

Summary
Buccal epithelial gene-expression reflects the physiological host response to household air pollution (HAP) from smoky versus smokeless coal. These transcriptional changes highlight coal type as an important determinant in shaping the relationship between HAP exposure and lung cancer risk.
Abstract
In China’s rural counties of Xuanwei and Fuyuan, lung cancer rates are among the highest in the world. While the elevated disease risk in this population has been linked to the usage of smoky (bituminous) coal as compared to smokeless (anthracite) coal, the underlying molecular changes associated with this exposure remains unclear. To understand the physiologic effects of smoky coal exposure, we analyzed the genome-wide gene-expression profiles in buccal epithelial cells collected from healthy, non-smoking female residents of Xuanwei and Fuyuan who burn smoky (n = 26) and smokeless (n = 9) coal. Gene-expression was profiled via microarrays, and changes associated with coal type were correlated to household levels of fine particulate matter (PM2.5) and polycyclic aromatic hydrocarbons (PAHs). Expression levels of 282 genes were altered with smoky versus smokeless coal exposure (P < 0.005), including the 2-fold increase of proinflammatory IL8 and decrease of proapoptotic CASP3. This signature was more correlated with carcinogenic PAHs (e.g. Benzo[a]pyrene; r = 0.41) than with non-carcinogenic PAHs (e.g. Fluorene; r = 0.08) or PM2.5 (r = 0.05). Genes altered with smoky coal exposure were concordantly enriched with tobacco exposure in previously profiled buccal biopsies of smokers and non-smokers (GSEA, q < 0.05). This is the first study to identify a signature of buccal epithelial gene-expression that is associated with smoky coal exposure, which in part is similar to the molecular response to tobacco smoke, thereby lending biologic plausibility to prior epidemiological studies that have linked this exposure to lung cancer risk.
Introduction
Approximately 3 billion people in the world use coal and biomass (e.g. charcoal, wood, animal dung and crop waste) to cook and heat their homes (1). This practice poses long-term risks for the development of cardiovascular and respiratory diseases including stroke, chronic obstructive pulmonary disease (COPD) and lung cancer (2–4). Consequently, the World Health Organization estimates that 4.3 million deaths in 2012 alone were attributable to household use of solid fuels (5).
Exposure to household air pollution (HAP) is especially prevalent in developing countries such as China, where a large proportion of the population still relies on solid fuel consumption (3). The rural counties of Xuanwei and Fuyuan in Yunnan Province, China have served as a particular focal point in large-scale epidemiological studies, in part due to the notably high lung cancer rates among its non-smoking female residents (6–8). Previous investigations within this population have highlighted fuel subtype as an important factor in lung cancer etiology, linking the high disease rates to the combustion of ‘smoky coal’ (bituminous) as compared to ‘smokeless coal’ (anthracite) (9,10). More recently, key compositional differences in hydrocarbon, elemental and quartz content between smoky and smokeless coals used in households across Xuanwei and Fuyuan (11,12) have been elucidated, further bolstering the premise that the use of different coal types contributes to the observed heterogeneity in disease risk.
Despite the wealth of insights gleaned from classic epidemiological studies performed in this region to date, the mechanisms by which smoky and smokeless coal usage can lead to widely different health risks remain poorly understood. The application of airway gene-expression profiling in HAP molecular epidemiology studies may help elucidate the biology behind observed health effects. We have previously shown that gene-expression profiling of the airway epithelium can be used to characterize the physiologic response to respiratory carcinogens and irritants such as cigarette smoke (13–15) and to generate clinically relevant biomarkers in smokers who develop lung cancer and COPD (16–18). Notably, we have also demonstrated that a subset of these smoking-related molecular changes are shared between the intrathoracic bronchial airway epithelium and the relatively accessible buccal epithelium (15).
In this study, we profiled the buccal epithelium of rural Chinese women with HAP exposure due to the burning of smoky and smokeless coal in order to characterize gene-expression changes that might offer insight to the physiologic response associated with the burning of smoky coal. We have identified a signature of genes that is differentially expressed in the buccal epithelium in response to smoky coal HAP exposure. Importantly, we found the enrichment of a number of proinflammatory mediators among these differentially expressed genes as well as the significant enrichment of this gene signature with that previously defined as changing within the upper and lower airway of tobacco smokers. These results shed new light on the molecular mechanisms associated with smoky coal exposure and may provide a biological basis for the increased risk of lung cancer.
Materials and methods
Subject recruitment
The subjects included in this analysis were enrolled as part of a larger HAP study that comprehensively characterized residential solid fuel usage and personal indoor exposure levels to HAP from residences in 30 rural villages throughout the counties of Xuanwei and Fuyuan in Yunnan Province, China (12,19). In order to best reflect historical stove usage, up to five households were preferentially selected from each village using the following criteria: (i) the household contains the presence of a non-smoking, healthy female aged 20–80 who is primarily responsible for cooking; (ii) the residence contains a stove using solid fuel; (iii) the resident has used predominantly the same cooking and heating equipment for the past 5 years; (iv) the residence is at least a decade old. The solid fuel type used at each residence was recorded based on self-report and further corroborated by petrochemical analysis of collected coal samples (11). All participants provided informed consent. This study was approved by the NIH’s Institutional Review Board and was conducted in accordance to the World Medical Association Declaration of Helsinki’s recommendations for human subject protection.
