

Abstract
Nephrolithiasis is a highly prevalent disorder affecting approximately one in eleven people and is associated with multiple complications including hypertension, cardiovascular disease, and chronic kidney disease. Significant epidemiologic associations with chronic kidney disease and ESRD have been noted and are reviewed herein, but debate persists in the literature as to whether kidney stone formation is a pathogenic process contributing to kidney disease. Corroborating evidence supporting the presence of kidney disease in stone formers includes the variability of renal function by stone type, the positive association of stone size with renal dysfunction, the presence of markers of renal injury in the urine of even asymptomatic stone formers, and direct evidence of renal tissue injury on histopathology. Proposed pathogenic mechanisms include recurrent obstruction and comorbid conditions such as recurrent urinary tract infections and structural abnormalities. Recent work evaluating the renal histopathology of different groups of stone formers adds further granularity, suggesting variability in mechanisms of renal injury by stone type and confirming the pathogenic effects of crystal formation. Genetic abnormalities leading to stone formation including cystinuria and primary hyperoxaluria, among others, contribute to the burden of disease in the stone-forming population.
Keywords: kidney stone, chronic kidney disease (CKD), histopathology
For many years nephrolithiasis has been viewed as a highly unpleasant nuisance by patients and doctors alike, but there had been little concern regarding long-term ramifications. Over the last several decades, however, there has been an increasing appreciation for the association of nephrolithiasis with negative long-term outcomes including cardiovascular morbidity1, 2, metabolic disturbances3, and renal complications including chronic kidney disease (CKD) and End Stage Renal Disease (ESRD)4–7. Guilt by association, however, places nephrolithiasis in a nebulous category – is it simply a bother, a risk factor for other disease states, or is kidney stone formation itself a disease? In the ensuing pages we will consider these issues through the panoramic lens of epidemiology, reviewing data supporting the relationship between nephrolithiasis and impaired kidney function, as well as through the microscopic lens of histopathology, potentially shedding light on the mechanisms leading to kidney injury and dysfunction.
Evidence for Impairment of Normal Kidney Function
While classically renal dysfunction has been thought of as a decrement in the glomerular filtration rate (GFR), renal disease states may present with normal GFR but an abnormality in one of its other functions such as maintenance of blood pressure (through salt and water handling and hormonal regulation) or maintenance of acid /base homeostasis. For example, patients with a renal tubular acidosis may have a normal GFR but have an inability to maintain acid/base homeostasis. Halperin et al8 have proposed that the human kidney is designed to maintain systemic acid-base balance while maintaining the “ideal” urine pH of 6 to prevent crystallization within the kidney. Within this framework, any stone former has failed the test of normal renal function, as stone formation has resulted from a failure of the kidney to prevent crystallization.
More recently, international guidelines9, 10 have expanded the definition of chronic kidney disease (CKD) from simply a decreased GFR to the presence of any of the following for more than 3 months: eGFR<60 ml/min/1.73 m2, albuminuria, urine sediment abnormalities, electrolyte abnormalities due to tubular disorders, structural abnormalities detected by imaging, or history of kidney transplantation, as these have been shown to be predictive of downstream complications. For the purposes of our discussion, we will focus on kidney stone formation as a parenchymal disease of the kidney that may be associated with decrements in GFR, rather than simply a nuisance within the urological tract.
The aforementioned guidelines specifically include abnormal histopathological findings in the renal parenchyma as its own category defining CKD, which, as we will demonstrate, is not an uncommon finding in stone-forming patients11–24. Furthermore, recent data highlight the increased prevalence of albuminuria and renal scarring even in asymptomatic stone formers – both considered diagnostic for CKD25. In healthy subjects being evaluated for kidney donation at the Mayo clinic, subjects noted to have asymptomatic kidney stones on computerized tomography (CT) imaging were significantly more likely than donors without a stone to have renal parenchymal thinning and focal scarring25. Furthermore, among subjects who had previously had a symptomatic stone event, 13% had evidence of albuminuria of >30 mg/24 hr, compared to 3.5% and 3.6% of subjects with no stone disease and asymptomatic stone disease, respectively.
While albuminuria is suggestive of glomerular injury, markers of tubular injury are also elevated in patients in nephrolithiasis. Sun et al26 have shown that in a series of 60 stone formers urinary angiotensinogen is significantly higher than in control subjects and is negatively correlated with eGFR. Urinary angiotensinogen concentrations are a marker of intrarenal angiotensin II levels, a critical modulator of renal injury via its role in potentiating glomerular capillary hypertension27, and activation of signaling pathways associated with inflammation, generation of reactive oxygen species, and endothelial dysfunction28. Urinary angiotensinogen levels were also significantly correlated with urinary α1-microglobulin, a marker of proximal tubular injury which is believed to be one of the earliest markers of tubular dysfunction29. Urinary excretion of 8-hydroxydeoxyguanosine (8-OHdG), a marker of oxidative DNA damage associated with glomerular and interstitial fibrosis30, is also increased in stone formers31. Furthermore, immunohistochemical analysis demonstrates increased 8-OHdG expression in the renal tissue adjacent to stone in subjects with nephrolithiasis32.
Epidemiologic Data
The earliest study to note the relationship between nephrolithiasis and chronic kidney disease in the general population was performed by Vupputuri et al33. In their case-control study performed in North Carolina, the authors queried 548 patients with newly diagnosed CKD (defined as a creatinine greater than 1.5 mg/dL) and 514 age, sex, and race-matched community dwelling adults without kidney disease regarding history of nephrolithiasis. Among patients, 16.8% of subjects reported a diagnosis of a kidney stone, compared to 6.4% of controls. After adjustment for comorbidities, CKD was nearly twice as likely (OR 1.9) in those with a history of nephrolithiasis.
The following year Gillen et al34 demonstrated that the relationship between estimated GFR and kidney stone history in more than 15,000 subjects (6% stone formers) from the Third National Health and Nutrition Examination Survey (NHANES III) was dependent on weight. Generally, those with a history of kidney stones were more likely to be older, non-African American, male, and have a history of coronary artery disease. After adjustment for potential confounders, overweight stone formers (BMI>27) had a mean eGFR 3.4 ml/min/1.73m2 lower than their non-stone forming counterparts. This difference was not noted in those with a BMI<27.
More recent data by Shoag et al using the NHANES 2007–2010 database confirms the association between kidney stones and CKD35. Among 5971 NHANES participants with data on stones and kidney function, 521 subjects admitted to a history of nephrolithiasis. In a multivariate analysis, history of stone disease was strongly associated with CKD (OR 1.5) and dialysis requirement (OR 2.37) in the cohort. Notably, this association appeared driven by women (OR 1.76 for CKD, OR 3.26 for dialysis), as it was not noted in men.
Stankus et al36 compared the incidence of pre-ESRD kidney stones in a cohort of African American hemodialysis patients. In the sample of 300 subjects, 8.2% had a history of nephrolithiasis prior to initiating dialysis, compared to 2.8% of patients matched for age, sex, and race in the NHANES III cohort. Generally, patients of African descent are at a lower risk for kidney stone disease37, but at a higher risk for ESRD38. This data suggest that those already at risk for CKD may have an additive burden in the face of nephrolithiasis.
Several studies out of the Mayo clinic shed further light on the association between stone disease and CKD. In a nested case-control study in residents of Olmstead County, Saucier et al39 compared the characteristics of stone formers with CKD (n=53) and those without CKD (n=106). Predictably, subjects with CKD were more likely to have diabetes (OR 4.27) and hypertension (OR 3.57), as well as recurrent UTIs (OR 5.81). CKD sufferers were also more likely to have had an ileal conduit (OR 7.69) and to have had a documented struvite stone (OR 15.61). Obesity and smoking were also more frequent in the CKD group, but this did not reach statistical significance.
Concurrently, the Mayo group also reported on a much larger population-based cohort study, identifying all incident stone formers (n=4066) in Olmstead County diagnosed between 1986 and 2003 and matched to control subjects (n=10,150) from the local area40. Incidence of CKD was determined using both diagnostic codes and lab confirmation of decreased GFR (eGFR<60ml/min/1.73 m2) sustained ≥ 3 months. Stone formers were more likely to have underlying hypertension, gout, diabetes, obesity, and coronary artery disease, but even in analyses adjusted for these CKD risk factors, risk of clinical CKD was 50–67% higher in stone formers than in controls.
