

Abstract
The objective of the present investigation was to optimize diazepam (Dzp)-loaded poly(lactic-co-glycolic acid) nanoparticles (NP) to achieve delivery in the brain through intranasal administration. Dzp nanoparticles (DNP) were formulated by nanoprecipitation and optimized using Box-Behnken design. The influence of various independent process variables (polymer, surfactant, aqueous to organic (w/o) phase ratio, and drug) on resulting properties of DNP (z-average and drug entrapment) was investigated. Developed DNP showed z-average 148–337 d.nm, polydispersity index 0.04–0.45, drug entrapment 69–92%, and zeta potential in the range of −15 to −29.24 mV. Optimized DNP were further analyzed by differential scanning calorimetry (DSC), Fourier transform infrared spectroscopy (FTIR), ex-vivo drug release, and in-vitro cytotoxicity. Ex-vivo drug release study via sheep nasal mucosa from DNP showed a controlled release of 64.4% for 24 h. 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay performed on Vero cell line showed less toxicity for DNP as compared to Dzp suspension (DS). Gamma scintigraphy and biodistribution study of DNP and DS was performed on Sprague-Dawley rats using technetium-99m-labeled (99mTc) Dzp formulations to investigate the nose-to-brain drug delivery pathway. Brain/blood uptake ratios, drug targeting efficiency, and direct nose-to-brain transport were found to be 1.23–1.45, 258, and 61% for 99mTc-DNP (i.n) compared to 99mTc-DS (i.n) (0.38–1.06, 125, and 1%). Scintigraphy images showed uptake of Dzp from nose-to-brain, and this observation was in agreement with the biodistribution results. These results suggest that the developed poly(D,L-lactide-co-glycolide) (PLGA) NP could serve as a potential carrier of Dzp for nose-to-brain delivery in outpatient management of status epilepticus.
KEY WORDS: controlled release, nanoparticles, process optimization, scintigraphy
INTRODUCTION
Diazepam (Dzp) is a benzodiazepine widely used as sedative-hypnotic, antianxiety, and antiepileptic drug. It is a lipophilic drug and can readily pass through the blood-brain barrier (BBB) and some other lipophilic tissues. However, due to its lipophilicity, it is rapidly redistributed out of the brain. Due to fast distribution, serum levels of diazepam fall down quickly in the brain leading to repeated dosing, accumulation in the body, and serious complications (1). Dzp is poorly soluble in water, and its intravenous formulation has to be prepared using cosolvents propylene glycol (40%) and ethanol (10%) or as emulsions. Use of polypropylene as a cosolvent causes discomfort and adverse reactions in patients. Dzp is also available as a rectal gel formulation, the only outpatient alternative therapy available, but it shows variable bioavailability, slower onset of action, and low patient compliance.
Besides Dzp being reported as a useful alternative to lorazepam for intravenous therapy in management of premonitory and early status epilepticus, there have been studies to explore the intranasal formulations of Dzp as a noninvasive and a more socially acceptable treatment option over either intravenous or rectal gel formulations (2). In an attempt to explore intranasal formulation of Diazepam, Henney et al. 2014 compared the pharmacokinetic parameters and tolerability of equal doses of intranasal and rectal diazepam in 24 healthy adults. The authors reported comparable bioavailability for intranasal, and the rectal gel formulations and both the formulations were well tolerated with mild to moderate adverse events (2). Ivaturi et al. 2009 studied and compared the pharmacokinetics and tolerability of intranasal and intravenous diazepam and midazolam in four-way crossover trail in healthy adults. The authors concluded that both formulations were rapidly absorbed via intranasal administration; however, diazepam showed extended duration of action (3).
As diazepam exhibits a longer elimination half life of (43 ± 13 h) compared to midazolam (1.9 ± 0.6 h) and lorazepam (14 ± 5 h), intranasal diazepam can provide extended duration of action with rapid onset at low dose, resulting in low adverse effects (1).
Nose-to-brain delivery of drugs has attracted a lot of attention as a potential route of drug delivery to the brain as it bypasses first-pass metabolism and prevents enzymatic/chemical degradation of drugs. Being noninvasive in nature, nasal route provides an alternative to injectable formulations and enhances patient compliance. The drug molecule permeates directly into the central region of the brain bypassing BBB via the olfactory and trigeminal nerves present in the nasal cavity (4–7). Several studies in literature support drug transport from nose-to-brain via olfactory regions of the nasal cavity and trigeminal nerve. Thorne et al. 2004 did a comparative study by administering insulin-like growth factor-I through intranasal and intravenous route in Sprague–Dawley rats and studied possible mechanism of nose-to-brain pathways. The results supported the fact of that intranasal administration of insulin-like growth factor-I can bypass the BBB via olfactory and trigeminal pathways. Gamma counting of the microdissected tissue and high-resolution phosphor imaging of tissue sections of the sacrificed animals showed that the tissue concentrations and distribution following intranasal administration led to rapid entry into the CNS. The authors proposed one route associated with the peripheral olfactory system connecting the nasal passages with the olfactory bulbs and rostral brain regions and another route associated with the peripheral trigeminal system connecting the nasal passages with brainstem and spinal cord regions (8). Westin et al. 2006 investigated the nose-to-brain pathway for morphine delivery on rats and compared with intravenous administration. The results showed brain hemisphere/plasma morphine area under the curve (AUC) ratio to be higher after intranasal administration as compared to intravenous route for 0–5 min period. A significantly higher ratio of morphine in the brain hemisphere was found via nasal administration supporting the nose-to-brain uptake of the drug. The authors concluded that the observed ratio suggests that morphine could be transferred via olfactory pathways to the brain (9).
Shingaki et al. compared the uptake of 5 fluorouracil intravenously and intranasally in the presence and absence of intravenously administered acetazolamide. Concentrations of 5 fluorouracil in plasma, CSF, and the cerebral cortex were measured. It was observed that in the presence of intravenous acetazolamide, there was a marked increase in the concentration of 5 fluorouracil in the cerebrospinal fluid and brain following the nasal perfusion of the said drug; although the plasma concentrations of the drug were found to be comparative with intravenous infusion and nasal perfusions; the apparent brain uptake clearance of 5 fluorouracil after the nasal perfusion with acetazolamide was significantly increased by 104 and 46% as compared to intravenous infusion and nasal perfusion without acetazolamide, respectively. There was a clear evidence of 5 fluorouracil delivery to the brain through a nose-to-brain pathway in the presence of acetazolamide (10).
Literature suggests that the drugs can be administered intranasally using nanocarriers or coadministered with absorption enhancers (11). Polymeric biodegradable nanoparticles have been extensively reported for encapsulation of drugs and nose-to-brain drug delivery. Poly(D,L-lactide-co-glycolide (PLGA) is a widely accepted and US Food and Drug Administration (FDA) approved polymer for the development of nanoparticle (NP). It has been widely used in the preparation of NP to encapsulate hydrophobic as well as hydrophilic drugs for controlled drug delivery (12–15). The polymer matrix prevents drug from degradation and helps in controlling drug release profile (16,17).
Characteristic properties of polymeric NP such as particle size (z-average) and percentage drug entrapment and surface charge (zeta potential) play an important role in efficient delivery of these nanoparticles across the mucosa (15,18).
To develop a robust and reproducible formulation, it is necessary to establish the relationship between process parameters and characteristics of NP. The number of studies for NP formulations has reported the importance of analyzing the process parameters by experimental designs (19,20). Budhian et al. studied the effect of various process variables in emulsion-solvent evaporation on haloperidol-loaded PLGA NP (21). Song et al. studied six independent process variables for dual loading of vincristine and quercetin in single o/w emulsification system (22). Feczko et al. studied the effect of process variables of bovine serum albumin-loaded PLGA NP by double emulsion-solvent evaporation technique (18). Songa et al. investigated the effect of organic phase on the characteristic properties of PLGA NP (23).
In the present investigation, it was hypothesized that intranasal PLGA nanoparticles could serve as a noninvasive carrier for diazepam and provide controlled release with enhanced bioavailability to the brain.
Dzp-loaded PLGA NP were developed by nanoprecipitation and optimized using Box-Behnken design (BBD) (24). Nanoprecipitation considers several process parameters such as polymer concentration, aqueous to organic phase volume ratio (w/o), surfactant/stabilizer concentration, and drug concentration (25,26). Varying these process parameters alters the outcome in terms of z-average, percentage drug entrapment, and percentage drug release.