HAP sampling
Two sequential personal 24-h air measurements were collected from each subject and analyzed as described previously (12). Briefly, particulate matter with an aerodynamic cut-off of 2.5 µm and less (PM2.5) was collected for 24h on a 37-mm SKC Teflon filter using a BGI cyclone model GK 2.05SH and an AFC400S air pump (BGI, Waltham, MA) operating at median flow rate of 3.3 l/min (interquartile range: 3.24−3.47 l/min). The cyclone was attached in the subject’s breathing zone. PM2.5 concentrations (µg/m3) were calculated by dividing the postminus preweight of the filters by the volume of air drawn through them. For a subset, the organic fraction of the particulate matter was solvent extracted, whereupon the concentration of particle bound polycyclic aromatic hydrocarbons (PAHs) was determined using chromatography-mass spectrometry. Gas phase PAHs were collected with XAD-2 sorbent tubes at a median air flow rate of 63ml/min (interquartile range: 47−80ml/min) and analyzed similarly as the particulate matter.
Buccal epithelial cell collection and RNA isolation
Buccal mucosa epithelial cell scrapings were collected on the morning commencing the 24-h HAP sampling measurements. Sample collection was largely performed as described previously (20). Briefly, a custom concave plastic tool with serrated edges (Plastronics Engineering, Hampstead, NH) was gently scraped against the buccal mucosa on the inside left and right cheeks and then placed immediately into 1ml of RNAlater (Qiagen, Valencia, CA). Cells were kept at room temperature for several days before being stored at −80°C until RNA isolation. Total RNA was isolated using miRNeasy Mini Kit (Qiagen, Valencia, CA). RNA integrity was assessed using an Agilent BioAnalyzer and RNA purity was confirmed using a NanoDrop spectrophotometer. RNA was isolated from 201 buccal brushings, of which 43 samples (21%) produced sufficient quality and yield (total RNA ≥100ng) for microarray processing. We observed no significant differences between the distribution of samples from smoky and smokeless coal users that met the criteria for subsequent microarray preprocessing (Supplementary Table 1, available at Carcinogenesis Online).
Microarray data acquisition and preprocessing
Between 100 and 300ng of total RNA was processed, labeled and hybridized to Affymetrix Human Gene 1.0 ST GeneChips (Affymetrix, Santa Clara, CA) according to the Affymetrix protocol as described previously (21). A custom Chip Definition File annotating 19 741 entrez genes (‘hugene10stv1hsentrezgcdf’ and ‘hugene10stv1hsentrezg.db’ packages) was used for Robust Multichip Average array normalization and probe-level summarization (22). Among the 43 samples profiled on arrays, 8 outliers were excluded based on principal components analysis, Relative Log Expression, and Normalized Unscaled Standard Error metrics (Supplementary Table 1B, available at Carcinogenesis Online).
Smoky versus smokeless coal gene-expression analysis
A Student’s t-test was used to identify buccal epithelial gene-expression changes significantly associated with exposure to the indoor burning of smoky versus smokeless coal (P < 0.005). Enrichr (23) was used to identify Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, Gene Ontology (GO) biological processes and oncogenic signatures from the Molecular Signatures Database (MSigDB) significantly enriched (P < 0.05) among specific gene clusters. Linear models examined the variability in gene-expression attributable to PAHs and PM2.5, both before and after adjusting for smoky or smokeless coal use. Prior to correlating our results to these exposure metrics, a composite metagene score was computed from the first principal component of the normalized 282-gene signature (55.3% explained variance).
Connecting the smoky coal signature to smoking datasets
Using raw gene-expression data from the NCBI’s Gene Expression Omnibus (24), we examined the behavior of these genes in the intra- and extrathoracic airways of current smokers and never smokers. Dataset GSE17913 (25), which contains expression data generated from mucosal biopsy samples of healthy current smokers (n = 39) and never smokers (n = 40) recruited by Weill Cornell Medical College, was Robust Multichip Average normalized with Chip Definition File ‘hgu133plus2hsentrezgcdf’. Applying a linear model adjusting for age, we ranked genes based on the t-statistic associated with smoking status. Gene Set Enrichment Analysis (GSEA) v2.0 (26) compared this ranked list to the genes significantly associated with smoky coal exposure. The behavior of our signature was also evaluated in nasal and bronchial brushing data of current smokers and never smokers from GSE8987 (15) and GSE994 (14), where the smoky coal signature was collapsed into a composite metagene score as described previously and subsequently projected into each external dataset.
