

Abstract
Perinatal environmental exposures are potentially important contributors to the increase in autoimmune diseases. Yet, the mechanisms by which these exposures increase self-reactive immune responses later in life are poorly understood. Autoimmune diseases require CD4+ T cells for initiation, progression, and/or clinical symptoms; thus, developmental exposures that cause durable changes in CD4+ T cells may play a role. Early life activation of the aryl hydrocarbon receptor (AHR) causes persistent changes in the response of CD4+ T cells to infection later in life but whether CD4+ T cells are affected by developmental exposure in the context of an autoimmune disease is unknown. Gnaq+/− mice develop symptoms of autoimmune disease similar to those measured clinically, and therefore can be used to evaluate gene-environment interactions during development on disease progression. Herein, we examined the effect of AHR activation in utero and via lactation, or solely via lactation, on disease onset and severity in adult Gnaq+/− offspring. Developmental activation of the AHR-accelerated disease in Gnaq+/− mice, and this correlates with increases in effector CD4+ T-cell populations. Increased symptom onset and cellular changes due to early life AHR activation were more evident in female Gnaq+/− mice compared with males. These observations suggest that developmental AHR activation by pollutants, and other exogenous ligands, may increase the likelihood that genetically predisposed individuals will develop clinical symptoms of autoimmune disease later in life.
Keywords: aryl hydrocarbon receptor; autoimmune disease; developmental exposure; CD4 T cell; gene-environment interactions; 2,3,7,8-tetrachlorodibenzo-p-dioxin; TCDD
The incidence and prevalence of autoimmune diseases are on the rise but the factors responsible for the increase of these diseases remain unclear (Parks et al., 2014). Genetics alone does not provide a sufficient explanation for this increase. For example, the concordance of systemic autoimmune disease development in monozygotic twins is < 50%, suggesting other factors contribute to disease (Bogdanos et al., 2012). Recent reviews of the current state of knowledge suggest that environmental exposures and gene-environment interactions likely contribute to the increased prevalence and incidence of autoimmune diseases (Ellis et al., 2014; Parks et al., 2014). In particular, there is growing evidence that environmental exposures contribute to the increased incidence of 2 common systemic autoimmune diseases: rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) (Karlson and Deane, 2012; Somers and Richardson, 2014). However, the impact of environmental exposures on the cellular mechanisms that cause autoimmune disease is not understood. Studies in animal models suggest that the timing of exposure is important; specifically, that exposures during early development affect disease incidence later in life (Gilbert et al., 2014; Gogal and Holladay, 2008). This suggests that the exposure and appearance of disease may be separated in time, a concept often referred to as the developmental origins of health and disease.
Evidence that early life exposures impact the function of the immune system later in life is increasing. Yet, how these exposures cause persistent immunological changes is unknown, partly because the molecules with which these chemicals interact in vivo are largely unknown. Environmentally derived ligands of the aryl hydrocarbon receptor (AHR) represent a group of chemicals to which we are regularly exposed and for which there is a known cellular receptor. The AHR is a ligand activated transcription factor that is a member of the environment sensing Per-Arnt-Sim (PAS) protein super family. The AHR binds many xenobiotics, and its prototypical ligand is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (Nguyen and Bradfield, 2008). Early life exposure to some AHR-binding pollutants is associated with lasting changes in the function of the immune system (Winans et al., 2011). Moreover, several studies have shown that AHR activation early in life influences the development of autoimmune pathologies in the kidney later in life (Holladay et al., 2011; Mustafa et al., 2011a,b, 2008). However, it is not known which immune cells are affected by developmental activation of the AHR in the context of autoimmune disease. Direct activation of the AHR in adult animals alters the progression of several autoimmune disease models, which are dependent upon AHR-mediated changes in CD4+ T-cell function (Quintana and Sherr, 2013). Developmental activation of the AHR, via maternal ligand treatment, impacts the function of CD4+ T cells later in life in the context of infection (Boule et al., 2014, 2015). However, whether developmental exposure also changes CD4+ T cells in the context of an autoimmune disease is unknown.
Upon activation, naïve CD4+ T cells differentiate into several functionally distinct effector subsets. Th1, Th2, and Th17 cells are considered conventional CD4+ T cells, and produce the immunostimulatory cytokines IFNγ, IL-4, and IL-17, respectively (Yamane and Paul, 2013). Th1 and Th17 cells are the 2 main CD4+ T-cell subsets implicated in the development of autoimmune disease symptoms (Raphael et al., 2014). CD4+ T cells can also develop into regulatory CD4+ T cells (Tregs), which maintain peripheral tolerance and dampen the response of other immune cells (Sakaguchi et al., 2006). Activation of the AHR during development skews the proportion of CD4+ T-cell subsets, resulting in tissue-specific alterations in the proportion of conventional CD4+ T cells and Tregs after infection (Boule et al., 2014, 2015). There are several pathophysiological implications of skewing CD4+ T-cell subsets because CD4+ T cells interact with many other cell types, including antibody-producing B cells (Raphael et al., 2014). Autoantibodies produced by B cells lead to pathology associated with autoimmune disease, such as joint swelling, kidney damage, and skin rashes, due to the deposition of immune complexes in tissues and the inflammation that this elicits (Jancar and Sanchez Crespo, 2005; Mayadas et al., 2009). Yet, it is unknown how environmental exposures, particularly those during development, influence CD4+ T cells and CD4+ T cell-mediated events responsible for the onset of symptoms in systemic autoimmune disease.