Further ex vivo release was carried out using sheep nasal mucosa, and in vitro cytotoxicity was assessed on Vero cells. In vivo studies were carried out by radiolabeling Dzp with 99mTc. The 99mTc-DNP were administered (intranasal (i.n) and intravenous (i.v)) to rats, and images were taken using a gamma camera. Dzp biodistribution was investigated in the brain and blood on Sprague-Dawley rats.
MATERIALS AND METHODS
Materials and Animals
Poly(D,L-lactide-co-glycolide) (PLGA) 50:50 (molecular weight 30,000–60,000) and poloxamer 407 were purchased from Sigma-Aldrich, St. Louis, USA. 99mTc was obtained from the Regional Center for Radiopharmaceuticals of the Board of Radiation and Isotope Technology (BRIT), Delhi, India. Diazepam was purchased from R L Fine Chem, Bangalore, Karnataka, India. HPLC grade acetone and water were purchased from Thermo Fisher Scientific, Mumbai, Maharashtra, India. All the other solvents were of HPLC grade.
All animal experiments were carried out in compliance with the Institute of Nuclear Medicine and Allied Sciences (INMAS) Institutional Animal Ethics Committee (IAEC), New Delhi, India, vide number INM/IAEC 2013/07/007, and their guidelines were followed throughout the study. Sprague-Dawley rats (male 2–3 months) weighing 180–200 g obtained were from the Central Animal House Facility of INMAS, Delhi, India. All animals were given normal feed and filtered drinking water ad libitum. Rats were kept at room temperature of 25 ± 5°C.
Nanoparticle Preparation
DNP were prepared by nanoprecipitation (21,27,28). Accurately weighed PLGA and Dzp were dissolved in organic phase (acetone). The organic phase was added dropwise at the rate of 1 ml/min into an aqueous phase containing poloxamer 407 under continuous magnetic stirring. The stirring speed was maintained at 325 rpm for 4 h at room temperature to evaporate organic solvent completely and to obtain a colloidal suspension of DNP. Colloidal suspension of DNP was centrifuged at 12,000 rpm for 30 min at 4°C (REMI, Mumbai, Maharashtra, India). The DNP pellet was collected and washed twice with HPLC water to remove unentrapped drug from the surface of DNP and subjected for estimation of percentage drug entrapment and drug loading.
Experimental Design
Box-Behnken statistical design was employed to optimize DNP and to investigate the main, quadratic, and interaction effects on z-average and percentage drug entrapment. A four-factor, two-level Box-Behnken design was employed to generate second-order polynomial equation (14). For the response surface methodology (RSM) consisting Box-Behnken design, 26 total confirmatory formulation runs were generated with 2 center points using Design-Expert software (version 8.0.0, Stat-Ease Inc., Minneapolis, Minnesota). Table I provides the level of independent and dependent variables. The effect of independent variables on dependent response was studied by second-order polynomial equation
where, y is the dependent variable, b0 is the intercept, and b1 to b14 are the regression coefficients. X1, X2, X3, and X4 are the coded values of the independent variables. XaXb (a,b = 1, 2, 3, 4) and Xi2 (i = 1,2,3,4) represent the interaction and quadratic terms, respectively.
Table I.
Different Levels of Variables in Box-Behnken Design
| Levels | |||
|---|---|---|---|
| Low | Medium | High | |
| Independent variables | |||
| X 1 = polymer concentration (w/v) | 10 | 35 | 60 |
| X 2 = surfactant concentration (w/v) | 2 | 8.50 | 15 |
| X3 = aqueous/organic phase ratio (v/v) | 2 | 4 | 6 |
| X4 = drug concentration (w/v) | 1 | 3 | 5 |
| Dependent variables | Desired constraints | ||
| Y1 = z-average (d.nm) | Minimize | ||
| Y2 = percentage drug entrapment | Maximize | ||
HPLC Method for Dzp Estimation
Reversed-phase (RP)-HPLC method was developed and validated as per USP monograph (29) using Waters HPLC isocratic system (Waters, Vienna, Austria) with UV detector for the analysis of Dzp in prepared NP (USP30-NF25). The data was collected using Breeze 2 software (Waters, Vienna, Austria). SunFire column C-18 (250 × 4.6 mm, 5 μm), column temperature was maintained at 30°C. Filtered (0.22 μ millipore filter) and degassed mixture of acetonitrile, water, and methanol (2:2:1) was used as mobile phase at the flow rate of 1 ml/min. The injection volume used was 20 μl, and detector wavelength was 254 nm.
Percentage Drug Entrapment and Drug Loading
The DNP suspension was centrifuged at 12,000 rpm, 4°C for 30 min (REMI, Mumbai, Maharashtra, India). The NP pellet was settled down and washed twice with HPLC water to remove unentrapped drug completely, and the supernatant was collected. The amount of unentrapped drug in the supernatant was determined by the RP-HPLC method. The percentage drug entrapment and drug loading of NP were calculated using the following formula:
| 1 |
| 2 |
Measurement of z-average and Zeta Potential
Z-average, polydispersity index (PDI), and zeta potential of the developed NP were determined using Malvern Zetasizer (Malvern, Worcestershire, UK). Particle size and zeta potential investigation was performed in triplicate by diluting DNP suspension to 1:50 v/v in HPLC water.
Differential Scanning Calorimetry (DSC) Analysis
DSC analysis was performed to study the phase behavior of optimized DNP and pure Dzp. Dried powder of Dzp and optimized DNP were placed individually and sealed in DSC pan with a DSC loading puncher. The samples were scanned between −20 and 200°C with a heating rate of 10°C/min, under nitrogen atmosphere using TA instruments Q-200 DSC (New Castle, USA).
Fourier Transform Infrared Spectroscopy (FTIR)
FTIR analysis of optimized DNP, PLGA, and Dzp was performed using PerkinElmer BX II (PerkinElmer Massachusetts, USA). Potassium bromide (KBr) pellets of samples were prepared and scanned at the resolution of 4 from 400 to 4000 cm−1.
Ex vivo Drug Release Study
Ex vivo drug release behavior of Dzp from NP was investigated using sheep nasal mucosa keeping DS as control. Sheep nasal mucosa was procured from a local slaughter house. Nasal mucosa with a contact area of 1.5 cm2 was mounted on a receptor compartment of the Franz diffusion cell (diameter 10 mm, 15 ml volume), with the mucosal face in contact with phosphate buffer (pH = 6.4). Two experimental sets for DNP and DS were performed keeping temperature 37 ± 0.5°C, 100 rpm. The DNP/DS 5 mg/ml (NP/drug resuspended in 2 ml phosphate buffer) was applied on the outer surface of the nasal mucosa. Two milliliters of sample was withdrawn after regular intervals of time and replaced with fresh PBS to maintain sink conditions. The samples were then analyzed using RP-HPLC method.
Cell Viability Analysis
NP systems have broad therapeutic applications, but being used in delicate administration routes such as intranasal route, there is a need for possible toxicity evaluation (30). Besides intravenous (i.v) injection, NP can enter systemic circulation and get accumulated in various organs including the liver and kidney (31,32). Cell viability analysis of optimized DNP was assessed on Vero cell line (green monkey kidney epithelial cells) using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay (33,34). The cells were maintained in Dulbecco’s Modified Eagle’s media (DMEM) supplemented with 10% fetal bovine serum, at 37°C in humidified 5% CO2/95% air incubator (New Brunswick, Germany). Vero cells were seeded at a concentration of 105 cells/ml in 96-well TC plate (HiMedia) and allowed to adhere overnight. Different concentrations of optimized DNP (3.12–100 μg/ml diluted in DMEM), DS (3.12–100 μg/ml), and placebo were added in triplicates and incubated under normal conditions for 24 h. After incubation, 20 μl of MTT (5 mg/ml in DPBSA) was added to each well and replaced after 4 h with 200 μl of dimethyl sulfoxide (DMSO) to dissolve the insoluble formazan. The absorbance was measured at 570 nm using an ELISA microplate reader (Bio-Rad, Hercules, CA). Untreated cells and media were taken as positive control and blank, respectively. Percentage cell viability was calculated using Eq. 3.
| 3 |
Radiolabeling of Dzp and Its NP with 99mTc
Dzp and DS were radiolabeled with 99mTc using direct labeling method (35,36). Five hundred microliters of d-Sol (3 mg Dzp in acetone) was taken and mixed with 200 μl of stannous chloride dihydrate solution (2 mg/ml in ethanol). To the resultant mixture (filtered through 0.22 μ nylon filter), 500 μl of 99mTc (1–2 mCi) was added with continuous mixing and incubated at 37 ± 0.5°C for 30 min. The radiochemical purity of 99mTc-Dzp, 99mTc-DNP, and 99mTc-DS was determined using ascending instant thin-layer chromatography (ITLC; Gelman Sciences, Inc., Ann Arbor, MI, USA) using acetone as mobile phase. The effect of incubation time and stannous chloride concentration on radiolabeling efficiency were studied to achieve optimum reaction conditions. In vitro stability of radiolabeled formulation in normal saline and in rat plasma was evaluated and optimized (37). Stable radiolabeled formulation was then subjected to gamma scintigraphy and biodistribution.