Real-time reverse transcription polymerase chain reaction (RT-PCR)
We performed RT-PCR of select genes on an independent set of smoky (n = 3) and smokeless (n = 3) coal-exposed subjects using SYBR Green-based RT2 qPCR Primer Assays (Qiagen, Valencia, CA). These samples had been excluded from the microarray analysis due to low yield. Primers for candidate gene (IL8, CASP3) and control gene (GAPDH) assays were designed and experimentally verified to ensure uniform and high PCR efficiencies. gDNA elimination buffer removed contaminating genomic DNA and samples were reverse transcribed with a mix of random hexamers and oligo-dT primer to generate first-strand cDNA using Qiagen’s RT2 First Strand Kit. PCR amplification mixtures (25 μl) contained 9ng of template cDNA, 12.5 μl of 2× RT2 SYBR Green master mix (Qiagen) and 400nM RT2 qPCR primers. Forty cycles of amplification and data acquisition were carried out in StepOnePlus Real-Time PCR systems (Applied Biosystems). StepOne Software (version 2.2.2; Applied Biosystems) automatically performed threshold determinations for each reaction. Relative gene-expression levels were calculated using the comparative CT method (27). Smoky versus smokeless fold changes were calculated from the average expression values obtained across each exposure group.
Additional information
All statistical analyses described were performed with R (http://r-project.org) 2–13.0 and Bioconductor (28). Microarray data from this study have been deposited in the Gene Expression Omnibus under accession GSE64277.
Results
Study population
We generated HAP metrics and buccal gene-expression profiles from 35 subjects who are smoky coal (n = 26) and smokeless coal (n = 9) users (Table 1). Specifically, we obtained 2-day averages of personal PAH and PM2.5 concentrations from healthy, non-smoking females who reside in villages across Xuanwei and Fuyuan counties. There were no significant differences in personal PM2.5 air concentrations between the two coal-user groups. Particle phase PAHs were detected at significantly higher levels for smoky coal users as compared to smokeless coal users, which is reflective of the larger study population from which these 35 subjects were derived (12). None of the subjects were active tobacco users but all of them reported to have a history of passive smoke exposure. This was expected in this population, as females traditionally do not smoke while males are predominantly smokers.
Table 1.
Overview of 35 subjects exposed to smoky and smokeless coal
| Smoky coal (n = 26) | Smokeless coal (n = 9) | ||
|---|---|---|---|
| Age | Mean ± SD | 57±15 | 59±14 |
| Secondhand smokea | n (%) | 26 (100.0) | 9 (100.0) |
| PM2.5 (µg/m3) | Median (IQR) | 177.2 (121.3) | 145.1 (171.5) |
| PAHsb (ng/m3) | Median (IQR) | ||
| Acenaphthylene | 620.0 (619.7) | 491.6 (323.1) | |
| Benz[a]anthracene* | 85 (81.3) | 9.4 (16.8) | |
| Benzo[a]pyrene* | 60.9 (59.4) | 10.6 (12.3) | |
| Benzo[b]fluoranthene | 96.7 (95.0) | 19.4 (28.9) | |
| Benzo[g,h,i]perylene* | 69.8 (66.0) | 12.7 (16.4) | |
| Benzo[k]fluoranthene* | 20.9 (22.8) | 5.2 (5.8) | |
| Chrysene | 69.7 (89.8) | 12.1 (16.4) | |
| Dibenz[a,h]anthracene* | 16.9 (28.9) | 1.8 (7.0) | |
| Fluoranthene | 29.8 (62.7) | 5.3 (4.9) | |
| Fluorene | 290.6 (380.0) | 250 (73.4) | |
| Indeno[1,2,3-cd]pyrene* | 37.4 (31.3) | 11.7 (12.5) | |
| Napthalene | 4416.7 (3743.6) | 4220 (1833.9) | |
| Phenanthrene | 464.2 (513.5) | 351.5 (172.5) | |
| Pyrene | 35.4 (77.4) | 6.6 (6.4) |
aStatus based on self-report.
bPersonal filters for PAH analysis available for all but one subject.
*Statistically significant (P < 0.01) between smoky and smokeless groups via Wilcoxon–Mann–Whitney test or Fisher’s exact test.
Gene-expression changes associated with smoky coal exposure
We identified 282 genes (Supplementary Table 2, available at Carcinogenesis Online) as differentially expressed (P < 0.005) in the buccal epithelium of subjects exposed to smoky versus smokeless coal (Figure 1), which was approximately three times more than the 98 genes expected by chance. This signature is comprised of two main clusters: genes with lower expression in smoky coal-exposed subjects (Cluster 1) and genes with higher expression in smoky coal-exposed subjects (Cluster 2), relative to smokeless coal users. Moreover, we found that including coal type in multiple linear regression models diminished the explanatory power associated with most PAHs (Supplementary Table 3, available at Carcinogenesis Online). Interestingly, our 282-gene signature also exhibited higher correlations with carcinogenic PAHs as compared to non-carcinogenic PAHs (Supplementary Figure 1, available at Carcinogenesis Online). In order to validate the differential behavior of the 282-gene signature, candidates exhibiting strong biological relevance (IL8, CASP3) were selected for RT-PCR within an independent set of buccal samples from smoky (n = 3) and smokeless (n = 3) coal users (Figure 2).