To examine the effect of developmental activation of the AHR on CD4+ T cells and CD4+ T cell-driven antibody responses in the context of autoimmunity, we used a mouse model that mirrors attributes of common systemic human autoimmune diseases. Mice that lack the Gαq protein, Gnaq−/− mice, develop an autoimmune disease with symptoms similar to SLE and RA (Misra et al., 2010). Disease penetrance is 100% in Gnaq−/− mice; however, only some heterozygous Gnaq+/− mice develop symptoms (Misra et al., 2010). Moreover, Gnaq−/− mice develop severe neurological problems (Offermanns et al., 1997), preventing their use in long-term studies examining symptom onset. Therefore, to determine the effects of AHR ligand exposure during development on disease progression later in life, we used heterozygous Gnaq+/− mice. These animals are genetically predisposed, but not guaranteed, to develop autoimmune symptoms as they age. Thus, they provide a model system to establish whether AHR activation during development alters the proportion of offspring that develop autoimmune symptoms, changes in the kinetics of disease development, and/or modulates the CD4+ T-cell subsets and autoantibodies that are thought to be central to disease. These assessments would not be possible using Gnaq−/− mice due to their rapid acquisition of immunological and neurological deficits. We characterized whether changes in disease onset correlated with alterations in CD4+ T-cell subsets, circulating anti-self antibodies, and whether there were differences when the exposure occurred during gestation and lactation (ie, throughout development of the immune system) or only via lactation (ie, after thymopoiesis was initiated). Understanding the impact of AHR activation during discreet windows of immunological development on the progression of many facets of systemic autoimmunity later in life expands our understanding of the implications of early life exposures on immune-mediated disease.
MATERIALS AND METHODS
Animal treatment
Nulliparous Gnaq+/− female mice (age 8–10 weeks) were housed with Gnaq+/− males, and checked daily for the presence of a vaginal plug (day 0 of gestation). Impregnated female mice were treated with 1 μg/kg body weight of TCDD (≥ 99% purity; Cambridge Isotope Laboratories, Woburn, Massachusetts) or peanut oil (vehicle) by gavage on days 0, 7, and 14 of gestation, and 2 days postparturition (Boule et al., 2014; Vorderstrasse et al., 2004). In some experiments, dams were given TCDD or vehicle only once, at 2 days after parturition. All offspring remained with their mother until they were weaned (age 21 days), at which time they were genotyped using PCR. Gnaq−/− and Gnaq+/+ offspring were culled, and the remaining mice were housed with same sex littermates. Blood samples were collected by either tail bleed or cardiac puncture. Mice were housed in a specific-pathogen free facility, with controlled light, temperature, and humidity, and were provided standard mouse chow (LabDiet, 5010) and water ad libitum. Initial breeding stock of Gnaq+/− mice was provided by Dr Frances Lund, and is on a C57Bl/6 (Ahrb/b) background (Misra et al., 2010). All animal treatments were conducted with prior approval of Institutional Animal Care and Use Committee and Institutional Biosafety Committee of the University of Rochester.
Joint measurement and histology
Starting at 12 weeks of age, 1 Gnaq+/− mouse of each sex from each separately treated dam was monitored weekly for development of joint swelling. Specifically, ankle joints on the left and right hind limbs were measured using digital calipers. Mice were considered symptomatic if either ankle swelled to > 2.1 mm for 2 consecutive weeks (1 standard deviation above the normal joint diameter). At 1 year of age, joints were examined by histology. The femur was severed above the knee and all tissue was removed from the bones. The tibia was cut to separate the ankle joint, and the ankle was placed in 10% neutral buffered formalin for 72 h. Ankles were then washed in PBS, and stored in 70% ethanol until embedded in paraffin. Sections were cut (5 μm) and slides were stained with hematoxylin and eosin (H&E). Images were collected using a 4X objective of a BX51 microscope (Olympus, Melville, New York) and SPOT RT camera and computer software (Sterling Heights, Michigan).
Cell preparation and flow cytometry
Spleens were removed at 1 year of age and single cell suspensions were made. Erythrocytes were removed by hypotonic lysis. Isolated cells were colabeled with fluorochrome-conjugated antibodies to specific cell surface molecules to define cell populations as previously described (Boule et al., 2014). Briefly, cells were incubated with antibodies for extracellular molecules, including CD4, CD25, CD44, and CD62L. For intracellular molecules, cells were fixed, permeabilized, and coincubated with antibodies against Foxp3 and TBet. Nonspecific staining was blocked using anti-mouse CD16/32 mAb. All antibodies were from BD Biosciences (San Diego, California) or eBiosciences (San Diego, California). Data were collected using an LSRII flow cytometer (BD Bioscience), and analyzed using FlowJo software (TreeStar, Ashland, Oregon). Fluorescence minus 1 controls were used to define gating parameters.