Most recently, the Mayo group focused on the association of urolithiasis with ESRD41. The authors identified newly diagnosed stone formers in Olmstead County between 1984 and 2008 and matched each individual by age and sex to up to four controls without stone disease. After adjusting for classic cardiovascular risk factors such as diabetes, gout, obesity, hyperlipidemia, and, notably, CKD, stone formers had more than twice the risk of developing ESRD compared to controls (HR 2.09). The findings were similar (HR 1.95) in a subset of patients who were validated to be symptomatic stone formers by chart review.
The association between stone disease and renal dysfunction does not appear to be limited to the United States. In a registry cohort study of adults living in Alberta, Canada between 1997–2009 encompassing nearly two million subjects, a significant association between one or more episodes of kidney stones and development of new stage 3b-5 CKD (HR 1.74), ESRD (HR 2.16) or a sustained doubling of serum creatinine from baseline was noted (HR 1.94) over a median follow up of 4 years42. Across the Atlantic, in a large British population-based cohort study of over 1.5 million designed to develop a prediction model for risk of moderate-to-severe CKD43, the adjusted risk for moderate-to-severe CKD was 1.27 in females, but not males, with a history of nephrolithiasis. The adjusted risk for ESRD was double in females with a history of stone disease (HR 2.1), but the association was not present in males.
Despite these strong associations, however, it is important to note that only 0.2% of incident ESRD patients from 2004–2008 had nephrolithiasis listed as a primary etiology on the CMS ESRD Medical Evidence Form44. The complications of CKD should not be discounted, however, as the associations between CKD and cardiovascular disease and mortality are well described4–7. Furthermore, while nephrolithiasis may not be the primary reason for an individual’s ESRD, evidence of distorted renal architecture in stone formers11–24 could be conceived to predispose to injury from other causes.
Is There a Dose Effect?
If kidney stones are to be linked to a decrement in kidney function, one might expect that greater stone burden would be inversely associated with renal function. This is indeed what Ahmadi et al45 noted in their study of 97 kidney stone formers presenting for ESWL. In patients with a total stone burden of <20 mm, each 1 mm increment in cumulative stone size (as determined by the sum of diameters of all stones present) was associated with a 20% increased risk of having chronic kidney disease in a multivariate model. This is particularly striking as urine calcium levels markedly decline with even a small decline in renal function46. Thus in the case of calcium stones, it would be likely that the stone formation preceded the decline in GFR, though certainly causality cannot be established by these data.
Determinants of Decreased Renal Function
Kidney loss
Historically, much of the ESRD ascribed to stone disease has been attributed to patients with solitary kidneys. In an elegant study by Jungers et al47, the authors reviewed the histories of 1391 consecutive patients with ESRD, of which 45 (3.2%) were attributed to nephrolithiasis. Struvite stones were noted in 19, calcium stones were noted in 12, and uric acid stones in eight, with the remainder due to hereditary stone disease (four with primary hyperoxaluria and two with cystinuria). Interestingly, 40% of subjects in this study had a solitary kidney prior the development of ESRD.
Worcester et al48 characterized the etiology for presentation with a solitary kidney in a cohort of 115 patients with nephrolithiasis and a single kidney evaluated at the University of Chicago. The most common cause for loss of a kidney in this group of stone formers was a staghorn calculus or high stone burden (29%), followed by infection (23%), ureteral obstruction (21%), and surgical removal (8%). It should be noted, however, that with advances in surgical techniques, loss of a kidney due to stone disease is likely becoming significantly less common.
Idiopathic stones versus stones associated with systemic disease
If the majority of the burden of kidney disease is not in those with a solitary kidney, is there evidence that renal function is not normal in the routine stone former? Worcester et al49 compared pretreatment CrCl values by stone type from 1856 stone formers presenting for their initial evaluation at the University of Chicago. Adjusted for anthropometric measures, CrCl was significantly lower in cystine stone formers compared to all others, while hydroxyapatite, calcium oxalate, and struvite stone formers had significantly lower CrCl compared to normal subjects. Notably, the mean CrCl values for all stone formers were essentially in the normal range (above 100 ml/min), but the discrepant values by stone type may point to variability in the injury mechanism. Similarly, when grouped by etiology of disease leading to kidney stones, patients with nephrolithiasis linked to bowel disease, bariatric surgery, and renal tubular acidosis had lower creatinine clearance than normal subjects.
Mechanisms of Renal Injury
Recurrent obstruction
A reasonable mechanism for renal injury in patients with stone disease is recurrent obstruction. While most often this represents an acute kidney injury (AKI), it is now clear that recurrent AKI episodes are a risk factor for development of CKD50. Being unilateral in most cases, stone passage may not produce obvious signs of kidney injury in patients with two kidneys. For example, GFR is not likely to fall and sodium retention may not occur unless the contralateral kidney has CKD and is unable to compensate. Notably, with only one kidney, passage of a stone produces an obvious fall in renal function, sometimes anuria with marked renal failure.
Animal models indicate that ureteral obstruction can lead to renal injury via several mechanisms. Increased intratubular pressure may lead to significant renal vasoconstriction, resulting in a drop in renal blood flow, reduction in GFR, and tissue ischemia51, 52. Persistence of the ischemic state can lead to glomerulosclerosis, tubular atrophy, and subsequent interstitial fibrosis53, 54. In rodent models, complete unilateral obstruction for 24 hours leads to irreversible dysfunction in 15% of the nephrons in the kidney55. Little effect on the overall kidney function would be noted at first, but with repeated episodes it is plausible that the remaining nephrons have to increase their single nephron GFR, with resultant hyperfiltration and potential subsequent injury. In a large human case series of 2073 ureteral stones, Wang et al found the incidence of AKI to be 0.72% (15 patients), with eight of those patients having a unilateral stone56. Of those eight subjects, however, seven had a single functioning renal unit, and the remaining subject had underlying CKD.
Associated diseases
In the Mayo study, risk factors identified among stone formers who developed CKD were hypertension and diabetes, diseases associated with both CKD and stone formation. Recurrent UTIs, struvite stones, and allopurinol therapy were other risk factors noted39.
Decreased renal function: importance of stone type and histology
While the epidemiological data certainly indicate a relationship between stone formation and a decrement in kidney function, they are limited by the heterogeneous populations included in the study samples and hampered by use of billing records and diagnostic codes. Additional limitations include the high prevalence of comorbid conditions such as diabetes and metabolic syndrome, as well as the retrospective nature of nearly all the current work57. In recent years, however, our group has pioneered advanced surgical and histopathological techniques to evaluate potential mechanisms of stone formation. These studies, performed on tissues obtained during percutaneous nephrolithotomy (PCNL) procedures, have revealed a surprising amount of injury in the renal papillae and cortices of many stone formers. It is plausible, however, that the degree of injury is accentuated by the selection bias of obtaining samples from stone formers with a high stone burden requiring PCNL.
Calcium Oxalate (CaOx)
Multiple etiologies exist for CaOx stone formation ranging from idiopathic hypercalciuria, to complications of bowel surgery, to inborn errors of metabolism. While each disorder leads to the clinical phenotype of the CaOx stone former, recent investigation suggests that the mechanisms of stone formation vary by the underlying etiology, and are associated with strikingly different papillary histopathology.
The most common stones are idiopathic CaOx stones, which are not associated with systemic disease, but may be associated with a number of metabolic abnormalities such as idiopathic hypercalciuria, hypocitraturia, or dietary hyperoxaluria. Stones form as overgrowths on interstitial deposits of calcium phosphate that have come to be known as Randall’s plaque, after Alexander Randall, who first described them. Histopathogical analyses going back to the time of Randall58 suggest that little interstitial inflammation is present surrounding these deposits (Figure 1, panel A). More recently, our group has confirmed the absence of overt inflammation or tissue injury in the interstitial compartment surrounding these deposits in the idiopathic calcium oxalate stone former15, 20, 22.