Gamma Scintigraphy Imaging
The Sprague-Dawley rats (male, aged 2–3 months) weighing between 180 and 200 g were selected for the study. Radiolabeled formulation 99mTc-DS (200 μCi/20 μl) with the concentration of 0.04–0.05 mg Dzp (equivalent to 0.2–0.25 mg/kg) was intravenously injected through the tail vein of the rat. Similarly, 20 μl of 99mTc-DS/DNP (5 mCi/ml) containing 0.040–0.050 mg Dzp (equivalent to 0.2–0.25 mg/kg body weight (B.W.)) was administered (10 μl) in each nostril. The rats were anesthetized using 0.4 ml ketamine hydrochloride intraperitoneal injection (50 mg/ml) prior to administration of formulations and placed on the imaging platform. Imaging was performed using a single-photon emission computerized tomography (SPECT; LC 75-005, Diacam, Siemens AG; Erlanger, Germany) gamma camera (36–39).
Biodistribution Studies
The Sprague-Dawley rats (male, aged 2–3 months) weighing between 180 and 200 g were selected for the study. Three rats for each formulation (99mTc-DS i.n, 99mTc-DNP i.n, and 99mTc-DS i.v) per time point (0.5, 1, 2, 4, 8 h) were used in the study. Prior to administration of the formulations, the rats were anesthetized using 0.4 ml ketamine hydrochloride intramuscular injection (50 mg/ml). 99mTc-DS (5 mCi/ml) containing 0.04–0.050 mg Dzp (equivalent to 0.2–0.25 mg/kg) was injected through the tail vein of rats. Similarly, 20 μl of radiolabeled complex of 99mTC-DS/DNP (5 mCi/ml) containing 0.040–0.050 mg Dzp (equivalent to 0.2–0.25 mg/kg B.W.) was administered (10 μl) in each nostril. The formulations were instilled into the nostrils with the help of micropipette (20 μl) attached with low-density polyethylene tube having 0.1 mm internal diameter. The rats were held from the back in slanted position during nasal administration of the formulations.
Blood and brain tissue samples were collected at predetermined time points (0.5, 1, 2, 4, 8 h) post-administration. Blood samples were collected through the retro orbital vein. Subsequently, brain was extracted and washed twice using normal saline solution to remove any adhering tissue/fluid and then weighed. Radioactivity present in blood and brain was measured using shielded well-type gamma scintillation counter (36–38). The radiopharmaceutical uptake per gram in brain/blood was calculated as a fraction of administered dose using GraphPad Prism 5 software, USA.
To evaluate the brain targeting efficiency, two indexes drug targeting efficiency (% DTE) and direct nose-to-brain transport (% DTP) were adopted as mentioned below (26,29).
| 4 |
| 5 |
Where,
| 6 |
- Bx
Brain AUC fraction contributed by systemic circulation through the blood-brain barrier (BBB) following intranasal administration.
- Bi.v.
AUC0→480 (brain) following intravenous administration.
- Pi.v.
AUC0→480 (blood) following intravenous administration.
- Bi.n.
AUC0→480 (brain) following intranasal administration.
- Pi.n.
AUC0→480 (blood) following intranasal administration.
- AUC
Area under the curve
Stability Studies of Optimized Formulation
Optimized DNP were subjected for accelerated stability studies by incubating at 25 ± 2°C and 60 ± 5% RH for 3 months (20). The optimized DNP were then studied for change in z-average and percentage drug entrapment. Shelf life analysis was calculated, and graph was plotted using SigmaPlot™ 13 (Systat Software Inc, USA).
Data Analysis
Results of ex vivo drug release and biodistribution data were reported as mean ± SD (n = 3), and the difference between the groups were tested using two-way ANOVA using GraphPad Prism 5.0 and data analysis tool in Microsoft Excel and the results were found to be significant at p < 0.05.
RESULTS
RSM was used in the present study to investigate the interaction between the four independent factors and their effect on dependent response (Table II). Polynomial equation was generated for all the response variables. 3D response surface plots were constructed using Design-Expert software (version 8.0.0, Stat-Ease Inc., Minneapolis, Minnesota). Maximum percentage drug loading was found to be 11.5% for run 5.
Table II.
Effect of Independent Process Variables on Dependent Variable
| Run | PLGA mg/ml | Poloxamer mg/ml | w/o phase volume ratio | Drug conc. mg/ml | z-average d.nm (±SD) | Percentage drug entrapment (±SD) | PDI (±SD) |
|---|---|---|---|---|---|---|---|
| 1. | 35 | 2 | 4 | 1 | 214 ± 0.8 | 82 ± 0.3 | 0.183 ± 0.02 |
| 2. | 35 | 2 | 4 | 5 | 211 ± 0.5 | 88 ± 0.2 | 0.150 ± 0.01 |
| 3. | 10 | 8.50 | 6 | 3 | 176 ± 1.2 | 77 ± 0.8 | 0.048 ± 0.04 |
| 4. | 35 | 8.50 | 2 | 1 | 194 ± 0.4 | 81 ± 1 | 0.17 ± 0.03 |
| 5. | 10 | 2 | 4 | 3 | 190 ± 0.5 | 82 ± 0.5 | 0.31 ± 0.04 |
| 6. | 10 | 8.50 | 4 | 5 | 148 ± 0.5 | 84 ± 0.8 | 0.11 ± 0.05 |
| 7. | 10 | 8.50 | 2 | 3 | 169 ± 1.5 | 82 ± 1.4 | 0.078 ± 0.003 |
| 8. | 35 | 8.50 | 4 | 3 | 183 ± 1.2 | 84 ± 0.5 | 0.11 ± 0.04 |
| 9. | 60 | 8.50 | 4 | 1 | 286 ± 1 | 87 ± 1.5 | 0.24 ± 0.03 |
| 10. | 35 | 15 | 4 | 5 | 180 ± 0.8 | 86 ± 0.6 | 0.19 ± 0.02 |
| 11. | 10 | 15 | 4 | 3 | 163 ± 1 | 79 ± 0.8 | 0.04 ± 0.003 |
| 12. | 60 | 2 | 4 | 3 | 337 ± 1.8 | 92 ± 1.4 | 0.45 ± 0.02 |
| 13. | 35 | 15 | 6 | 3 | 175 ± 0.5 | 85 ± 1 | 0.17 ± 0.04 |
| 14. | 60 | 15 | 4 | 3 | 226 ± 0.5 | 86 ± 1.3 | 0.15 ± 0.02 |
| 15. | 60 | 8.50 | 2 | 3 | 257 ± 0.7 | 88 ± 0.8 | 0.30 ± 0.05 |
| 16. | 35 | 15 | 4 | 1 | 169 ± 1 | 76 ± 0.5 | 0.09 ± 0.004 |
| 17. | 35 | 2 | 2 | 3 | 201 ± 1.2 | 89 ± 0.5 | 0.15 ± 0.03 |
| 18. | 60 | 8.50 | 4 | 5 | 276 ± 0.5 | 92 ± 0.8 | 0.2 ± 0.05 |
| 19. | 35 | 8.50 | 2 | 5 | 177 ± 0.7 | 87 ± 1 | 0.1 ± 0.03 |
| 20. | 10 | 8.50 | 4 | 1 | 149 ± 0.5 | 69 ± 0.5 | 0.21 ± 0.05 |
| 21. | 35 | 8.50 | 6 | 1 | 180 ± 1.2 | 73 ± 0.2 | 0.28 ± 0.02 |
| 22. | 35 | 2 | 6 | 3 | 258 ± 0.4 | 91 ± 1.2 | 0.21 ± 0.06 |
| 23. | 35 | 8.50 | 6 | 5 | 189 ± 0.6 | 89 ± 0.5 | 0.19 ± 0.05 |
| 24. | 35 | 15 | 2 | 3 | 174 ± 1.5 | 88 ± 0.7 | 0.15 ± 0.04 |
| 25. | 60 | 8.50 | 6 | 3 | 298 ± 1.2 | 91 ± 0.5 | 0.15 ± 0.03 |
| 26. | 35 | 8.50 | 4 | 3 | 183 ± 1 | 84 ± 1.5 | 0.11 ± 0.04 |
PLGA poly(D,L-lactide-co-glycolide), conc. concentration, SD standard deviation,PDI polydispersity index
Effect on z-average
Polynomial equation was constructed for the measured response:
The polynomial equation shows the quantitative effect of process variables and their interaction on z-average of the developed DNP. From the above equation, positive coefficient for X1 and X3 and the positive coefficient found for the interaction of X1X3, X2X4, and X3X4, suggested that z-average is directly proportional to X1 and X3. The negative sign on coefficient for X2 and X4 is attributed to the opposite effect of variable on the response. The overall effects of all four responses are shown in Fig. 1.