Figure 1.
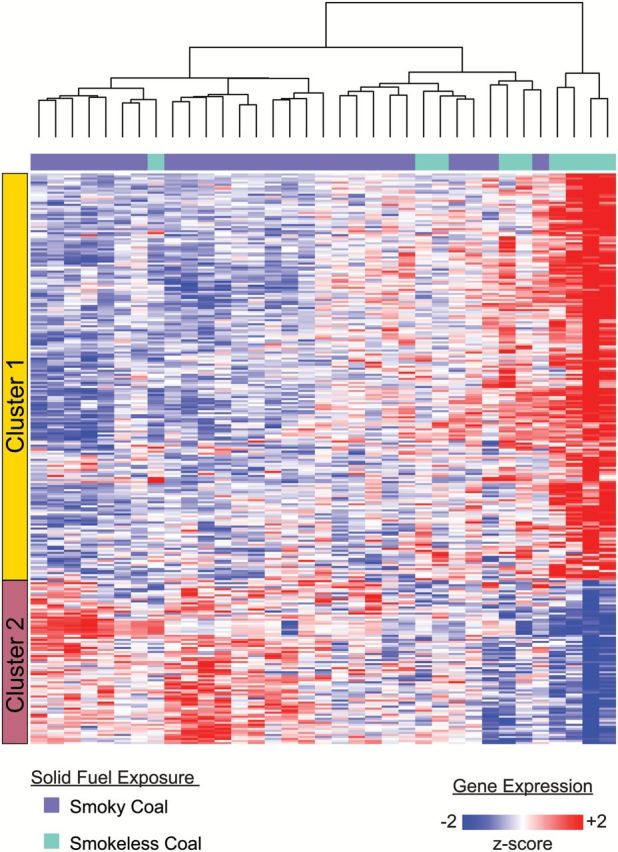
Buccal gene-expression changes in women exposed to household air pollution from smoky versus smokeless coal. Unsupervised hierarchical clustering of 282 genes with significantly different means (Student’s t test; P < 0.005) between smoky (n = 26) and smokeless (n = 9) coal-exposed subjects. The left-most color bar corresponds to the two main clusters of genes (rows) that separate the samples (columns) based on their relative expression with respect to fuel type. Red and blue intensities correspond to higher and lower expression, respectively.
Figure 2.

Real-time RT-PCR and microarray expression levels of select candidates from the 282-gene signature. The log2 fold change of IL8 and CASP3 in buccal epithelial epithelium of smoky versus smokeless subjects as computed from microarrays (black) and RT-PCR (striped). Microarray results were averaged across smoky (n = 26) and smokeless (n = 9) coal users. RT-PCR results were averaged across an independent set of smoky coal (n = 3) and smokeless (n = 3) coal users.
Biological enrichment and pathway analysis
We conducted functional enrichment analysis on each of the two gene clusters (Table 2). The top biological categories enriched among the genes expressed at lower levels in smoky coal users (Cluster 1) include regulatory processes such as the regulation of transcription and regulation of the cell cycle. Cluster 1 is also enriched for genes involved in the vascular endothelial growth factor (VEGF) pathway, which has been associated with lung injury and wound repair (29,30). In contrast, the genes expressed at higher levels in smoky coal users (Cluster 2) are dominantly enriched for inflammatory pathways such as hedgehog signaling, Toll-like receptor signaling and cytokine–cytokine receptor interactions. In particular, we observed increased expression of proinflammatory mediators (e.g. IL8, Iβ and WNT5B) in subjects who burned smoky coal. We found significant overlap between Cluster 2 and a signature that was generated in immortalized human lung epithelial cells following oncogenic KRAS overexpression (P <0 .05), which included genes LRIG1, G0S2 and IL8 (31). This is noteworthy given that lung tumors of non-smokers exposed to smoky coal emissions have distinct KRAS mutations that differ from those found in other non-smoker lung tumors (32).
Table 2.
Functional enrichment within gene clusters whose behavior is associated with household air pollution of burning smoky versus smokeless coal
| Gene Cluster 1 (lower in smoky versus smokeless, n = 201 genes) | |
| Enrichment Category: KEGG | P value |
| Axon guidance (HSA04360) | 0.016 |
| VEGF signaling pathway (HSA04370) | 0.016 |
| Colorectal cancer (HSA05210) | 0.027 |
| Valine Leucine and Isoleucine degradation (HSA00280) | 0.045 |
| Enrichment Category: GO biological process | P value |
| Response to heat (GO:0009408) | 0.008 |
| Response to temperature stimulus (GO:0009266) | 0.016 |
| Regulation of transcription (GO:0045449) | 0.018 |
| Regulation of cell cycle (GO:0051726) | 0.020 |
| Nucleobase, nucleoside, nucleotide and nucleic acid metabolic process (GO:0006139) | 0.022 |
| Regulation of endocytosis (GO:0030100) | 0.029 |
| Gene Cluster 2 (higher in smoky versus smokeless, n = 81 genes) | |
| Enrichment Category: KEGG | P value |
| Hedgehog signaling pathway (HSA04340) | 0.012 |
| Hematopoietic cell lineage (HSA04640) | 0.027 |
| Toll-like receptor signaling pathway (HSA04620) | 0.035 |
| Cytokine-cytokine receptor interaction (HSA04060) | 0.035 |
| Enrichment Category: GO biological process | P value |
| Glycerophospholipid metabolic process (GO:0006650) | 0.014 |
| Cellular amino acid derivative metabolic process (GO:0006575) | 0.015 |
| Negative regulation of cell proliferation (GO:0008285) | 0.018 |
| Phospholipid metabolic process (GO:0006644) | 0.029 |
| Anatomical structure morphogenesis (GO:0009653) | 0.043 |
| Positive regulation of cellular protein metabolic process (GO:0032270) | 0.044 |
| Positive regulation of protein metabolic process (GO:0051247) | 0.047 |
Within Enrichr, P values were calculated using Fisher’s exact tests under the assumptions of a binomial distribution and independence for probability of any gene belonging to any set.