Figure 1.
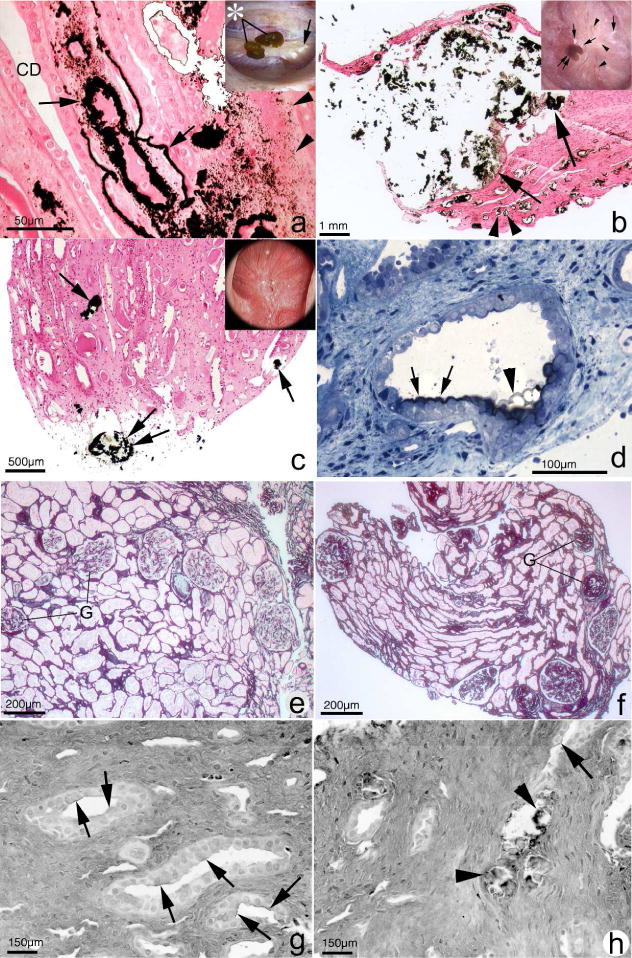
Histopathology of renal papilla and cortex from CaOx stone formers. Papillary tissue of ICSF patients (panel A) shows initial sites of individual calcium phosphate deposits (arrowheads) by Yasue stain (brown-black deposits) in the basement membranes of thin loops of Henle when viewed by light microscopy. These individual deposits coalesce in the interstitial space to form islands of mineral encased in matrix material (arrows) that can extend to the basal side of the urothelium. No evidence of cell injury or inflammation has been noted in the collecting ducts (CD) or interstitium of these papilla. Endoscopic examination of the papilla of ICSF patients reveals small stones (insert panel A, asterisk) attached to the papilla by way of white or interstitial plaque (arrow). In addition to sites of interstitial plaque (panel B, double arrowhead), the papilla of patients with small bowel resection show intraluminal plugs of varying size in the IMCD (arrows). These deposits contained a mixture of CaOx, apatite and ammonium acid urate. Note the extensive interstitial fibrosis surrounding the IMCD plug. These patients also have attached stones (insert panel B, double arrow) at sites of white plaque (arrows). Areas of yellow plaque are also seen (arrowheads). Papilla of intestinal bypass patients for obesity possesses little to no interstitial plaque (panel C) but have a few intraluminal plugs in IMCD (arrow) and ducts of Bellini (double arrow) composed of apatite and CaOx. The insert in panel C shows a dilated opening to a BD (arrowhead). The initial layer of mineral to coat the apical surface of the lining cells of IMCD of these patients is Yasue positive (panel D, arrows) and particularly clear at the arrowhead. Panel E shows a cortical biopsy from an ICSF patient revealing only minimal interstitial fibrosis and glomerular (G) changes. A moderate level of change is seen panel F from a patient with small bowel resection. Panels G and H show sites of hyaluronan staining (arrows) in papillary tissue from intestinal bypass patients revealing areas of cell surface changes at sites of crystal deposits (panel H, arrowheads) or away from deposits (panel G).
In contrast, patients with CaOx stones stemming from other causes have calcium deposits in Bellini ducts (BD) and inner medullary collecting ducts (IMCD) which are associated with significant inflammatory changes and evidence of subsequent fibrosis. For example, patients with small bowel resections with subsequent CaOx nephrolithiasis have been shown to have the interstitial deposits classically seen in idiopathic CaOx stone formers, but also to have IMCD plugging with calcium phosphate21 (Figure 1, panel B). As in the idiopathic CaOx stone formers, no inflammation is seen in areas of interstitial plaque despite substantial interstitial plaque burden, but significant inflammation and fibrosis was noted surrounding the plugged IMCD. Perhaps more notably, cortical changes including moderate to severe glomerular sclerosis and mild to moderate interstitial fibrosis were noted in the majority of studied patients21 (Figure 1, panel F), whereas the cortices of idiopathic stone formers were without abnormalities (Figure 1, panel E).
Not all patients with nephrolithiasis related to bowel pathology show the same histopathological characteristics. Patients with CaOx nephrolithiasis as sequela of bariatric bypass procedures show IMCD plugging with the inflammation and fibrosis similar to those with small bowel resections18 (Figure 1, panel C). However, these patients do not show evidence of Randall’s plaque. Notably, some bypass patients had tiny focal crystal deposits within IMCD lumens (Figure 1, panel D), seemingly attached to cell membranes, whereas all patients demonstrated intratubular IMCD plugging. In addition, hyaluronan expression, a marker of cell injury, was noted in the IMCD tubule epithelium of tubules that were not involved with crystal deposits (Figure 1, panel G), suggesting that injury mediators may have influenced cells far away from actual crystal plugs18 (Figure 1, Panel H).
Mechanisms of crystal induced tubular damage have been pursued in animal models which most resemble primary hyperoxaluria. In these models, injury is thought to be either direct by crystal induced cellular death or indirect, via the inflammatory cascade59. Crystals stimulate innate immunity via the NACHT, LRR, and PYD domains-containing-protein (NLRP)3 inflammasome59. CaOx monohydrate (COM) crystals attach to MDCK cells60. Once internalized, the crystals interfere with proteins responsible for energy production, resulting in the increased production of reactive oxygen species which are believed to stimulate inflammation61. Of note, oxalate ion itself, in high concentrations, has been associated with adverse biologic effects and loss of cell viability in in-vitro experiments with various renal tubular cell lines,62, 63 but relevance of these findings to human disease is unknown at this time.
Calcium Phosphate
Calcium phosphate stones come in two varieties: brushite (BR) and hydroxyapatite (HA). Data from our group demonstrate that both stone types are associated with BD tubular plugging, as well as variable stone growth over plaque22 (Figure 2, Panels A and B). BD can be dilated nearly 20-fold. Epithelium of plugged tubules is injured and in many cases lost altogether so that crystals reach the basement membrane. Peritubular fibrosis is invariable. Cortical injury is present in both HA and BR calcium phosphate stone formers at levels above what is found in ICSF (Figure 2, panels E and F).
Figure 2.