Fig. 1.
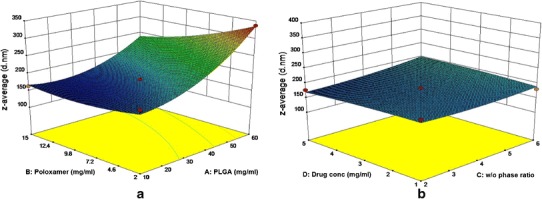
3D response surface plot showing effect of polymer and poloxamer concentration on z-average (a) and effect of drug concentration and w/o phase ratio on z-average (b)
Z-average for the developed NP varied in the range of 148 (F-6) to 337 d.nm (F-12). Polymer concentration positively affected the particle size, i.e., with increase in polymer concentration z-average value also increased.
To assess the effect of polymer concentration on stability of developed NP, photon correlation spectroscopy was used, and the zeta potential was found in the range of −15 to −29.24 mV. PDI values were found in the range of 0.04–0.45, indicating uniform-size distribution.
The slight positive sign on the coefficient of ratio of w/o phase variable indicated positive effect on z-average.
Polynomial equation shows the effect of poloxamer concentration (X2) in the aqueous phase on the z-average. The large negative sign on the coefficient of factor X2 indicated that with increase in surfactant concentration, z-average of the developed DNP decreased and vice versa. Amount of the drug that can be encapsulated in DNP is an important parameter to be studied; however, the slight negative sign on the coefficient for drug concentration (X4) indicated no significant effect on z-average.
Effect on Percentage Drug Entrapment
To investigate the impact of each variable on response Y2 (i.e., percentage drug entrapment), polynomial equation was constructed by Box-Behnken design.
As indicated in the above polynomial equation, the positive sign on coefficient for factor X1 and X4 shows positive impact on percentage drug entrapment. Whereas, the negative sign on coefficient for factor X2 and X3 indicates negative impact on response Y2.
Figure 2 showed the effect of PLGA and poloxamer concentration on percentage drug entrapment. There was an increase in the percentage drug entrapment with increasing PLGA concentration. The negative sign on coefficient of factor poloxamer concentration (X2) indicated that with increase in poloxamer concentration, the percentage drug entrapment decreased. The slight negative sign on coefficient for factor X3 indicated no significant effect of w/o phase ratio on percentage drug entrapment.
Fig. 2.
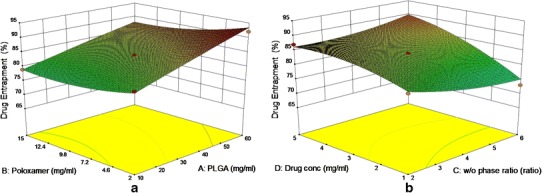
3D response surface plots showing effect of PLGA and poloxamer on percentage drug entrapment (a) and effect of w/o phase ratio and drug concentration on percentage drug entrapment (b)
3D response surface plot (Fig. 2) and polynomial equation showed that with increase in drug concentration, the response Y2 also increased. The results are in agreement with Budhian et al. and Panyam et al. (21,40).
Validation of RSM and Optimization of DNP
Optimum formulation combination of DNP was selected based on desired constraints within range for independent variables and minimized and maximized constraints for z-average and percentage drug entrapment, respectively. RSM generated various solutions, and the optimized formulation (X1 = 32 mg/ml, X2 = 15 mg/ml, X3 = 6, and X4 = 5 mg/ml with predictable response value for Y1 = 170.15 d.nm and Y2 = 88.7%) was selected on the basis of desirability factor. The experimental value for response Y1 (183.2 d.nm) and Y2 (87.8%) of optimized formulation was found in good agreement with the predicted values generated by RSM, and the result assured the validity of RSM model.
Ex vivo Drug Release
Ex vivo drug release behavior of Dzp from optimized DNP was accessed via sheep nasal mucosa. Figure 3 showed the drug release behavior from DS and DNP (studied up to 24 h). DS showed maximum 78.5 ± 1.03% release within 4 h, whereas DNP showed initial drug release of 18.2 ± 2.2% in 2 h, and drug release was sustained (64.4 ± 1.8%) up to 24 h. Ex vivo drug release data showed that the best-fit model for NP was Korsmeyer-Peppas model with correlation coefficient (r2) 0.947 and release exponent value (n) 0.460. The release exponent value (n) was below 0.5, which suggested that the release of Dzp from NP followed Fickian diffusion.
Fig. 3.
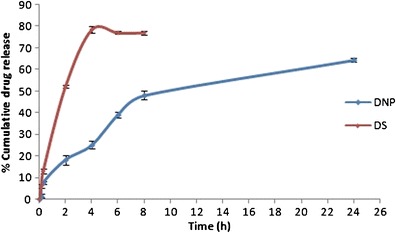
Ex vivo drug release data of DS and DNP
Differential Scanning Calorimetry (DSC)
DSC thermograms of Dzp and DNP are shown in Fig. 4. Dzp has characteristic endothermic peak at 132.56°C; however, melting point of Dzp in the formulation shifted towards lower temperature 100.37°C.
Fig. 4.
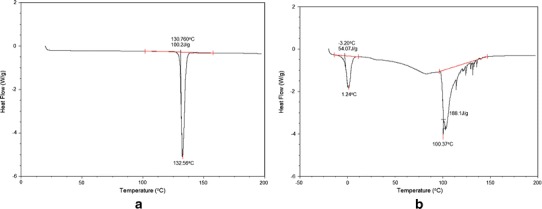
DSC thermogram a Dzp and b DNP
FTIR Analysis
FTIR analysis of Dzp, PLGA, and DNP was performed to investigate the interaction between drug and polymer. Figure 5 showed FTIR spectra of Dzp, PLGA, and DNP. Dzp showed characteristics peaks of –CH stretching (3354 cm−1), –CH2 stretching (2972 cm−1), –C = C (1683 cm−1), CH2 deformation (1559 cm−1), C = C (1604 cm−1), CH3 bending (1483 cm−1), and CN (1350 cm−1). FTIR spectra of PLGA showed significant peaks such as stretching –OH stretching (3200–3500 cm−1), –CH, –CH2, –CH3 (2800–3000 cm−1), carbonyl –C = O stretching (1760 cm−1), and C–O stretching (1090 cm−1).
Fig. 5.
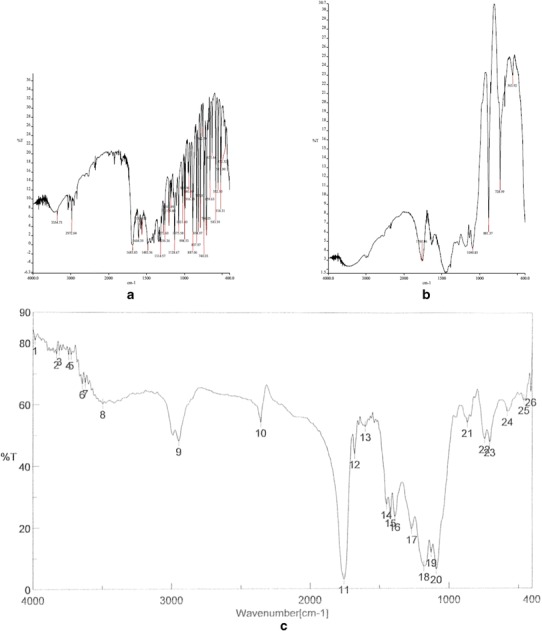
FTIR spectra of pure diazepam (a), PLGA (b), and diazepam-loaded PLGA nanoparticles (c)
Cell Viability Analysis
In vitro cell viability of the optimized DNP, DS, and corresponding placebo was performed on Vero cells (monkey kidney epithelial cell line) using MTT assay. The results of MTT cell viability assay on Vero cell line are shown in Fig. 6. A dose-dependent increase in cytotoxicity was observed with increasing concentration of DNP and DS. DNP, DS, and placebo exhibited 79 ± 1.2, 74 ± 1, and 95.5 ± 0.8% cell viability at 100 μg/ml respectively, with respect to control. The results showed that PLGA NP reduces cytotoxicity of Dzp.