Enzyme-linked immunosorbent assay
Anti-dsDNA antibodies were detected in serum using isotype-specific enzyme-linked immunosorbent assay as described (Misra et al., 2010). Briefly, plates were coated with 0.01% poly-L-lysine followed by salmon sperm DNA (Life Technologies, Grand Island, New York). After blocking, a dilution series of serum was added to the plate. Biotinylated anti-mouse IgG (Southern Biotech, Birmingham, Alabama) was added followed by avidin-peroxidase (Sigma Aldrich, Milwaukee, Wisconsin). Addition of 2,2′-azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt substrate (MP Biomedicals, Santa Ana, California) led to a colorimetric change, which was read at 405 nm using a SpectraMax Plate reader (Molecular Devices, Sunnyvale, California).
ANA assay
A previously optimized 1:200 dilution of serum was added to fixed HepG2 cells, and anti-nuclear antibodies (ANAs) were detected by anti-mouse IgG conjugated to fluorescein isothiocyanate (FITC) (Southern Biotech) (Misra et al., 2010). Fluorescence was visualized using an Olympus BX43 conventional fluorescence microscope (Olympus America, Center Valley, Pennsylvania), Retiga 1300 camera (QImaging, Surrey, British Columbia), and ImagePro Plus Software (Media Cybernetics, Rockville, Maryland). Brightfield and fluorescent images for each sample were taken using the same magnification and exposure settings. A macro using ImagePro Plus Software was designed to quantify each image. The macro opened a brightfield image, duplicated it, and performed a Gaussian smoothing operation. This image was then background-subtracted from the original image. The resulting image was Sobel filtered and auto-bright thresholded to produce a binary mask, which was then area filtered to a threshold of 9 square pixels. This mask was next applied to the fluorescent image using an AND operation, and auto-bright counts were performed on the fluorescent image to obtain the sum of the object areas and the average FITC fluorescent intensity within the cells.
Statistical analyses
The dam was defined as the statistical unit for all experiments; therefore, offspring in each treatment group were from a different treated dam. Data were analyzed using JMP software (SAS Software, Cary, North Carolina). Differences between 2 groups were evaluated using a Student’s t test. A one-way analysis of variance was used to evaluate differences over time or within a dilution series. The onset of symptoms in Gnaq+/− mice was presented using a Kaplan Meier curve; the difference between treatment groups was tested using the Wilcoxon test. Differences were considered statistically significant when P values were ≤ .05. In addition, to provide a measure of the magnitude of effect of developmental exposure to TCDD on disease onset, we estimated a relative risk (RR). This was estimated as the percentage of symptomatic offspring of TCDD-treated dams divided by the percentage of symptomatic control offspring (ie, the cumulative incidence of symptoms in each treatment group). A RR > 1 indicated a greater risk of symptoms among mice developmentally exposed to TCDD relative to offspring of dams treated with the vehicle control, a RR < 1 indicated a lower risk of symptoms among animals developmentally exposed to TCDD relative to controls, and a RR = 1 indicated no association between treatment group and symptom development (Weiss and Koepsell, 2014). All other data were presented as the mean ± SEM.
RESULTS
Developmental Activation of the AHR Expedites Disease Onset in Gnaq+/− Mice
Recent evidence suggests that certain developmental exposures may alter autoimmune disease progression later in life (Parks et al., 2014). In this study, we used Gnaq+/− mice to examine the effect of early life activation of the AHR by its prototypical ligand TCDD on the progression of autoimmune symptoms in a mouse model of genetic predisposition to autoimmune disease. Gnaq+/− mice were developmentally exposed to TCDD or peanut oil vehicle control and monitored weekly for joint swelling (Figure 1A). The onset of joint swelling was not different in male Gnaq+/− mice (Figure 1B) (RR = 1.07) but female Gnaq+/− offspring of TCDD-treated dams developed disease sooner and at a higher frequency than control offspring (RR = 2.20), although this change in disease progression was not statistically significant (Figure 1C). We did not observe any joint swelling in wild-type Gnaq+/+ littermates of either sex in either treatment group (data not shown). The maximum joint diameter was significantly increased in male (Figure 1D) but not female (Figure 1E), Gnaq+/− mice developmentally exposed to TCDD. Moreover, differences between asymptomatic and symptomatic animals, defined using joint diameter measurements, were consistent with observed joint pathology (Figs. 1F and 1G). Asymptomatic male and female mice (left columns, Figs. 1F and 1G) showed little joint inflammation, and only few immune cells were present. Conversely, symptomatic mice (right columns, Figs. 1F and 1G) had immune infiltrates in the interstitial spaces of their joints, with the highest degree of inflammation present in male and female Gnaq+/− mice that were developmentally exposed to TCDD (bottom right images, Figs. 1F and 1G). Together, the histology and joint swelling measurements demonstrated that developmental activation of the AHR advanced disease in both male and female Gnaq+/− mice, although the more affected metric of enhanced disease varied between sexes.
FIG. 1.