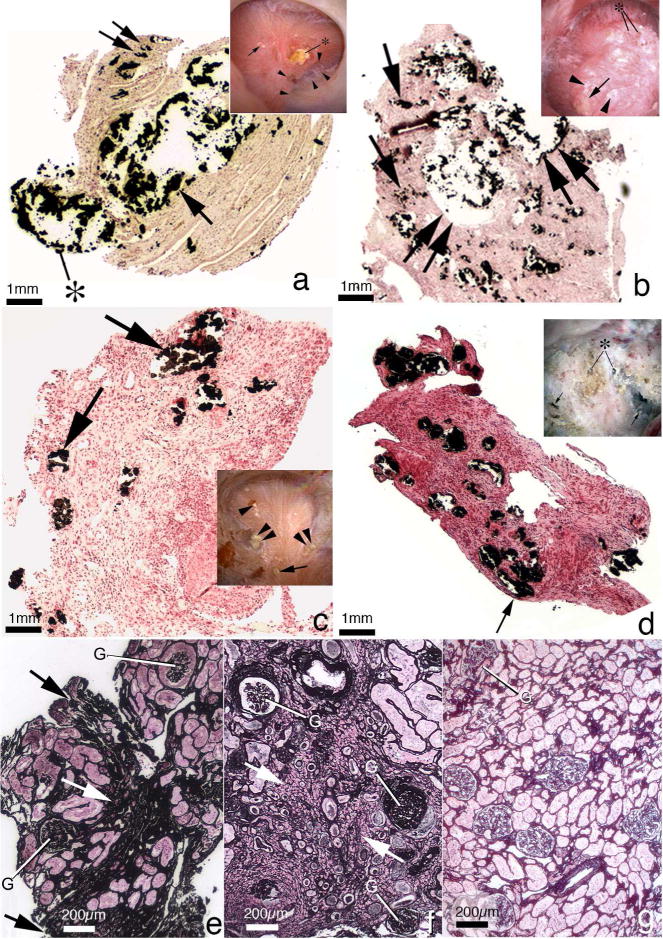
Histopathology of renal papilla and cortex from apatite and brushite stone formers. Panel A is a low magnification light microscopic image of a renal papilla from a brushite stone former showing an enormous amount of Yasue-positive deposits (arrow) filling a grossly dilated IMCD and BD. The crystalline material is protruding (asterisk) from the dilated opening of a BD. Sites of interstitial plaque are noted through out papilla (double arrow). Insert to panel A shows endoscopic view of papillum from brushite patients characterized by areas of white plaque (arrows), yellowish crystalline material protruding from dilated BD (asterisk), pitting (arrowheads) and sites of yellow plaque. Panel B is a histologic section from an apatite stone former with large fields of interstitial plaque termed “novel interstitial plaque structures” (NIPS, arrows) located near greatly dilated IMCD and BD filled with Yasue-positive deposits (double arrows) which are hydroxyapatite. Extensive regions of interstitial fibrosis surround the dilated IMCD and NIPS. The insert in panel B shows endoscopic view of a papilla from an apatite stone former characterized by sites of white plaque (arrowheads), protruding plugs for dilated opening of BD (asterisk) and an occasional site of yellow plaque (arrow). Panel C is a histologic section from a patient with primary hyperparathyroidism with stones. These patients show numerous IMCD with apatite plugs (arrows), extensive interstitial fibrosis at sites of intraluminal plugs and areas of interstitial plaque. The insert in panel C shows an endoscopic view of a papilla from a patient with primary hyperparathyroidism characterized by attached stone to white plaque, sites of yellow plaque (double arrows), interstitial plaque (arrowhead), and dilated BD with protruding plug (double arrowhead). Panel D is a histologic section from a patient with distal renal tubular acidosis presenting with diffuse IMCD plugging (arrows), severe and diffuse interstitial fibrosis and little to no interstitial plaque. The insert to panel D shows endoscopic view of a papilla from a patient with distal renal tubular acidosis characterized extensive papillary damage with retraction, loss of normal architecture, multiple dilated BD that produce a pitted appearance (arrows), some with protruding mineral deposits (asterisks) and a whitish thickened membrane appearance. Panel E shows a cortical biopsy from an apatite stone former revealing a moderate level of interstitial fibrosis (arrow) and glomerular (G) changes while panel F shows a severe level of interstitial fibrosis (arrow) and glomerular (G) change in a brushite stone former. Panel H shows a cortical biopsy from a patient with distal renal tubular acidosis revealing only a minimal level of change.
While the most common stone type in patients with primary hyperparathyroidism is CaOx, as in the general population, the proportion of patients with calcium phosphate stone disease is much higher in those with the endocrine disorder64. In our surgical biopsy series, patients with calcium phosphate stone disease stemming from hyperparathyroidism demonstrated findings similar to that of brushite stone formers with ICMD and BD plugging composed of apatite crystals and accompanied by loss of epithelial cells11 (Figure 2, panel C). There was surrounding peritubular interstitial inflammation and fibrosis, with significant retraction of papillae. However, the findings were less pronounced than those seen in BR stone disease and there was more variability in the degree of papillary distortion among patients. Patients with primary hyperparathyroidism demonstrated stone growth on Randall’s plaque, more often than idiopathic BR or HA patients64.
Another singular group of stone formers with calcium phosphate stones are those with distal tubular acidosis (dRTA). Characterized by calcium apatite stone formation and diffuse nephrocalcinosis, patients in our surgical series also demonstrated plugging of BD and IMCD16 like their counterparts without systemic illness, but the degree and distribution of plugging was far greater (Figure 2, panel D). The crystals within the plugs were HA and there was a marked degree of associated epithelial cell injury and loss. Furthermore, the degree of fibrosis surrounding the plugged tubules was significantly greater in those with dRTA, and was detected even in the absence of tubular plugging. It is worth noting that while there are genetic causes of dRTA, there is no reason to believe that the histological findings would differ.
Struvite
Struvite (magnesium ammonium phosphate) stones are formed exclusively in the presence of bacteria possessing the urease enzyme to hydrolyze urea to ammonia. The resultant high urine pH and ammonium concentrations predispose to the growth of struvite either alone or admixed with preexisting calcium stones65. Struvite stones are particularly hazardous to kidney function as they grow rapidly and can form staghorn stones filling the entire collecting system of the kidney, increasing the risk for significant obstruction. Large staghorn stones may also predispose the kidney to papillary necrosis.
Patients with recurrent urinary tract infections, instrumentation, anatomic abnormalities of the urinary tract, and urinary diversion are at particularly high risk for struvite stones and renal dysfunction66. However, even patients without such underlying risk factors who form struvite stones are at risk for reduced renal function. In a series of 75 patients with struvite stones from our center that excluded those with underlying anatomic abnormalities, 25 had serum creatinines that were more than 2 SD above normal for sex67. Twenty four of these patients had staghorn stones and 11 had undergone a nephrectomy. Creatinine clearance was significantly reduced in both men and women. The mechanism of injury is almost certainly obstruction and local injury, with loss of functional nephron mass.
Uric Acid
Uric acid nephrolithiasis, accounting for 8–10% of stone disease in the Western world, is becoming increasingly common as populations grow older and more obese68. Associated risk factors include diabetes mellitus type 2, hypertension, and obesity with the metabolic syndrome, which is known to be associated with low urinary pH68. Low urinary pH is a requirement for the precipitation of uric acid crystals.
Indirect evidence from epidemiologic data suggests that uric acid stone formers are particularly prone to the development of CKD. For example, in the report from Olmstead county39, patients with a documented uric acid stone were three times more likely to develop CKD, though this association was not significant. Subjects receiving allopurinol therapy, presumably for gout and with a likely predisposition to uric acid nephrolithiasis, were 10-fold more likely to develop CKD, which was highly significant. Of course, they may have also been receiving allopurinol due to hyperuricemia stemming from CKD. From a histopathological perspective, uric acid crystals are known to deposit in BD and IMCD69, whereas sodium mono-hydrogen urate crystals deposit in the renal interstitial space with associated inflammatory response and fibrosis70.
One group of patients particularly prone to uric acid stone disease is those with ileostomies. Due to excessive enteric losses of water and alkali, patients with ileostomies have acidic urine and low urine volumes, optimal conditions for uric acid crystallization. Interestingly, in our surgical series19 patients with ileostomies were also found to have BD and IMCD plugging with apatite, as well as interstitial apatite plaque, a constellation of findings more characteristic of those with intestinal bypass surgery.
Comparison of Histopathologic Findings with Other Series
Others have begun to explore papillary histopathology in a variety of stone formers. Linnes et al enrolled 78 consecutive stone formers undergoing PCNL at Mayo to evaluate the correlation of stone type to Randall’s plaque burden, tubular plugging, and metabolic parameters71. Generally, findings were similar to our group’s with the greatest amount of plaque burden in those with CaOx stone disease and the least in those with uric acid stones, while tubular plugging was greatest in those with brushite nephrolithiasis. Overall the amount of plaque seen in CaOx stone formers was less than seen in our cohort, but the study population was not selected for hypercalciuria as in our series and included subjects with primary hyperoxaluria, primary hyperparathyroidism, and bowel disease. More recently, the Mayo group compared the histopathology of patients with uric acid stones to those with CaOx disease69. As in our series, their subjects with UA stones had a high degree of IMCD plugging, despite a low percentage of patients (<10%) with small bowel resection.