Fig. 6.
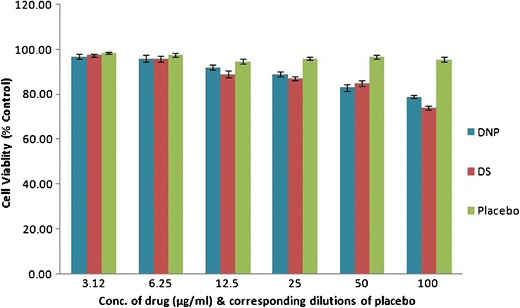
Cell viability assay of DNP, DS, and corresponding placebo in Vero cells. The experiment was performed in triplicates, and ±SD are shown as error bars
Gamma Scintigraphy Studies
Dzp was effectively radiolabeled using 99mTc. Optimum SnCl2·2H2O concentration was found to be 2 mg/ml with an incubation time of 30 min. Maximum labeling efficiency and stability of DNP and DS was found to be 96.5 and 98.3%, respectively.
Gamma scintigraphy images of Sprague-Dawley rats 0.5 h post intranasal and intravenous administration are shown in Fig. 7. Presence of high radioactivity was observed in rat brain after administration of 99mTc-DNP (i.n) compared to 99mTc-DS (i.v) and 99mTc-DS (i.n). The scintigraphy images clearly indicated the high uptake of 99mTc-DNP into the brain.
Fig. 7.
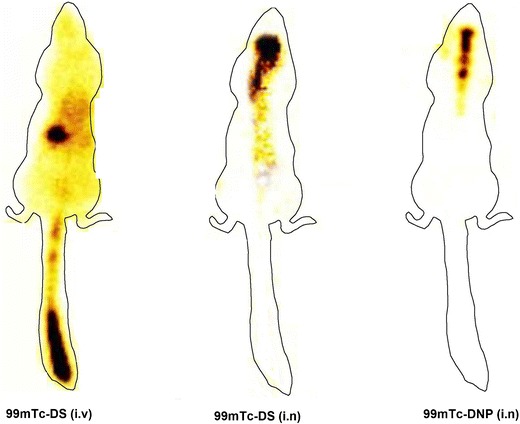
Gamma scintigraphy images of the rat after administration of 99mTc-DS (i.v), 99mTc-DS (i.n), and 99mTc-DNP (i.n) showing presence of radioactivity in different organs
Biodistribution
Biodistribution studies of 99mTc-Dzp following i.v administration (99mTc-DS) and intranasal (99mTc-DS and 99mTc-DNP) administration on Sprague-Dawley rats were performed, and the radioactivity was estimated at predetermined time intervals up to 8 h. The brain/blood ratio of the drug at all time points for different formulations and the results obtained are shown in Tables III, IV, and V and Fig. 8. The brain/blood ratios of the drug were found to be higher for 99mTc-DNP when administered intranasally. This further confirmed direct nose-to-brain transport. The concentration of the drug in the brain after intranasal administration of 99mTc-DNP was found to be higher at all sampling time points as compared to 99mTc-DS (i.v) and 99mTc-DS (i.n) up to 8 h post-administration. The results were found to be significant at p < 0.05.
Table III.
Distribution of 99mTc-Dzp from 99mTc-DS (i.v), 99mTc-DS (i.n), and 99mTc-DNP (i.n) at Different Time Intervals in Sprague-Dawley Rats
| Formulation and route of administration | Distribution of diazepam in blood and brain compartments at different sampling time points | |||||
|---|---|---|---|---|---|---|
| Organ/tissue | 0.5 h | 1 h | 2 h | 4 h | 8 h | |
| 99mTc-DS (i.v) | Blood | 3.09 ± 0.45 | 2.97 ± 0.3 | 2.13 ± 0.7 | 1.74 ± 0.5 | 1.44 ± 0.3 |
| Brain | 1.19 ± 0.2 | 1.28 ± 0.2 | 1.73 ± 0.3 | 1.09 ± 0.5 | 0.25 ± 0.12 | |
| 99mTc-DS (i.n) | Blood | 1.7 ± 0.5 | 1.83 ± 0.16 | 1.65 ± 0.4 | 1.46 ± 0.4 | 0.96 ± 0.15 |
| Brain | 1.81 ± 0.5 | 1.73 ± 0.2 | 1.19 ± 0.2 | 0.85 ± 0.05 | 0.37 ± 0.1 | |
| 99mTc-DNP (i.n) | Blood | 1.77 ± 0.3 | 2.26 ± 0.45 | 2.35 ± 0.3 | 1.67 ± 0.5 | 1.4 ± 0.4 |
| Brain | 2.39 ± 0.5 | 3.07 ± 0.6 | 2.9 ± 0.4 | 2.34 ± 0.5 | 2.02 ± 0.8 | |
| 99mTc-DS (i.v) | Brain/blood | 0.38 ± 0.25 | 0.43 ± 0.15 | 0.81 ± 0.12 | 0.62 ± 0.4 | 0.17 ± 0.1 |
| 99mTc-DS (i.n) | Brain/blood | 1.06 ± 0.4 | 0.94 ± 0.2 | 0.72 ± 0.2 | 0.58 ± 0.1 | 0.38 ± 0.1 |
| 99mTc-DNP (i.n) | Brain/blood | 1.35 ± 0.5 | 1.35 ± 0.3 | 1.23 ± 0.3 | 1.4 ± 0.5 | 1.4 ± 0.3 |
The rats were administered 100 μCi 99mTc-Dzp, and the radioactivity was measured in percent per gram of tissue of the administered dose. Each value is the mean ± SD of three estimations. Radioactivity was measured at 0 h, and all the measurements were performed using 0 h sample corresponding the tissue/organ as blank sample. Only statistically significant outcomes at p < 0.05 have been reported
99m Tc technetium-99m, DS Dzp suspension, DNP Dzp nanoparticles, i.v intravenous, i.n intranasal
Table IV.
Pharmacokinetics of 99mTc-DNP (i.n), 99mTc-DS (i.n), and 99mTc-DS (i.v) at Different Time Intervals Sprague Dawley Rats
| Organ/tissue | C max (%/g) | T max (h) | AUC0→480 | |
|---|---|---|---|---|
| 99mTc-DS (i.v) | Blood | 3.09 | 0.5 | 15.10 |
| Brain | 1.73 | 2 | 7.95 | |
| 99mTc-DS (i.n) | Blood | 1.83 | 1 | 11.02 |
| Brain | 1.81 | 0.5 | 7.30 | |
| 99mTc-DNP (i.n) | Blood | 2.35 | 2 | 13.92 |
| Brain | 3.07 | 1 | 18.94 |
The rats were administered 100 μCi 99mTc-Dzp, and the radioactivity was measured in percent per gram of tissue of the administered dose. Each value is the mean ± SD of three estimations. Only statistically significant outcomes at p < 0.05 have been reported
AUC area under the curve, DS Dzp suspension, DNP Dzp nanoparticles, i.v intravenous, i.n intranasal
Table V.
Brain Targeting Efficiency and Direct Nose-to-Brain Transport Following Intranasal Administration of 99mTc-DNP and 99mTc-DS
| Formulation and route of administration | Brain targeting efficiency (DTE %) | Direct nose-to-brain transport (DTP %) |
|---|---|---|
| 99mTc-DNP (i.n) | 258 | 61.3 |
| 99mTc-DS (i.n.) | 125 | 1 |
DNP Dzp nanoparticles, DTE % drug targeting efficiency percentage, DTP % direct nose-to-brain transport percentage
Fig. 8.
