435–49. doi: 10.1093/nar/gku088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cui X, Yu Y, Gupta S, Cho YM, Lees-Miller SP, Meek K. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol Cell Biol. 2005;25:10842–52. doi: 10.1128/MCB.25.24.10842-10852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Convery E, Shin EK, Ding Q, Wang W, Douglas P, Davis LS, Nickoloff JA, Lees-Miller SP, Meek K. Inhibition of homologous recombination by variants of the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs) Proc Natl Acad Sci U S A. 2005;102:1345–50. doi: 10.1073/pnas.0406466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chan DW, Lees-Miller SP. The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J Biol Chem. 1996;271:8936–41. doi: 10.1074/jbc.271.15.8936. [DOI] [PubMed] [Google Scholar]
- 54.Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, Rambo RP, Hura GL, Pelikan M, So S, Abolfath RM, Chen DJ, Lees-Miller SP, Tainer JA. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J Biol Chem. 2010;285:1414–23. doi: 10.1074/jbc.M109.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Merkle D, Douglas P, Moorhead GB, Leonenko Z, Yu Y, Cramb D, Bazett-Jones DP, Lees-Miller SP. The DNA-dependent protein kinase interacts with DNA to form a protein-DNA complex that is disrupted by phosphorylation. Biochemistry. 2002;41:12706–14. doi: 10.1021/bi0263558. [DOI] [PubMed] [Google Scholar]
- 56.Dobbs TA, Tainer JA, Lees-Miller SP. A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair (Amst) 2010;9:1307–14. doi: 10.1016/j.dnarep.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morris EP, Rivera-Calzada A, da Fonseca PC, Llorca O, Pearl LH, Spagnolo L. Evidence for a remodelling of DNA-PK upon autophosphorylation from electron microscopy studies. Nucleic Acids Res. 2011;39:5757–67. doi: 10.1093/nar/gkr146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daley JM, Palmbos PL, Wu D, Wilson TE. Nonhomologous end joining in yeast. Annu Rev Genet. 2005;39:431–51. doi: 10.1146/annurev.genet.39.073003.113340. [DOI] [PubMed] [Google Scholar]
- 59.Waters CA, Strande NT, Wyatt DW, Pryor JM, Ramsden DA. Nonhomologous end joining: a good solution for bad ends. DNA Repair (Amst) 2014;17:39–51. doi: 10.1016/j.dnarep.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Betermier M, Bertrand P, Lopez BS. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014;10:e1004086. doi: 10.1371/journal.pgen.1004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raymond AC, Staker BL, Burgin AB., Jr Substrate specificity of tyrosyl-DNA phosphodiesterase I (Tdp1) J Biol Chem. 2005;280:22029–35. doi: 10.1074/jbc.M502148200. [DOI] [PubMed] [Google Scholar]
- 62.Pouliot JJ, Robertson CA, Nash HA. Pathways for repair of topoisomerase I covalent complexes in Saccharomyces cerevisiae. Genes Cells. 2001;6:677–87. doi: 10.1046/j.1365-2443.2001.00452.x. [DOI] [PubMed] [Google Scholar]
- 63.Li G, Alt FW, Cheng HL, Brush JW, Goff PH, Murphy MM, Franco S, Zhang Y, Zha S. Lymphocyte-specific compensation for XLF/cernunnos end-joining functions in V(D)J recombination. Mol Cell. 2008;31:631–40. doi: 10.1016/j.molcel.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zha S, Guo C, Boboila C, Oksenych V, Cheng HL, Zhang Y, Wesemann DR, Yuen G, Patel H, Goff PH, Dubois RL, Alt FW. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature. 2011;469:250–4. doi: 10.1038/nature09604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gapud EJ, Dorsett Y, Yin B, Callen E, Bredemeyer A, Mahowald GK, Omi KQ, Walker LM, Bednarski JJ, McKinnon PJ, Bassing CH, Nussenzweig A, Sleckman BP. Ataxia telangiectasia mutated (Atm) and DNA-PKcs kinases have overlapping activities during chromosomal signal joint formation. Proc Natl Acad Sci U S A. 2011;108:2022–7. doi: 10.1073/pnas.1013295108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gapud EJ, Sleckman BP. Unique and redundant functions of ATM and DNA-PKcs during V(D)J recombination. Cell Cycle. 