Genetic Diseases with Nephrolithiasis and Kidney Disease
Primary Hyperoxaluria
A small portion of patients presenting with CaOx stones will have underlying Primary Hyperoxaluria (PH). A group of inborn metabolic disorders, the primary hyperoxalurias are characterized by liver enzyme deficiencies that result in overproduction of oxalate, which must be excreted through the kidney.
Patients typically present early in childhood, with 85% of patients diagnosed before age 2072. Recurrent stone formation throughout childhood and adolescence, along with nephrocalcinosis, is typical. The median age of progression to ESRD is 3373. While it has been widely accepted that CaOx crystallization can be harmful to cells74, 75, only recently the histopathological changes through the cycle of PH Type 1 from normal renal function to ESRD on dialysis have been demonstrated.
Evaluating differences in the patterns of injury between kidneys of PH1 patients with normal renal function (obtained during PCNL for stone), those just started on renal replacement therapy, and those on long term dialysis, Worcester et al75 demonstrated that while renal function is preserved, CaOx crystals in the cortical S1 and S2 proximal tubule segments are present but not accompanied by inflammation or scarring. Papillary findings early in the course of the disease are characterized by IMCD plugging with CaOx crystals and extensive interstitial fibrosis of the papillary tip (Figure 3, panels A and B). By the time the PH1 patient is started on dialysis, intratubular deposits of CaOx are predominately found at the proximal tubule’s S3 segment at the corticomedullary junction, as well as in cells and in the surrounding interstitial space. In advanced disease on long term dialysis, intratubular deposits were prevalent throughout the cortex.
Figure 3.
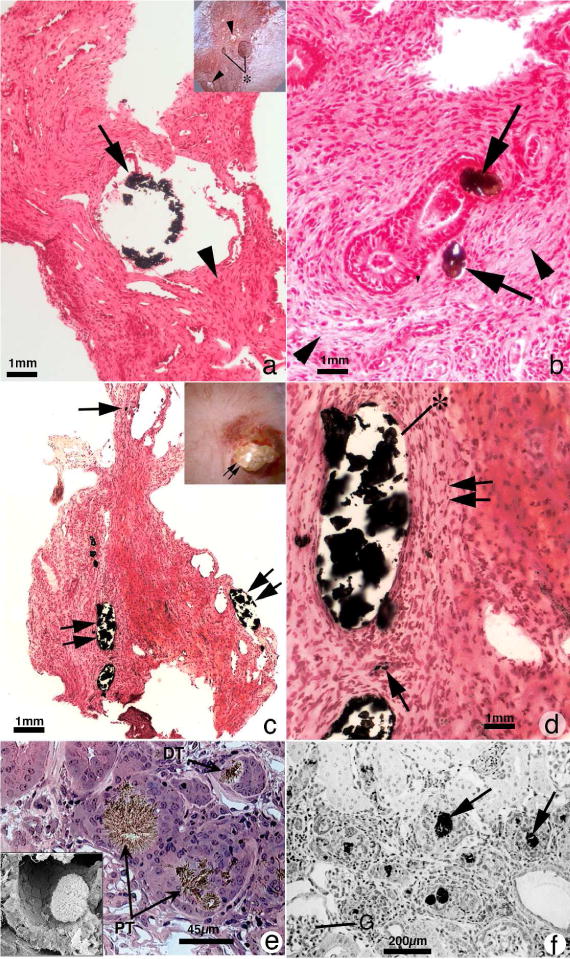
Histopathology of renal papilla from primary hyperoxaluria, cystine and DHA stone formers. Panel A is a histologic section from a primary hyperoxaluric stone former with normal renal function showing isolated dilated IMCD filled with CaOx mineral (arrow) encased in regions of extensive interstitial fibrosis (arrowhead). Sites of interstitial plaque were rare. Insert to panel A shows endoscopic view of papillum from a primary hyperoxaluric stone former characterized by a few dilated ducts of Bellini with and without (asterisk) protruding plugs were seen scattered between sites of yellow plaque (arrowheads). Panel B is a higher magnification Yasue-stained tissue section showing regions of extensive interstitial fibrosis adjacent to and away from (arrowheads) sites of intraluminal IMCD deposits. Panel C is a Yasue-stained histologic section from a patient with cystine stones revealing numerous dilated IMCD (double arrows) and BD plugged primarily apatite with a few BD containing cystine deposits. Occasionally a thin loop of Henle was noted plugged with apatite deposits (arrow). Panel D is a higher magnification image of a region from panel C showing extensive interstitial fibrosis (double arrow) adjacent to sites of intraluminal IMCD deposits (asterisk) and an occasional site of interstitial plaque (arrow). Panels E and F show the morphology of 2,8-dihydroxyadenine (DHA) crystal in kidneys from adenine phosphoribosyltranserase (Aprt)-deficient mice that resembles human Aprt deficiency. In panel E two proximal (PT, arrows) and a distal tubule (DT, arrow) is seen, each with large brownish-red crystals in the tubular lumens. Interstitial fibrosis is noted around glomeruli and various tubular segments. The insert in panel E shows by scanning electron microscopy an individual DHA crystal in the lumen of a proximal tubule. Panel F shows numerous DHA crystals (arrows) in the cortex of an Aprt-deficient mouse with advanced pathology (score of 4) characterized by interstitial fibrosis and glomerulosclerosis (G).
Cystinuria
A stone type commonly associated with kidney disease is cystine. While only 5% of patients with cystinuria progress to ESRD, up to 70% of patients develop evidence of renal dysfunction76. Biopsies from patients with cystine stones show a variety of histopathologies ranging from normal to tubular plugging, severe injury to BD, and plugging of the IMCD14 (Figure 3, panel C). Plugging within the BD is typically with cystine aggregates, while the loop of Henle and the IMCD are typically filled with HA crystals (Figure 3, panel D). The mechanism of injury is thought to be secondary to tubular obstruction with the secondary cell injury, interstitial inflammation, glomerular loss, and cortical fibrosis14.
Adenine Phosphoribosyltranferase (APRT) Deficiency and 2,8-dihydroxyadeninuria
APRT deficiency is a rare autosomal recessive inborn error of adenine metabolism that leads to accumulation of the very poorly soluble dihydroxyadenine (DHA)77. Prevalent in Japan, France, and Iceland, among others, this disorder resulting in DHA accumulation can lead to both discrete stone formation and a crystal mediated nephropathy78, 79 (Figure 3, panels E and F). Due to its similarity to uric acid stone disease, with radiolucent stones and a low index of suspicion, stones are often mistakenly identified as uric acid. This is problematic as early identification of the disorder with timely initiation of therapy with a xanthine oxidase inhibitor can prevent the onset of renal failure. Currently, APRT deficiency appears to be a highly unrecognized cause of kidney stones with CKD, which can progress to ESRD in a large proportion of affected patients that are untreated72. While recurrent obstruction, hematuria, and recurrent urinary tract infections are common presentations in children, adults are more likely to present with ESRD secondary to crystalline nephropathy78, 79. Several publications report cases of the diagnosis being made only after renal transplantation80–82.
Dent Disease
Dent disease is an X-linked disorder caused by mutations in either the CLCN5 or OCRL1 genes and is characterized by low molecular weight proteinuria and the presence of hypercalciuria, nephrolithiasis, or nephrocalcinosis72. Typically patients present in childhood with nephrocalcinosis, proteinuria, or unexplained CKD, with subsequent progression to CKD noted by the 3rd to 5th decade in 30–80% of male subjects83. Biopsy findings of nephrocalcinosis and interstitial fibrosis are typical in patients with Dent disease72. In recent years, a subset of patients with nephrotic range proteinuria and biopsy findings of focal segmental glomerulosclerosis (FSGS) has been identified84, 85, but the contribution of glomerular disease to renal dysfunction in Dent Disease remains unclear.
Conclusion
The posed question is whether patients with nephrolithiasis have a kidney disease? Using epidemiologic data, the answer is a resounding “yes” given the strong associations with CKD and ESRD.