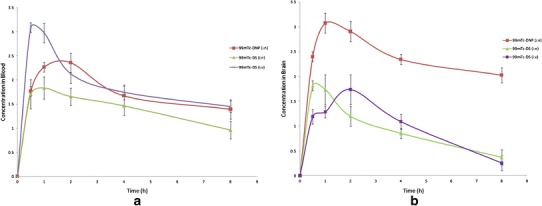
a 99mTc-Dzp concentration in rat blood at different time intervals following 99mTc-DS (i.v), 99mTc-DS (i.n), and 99mTc-DNP (i.n) administration. b 99mTc-Dzp concentration in rat brain at different time intervals following 99mTc-DS (i.v), 99mTc-DS (i.n), and 99mTc-DNP (i.n) administration
Stability Study of Optimized Formulation
Stability study results of optimized DNP showed slight increase in z-average from 183.5 ± 4 to 196.5 ± 2.5 d.nm. Sigma plot was constructed using SigmaPlot™ 13 software (Systat Software Inc, USA) and is depicted in Fig. 9. Further, the shelf life of the optimized DNP was found to be 18 months.
Fig. 9.
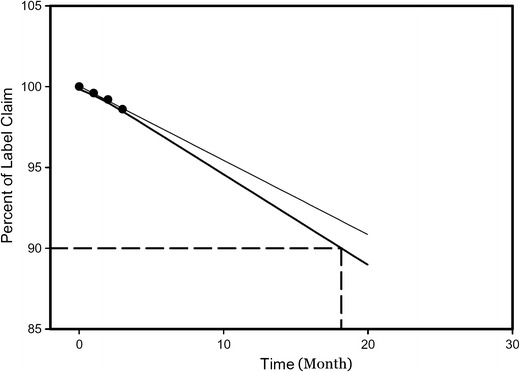
Shelf life analysis of optimized DNP
DISCUSSION
Brain drug delivery in the management of neurological disorders is always a tough challenge due to the presence of biological membranes such as blood-brain barrier (BBB), nature of drug molecule (molecular weight, hydrophilic/hydrophobic) (41–43). There have been several attempts by scientists to enhance delivery of drugs to brain via BBB. Literature suggests a number of investigations have been performed to investigate an alternate, viable, and feasible mode of drug delivery over the conventional techniques. One of them is administration of drugs via nasal route, bypassing the BBB. Diazepam, being lipophilic in nature, rapidly permeates across the membranes and shows fast onset of action; however, it redistributes in the body tissues resulting in decreased serum levels quickly. Diazepam is available in market as conventional dosage forms, i.e., tablets and injectable formulation. Intravenous diazepam is used for the management of status epilepticus. In addition to being an invasive technique, local pain, thrombosis, etc. have been reported after i.v. administration. The US FDA has also approved rectal gel of diazepam; however, its use is further limited in emergency conditions and there is a low patient compliance owing to socioculture issues.
To investigate an alternate route, Ivaturi et al. 2013 studied the bioavailability of intranasal diazepam and compared it with rectal diazepam in 12 healthy volunteers, and it was found that diazepam showed higher Cmax (181.8 ± 84 ng/ml) after intranasal administration compared to rectal gel (160.9 ± 109.4 ng/ml) with median Tmax of 0.75 h. The intranasal formulation showed relatively rapid but variable absorption with bioavailability of 70–90% relative to diazepam rectal gel. It was concluded that intranasal diazepam formulation can offer feasible alternate to rectal administration of diazepam in the management of epilepsy seizures (44). Similar studies were reported by Agarwal et al. 2013 where the bioavailability and pharmacokinetic parameters of diazepam rectal gel (Diastat) with intranasal diazepam solution and diazepam suspension were compared. The results showed that intranasal diazepam solution showed an absolute bioavailability of 97% as compared to 67% absolute bioavailability by intranasal suspension. Mean Cmax values for the suspension and solution formulations were 221 and 272 ng/ml with Tmax of 1 and 1.5 h, respectively. From the results, the authors concluded that intranasal diazepam formulations can be developed with high bioavailability and good tolerability (45).
In the present study, we have developed diazepam-loaded PLGA NP by nanoprecipitation method and optimized using response surface methodology (RSM) (46–49). RSM is a widely used tool to investigate the effect of independent process variables on dependent characteristic response. RSM consists of different designs such as central composite design, Box-Behnken design, 3-level factorial designs, etc. Box-Behnken design was used to optimize DNP keeping PLGA, poloxamer, w/o phase ratio, drug concentration as independent factors and z-average, and percentage drug entrapment as dependent factor.
Polynomial equations were generated by the Box-Behnken design to investigate the results, and it was found that with increase in PLGA concentration, particle size of the developed NP increased, which could be due to formation of large coacervates formed as a result of increase in polymer concentration. This is in agreement with Mainardes et al. where the similar effect was observed on particle size and size distribution with the increase in PLGA concentration (19). Increase in aqueous to organic phase ratio showed an increase in z-average, which could be attributed to small amount of organic phase volume available for solubilization of drug, leading to rapid emulsification with aqueous phase and hence the formation of larger coacervates.
Surfactants play an important role in the preparation of NP by nanoprecipitation. Concentration of surfactant in the colloidal suspension governs the stability and solubility of drug in the aqueous phase and affects the resulting entrapment and z-average (25). Polynomial equations indicated that with increase in surfactant concentration, z-average of the developed NP decreased and vice versa. This could be due to insufficient concentration of surfactant that failed to stabilize the interfacial layer leading to aggregated large-size particles.
There was an increase in the percentage drug entrapment with increasing PLGA concentration. This could be due to the lipophilic drug that has increased tendency to remain in the viscous organic phase. Large z-average of NP poses further hindrance for the movement of the drug (due to increase in PLGA concentration) as the diffusional length for the drug entering into the aqueous phase increases with enhanced viscosity of the organic phase. The negative sign on coefficient of factor poloxamer concentration (X2) indicated that with increase in poloxamer concentration, the percentage drug entrapment decreased. This could be due to the fact that with increase in surfactant concentration, there is an increase in solubility of diazepam in aqueous phase, resulting in less percentage of the drug entrapped in NP. This observation is in agreement with Seju et al. for olanzapine-loaded PLGA NP for intranasal delivery (25). With increase in drug concentration, the percentage drug entrapment also increased due to enhanced availability of drug to the polymer solution owing to high polymer-drug interaction. The findings are in agreement with Budhian et al. and Panyam et al. (21,40).
The surface charge of the optimized DNP (−15 to −29.24 mV) was found to be negative, indicating dominating negative charge of PLGA. The high negative surface charge will also oppose the aggregation of particle and results in better stability. Surface charge is a critical parameter on stability of nanoparticles and permeation across biological membranes.
Ex vivo release of diazepam from optimized DNP was performed across sheep nasal mucosa, and results showed that Dzp showed maximum 78.5 ± 1.03% release within 4 h from DS, which could be due to its lipophilic nature. However, initial burst release of Dzp was observed from DNP in 2 h and sustained drug release thereafter. The initial burst release of Dzp from DNP could be due to adsorption of Dzp on NP surface, while at later stage, Dzp may be constantly released from the core of DNP as a result of polymer matrix erosion due to hydration. Once transported to the brain, this will help achieve a prolonged stay of the drug inside the brain obviating the need of repeated administrations because of short half life of Dzp.
Dzp has characteristic endothermic peak at 132.56°C; however, melting point of Dzp in the formulation shifted towards lower temperature 100.37°C. This shows change of crystalline form of Dzp to amorphous form upon encapsulation in PLGA NP. Similar results were reported by Averineni et al. with paclitaxel as model drug. Encapsulated paclitaxel showed decrease in melting temperature as compared to pure paclitaxel (42). FTIR spectra of DNP showed presence of characteristic peaks of both PLGA and Dzp that suggests no significant molecular interaction between drug and polymer. Spectra for Dzp and PLGA were found in agreement with the spectra reported by Sylaja et al. and Mainardes et al., respectively (50,51).
Further, the optimized nanoparticles of diazepam were tested in vivo in Sprague-Dawley rats (52–55). Gamma scintigraphy was used to assess the nose-to-brain uptake of drug. Dzp was radiolabeled using 99mtechnetium by direct labeling method. The brain/blood ratios of the drug were found to be higher for 99mTc-DNP when administered intranasally, and the presence of high radioactivity was observed in rat brain after administration of 99mTc-DNP (i.n) compared to 99mTc-DS (i.v) and 99mTc-DS (i.n). This is a clear evidence of a nose-to-brain uptake of Dzp following nasal administration in rats. 99mTc-DNP (i.n) was more efficient than 99mTc-DS (i.n) due to the nanometric size range and mucoadhesive nature of PLGA used in nanoparticles, which resulted in better permeation and residence contact time with nasal mucosa, whereas DS could be rapidly washed out from the nasal tract. The results were found in agreement of Kumar et al. 2008 and Vyas et al. 2006 (36–38).