2011;10:1928–35. doi: 10.4161/cc.10.12.16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zha S, Jiang W, Fujiwara Y, Patel H, Goff PH, Brush JW, Dubois RL, Alt FW. Ataxia telangiectasia-mutated protein and DNA-dependent protein kinase have complementary V(D)J recombination functions. Proc Natl Acad Sci U S A. 2011;108:2028–33. doi: 10.1073/pnas.1019293108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hawkins AJ, Subler MA, Akopiants K, Wiley JL, Taylor SM, Rice AC, Windle JJ, Valerie K, Povirk LF. In vitro complementation of Tdp1 deficiency indicates a stabilized enzyme-DNA adduct from tyrosyl but not glycolate lesions as a consequence of the SCAN1 mutation. DNA repair. 2009;8:654–63. doi: 10.1016/j.dnarep.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Katyal S, el-Khamisy SF, Russell HR, Li Y, Ju L, Caldecott KW, McKinnon PJ. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 2007;26:4720–31. doi: 10.1038/sj.emboj.7601869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ruscetti T, Lehnert BE, Halbrook J, Le Trong H, Hoekstra MF, Chen DJ, Peterson SR. Stimulation of the DNA-dependent protein kinase by poly(ADP-ribose) polymerase. J Biol Chem. 1998;273:14461–7. doi: 10.1074/jbc.273.23.14461. [DOI] [PubMed] [Google Scholar]
- 71.Spagnolo L, Barbeau J, Curtin NJ, Morris EP, Pearl LH. Visualization of a DNA-PK/PARP1 complex. Nucleic Acids Res. 2012;40:4168–77. doi: 10.1093/nar/gkr1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beck C, Robert I, Reina-San-Martin B, Schreiber V, Dantzer F. Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp Cell Res. 2014;329:18–25. doi: 10.1016/j.yexcr.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 73.Schneider L, Fumagalli M, d’Adda di Fagagna F. Terminally differentiated astrocytes lack DNA damage response signaling and are radioresistant but retain DNA repair proficiency. Cell Death Differ. 2012;19:582–91. doi: 10.1038/cdd.2011.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Daley JM, Sung P. 53BP1, BRCA1 and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol. 2014 doi: 10.1128/MCB.01639-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koch CA, Agyei R, Galicia S, Metalnikov P, O’Donnell P, Starostine A, Weinfeld M, Durocher D. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J. 2004;23:3874–85. doi: 10.1038/sj.emboj.7600375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weinfeld M, Mani RS, Abdou I, Aceytuno RD, Glover JN. Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair. Trends Biochem Sci. 2011;36:262–71. doi: 10.1016/j.tibs.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mani RS, Yu Y, Fang S, Lu M, Fanta M, Zolner AE, Tahbaz N, Ramsden DA, Litchfield DW, Lees-Miller SP, Weinfeld M. Dual modes of interaction between XRCC4 and polynucleotide kinase/phosphatase: implications for nonhomologous end joining. J Biol Chem. 2010;285:37619–29. doi: 10.1074/jbc.M109.058719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pawelczak KS, Bennett SM, Turchi JJ. Coordination of DNA-PK activation and nuclease processing of DNA termini in NHEJ. Antioxid Redox Signal. 2011;14:2531–43. doi: 10.1089/ars.2010.3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li S, Kanno S, Watanabe R, Ogiwara H, Kohno T, Watanabe G, Yasui A, Lieber MR. Polynucleotide kinase and aprataxin-like forkhead-associated protein (PALF) acts as both a single-stranded DNA endonuclease and a single-stranded DNA 3′ exonuclease and can participate in DNA end joining in a biochemical system. J Biol Chem. 2011;286:36368–77. doi: 10.1074/jbc.M111.287797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mohapatra S, Yannone SM, Lee SH, Hromas RA, Akopiants K, Menon V, Ramsden DA, Povirk LF. Trimming of damaged 3′ overhangs of DNA double-strand breaks by the Metnase and Artemis endonucleases. DNA Repair (Amst) 2013;12:422–32. doi: 10.1016/j.dnarep.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roberts SA, Strande N, Burkhalter MD, Strom C, Havener JM, Hasty P, Ramsden DA. Ku is a 5′-dRP/AP lyase that excises nucleotide damage near broken ends. Nature. 2010;464:1214–7. doi: 10.1038/nature08926. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.