Shared response to tobacco smoke exposure
We have previously shown that tobacco smoke induces gene-expression changes throughout the epithelium of the respiratory tract (33). Since tobacco smoke, like smoky coal, is an established risk factor for lung cancer and other non-malignant respiratory diseases, we were interested to examine whether there are similarities between the effects of smoky coal and tobacco smoke exposure. We first re-examined the smoking-associated transcriptomic changes that were previously detailed in buccal mucosal biopsies from current smokers and never smokers (25). By GSEA we find that a significant number of the genes that were induced in smoky coal users are enriched among the genes induced in current smokers from the buccal biopsy dataset, and that a similar relationship exists for genes repressed in smoky coal users and in current smokers (Figure 3; q < 0.05). Furthermore, our smoky coal signature appears to be modulated throughout the buccal, nasal and bronchial epithelium of current smokers and never smokers (Figure 4).
Figure 3.
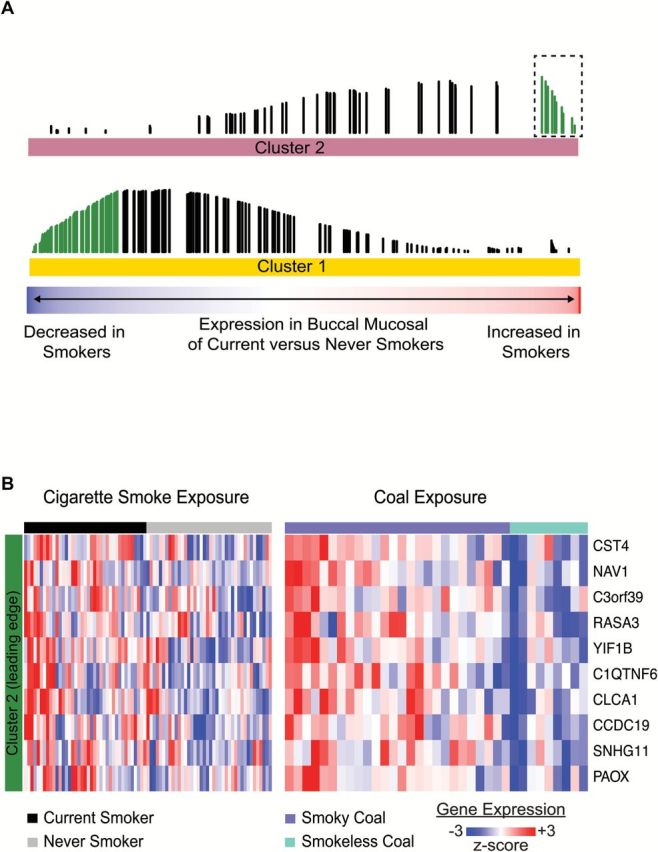
The buccal gene-expression pattern associated with household air pollution has significant similarities to the pattern associated with cigarette smoking. (A) Distribution of 282 genes associated with smoky versus smokeless coal exposure among all genes ranked according to their differential expression in current smokers versus never smokers from dataset GSE17913. Genes expressed at higher levels in the buccal mucosa of smoky coal users are significantly enriched (GSEA, q < 0.05) among the genes most highly induced in the buccal mucosa of current smokers (top). There is a similar enrichment between genes that are repressed by smoky coal and cigarette smoke (bottom). The bottom color bar represents the degree to which the gene is altered in current smokers (red: increased in smokers, blue: decreased in smokers). Each vertical line represents one of the 282 genes, the height of which represents the running GSEA enrichment score. Green lines represent the leading edge genes or subset of genes most concordantly up- or down-regulated with respect to dataset GSE17913. (B) Supervised heatmap of 10 leading edge genes from Cluster 2 (boxed green lines in A) generated across current smokers and never smokers (left), and subjects exposed to smoky or smokeless coal (right).
Figure 4.