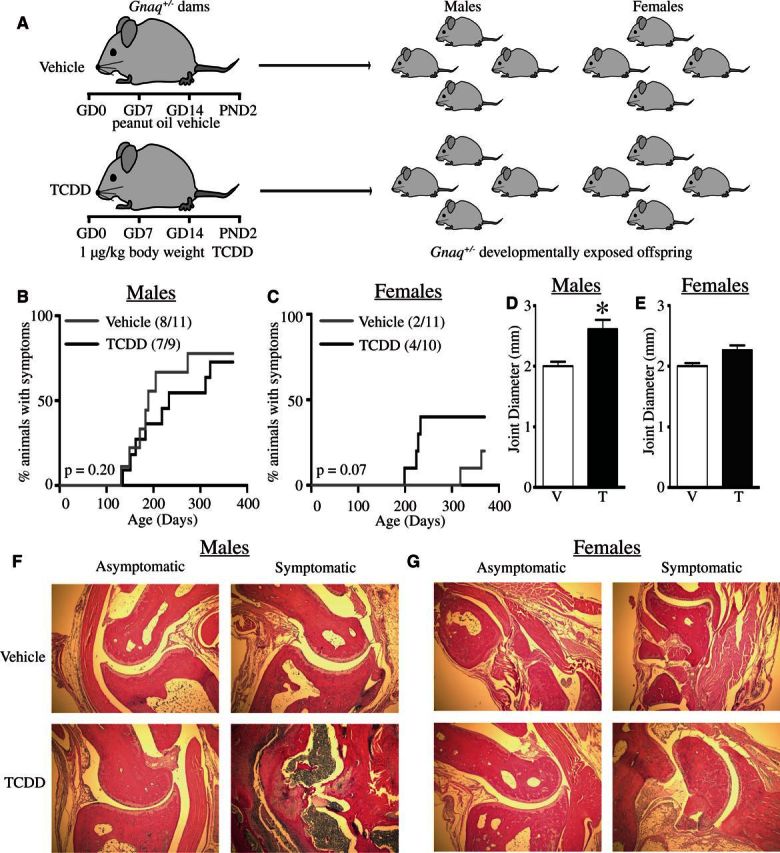
Developmental exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) advances disease progression in Gnaq+/− mice. A, Pregnant Gnaq+/− mice were gavaged with 1 μg TCDD/kg body weight (TCDD) or peanut oil control (Vehicle) on gestational day 0, 7, 14, and postnatal day (PND) 2. B and C, Hind ankle joint diameter of 1 male and 1 female Gnaq+/− offspring from each treated dam was measured weekly starting at 12 weeks of age until they were 1 year old. The percentage of male (B) or female (C) Gnaq+/− offspring that demonstrate swelling (diameter > 2.1 mm for at least 2 consecutive weeks) is shown. D and E, Bar graphs show the maximum joint diameter of Gnaq+/− male (D) and female (E) mice developmentally exposed to vehicle control (V) or TCDD (T). F and G, Representative images of ankle joints stained with H&E are shown for male (F) and female (G) Gnaq+/− asymptomatic (left) and symptomatic (right) mice after developmental exposure to vehicle (top row) or TCDD (bottom row). All graphs show the mean ± SEM and an * signifies a P value ≤ .05.
Activation of the AHR During Development Increases the Circulating Level of Anti-dsDNA Antibodies, but Not ANA, in Female Gnaq+/− Mice
Autoimmune symptom progression is often coupled with an increase in circulating autoreactive antibodies. Two types of self-reactive antibodies typically measured are anti-dsDNA and ANAs (Satoh et al., 2007). We measured the levels of circulating anti-dsDNA antibodies at different dilutions of serum in Gnaq+/− mice that were 1 year old. There were no differences in anti-dsDNA antibodies at any dilution tested in male Gnaq+/− mice (Figure 2A). However, 1-year-old female Gnaq+/− mice that were developmentally exposed to TCDD had approximately 30% higher levels of circulating anti-dsDNA IgG compared with age-matched offspring of vehicle treated dams (Figure 2B). Given that anti-dsDNA antibodies may precede disease onset, we also measured anti-dsDNA antibodies in the serum of developmentally exposed Gnaq+/− mice over time (Figs. 2C and 2D). There was not an increase in anti-dsDNA antibody levels in male Gnaq+/− offspring of vehicle and TCDD-treated dams at any point in time examined (Figure 2C). In developmentally exposed female Gnaq+/− mice, circulating anti-dsDNA antibodies were the same at 3 months. After 3 months of age, female Gnaq+/− mice developmentally exposed to TCDD had increased amounts of circulating anti-dsDNA antibodies compared with controls, with the difference between the 2 groups becoming statistically significant at 1 year of age (Figure 2D). Moreover, the overall levels of anti-dsDNA antibodies in female offspring of TCDD-treated dams were significantly higher over time than vehicle offspring (Figure 2D), yet these levels were not increased in male offspring (Figure 2C).
FIG. 2.