A more nuanced argument requires a clearer definition of disease. If kidney disease is thought of as a decrement in GFR, indirect evidence using eGFR and creatinine clearance would suggest that some stone types can be thought of as disease states. For example, given the association of PH1, cystinuria, and ileostomy with stone disease with decreased GFR, these states can be thought of as having renal manifestations.
On a broader platform, if disease is defined as a tissue abnormality, nearly all stone formers, with perhaps the exception of idiopathic CaOx stone formers, can be thought of as afflicted.
Perhaps the bigger question in the age of personalized medicine is how we can use our current knowledge to better identify individual stone patients at risk for kidney complications downstream. Does every first time CaOx stone former have a disease? How can we best utilize the metabolic data typically collected during evaluation, in conjunction with surgical imaging, to identify those at risk for CKD down the line? Do histological abnormalities seen in the kidneys of some stone formers portend the same associated risk factors of cardiovascular and bone disease as that in patients with CKD from other causes? Unfortunately at this time, we have more questions than answers – though without a doubt the answer to the question of kidney stone formers having a disease is certainly “yes.”
Acknowledgments
NIH P01 DK 56788
Footnotes
Disclosures: No disclosures relevant to the subject matter of this review
Reference List
- 1.Ferraro PM, Taylor EN, Eisner BH, et al. History of kidney stones and the risk of coronary heart disease. JAMA. 2013;310(4):408–415. doi: 10.1001/jama.2013.8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu Y, Li S, Zeng Z, et al. Kidney stones and cardiovascular risk: a meta-analysis of cohort studies. Am J Kidney Dis. 2014;64(3):402–410. doi: 10.1053/j.ajkd.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 3.Domingos F, Serra A. Metabolic syndrome: a multifaceted risk factor for kidney stones. Scand J Urol. 2014;48(5):414–419. doi: 10.3109/21681805.2014.903513. [DOI] [PubMed] [Google Scholar]
- 4.Sarnak MJ, Levey AS, Schoolwerth AC, et al. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Hypertension. 2003;42(5):1050–1065. doi: 10.1161/01.HYP.0000102971.85504.7c. [DOI] [PubMed] [Google Scholar]
- 5.Schiffrin EL, Lipman ML, Mann JF. Chronic kidney disease: effects on the cardiovascular system. Circulation. 2007;116(1):85–97. doi: 10.1161/CIRCULATIONAHA.106.678342. [DOI] [PubMed] [Google Scholar]
- 6.Shavit L, Girfoglio D, Vijay V, et al. Vascular calcification and bone mineral density in recurrent kidney stone formers. Clin J Am Soc Nephrol. 2015;10(2):278–285. doi: 10.2215/CJN.06030614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tonelli M, Wiebe N, Culleton B, et al. Chronic kidney disease and mortality risk: a systematic review. J Am Soc Nephrol. 2006;17(7):2034–2047. doi: 10.1681/ASN.2005101085. [DOI] [PubMed] [Google Scholar]
- 8.Halperin ML, Cheema DS, Kamel KS. Physiology of acid-base balance: links with kidney stone prevention. Semin Nephrol. 2006;26(6):441–446. doi: 10.1016/j.semnephrol.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 9.KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Kidney Int Suppl. 2009;(113):S1–130. doi: 10.1038/ki.2009.188. [DOI] [PubMed] [Google Scholar]
- 10.Levey AS, Coresh J. Chronic kidney disease. Lancet. 2012;379(9811):165–180. doi: 10.1016/S0140-6736(11)60178-5. [DOI] [PubMed] [Google Scholar]
- 11.Evan AE, Lingeman JE, Coe FL, et al. Histopathology and surgical anatomy of patients with primary hyperparathyroidism and calcium phosphate stones. Kidney Int. 2008;74(2):223–229. doi: 10.1038/ki.2008.161. [DOI] [PubMed] [Google Scholar]
- 12.Evan AP, Coe FL, Rittling SR, et al. Apatite plaque particles in inner medulla of kidneys of calcium oxalate stone formers: osteopontin localization. Kidney Int. 2005;68(1):145–154. doi: 10.1111/j.1523-1755.2005.00388.x. [DOI] [PubMed] [Google Scholar]
- 13.Evan AP, Lingeman JE, Coe FL, et al. Crystal-associated nephropathy in patients with brushite nephrolithiasis. Kidney Int. 2005;67(2):576–591. doi: 10.1111/j.1523-1755.2005.67114.x. [DOI] [PubMed] [Google Scholar]
- 14.Evan AP, Coe FL, Lingeman JE, et al. Renal crystal deposits and histopathology in patients with cystine stones. Kidney Int. 2006;69(12):2227–2235. doi: 10.1038/sj.ki.5000268. [DOI] [PubMed] [Google Scholar]
- 15.Evan AP, Coe FL, Lingeman JE, et al. Mechanism of formation of human calcium oxalate renal stones on Randall’s plaque. Anat Rec (Hoboken) 2007;290(10):1315–1323. doi: 10.1002/ar.20580. [DOI] [PubMed] [Google Scholar]
- 16.Evan AP, Lingeman J, Coe F, et al. Renal histopathology of stone-forming patients with distal renal tubular acidosis. Kidney Int. 2007;71(8):795–801. doi: 10.1038/sj.ki.5002113. [DOI] [PubMed] [Google Scholar]
- 17.Evan AP, Lingeman JE, Coe FL, Worcester EM. Role of interstitial apatite plaque in the pathogenesis of the common calcium oxalate stone. Semin Nephrol. 2008;28(2):111–119. doi: 10.1016/j.semnephrol.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evan AP, Coe FL, Gillen D, Lingeman JE, Bledsoe S, Worcester EM. Renal intratubular crystals and hyaluronan staining occur in stone formers with bypass surgery but not with idiopathic calcium oxalate stones. Anat Rec (Hoboken) 2008;291(3):325–334. doi: 10.1002/ar.20656. [DOI] [PubMed] [Google Scholar]
- 19.Evan AP, Lingeman JE, Coe FL, et al. Intra-tubular deposits, urine and stone composition are divergent in patients with ileostomy. Kidney Int. 2009;76(10):1081–1088. doi: 10.1038/ki.2009.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evan AP, Weinman EJ, Wu XR, Lingeman JE, Worcester EM, Coe FL. Comparison of the pathology of interstitial plaque in human ICSF stone patients to NHERF-1 and THP-null mice. Urol Res. 2010;38(6):439–452. doi: 10.1007/s00240-010-0330-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evan AP, Lingeman JE, Worcester EM, et al. Renal histopathology and crystal deposits in patients with small bowel resection and calcium oxalate stone disease. Kidney Int. 2010;78(3):310–317. doi: 10.1038/ki.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evan AP, Lingeman JE, Worcester EM, et al. Contrasting histopathology and crystal deposits in kidneys of idiopathic stone formers who produce hydroxy apatite, brushite, or calcium oxalate stones. Anat Rec (Hoboken) 2014;297(4):731–748. doi: 10.1002/ar.22881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evan AP, Worcester EM, Williams JC, Jr, et al. Biopsy Proven Medullary Sponge Kidney: Clinical findings, histopathology, and role of osteogenesis in stone and plaque formation. Anat Rec (Hoboken) 2015 doi: 10.1002/ar.23105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evan AP, Worcester EM, Coe FL, Williams J, Jr, Lingeman JE. Mechanisms of human kidney stone formation. Urolithiasis. 2015;43(Suppl 1):19–32. doi: 10.1007/s00240-014-0701-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lorenz EC, Lieske JC, Vrtiska TJ, et al. Clinical characteristics of potential kidney donors with asymptomatic kidney stones. Nephrol Dial Transplant. 2011;26(8):2695–2700. doi: 10.1093/ndt/gfq769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun W, Feng Y, Yao XD, et al. Urinary angiotensinogen is elevated in patients with nephrolithiasis. Biomed Res Int. 2014;2014:349602. doi: 10.1155/2014/349602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siragy HM, Carey RM. Role of the intrarenal renin-angiotensin-aldosterone system in chronic kidney disease. Am J Nephrol. 2010;31(6):541–550. doi: 10.1159/000313363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu TW, Liu JS, Hung SC, et al. Renoprotective effect of renin-angiotensin-aldosterone system blockade in patients with predialysis advanced chronic kidney disease, hypertension, and anemia. JAMA Intern Med. 2014;174(3):347–354. doi: 10.1001/jamainternmed.2013.12700. [DOI] [PubMed] [Google Scholar]