The scintigraphy images clearly indicated the high uptake of 99mTc-DNP into the brain (Fig. 7). Whereas, presence of 99mTc-Dzp was found higher in the liver after 99mTc-DS i.v administration compared to 99mTc-DS i.n and 99mTc-DNP i.n. It is suggestive that intranasal route also helps in reduction of drug to peripheral circulation and hence would result in less adverse effects.
As hypothesized, intranasal PLGA nanoparticles of Diazepam could be a promising and feasible approach for outpatient management of status epilepticus.
CONCLUSION
In the present investigation, Dzp-loaded PLGA NP were successfully developed and optimized using Box-Behnken design. Ex vivo drug release across sheep nasal mucosa and cell viability assay on Vero cell line supported the controlled drug release and safety of developed nanoparticles for intranasal administration. As hypothesized, biodistribution studies of the optimized 99mTc-DNP when administered intranasally showed significantly higher brain uptake of Dzp as compared to intranasal 99mTc-DS and intravenous 99mTc-DS in Sprague-Dawley rats. Moreover, the biodistribution results were in agreement with scintigraphy imaging in Sprague-Dawley rats, and it can be concluded from the results that intranasal administration of Dzp-loaded PLGA NP delivers Dzp rapidly and more effectively than 99mTc-DS administered via intranasal and intravenous route. The present investigation demonstrates that intranasal DNP can potentially transport Dzp via nose-to-brain and can serve as a noninvasive alternative for the delivery of Dzp to brain.
Acknowledgments
The authors would like to thank the Department of Biotechnology, Government of India for providing financial support to conduct the research work (DBT project No. BT/PR1891/MED/30/626/2011). The authors are grateful to the Jaypee Institute of Information Technology, Noida, UP (India), for the infrastructural support, Director of INMAS and Dr. Aseem Bhatnagar, Nuclear Medicine Department, for providing nuclear scintigraphy facility, and Dr. A. K. Panda from the National Institute of Immunology, New Delhi (India), for providing the facility for particle size analysis and zeta potential.
Conflict of Interest
The authors declare that this paper content has no conflict of interests.
References
- 1.Shorvon S, Perucca E, Engel J., Jr . The treatment of epilepsy. 3. UK: Wiley-Blackwell West Sussex; 2009. [Google Scholar]
- 2.Henney HR, 3rd, Sperling MR, Rabinowicz AL, Bream G, Carrazana EJ. Assessment of pharmacokinetics and tolerability of intranasal diazepam relative to rectal gel in healthy adults. Epilepsy Res. 2014;108:1204–11. doi: 10.1016/j.eplepsyres.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Ivaturi VD, Riss JR, Kriel RL, Cloyd JC. Pharmacokinetics and tolerability of intranasal diazepam and midazolam in healthy adult volunteers. Acta Neurol Scand. 2009;120:353–7. doi: 10.1111/j.1600-0404.2009.01170.x. [DOI] [PubMed] [Google Scholar]
- 4.Sperling MR, Haas KF, Krauss G, Seif Eddeine H, Henney HR, 3rd, Rabinowicz AL, et al. Dosing feasibility and tolerability of intranasal diazepam in adults with epilepsy. Epilepsia. 2014;55:1544–50. doi: 10.1111/epi.12755. [DOI] [PubMed] [Google Scholar]
- 5.Djupesland PG, Messina JC, Mahmoud RA. The nasal approach to delivering treatment for brain diseases: an anatomic, physiologic, and delivery technology overview. Ther Deliv. 2014;5:709–33. doi: 10.4155/tde.14.41. [DOI] [PubMed] [Google Scholar]
- 6.Masserini M. Nanoparticles for brain drug delivery. ISRN Biochem. 2013;2013:18. doi: 10.1155/2013/238428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gizurarson S. Anatomical and histological factors affecting intranasal drug and vaccine delivery. Curr Drug Deliv. 2012;9:566–82. doi: 10.2174/156720112803529828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thorne RG, Pronk GJ, Padmanabhan V, Frey WH. Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience. 2004;127:481–96. doi: 10.1016/j.neuroscience.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 9.Westin UE, Boström E, Gråsjö J, Hammarlund-Udenaes M, Björk E. Direct nose-to-brain transfer of morphine after nasal administration to rats. Pharm Res. 2006;23:565–72. doi: 10.1007/s11095-006-9534-z. [DOI] [PubMed] [Google Scholar]
- 10.Shingaki T, Hidalgo IJ, Furubayashi T, Katsumi H, Sakane T, Yamamoto A, et al. The transnasal delivery of 5-fluorouracil to the rat brain is enhanced by acetazolamide (the inhibitor of the secretion of cerebrospinal fluid) Int J Pharm. 2009;377:85–91. doi: 10.1016/j.ijpharm.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Patil SB, Sawant KK. Development, optimization and in vitro evaluation of alginate mucoadhesive microspheres of carvedilol for nasal delivery. J Microencapsul. 2009;26:432–43. doi: 10.1080/02652040802456726. [DOI] [PubMed] [Google Scholar]
- 12.Reis CP, Neufeld RJ, Ribeiro ANJ, Veiga F. Nanoencapsulation I. methods for preparation of drug-loaded polymeric NP. Nanomedicine Nanotech Biol Med. 2006;2:8–21. doi: 10.1016/j.nano.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Danhier F, Ansorena E, Silva JM, Coco R, Breton AL, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161:505–22. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 14.Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B. 2010;75:1–18. doi: 10.1016/j.colsurfb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 15.Lu JM, Wang X, Muller CM, Wang H, Lin PH, Yao Q, et al. Current advances in research and clinical applications of PLGA based Nanotechnology. Expert Rev Mol Diagn. 2009;9:325–41. doi: 10.1586/erm.09.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3:1377–97. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vergoni AV, Tosi G, Tacchi R, Vandelli MA, Bertolini A, Costantino L. Nanoparticles as drug delivery agents specific for CNS: in vivo biodistribution. Nanomedicine Nanotech Biol Med. 2009;5:369–77. doi: 10.1016/j.nano.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Feczko T, Toth J, Dosa G, Gyenis J. Influence of process conditions on the mean size of PLGA nanoparticles. Chem Eng Process. 2011;50:846–53. doi: 10.1016/j.cep.2011.05.006. [DOI] [Google Scholar]
- 19.Mainardes RM, Evangelista RC. PLGA nanoparticles containing praziquantel: effect of formulation variables on size distribution. Int J Pharm. 2005;290:7–144. doi: 10.1016/j.ijpharm.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 20.Sharma D, Maheshwari D, Philip G, Rana R, Bhatia S, Singh M, et al. Formulation and optimization of polymeric nanoparticles for intranasal delivery of lorazepam using Box-Behnken design: in vitro and in vivo evaluation. Biomed Res Int. 2014 doi: 10.1155/2014/156010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Budhian A, Siegel SJ, Winey KI. Haloperidol-loaded PLGA nanoparticles: systematic study of particle size and drug content. Int J Pharm. 2007;336:367–75. doi: 10.1016/j.ijpharm.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 22.Song X, Zhao Y, Hou S, Xu F, Zhao R, He J, et al. Dual agents loaded PLGA nanoparticles: systematic study of particle size and drug entrapment efficiency. Eur J Pharm Biopharm. 2008;69:445–53. doi: 10.1016/j.ejpb.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 23.Songa KC, Lee HS, Chounga IY, Choa KI, Ahnb Y, Choi EJ. The effect of type of organic phase solvents on the particle size of poly(d, l-lactide-co-glycolide) nanoparticles. Colloids Surf A. 2006;276:162–7. doi: 10.1016/j.colsurfa.2005.10.064. [DOI] [Google Scholar]
- 24.Hao J, Fang X, Wang J, Guo F, Li F, Peng X. Development and optimization of solid lipid nanoparticle formulation for ophthalmic delivery of chloramphenicol using a Box-Behnken design. Int J Nanomedicine. 2011;6:683–92. doi: 10.2147/IJN.S17386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seju U, Kumar A, Sawant KK. Development and evaluation of olanzapine-loaded PLGA nanoparticles for nose-to-brain delivery: in vitro and in vivo studies. Acta Biomater. 2011;7:4169–76. doi: 10.1016/j.actbio.2011.07.025. [DOI] [PubMed] [Google Scholar]
- 26.Fessi et al. Process for the preparation of dispersible colloidal systems of a substance in the form of nanoparticles. US Patent 1992; 5,118,528.