Activation of the aryl hydrocarbon receptor (AHR) during development increases the circulating level of anti-dsDNA IgG in adult female Gnaq+/− mice. A and B, Serum from 1-year-old male and female developmentally exposed Gnaq+/− mice was serially diluted and used in an enzyme-linked immunosorbent assay to compare relative levels of anti-dsDNA IgG, as described in the Materials and Methods. The mean ± SEM absorbance is shown for serum from male (A) and female (B) vehicle or TCDD groups at serum dilutions from 1:8 to 1:1024. C and D, Relative levels of circulating anti-dsDNA IgG were compared over time. A representative dilution (1:64) is shown for male (C) and female (D) Gnaq+/− offspring. The number in the bottom left corner of each graph denotes the P value comparing mean values for vehicle versus TCDD exposed offspring at all dilutions (A, B) or points in time (C, D). Male and female Gnaq+/− offspring from separate dams (n = 3–11) were used for each assay, and an * denotes a P value of ≤ .05.
Another commonly used diagnostic tool for autoimmune disease is the relative level of ANAs, which is often measured using immunohistochemistry (Moroni et al., 2012; Satoh et al., 2007; Tan, 1989). Representative images of ANA staining using serum from male (Figure 3A) and female (Figure 3C) developmentally exposed Gnaq+/− mice are shown. Morphometric quantification revealed no differences in the relative ANA staining frequency or intensity level in serum from male (Figure 3B) or female (Figure 3D) offspring of vehicle and TCDD-treated dams. In addition to examining relative ANA at 1 year, ANA levels were quantified at the same time points described in Figure 2, and no differences were detected after developmental activation of the AHR by TCDD in either sex (data not shown). Therefore, despite differences in joint swelling, male Gnaq+/− mice had no differences in circulating auto-reactive antibodies, yet female Gnaq+/− mice had higher circulating anti-dsDNA antibody, but not ANA, levels at 1 year of age after developmental exposure to TCDD.
FIG. 3.

Developmental activation of the AHR does not change the circulating anti-nuclear antibody (ANA) levels in Gnaq+/− mice. Serum from 1-year-old developmentally exposed Gnaq+/− mice was applied to fixed HepG2 cells at a dilution of 1:200. ANA levels were detected with FITC conjugated anti-mouse IgG. A and C, Representative brightfield (left) and fluorescent images (right) are shown for male (A) and female (C) Gnaq+/− mice developmentally exposed to vehicle or TCDD. B and D, The total FITC intensity within the cells was calculated as described in the Materials and Methods. Graphs depict the average ANA staining intensity for serum from male (B) and female (D) Gnaq+/− mice developmentally exposed to vehicle or TCDD. Offspring from 3–11 Gnaq+/− dams were used. All graphs show the mean ± SEM.
Activation of the AHR During Development Leads to an Increase in Effector CD4+ T Cells in Female Gnaq+/− Mice
Production of autoantibodies is dependent upon CD4+ T cells. Therefore, we enumerated key CD4+ T-cell subsets in developmentally exposed Gnaq+/− mice at 1 year of age. Compared with age-matched offspring of vehicle treated dams, there was no difference in the total number of CD4+ T cells in male or female Gnaq+/− mice after developmental exposure to TCDD (Figs. 4A and 4E). Activated CD4+ T cells are a measure of CD4+ T cells that have reacted to antigen and are defined as CD44hiCD62LloCD4+ cells (Dailey, 1998). At least 50% of the CD4+ T cells were activated in spleens of Gnaq+/− mice (Figs. 4B and 4F). Female Gnaq+/− mice had an even higher proportion of activated CD4+ T cells, with this number increasing to over 75% in those that were developmentally exposed to TCDD (Figure 4F). However, there were no differences in the frequency of activated CD4+ T cells between exposure groups in male Gnaq+/− mice (Figure 4B). In addition, there were no changes in the percentage of Th1 cells (Figure 4C) or Tregs (Figure 4D) in male Gnaq+/− mice after developmental exposure to TCDD. Female developmentally exposed Gnaq+/− mice had no changes in Th1 cells (Figure 4G) but an increased percentage of Tregs in Gnaq+/− offspring of TCDD-treated dams compared with control offspring (Figure 4H). There were no changes in the percentage of Th17 cells in the spleen between treatment groups in male or female developmentally exposed Gnaq+/− mice (data not shown). Together, these data show developmental exposure to TCDD increases both activated and regulatory CD4+ T cells in female, but not male, Gnaq+/− mice.
FIG. 4.

Developmental exposure to TCDD increases the frequency of activated and regulatory CD4+ T cells in female Gnaq+/− mice. Spleen cells were isolated from 1-year-old Gnaq+/− mice that were developmentally exposed to vehicle or TCDD. Cells were stained for flow cytometry and CD4+ T-cell populations were enumerated. Activated CD4+ T cells were defined as CD44hiCD62LloCD4+ cells, Th1 cells as TBet+CD4+ cells, and Tregs as Foxp3+CD25+CD4+ cells. A and E, The graphs depict the average number (± SEM) of CD4+ T cells in the spleen of male (A) and female (E) Gnaq+/− offspring of TCDD-treated or control dams. B–D and F–H, Representative dot plots show the percentage (± SEM) of CD4+ T cells that are activated (B, F), Th1 cells (C, G), or Tregs (D, H) in Gnaq+/− mice developmentally exposed to vehicle (top row) or TCDD (bottom row; B–D male, F–H female). Four to 11 Gnaq+/− offspring from separately treated dams were used, and an * denotes a P value ≤ .05.