- 29.Devarajan P, Krawczeski CD, Nguyen MT, Kathman T, Wang Z, Parikh CR. Proteomic identification of early biomarkers of acute kidney injury after cardiac surgery in children. Am J Kidney Dis. 2010;56(4):632–642. doi: 10.1053/j.ajkd.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsai JP, Liou JH, Yeh KT, Tai HC, Cheng YW, Chang HR. Intensity of cytosol expression of 8-OHdG in normal renal tubules is associated with the severity of renal fibrosis. Swiss Med Wkly. 2011;141:w13268. doi: 10.4414/smw.2011.13268. [DOI] [PubMed] [Google Scholar]
- 31.Boonla C, Wunsuwan R, Tungsanga K, Tosukhowong P. Urinary 8-hydroxydeoxyguanosine is elevated in patients with nephrolithiasis. Urol Res. 2007;35(4):185–191. doi: 10.1007/s00240-007-0098-0. [DOI] [PubMed] [Google Scholar]
- 32.Kittikowit W, Waiwijit U, Boonla C, et al. Increased oxidative DNA damage seen in renal biopsies adjacent stones in patients with nephrolithiasis. Urolithiasis. 2014;42(5):387–394. doi: 10.1007/s00240-014-0676-x. [DOI] [PubMed] [Google Scholar]
- 33.Vupputuri S, Soucie JM, McClellan W, Sandler DP. History of kidney stones as a possible risk factor for chronic kidney disease. Ann Epidemiol. 2004;14(3):222–228. doi: 10.1016/S1047-2797(03)00126-1. [DOI] [PubMed] [Google Scholar]
- 34.Gillen DL, Worcester EM, Coe FL. Decreased renal function among adults with a history of nephrolithiasis: a study of NHANES III. Kidney Int. 2005;67(2):685–690. doi: 10.1111/j.1523-1755.2005.67128.x. [DOI] [PubMed] [Google Scholar]
- 35.Shoag J, Halpern J, Goldfarb DS, Eisner BH. Risk of chronic and end stage kidney disease in patients with nephrolithiasis. J Urol. 2014;192(5):1440–1445. doi: 10.1016/j.juro.2014.05.117. [DOI] [PubMed] [Google Scholar]
- 36.Stankus N, Hammes M, Gillen D, Worcester E. African American ESRD patients have a high predialysis prevalence of kidney stones compared to NHANES III. Urol Res. 2007;35(2):83–87. doi: 10.1007/s00240-007-0079-3. [DOI] [PubMed] [Google Scholar]
- 37.Stamatelou KK, Francis ME, Jones CA, Nyberg LM, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976–1994. Kidney Int. 2003;63(5):1817–1823. doi: 10.1046/j.1523-1755.2003.00917.x. [DOI] [PubMed] [Google Scholar]
- 38.Hsu CY, Lin F, Vittinghoff E, Shlipak MG. Racial differences in the progression from chronic renal insufficiency to end-stage renal disease in the United States. J Am Soc Nephrol. 2003;14(11):2902–2907. doi: 10.1097/01.asn.0000091586.46532.b4. [DOI] [PubMed] [Google Scholar]
- 39.Saucier NA, Sinha MK, Liang KV, et al. Risk factors for CKD in persons with kidney stones: a case-control study in Olmsted County, Minnesota. Am J Kidney Dis. 2010;55(1):61–68. doi: 10.1053/j.ajkd.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rule AD, Bergstralh EJ, Melton LJ, III, Li X, Weaver AL, Lieske JC. Kidney stones and the risk for chronic kidney disease. Clin J Am Soc Nephrol. 2009;4(4):804–811. doi: 10.2215/CJN.05811108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El-Zoghby ZM, Lieske JC, Foley RN, et al. Urolithiasis and the risk of ESRD. Clin J Am Soc Nephrol. 2012;7(9):1409–1415. doi: 10.2215/CJN.03210312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alexander RT, Hemmelgarn BR, Wiebe N, et al. Kidney stones and kidney function loss: a cohort study. BMJ. 2012;345:e5287. doi: 10.1136/bmj.e5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hippisley-Cox J, Coupland C. Predicting the risk of chronic Kidney Disease in men and women in England and Wales: prospective derivation and external validation of the QKidney Scores. BMC Fam Pract. 2010;11:49. doi: 10.1186/1471-2296-11-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collins AJ, Foley RN, Herzog C, et al. US Renal Data System 2010 Annual Data Report. Am J Kidney Dis. 2011;57(1 Suppl 1):A8, e1–A8,526. doi: 10.1053/j.ajkd.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 45.Ahmadi F, Etemadi SM, Lessan-Pezeshki M, et al. Contribution of stone size to chronic kidney disease in kidney stone formers. Int J Urol. 2015;22(1):104–108. doi: 10.1111/iju.12606. [DOI] [PubMed] [Google Scholar]
- 46.Craver L, Marco MP, Martinez I, et al. Mineral metabolism parameters throughout chronic kidney disease stages 1–5–achievement of K/DOQI target ranges. Nephrol Dial Transplant. 2007;22(4):1171–1176. doi: 10.1093/ndt/gfl718. [DOI] [PubMed] [Google Scholar]
- 47.Jungers P, Joly D, Barbey F, Choukroun G, Daudon M. ESRD caused by nephrolithiasis: prevalence, mechanisms, and prevention. Am J Kidney Dis. 2004;44(5):799–805. [PubMed] [Google Scholar]
- 48.Worcester E, Parks JH, Josephson MA, Thisted RA, Coe FL. Causes and consequences of kidney loss in patients with nephrolithiasis. Kidney Int. 2003;64(6):2204–2213. doi: 10.1046/j.1523-1755.2003.00317.x. [DOI] [PubMed] [Google Scholar]
- 49.Worcester EM, Parks JH, Evan AP, Coe FL. Renal function in patients with nephrolithiasis. J Urol. 2006;176(2):600–603. doi: 10.1016/j.juro.2006.03.095. [DOI] [PubMed] [Google Scholar]
- 50.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 2012;81(5):442–448. doi: 10.1038/ki.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hsu CH, Kurtz TW, Rosenzweig J, Weller JM. Intrarenal hemodynamics and renal function in postobstructive uropathy. Invest Urol. 1978;15(4):348–351. [PubMed] [Google Scholar]
- 52.Klahr S. New insights into the consequences and mechanisms of renal impairment in obstructive nephropathy. Am J Kidney Dis. 1991;18(6):689–699. doi: 10.1016/s0272-6386(12)80611-1. [DOI] [PubMed] [Google Scholar]
- 53.Tanner GA, Evan AP. Glomerular and proximal tubular morphology after single nephron obstruction. Kidney Int. 1989;36(6):1050–1060. doi: 10.1038/ki.1989.300. [DOI] [PubMed] [Google Scholar]
- 54.Ophascharoensuk V, Giachelli CM, Gordon K, et al. Obstructive uropathy in the mouse: role of osteopontin in interstitial fibrosis and apoptosis. Kidney Int. 1999;56(2):571–580. doi: 10.1046/j.1523-1755.1999.00580.x. [DOI] [PubMed] [Google Scholar]
- 55.Bander SJ, Buerkert JE, Martin D, Klahr S. Long-term effects of 24-hr unilateral ureteral obstruction on renal function in the rat. Kidney Int. 1985;28(4):614–620. doi: 10.1038/ki.1985.173. [DOI] [PubMed] [Google Scholar]