The buccal signature of smoky coal exposure is modulated throughout the intra- and extra-thoracic airway epithelium of current smokers versus never smokers. The behavior of the 282-gene smoky coal signature derived from the 35 Xuanwei and Fuyuan females was evaluated in independent gene-expression datasets generated from buccal biopsies (GSE17913), nasal brushings (GSE8987) and bronchial airway brushings (GSE994) collected from current smokers and never smokers. For each dataset, the smoky coal signature was collapsed into a composite metagene score and projected into never smokers and current smokers. *P < 0.05 via one-tailed t test.
Discussion
It has been almost three decades since Mumford et al. (34). published their seminal study linking the high lung cancer mortality rates in rural Xuanwei County, China to the domestic burning of smoky coal. While several studies have examined the relative etiologic importance of different solid fuel emissions and identified distinct PAH-DNA adduct levels, mutational spectra, and polymorphisms associated with smoky coal exposure (10,32,35), little is known regarding the underlying physiologic responses that smoky coal induces in comparison to other fuel types. To this end, the results from our study lend valuable insight to the differential host response associated with smoky versus smokeless coal exposure by comprehensively examining the landscape of gene-expression changes present in the buccal epithelium.
We have identified a set of genes with altered expression between healthy, non-smoking women who are exposed to smoky or smokeless coal emissions. Notably, we observe the significant activation of inflammatory mediators (IL8, IL1β and WNT5B) and pathways (cytokine–cytokine interaction, Toll-like receptor signaling) in the buccal epithelium of subjects who burned smoky coal as compared to smokeless coal. Although exposure to particulate matter is known to activate these pathways in airway epithelial cells (36), the comparable levels of PM2.5 detected in smoky and smokeless coal burning residences suggests that other constituents may be responsible for triggering this molecular response.
Specifically, we have validated the greater than 2-fold activation of IL8, a neutrophil chemoattractant involved in the TLR pathway that has previously served as a marker to evaluate the inflammatory effects of ambient particulate matter, ozone, and vehicle emissions on respiratory epithelial cells (37–39). This differential human response parallels the elevated IL8 serum levels found in rural Indian women who cook with biomass compared to those who cook with liquefied petroleum gas (40). The upregulation of IL8 has also been observed in the bronchial airway epithelium of smokers with lung cancer (41). Overall, these molecular observations show that exposure to smoky coal emissions mounts a strong inflammatory host response.
Our results also suggest that the physiologic response to smoky coal exposure alters gene-expression involved in apoptosis and cell proliferation. For instance, we observed that the proapoptotic gene CASP3 exhibits lower expression levels in smoky coal users. Activated downstream of initiator caspases as part of the intrinsic apoptosis pathway, caspase-3 plays a central role in orchestrating programmed cell death (42). Decreased levels of CASP3 have been associated with apoptosis resistance, a hallmark of carcinogenesis and tumor progression (43). Furthermore, tumor cells in non-small cell lung cancer are highly apoptosis resistant and in vivo expression levels of CASP3 have been shown to correlate with lung cancer survival (44). Thus, our observation that smoky coal users have lower CASP3 levels biologically supports the high rates of lung cancer and lung cancer mortality rates observed in smoky coal users.
We also compared our smoky coal signature to gene-expression changes in tobacco users. By GSEA we observed that our 282-gene signature was concordantly enriched in tobacco smoke-associated gene-expression profiles derived from the buccal mucosa biopsies of current smokers and never smokers, suggesting that components of smoky versus smokeless coal emissions may elicit similar physiologic effects as those induced by tobacco smoke. Among the 10 ‘leading edge’ genes or subset that was concordantly activated in both datasets and accounted for the core enrichment signal, polyamine oxidase PAOX has been recognized to play a role in catalyzing the first step of the xenobiotic response to inhaled toxicants (45). CLCA1, another leading edge gene, has been demonstrated to regulate airway mucous production in inflammatory conditions such as asthma and COPD (46,47). CLCA1 activation is also associated with mucin production in cigarette smoke-exposed human bronchial epithelial cell lines and murine models (48). It has been observed that smoky coal exposure in Xuanwei reduces risk of lung cancer from tobacco use (49). This phenomenon has also been observed in a cohort of workers exposed to high levels of diesel engine exhaust (50). One possible mechanism suggested for these effects is the increased production of mucous airway levels by these environmental exposures, potentially providing some protection against tobacco’s carcinogenic effects. Our observation that smoky coal exposure induces genomic changes consistent with high levels of mucous production provides some support for this hypothesis.
We have demonstrated that the exposure to smoky versus smokeless coal may induce an airway-wide field of genomic changes that are present throughout the airway epithelium. This phenomenon has been consistently observed in the response to tobacco smoke and thereby enabled the development of airway-based gene-expression biomarkers for the early detection of lung cancer and for guiding therapy in COPD. We believe that by extending this genomic profiling approach to assess the biologic response to solid fuel emissions, this work will similarly open additional avenues for the future development of clinically relevant biomarkers in this population.