- 27.Bilati U, Emann EA, Doelker E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur J Pharm Sci. 2005;24:67–75. doi: 10.1016/j.ejps.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 28.Hornig S, Heinze T, Becer CR, Schubertb US. Nanoprecipitation and nanoformulation of polymers: from history to powerful possibilities beyond poly(lactic acid) Soft Matter. 2011;7:1581–8. doi: 10.1039/C0SM00862A. [DOI] [Google Scholar]
- 29.USP30-NF25, Diazepam, 1912.
- 30.Semete B, Booysen L, Lemmer Y, Kalombo L, Katata L, Jan V, et al. In vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems. Nanomedicine Nanotech Biol Med. 2010;6:662–71. doi: 10.1016/j.nano.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 31.El-Ansary A, Al-Daihan S. On the toxicity of therapeutically used nanoparticles: an overview. J Toxicol 2009; 754810. doi:10.1155/2009/754810. [DOI] [PMC free article] [PubMed]
- 32.Lei R, Wu C, Yang B, Ma H, Shi C, Wang Q, et al. Integrated metabolomic analysis of the nano-sized copper particle-induced hepatotoxicity and nephrotoxicity in rats: a rapid in vivo screening method for nanotoxicity. Toxicol Appl Pharmacol. 2008;232:292–301. doi: 10.1016/j.taap.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 33.Campos EVR, Melo NFSD, Guilherme VA, Paula ED, Rosa AH, ArauJo DRD, et al. Preparation and characterization of poly(ε-Caprolactone) nanospheres containing the local anesthetic lidocaine. J Pharm Sci. 2013;102:215–26. doi: 10.1002/jps.23350. [DOI] [PubMed] [Google Scholar]
- 34.Mathew A, Takahiro F, Yutaka N, Takashi H, Hisao M, Yasuhiko Y, et al. Curcumin loaded-PLGA nanoparticles conjugated with Tet-1 peptide for potential use in Alzheimer’s disease. PLoS ONE. 2012;12(7):e32616. doi: 10.1371/journal.pone.0032616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snehalatha M, Kolachina V, Saha RN, Babbar AK, Sharma N, Sharma RK. Enhanced tumor uptake, biodistribution and pharmacokinetics of etoposide loaded nanoparticles in Dalton’s lymphoma tumor bearing mice. J Pharm Bioallied Sci. 2013;5:38–45. doi: 10.4103/0975-7406.120081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vyas TK, Babbar AK, Sharma RK, Misra A. Intranasal mucoadhesive microemulsions of zolmitriptan: preliminary studies on brain-targeting. J Drug Target. 2005;13:317–24. doi: 10.1080/10611860500246217. [DOI] [PubMed] [Google Scholar]
- 37.Kumar M, Misra A, Babbar AK, Mishra AK, Mishra P, Pathak K. Intranasal nanoemulsion based brain targeting drug delivery system of risperidone. Int J Pharm. 2008;358:285–91. doi: 10.1016/j.ijpharm.2008.03.029. [DOI] [PubMed] [Google Scholar]
- 38.Kumar M, Misra A, Mishra AK, Mishra P, Pathak K. Mucoadhesive nanoemulsion-based intranasal drug delivery system of olanzapine for brain targeting. J Drug Target. 2008;16:806–14. doi: 10.1080/10611860802476504. [DOI] [PubMed] [Google Scholar]
- 39.Alam S, Khan ZI, Mustafa G, Kumar M, Islam F, Bhatnagar A, et al. Development and evaluation of thymoquinone- encapsulated chitosan nanoparticles for nose-to-brain targeting: a pharmacoscintigraphic study. Int J Nanomedicine. 2012;7:5705–18. doi: 10.2147/IJN.S35329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panyam J, Williams D, Dash A, Leslie-Pelecky D, Labhasetwar V. Solid-state solubility influences encapsulation and release of hydrophobic drugs from PLGA/PLA nanoparticles. J Pharm Sci. 2004;93:1804–14. doi: 10.1002/jps.20094. [DOI] [PubMed] [Google Scholar]
- 41.Hanson LR, Frey WH. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008;9:1–4. doi: 10.1186/1471-2202-9-S3-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Averineni RK, Shavi GV, Gurram AK, Deshpande PB, Arumugam K, Maliyakkal N, et al. PLGA 50:50 nanoparticles of paclitaxel: development, in vitro anti-tumor activity in BT-549 cells and in vivo evaluation. Bull Mater Sci. 2012;35:319–26. doi: 10.1007/s12034-012-0313-7. [DOI] [Google Scholar]
- 43.Nah JW, Y-Il J, Koh JJ. Drug release from nanoparticles of poly(dl-lactide-co-glycolide) Korean J Chem Eng. 2006;17:230–6. doi: 10.1007/BF02707148. [DOI] [Google Scholar]
- 44.Ivaturi V, Kriel R, Brundage R, Gordon L, Mansbach H, Cloyd J. Bioavailability of intranasal vs. rectal diazepam. Epilepsy Res. 2013;103:254–61. doi: 10.1016/j.eplepsyres.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 45.Agarwal SK, Kriel RL, Brundage RC, Ivaturi VD, Cloyd JC. A pilot study assessing the bioavailability and pharmacokinetics of diazepam after intranasal and intravenous administration in healthy volunteers. Epilepsy Res. 2013;105:362–7. doi: 10.1016/j.eplepsyres.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 46.Betancourt T, Brown B, Brannon-Peppas L. Doxorubicin-loaded PLGA nanoparticles by nanoprecipitation: preparation, characterization and in vitro evaluation. Nanomedicine. 2007;2:219–32. doi: 10.2217/17435889.2.2.219. [DOI] [PubMed] [Google Scholar]
- 47.Sharma D, Gabrani R, Sharma SK, Ali J, Dang S. Development of midazolam loaded PLGA nanoparticles for treatment of status epilepticus. Adv Sci Lett. 2014;20:1526–30. doi: 10.1166/asl.2014.5520. [DOI] [Google Scholar]
- 48.Prajapati RK, Mahajan HS, Surana SJ. PLGA based mucoadhesive microspheres for nasal delivery: in vitro/ex vivo studies. Indian J Novel Drug Deliv. 2011;3:9–16. [Google Scholar]
- 49.Quintanar-Guerrero D, Allemann E, Fessi H, Doelker E. Preparation techniques and mechanisms of formation of biodegradable nanoparticles from preformed polymers. Drug Dev Ind Pharm. 1998;24:1113–28. doi: 10.3109/03639049809108571. [DOI] [PubMed] [Google Scholar]
- 50.Sylaja B, Srinivasan S. Experimental and theoretical investigation of spectroscopic properties of diazepam. Int J Chem Tech Res. 2012;4:361–76. [Google Scholar]
- 51.Mainardes RM, Gremiao MPD, Evangelista RC. Thermoanalytical study of praziquantel-loaded PLGA nanoparticles. Braz J Pharm Sci. 2006;42:523–30. [Google Scholar]
- 52.van Woensel M, Wauthoz N, Rosière R, Amighi K, Mathieu V, Lefranc F, et al. Formulations for intranasal delivery of pharmacological agents to combat brain disease: a new opportunity to tackle GBM? Cancers. 2013;5:1020–48. doi: 10.3390/cancers5031020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mukerjee A, Vishwanatha JK. Formulation, characterization and evaluation of curcumin-loaded PLGA nanospheres for cancer therapy. Anticancer Res. 2009;29:3867–76. [PubMed] [Google Scholar]
- 54.Sharma G, Mishra AK, Mishra P, Misra A. Intranasal cabergoline: pharmacokinetic and pharmacodynamic studies. AAPS PharmSciTech. 2009;10:1321–30. doi: 10.1208/s12249-009-9329-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Babu RJ, Dayal PP, Pawar K, Singh M. Nose-to-brain transport of melatonin from polymer gel suspensions: a microdialysis study in rats. J Drug Target. 2011;19:731–40. doi: 10.3109/1061186X.2011.558090. [DOI] [PMC free article] [PubMed] [Google Scholar]