- 56.Wang SJ, Mu XN, Zhang LY, Liu QY, Jin XB. The incidence and clinical features of acute kidney injury secondary to ureteral calculi. Urol Res. 2012;40(4):345–348. doi: 10.1007/s00240-011-0414-6. [DOI] [PubMed] [Google Scholar]
- 57.Keddis MT, Rule AD. Nephrolithiasis and loss of kidney function. Curr Opin Nephrol Hypertens. 2013;22(4):390–396. doi: 10.1097/MNH.0b013e32836214b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Randall A. THE ORIGIN AND GROWTH OF RENAL CALCULI. Ann Surg. 1937;105(6):1009–1027. doi: 10.1097/00000658-193706000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mulay SR, Kulkarni OP, Rupanagudi KV, et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1beta secretion. J Clin Invest. 2013;123(1):236–246. doi: 10.1172/JCI63679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thongboonkerd V, Semangoen T, Sinchaikul S, Chen ST. Proteomic analysis of calcium oxalate monohydrate crystal-induced cytotoxicity in distal renal tubular cells. J Proteome Res. 2008;7(11):4689–4700. doi: 10.1021/pr8002408. [DOI] [PubMed] [Google Scholar]
- 61.Chaiyarit S, Thongboonkerd V. Changes in mitochondrial proteome of renal tubular cells induced by calcium oxalate monohydrate crystal adhesion and internalization are related to mitochondrial dysfunction. J Proteome Res. 2012;11(6):3269–3280. doi: 10.1021/pr300018c. [DOI] [PubMed] [Google Scholar]
- 62.Maroni PD, Koul S, Meacham RB, Chandhoke PS, Koul HK. Effects of oxalate on IMCD cells: a line of mouse inner medullary collecting duct cells. Ann N Y Acad Sci. 2004;1030:144–149. doi: 10.1196/annals.1329.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Umekawa T, Chegini N, Khan SR. Oxalate ions and calcium oxalate crystals stimulate MCP-1 expression by renal epithelial cells. Kidney Int. 2002;61(1):105–112. doi: 10.1046/j.1523-1755.2002.00106.x. [DOI] [PubMed] [Google Scholar]
- 64.Parks JH, Coe FL, Evan AP, Worcester EM. Clinical and laboratory characteristics of calcium stone-formers with and without primary hyperparathyroidism. BJU Int. 2009;103(5):670–678. doi: 10.1111/j.1464-410X.2008.08064.x. [DOI] [PubMed] [Google Scholar]
- 65.Griffith DP. Struvite stones. Kidney Int. 1978;13(5):372–382. doi: 10.1038/ki.1978.55. [DOI] [PubMed] [Google Scholar]
- 66.Gupta M, Bolton DM, Gupta PN, Stoller ML. Improved renal function following aggressive treatment of urolithiasis and concurrent mild to moderate renal insufficiency. J Urol. 1994;152(4):1086–1090. doi: 10.1016/s0022-5347(17)32509-0. [DOI] [PubMed] [Google Scholar]
- 67.Kristensen C, Parks JH, Lindheimer M, Coe FL. Reduced glomerular filtration rate and hypercalciuria in primary struvite nephrolithiasis. Kidney Int. 1987;32(5):749–753. doi: 10.1038/ki.1987.270. [DOI] [PubMed] [Google Scholar]
- 68.Sakhaee K. Epidemiology and clinical pathophysiology of uric acid kidney stones. J Nephrol. 2014;27(3):241–245. doi: 10.1007/s40620-013-0034-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Viers BR, Lieske JC, Vrtiska TJ, et al. Endoscopic and Histologic Findings in a Cohort of Uric Acid and Calcium Oxalate Stone Formers. Urology. 2015 doi: 10.1016/j.urology.2014.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Johnson RJ, Kivlighn SD, Kim YG, Suga S, Fogo AB. Reappraisal of the pathogenesis and consequences of hyperuricemia in hypertension, cardiovascular disease, and renal disease. Am J Kidney Dis. 1999;33(2):225–234. doi: 10.1016/s0272-6386(99)70295-7. [DOI] [PubMed] [Google Scholar]
- 71.Linnes MP, Krambeck AE, Cornell L, et al. Phenotypic characterization of kidney stone formers by endoscopic and histological quantification of intrarenal calcification. Kidney Int. 2013;84(4):818–825. doi: 10.1038/ki.2013.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Edvardsson VO, Goldfarb DS, Lieske JC, et al. Hereditary causes of kidney stones and chronic kidney disease. Pediatr Nephrol. 2013;28(10):1923–1942. doi: 10.1007/s00467-012-2329-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lieske JC, Monico CG, Holmes WS, et al. International registry for primary hyperoxaluria. Am J Nephrol. 2005;25(3):290–296. doi: 10.1159/000086360. [DOI] [PubMed] [Google Scholar]
- 74.Riese RJ, Mandel NS, Wiessner JH, Mandel GS, Becker CG, Kleinman JG. Cell polarity and calcium oxalate crystal adherence to cultured collecting duct cells. Am J Physiol. 1992;262(2 Pt 2):F177–F184. doi: 10.1152/ajprenal.1992.262.2.F177. [DOI] [PubMed] [Google Scholar]
- 75.Worcester EM, Evan AP, Coe FL, et al. A test of the hypothesis that oxalate secretion produces proximal tubule crystallization in primary hyperoxaluria type I. Am J Physiol Renal Physiol. 2013;305(11):F1574–F1584. doi: 10.1152/ajprenal.00382.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lindell A, Denneberg T, Granerus G. Studies on renal function in patients with cystinuria. Nephron. 1997;77(1):76–85. doi: 10.1159/000190250. [DOI] [PubMed] [Google Scholar]
- 77.Kelley WN, Levy RI, Rosenbloom FM, Henderson JF, Seegmiller JE. Adenine phosphoribosyltransferase deficiency: a previously undescribed genetic defect in man. J Clin Invest. 1968;47(10):2281–2289. doi: 10.1172/JCI105913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bollee G, Dollinger C, Boutaud L, et al. Phenotype and genotype characterization of adenine phosphoribosyltransferase deficiency. J Am Soc Nephrol. 2010;21(4):679–688. doi: 10.1681/ASN.2009080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Edvardsson V, Palsson R, Olafsson I, Hjaltadottir G, Laxdal T. Clinical features and genotype of adenine phosphoribosyltransferase deficiency in iceland. Am J Kidney Dis. 2001;38(3):473–480. doi: 10.1053/ajkd.2001.26826. [DOI] [PubMed] [Google Scholar]
- 80.Benedetto B, Madden R, Kurbanov A, Braden G, Freeman J, Lipkowitz GS. Adenine phosphoribosyltransferase deficiency and renal allograft dysfunction. Am J Kidney Dis. 2001;37(5):E37. doi: 10.1016/s0272-6386(05)90001-2. [DOI] [PubMed] [Google Scholar]
- 81.Cassidy MJ, McCulloch T, Fairbanks LD, Simmonds HA. Diagnosis of adenine phosphoribosyltransferase deficiency as the underlying cause of renal failure in a renal transplant recipient. Nephrol Dial Transplant. 2004;19(3):736–738. doi: 10.1093/ndt/gfg562. [DOI] [PubMed] [Google Scholar]
- 82.Nasr SH, Sethi S, Cornell LD, et al. Crystalline nephropathy due to 2,8-dihydroxyadeninuria: an under-recognized cause of irreversible renal failure. Nephrol Dial Transplant. 2010;25(6):1909–1915. doi: 10.1093/ndt/gfp711. [DOI] [PubMed] [Google Scholar]
- 83.Wrong OM, Norden AG, Feest TG. Dent’s disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. QJM. 1994;87(8):473–493. [PubMed] [Google Scholar]
- 84.Copelovitch L, Nash MA, Kaplan BS. Hypothesis: Dent disease is an underrecognized cause of focal glomerulosclerosis. Clin J Am Soc Nephrol. 2007;2(5):914–918. doi: 10.2215/CJN.00900207. [DOI] [PubMed] [Google Scholar]
- 85.Frishberg Y, Dinour D, Belostotsky R, et al. Dent’s disease manifesting as focal glomerulosclerosis: Is it the tip of the iceberg? Pediatr Nephrol. 2009;24(12):2369–2373. doi: 10.1007/s00467-009-1299-2. [DOI] [PubMed] [Google Scholar]