Exposure to TCDD Shortly After Birth Worsens Disease Symptoms in Adult Gnaq+/− Offspring
In both mice and humans, much of T-cell development, particularly the establishment of peripheral tolerance by Tregs, occurs shortly after birth (Debock and Flamand, 2014; Sauce and Appay, 2011; Wang et al., 2010). We investigated whether inappropriate AHR activation during this time period altered symptom onset in Gnaq+/− mice by treating dams with vehicle or TCDD 2 days after parturition (Figure 5A). This results in delivery of TCDD via lactation and activates the AHR in the pups (Winans et al., 2015). Both male (Figure 5B) and female (Figure 5C) Gnaq+/− offspring exposed postnatally to TCDD exhibited expedited disease onset, as evidenced by joint swelling (RRmale = 1.38; RRfemale = 4.13). The average joint diameter was significantly increased by lactational exposure to TCDD in male Gnaq+/− mice (Figure 5D). Yet, the average size of the joints of female Gnaq+/− mice exposed to TCDD soon after birth was not significantly larger than control Gnaq+/− mice (Figure 5E). Both male and female Gnaq+/− mice exposed to TCDD after birth had a significant increase in the development of autoimmune symptoms (Figs. 5B and 5C), compared with exposure to TCDD throughout development which led to no statistically significant changes (Figs. 1B and 1C).
FIG. 5.

Early postnatal exposure to TCDD increases disease progression in both female and male Gnaq+/− mice. A, Gnaq+/− dams were gavaged with 1 μg TCDD/kg body weight (TCDD) or peanut oil vehicle (Vehicle) on PND 2. B and C, Gnaq+/− offspring were monitored for joint swelling starting at 12 weeks of age until they were 1 year old as described in Figure 1. Graphs show the percentage of male (B) and female (C) Gnaq+/− offspring that exhibit disease over time. The proportion of offspring with disease is denoted on each graph. D and E, The graphs show the maximum joint diameter of Gnaq+/− male (D) and female (E) mice exposed to vehicle control (V) or TCDD (T) via lactaction. All graphs show the mean ± SEM and an * signifies a P value ≤ .05.
Male Gnaq+/− mice that were exposed to TCDD after birth had reduced circulating anti-dsDNA antibody (Figure 6A) and ANA levels (Figure 6B) at 1 year of age. In contrast, circulating anti-dsDNA antibody and ANA levels in female Gnaq+/− mice exposed to TCDD only during the early postnatal period were not different than offspring of lactating dams given the vehicle control at any dilution of serum or at any point in time between 12 weeks to 1 year of age (Figs. 6C and 6D, and data not shown). Unlike Gnaq+/− mice that were exposed to TCDD throughout gestation and via lactation, male and female Gnaq+/− mice exposed to TCDD after birth had significantly fewer CD4+ T cells in their spleens (Figs. 7A and 7E as compared with Figs. 4A and 4E). Although this early postnatal exposure to TCDD did not change the percentage of activated CD4+ T cells (Figure 7B), it increased the proportion of Th1 cells (Figure 7C) and Tregs (Figure 7D) in male Gnaq+/− mice. Female Gnaq+/− mice exposed to TCDD only via lactation had an increased proportion of activated CD4+ T cells, Th1 cells, and Tregs (Figs. 7F–H). Therefore, in contrast to when developmental exposure begins in the womb, inappropriate AHR activation soon after birth skewed the proportion of CD4+ T-cell subsets in both male and female Gnaq+/− mice.
FIG. 6.

Exposure to TCDD soon after birth does not increase the circulating anti-dsDNA IgG or ANA levels in Gnaq+/− mice. A and C, Circulating anti-dsDNA IgG levels were measured in Gnaq+/− offspring as in Figure 2, except the dams were treated only on PND2. The graphs depict the relative level of anti-dsDNA IgG in lactationally exposed male (A) and female (C) Gnaq+/ − mice. B and D, ANA levels were measured in the serum as in Figure 3. The graphs show the average ANA intensity of male (B) and female (D) Gnaq+/− offspring of TCDD or vehicle dams treated on PND2. The number on the bottom left corner of graphs A and C denotes the P value for all dilutions, comparing vehicle to TCDD exposed Gnaq+/− offspring. All data are mean ± SEM and an * signifies a P value ≤ .05.
FIG. 7.

Male and female Gnaq+/− mice exposed postnatally to TCDD have increased proportions of Th1 and Tregs. Spleen cells were isolated at 1 year of age from male and female Gnaq+/− exposed to TCDD or vehicle peanut oil control (Vehicle) via lactation only, as in Figure 5. Cells were stained for flow cytometry and CD4+ T-cell populations were quantified as in Figure 4. A and E, The graphs depict the average number (± SEM) of CD4+ T cells in male (A) and female (E) Gnaq+/− offspring. B–D and F–H, Representative flow plots show the proportion of CD4+ T-cell subsets in Gnaq+/− mice lactationally exposed to vehicle (top row) and TCDD (bottom row), with the average percentage (± SEM) of each subset denoted on each plot. CD4+ T-cell populations in male mice are represented in B–D and female mice in F–H. Four to 11 Gnaq+/− offspring from separately treated dams were used, and an * denotes a P value ≤ .05.