There are a number of limitations to our study. The sample size was relatively small and the findings require future replication. In spite of this limitation, we note that our report represents the first effort to characterize the transcriptome in people who experience this highly carcinogenic exposure and the overlap to some extent with the gene-signature associated with tobacco smoking provides some external validity to the findings. In addition, a substantial number of samples were found to have mRNA that was not analyzable by microarray. However, the characteristics of subjects with and without analyzable samples were not materially different and as such it is unlikely that this would have resulted in bias to our findings.
In summary, this is the first study to employ whole-genome expression profiling of the buccal epithelium to measure the physiological response to HAP. Applying this ‘field of injury’ paradigm to populations in China with high levels of HAP has enabled us to demonstrate differences in the physiologic response to smoky versus smokeless coal exposure. Specifically, our observation of increased IL8 expression and decreased CASP3 expression in smoky coal users suggests that the physiologic response to smoky coal modulates proinflammatory and apoptotic responses. Our results also suggest a shared molecular response in the airway epithelium to tobacco smoke and smoky coal exposure. Together, these findings lend mechanistic insight and biologic plausibility to prior epidemiological studies that have strongly linked the variability in lung cancer risk within this region to the exposure of smoky coal.
Supplementary material
Supplementary Table 1–3 and Figure 1 can be found at http://carcin.oxfordjournals.org/
Funding
National Institutes of Health/National Institute of Environmental Health Sciences (3U01ES16035-03S1); Intramural Research Program of the US National Cancer Institute.
Conflict of Interest Statement: None declared.
Supplementary Material
Glossary
Abbreviations
- COPD
chronic obstructive pulmonary disease
- GSEA
gene set enrichment analysis
- HAP
household air pollution
- PAHs
polycyclic aromatic hydrocarbons
References
- 1. Ezzati M., et al. (2002) Household energy, indoor air pollution, and health in developing countries: knowledge base for effective interventions. Annu. Rev. Energy Environ., 27, 233–270. [Google Scholar]
- 2. Ezzati M., et al. (2002) The health impacts of exposure to indoor air pollution from solid fuels in developing countries: knowledge, gaps, and data needs. Environ. Health Perspect., 110, 1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang J.J., et al. (2007) Household air pollution from coal and biomass fuels in China: measurements, health impacts, and interventions. Environ. Health Perspect., 115, 848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang Z.F., et al. (1988) Indoor air pollution of coal fumes as a risk factor of stroke, Shanghai. Am. J. Public Health, 78, 975–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. WHO. Burden of disease from household air pollution for 2012. http://www.who.int/phe/health_topics/outdoorair/databases/FINAL_HAP_AAP_BoD_24March2014.pdf (5 April 2015, date last accessed). [Google Scholar]
- 6. Chapman R.S., et al. (1988) The epidemiology of lung cancer in Xuan Wei, China: current progress, issues, and research strategies. Arch. Environ. Health, 43, 180–185. [DOI] [PubMed] [Google Scholar]
- 7. Hosgood H.D. 3rd et al. (2008) Portable stove use is associated with lower lung cancer mortality risk in lifetime smoky coal users. Br. J. Cancer, 99, 1934–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lan Q., et al. (2002) Household stove improvement and risk of lung cancer in Xuanwei, China. J. Natl. Cancer Inst., 94, 826–835. [DOI] [PubMed] [Google Scholar]
- 9. Barone-Adesi F., et al. (2012) Risk of lung cancer associated with domestic use of coal in Xuanwei, China: retrospective cohort study. BMJ, 345, e5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lan Q., et al. (2005) Smoky coal exposure, NBS1 polymorphisms, p53 protein accumulation, and lung cancer risk in Xuan Wei, China. Lung Cancer, 49, 317–323. [DOI] [PubMed] [Google Scholar]
- 11. Downward G.S., et al. (2014) Heterogeneity in coal composition and implications for lung cancer risk in Xuanwei and Fuyuan counties, China. Environ. Int., 68, 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Downward G.S., et al. (2014) Polycyclic aromatic hydrocarbon exposure in household air pollution from solid fuel combustion among the female population of Xuanwei and Fuyuan counties, China. Environ. Sci. Technol., 48, 14632–14641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beane J., et al. (2007) Reversible and permanent effects of tobacco smoke exposure on airway epithelial gene expression. Genome Biol., 8, R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spira A., et al. (2004) Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc. Natl. Acad. Sci. USA, 101, 10143–10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sridhar S., et al. (2008) Smoking-induced gene expression changes in the bronchial airway are reflected in nasal and buccal epithelium. BMC Genomics, 9, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Spira A., et al. (2007) Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat. Med., 13, 361–366. [DOI] [PubMed] [Google Scholar]