DISCUSSION
It is becoming recognized that environmental factors may contribute to autoimmune diseases but how they do so remains enigmatic. A recent report from an expert panel convened by the National Institutes of Environmental Health Sciences (U.S. National Institutes of Health), noted that more studies are needed to elucidate which environmental agents contribute to autoimmune diseases, and to establish causal mechanisms that underlie these associations (Parks et al., 2014). We report here 2 major findings: (1) exposure to the AHR ligand TCDD during development, particularly via lactation, expedites the onset and increases the severity of disease in mice genetically predisposed to systemic CD4+ T cell-dependent autoimmune disease and (2) that symptom manifestation differs in male versus female Gnaq+/− mice developmentally exposed to TCDD. Differences in symptom manifestation have also been reported in the human population (Fairweather et al., 2008), suggesting that multiple measures of symptom onset will be important for studies examining autoimmune diseases in exposed human populations. Together, these data indicate that early life exposure to environmental chemicals that bind the AHR may expedite or exacerbate autoimmune disease in genetically predisposed individuals.
The establishment of the CD4+ T-cell repertoire, including important central and peripheral tolerance mechanisms involving Tregs, starts in the womb, but continues after birth in both mice and humans (Bommhardt et al., 2004; Debock and Flamand, 2014; Klein et al., 2009; Sauce and Appay, 2011; Wang et al., 2010). Exposure to TCDD during development increased the frequency of effector CD4+ T-cell populations in male and female Gnaq+/− mice, and these changes were the most profound when exposure happened during the early postnatal period. Therefore, the developmental window in which AHR activation occurs may have differential effects on the function of CD4+ T cells later in life. This agrees with prior work examining critical windows of exposure to TCDD for other altered immune responses. For instance, TCDD exposure via lactation only was sufficient to impair the virus-specific CD8+ T-cell response to infection. However, developmental exposure only perturbed neutrophil responses during viral infection when exposure occurred during both gestation and lactation (Hogaboam et al., 2008). This suggests that while the later in life responses of myeloid (neutrophils) and lymphoid (T cells) lineages are perturbed by AHR activation during development, the window of time in which the persistent changes are established differs for different cell types. This may be due to many factors, including when a particular cell lineage initially becomes present in the developing offspring, AHR-mediated changes in other important cell types that interact with these lineages or their precursors, and/or effects on critical epigenetic program established during specific periods of development.
The importance of AHR activation on CD4+ T-cell differentiation and function is becoming increasingly evident. “Direct” AHR activation in adult animals changes the differentiation pattern of CD4+ T cells, leading to profound alterations in autoimmune disease progression (Quintana and Sherr, 2013). We have extended these observations by showing that developmental AHR activation also affects peripheral CD4+ T cells in the context of autoimmune disease. Yet, at least in the model system used, the changes observed are not precisely the same as the effects of “direct” AHR ligand treatment, suggesting different underlying mechanisms may be at play. Nevertheless, when viewed in their totality, these studies suggest that AHR activation is an important consideration in understanding CD4+ T-cell function in a variety of immunological settings. For instance, the timing of AHR activation is of critical importance in establishing the durability of AHR-mediated changes in this cell lineage. Changes observed after “direct” AHR activation in adult animals are transient (Kerkvliet et al., 2009; Quintana et al., 2008), whereas the increase in effector CD4+ T-cell populations observed in this study occurred 1 year after the developmental exposure. This suggests that there are persistent modifications in the differentiation capacity of CD4+ T cells that occur as a result of developmental AHR activation by an exogenous ligand. Furthermore, alterations in CD4+ T-cell differentiation imparted by developmental activation of the AHR can work both independently and in conjunction with other changes in CD4+ T cells. For example, lack of the Gαq protein enhances the differentiation of CD4+ T cells into Th17 cells, which is likely due to direct Gαq-mediated changes in CD4+ T cells, and changes in important antigen presenting cells (Liu et al., 2015). Moreover, it has been shown that activation of the AHR in adult animals alters the proportion of Th17 cells in several inducible models of autoimmunity in mice (Quintana and Sherr, 2013). Yet, we observed no changes in Th17 cells in Gnaq+/− mice after developmental activation of the AHR (data not shown), despite increases in other CD4+ T cell subsets in offspring developmentally exposed to TCDD compared with controls. Thus, many pathways are important for defining the differentiation of CD4+ T cells in the context of an immune response. Activation of the AHR is an important consideration, as is the interaction of AHR-mediated changes with other pathways critical for CD4+ T-cell differentiation.
In addition to establishing that, in the context of an autoimmune disease, CD4+ T cells are altered by developmental AHR activation, these data add to the growing information that suggest context-dependent changes in CD4+ T cells are imparted by developmental exposure to AHR ligands. Upon viral infection, there was a decrease in the frequency of conventional effector CD4+ T cells in lymph nodes of developmentally exposed mice (Boule et al., 2014), yet an increase in the same conventional CD4+ effectors in the infected lung (Boule et al., 2015). This increased proportion of conventional CD4+ T cells also occurred in the spleen of developmentally exposed Gnaq+/− mice reported in this study. In contrast to conventional helper cells, developmental activation of the AHR increased the proportion of Tregs in both the lymph nodes and lungs of virally infected mice (Boule et al., 2014, 2015), and in the spleen of Gnaq+/− mice. Together, these observations suggest that these persistent changes in CD4+ T cells are revealed in a context-dependent fashion.