- 17. Silvestri G.A., et al. (2015) A bronchial genomic classifier for the diagnostic evaluation of lung cancer. N. Engl. J. Med., 373, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Steiling K., et al. (2013) A dynamic bronchial airway gene expression signature of chronic obstructive pulmonary disease and lung function impairment. Am. J. Respir. Crit. Care Med., 187, 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu W., et al. (2014) Personal and indoor PM2.5 exposure from burning solid fuels in vented and unvented stoves in a rural region of China with a high incidence of lung cancer. Environ. Sci. Technol., 48, 8456–8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spira A., et al. (2004) Noninvasive method for obtaining RNA from buccal mucosa epithelial cells for gene expression profiling. Biotechniques, 36, 484–487. [DOI] [PubMed] [Google Scholar]
- 21. Zhang X., et al. (2007) Comparison of smoking-induced gene expression on Affymetrix exon and 3’-based expression arrays. Genome Informatics Int. Conf. Genome Informatics, 18, 247–257. [PubMed] [Google Scholar]
- 22. Dai M., et al. (2005) Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res., 33, e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen E.Y., et al. (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics, 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Edgar R., et al. (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res., 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boyle J.O., et al. (2010) Effects of cigarette smoke on the human oral mucosal transcriptome. Cancer Prev. Res., 3, 266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Subramanian A., et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmittgen T.D., et al. (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc., 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- 28. Gentleman R.C., et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol., 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bao P., et al. (2009) The role of vascular endothelial growth factor in wound healing. J. Surg. Res., 153, 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hicklin D.J., et al. (2005) Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol., 23, 1011–1027. [DOI] [PubMed] [Google Scholar]
- 31. Barbie D.A., et al. (2009) Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature, 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DeMarini D.M., et al. (2001) Lung tumor KRAS and TP53 mutations in nonsmokers reflect exposure to PAH-rich coal combustion emissions. Cancer Res., 61, 6679–6681. [PubMed] [Google Scholar]
- 33. Zhang X., et al. (2010) Similarities and differences between smoking-related gene expression in nasal and bronchial epithelium. Physiol. Genomics, 41, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mumford J.L., et al. (1987) Lung cancer and indoor air pollution in Xuan Wei, China. Science, 235, 217–220. [DOI] [PubMed] [Google Scholar]
- 35. Mumford J.L., et al. (1993) DNA adducts as biomarkers for assessing exposure to polycyclic aromatic hydrocarbons in tissues from Xuan Wei women with high exposure to coal combustion emissions and high lung cancer mortality. Environ. Health Perspect., 99, 83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Becker S., et al. (2005) TLR-2 is involved in airway epithelial cell response to air pollution particles. Toxicol. Appl. Pharmacol., 203, 45–52. [DOI] [PubMed] [Google Scholar]
- 37. Jaspers I., et al. (1997) Ozone-induced IL-8 expression and transcription factor binding in respiratory epithelial cells. Am. J. Physiol., 272(3 Pt 1), L504–L511. [DOI] [PubMed] [Google Scholar]
- 38. Lu Y., et al. (2014) Characteristics and cellular effects of ambient particulate matter from Beijing. Environ. Pollut., 191, 63–69. [DOI] [PubMed] [Google Scholar]
- 39. Scarpa M.C., et al. (2014) The role of non-invasive biomarkers in detecting acute respiratory effects of traffic-related air pollution. Clin. Exp. Allergy, 44, 1100–1118. [DOI] [PubMed] [Google Scholar]
- 40. Dutta A., et al. (2012) Systemic inflammatory changes and increased oxidative stress in rural Indian women cooking with biomass fuels. Toxicol. Appl. Pharmacol., 261, 255–262. [DOI] [PubMed] [Google Scholar]
- 41. Beane J., et al. (2011) C Characterizing the impact of smoking and lung cancer on the airway transcriptome using RNA-seq. Cancer Prev. Res., 4, 803–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McIlwain D.R., et al. (2013) Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol., 5, a008656–a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fulda S. (2009) Tumor resistance to apoptosis. Int. J. Cancer, 124, 511–515. [DOI] [PubMed] [Google Scholar]
- 44. Fennell D.A. (2005) Caspase regulation in non-small cell lung cancer and its potential for therapeutic exploitation. Clin. Cancer Res., 11, 2097–2105. [DOI] [PubMed] [Google Scholar]
- 45. Courcot E., et al. (2012) Xenobiotic metabolism and disposition in human lung cell models: comparison with in vivo expression profiles. Drug Metab. Dispos. Biol. Fate Chem., 40, 1953–1965. [DOI] [PubMed] [Google Scholar]
- 46. Hegab A.E., et al. (2004) CLCA1 gene polymorphisms in chronic obstructive pulmonary disease. J. Med. Genet., 41, e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wilk J.B., et al. (2003) A genome-wide scan of pulmonary function measures in the National Heart, Lung, and Blood Institute Family Heart Study. Am. J. Respir. Crit. Care Med., 167, 1528–1533. [DOI] [PubMed] [Google Scholar]
- 48. Hegab A.E., et al. (2007) Niflumic acid and AG-1478 reduce cigarette smoke-induced mucin synthesis: the role of hCLCA1. Chest, 131, 1149–1156. [DOI] [PubMed] [Google Scholar]
- 49. Kim C., et al. (2014) Smoky coal, tobacco smoking, and lung cancer risk in Xuanwei, China. Lung Cancer, 84, 31–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Silverman D.T., et al. (2012) The Diesel Exhaust in Miners study: a nested case-control study of lung cancer and diesel exhaust. J. Natl. Cancer Inst., 104, 855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.