Along with context and tissue-specific changes in cellular responses, this study also suggests that interactions between genetics and environmental exposures early in life are another central consideration. Our observations suggest that the genetic alteration in Gnaq+/− mice may enhance any context-dependent changes in CD4+ T cells that are imparted by developmental exposure. For example, given that lack of the Gαq protein increases B cell and CD4+ T-cell survival (Misra et al., 2010; Wang et al., 2014), lower levels of Gαq may alter mechanisms that promote the death of autoreactive lymphocytes. Direct AHR activation in adult animals alters apoptotic mechanisms; thus, changes in apoptosis that are critical for selection processes that maintain immunological tolerance may also be modified by developmental activation of the AHR (Camacho et al., 2005; Esser et al., 1994; Ishimaru et al., 2009; Lai et al., 1994; Laiosa et al., 2010; Nagai et al., 2006). Yet, the decrease in total CD4+ T cells, despite increases in effector populations, suggests the involvement of other immune cell types in propagating disease in Gnaq+/− mice. One cell type could be B cells. Both B and T cells drive disease development in Gnaq+/− mice, as either cell type is sufficient to recapitulate symptoms in asymptomatic control mice, and both cell types exhibit functional changes in Gnaq+/− mice (Misra et al., 2010; Wang et al., 2014). However, additional observations suggest that the mechanism is likely more complex than deregulation of apoptosis and changes in B-T cell interactions. In particular, we did not observe any changes in disease onset, severity, or CD4+ T-cell subset proportions in 2 different inducible models of autoimmune disease after developmental exposure to TCDD: experimental autoimmune encephalomyelitis and streptozotocin-induced diabetes (data not shown). This suggests that gene-environment interactions early in life are important for the increased progression of autoimmune disease in developmentally exposed animals, likely due to persistent changes that are later revealed in specific contexts. Therefore, deviations in immune cells prompted by early life AHR activation and a genetic predisposition to altered tolerance and other immunoregulatory mechanisms may synergize in specific contexts, resulting in an increase in the incidence and severity of systemic autoimmune diseases. Consequently, a combination of signals derived from gene-environment interactions, immunogens, and the tissues involved are important considerations in defining how of environmental exposures during development influence immune-mediated disease later in life.
Biomarkers measured in human cohorts are important for establishing links between developmental exposures and immune-mediated diseases later in life. Currently, specific predictive biomarkers remain poorly defined. The data presented here provide important pieces of evidence regarding what biomarkers are relevant to autoimmune diseases. Although circulating anti-dsDNA antibodies and ANAs are commonly used in clinical diagnostics, neither metric was overtly changed in Gnaq+/− mice exposed to TCDD during development, despite the observed alterations in disease symptoms. Consistent with these findings in mice, a recent study examining exposures to AHR ligands in the U.S. population also found no relationship between ANAs and exposure levels (Dinse et al., 2015). However, the relationship between AHR ligands and diagnosis of clinical autoimmune disease in that study population has not been explored. Our data suggest the measurement of CD4+ T cells in human cohorts should be further investigated as a biomarker of developmental exposure to AHR ligands. CD4+ T-cell number alone is not a sufficient measure, as Gnaq+/− mice developmentally exposed to TCDD either had no changes or a decrease in total CD4+ T cells; however, we observed changes in the proportion of different CD4+ T-cell subsets. Examining the phenotype of CD4+ T cells will be more informative in understanding the connection between developmental exposures and autoimmune disease later in life.
Work in this study, and in other animal models examining early life exposures and autoimmunity, suggests examination of specific tissues is an important consideration in linking exposures with disease onset (Holladay et al., 2011; Mustafa et al., 2011a,b, 2008; Parks et al., 2014). Analysis of tissues important in autoimmune disease may be difficult to accomplish in human cohorts, but with the rapid development of technology, studies examining organs using noninvasive techniques may become feasible in the future. For example, early studies examining thymic involution after developmental exposure to TCDD (Faith and Moore, 1977; Thomas and Hinsdill, 1979; Vos and Moore, 1974) have been recently corroborated in humans through the use of ultrasonography (Jusko et al., 2012; Park et al., 2008). In addition, new methodologies for the early detection of immune-mediated events leading to arthritis that use noninvasive techniques are being developed and validated (Bouta et al., 2013; Ju et al., 2012).
The work presented here furthers our understanding of how developmental exposures lead to lasting changes in the immune system. Previous work examining early life exposures to ligands of the AHR and autoimmune disease have suggested that there is an increase in immune complex deposition after developmental exposure (Holladay et al., 2011; Mustafa et al., 2011a,b, 2008; Parks et al., 2014). We have extended these observations by showing that early life exposure to TCDD expedites disease onset, severity, and alters CD4+ T-cell cellularity in mice predisposed to the development of systemic autoimmunity, providing evidence for immunological biomarkers that could be important for associations between exposed human populations and autoimmune disease.
FUNDING
This work was supported by research and training grants from the National Institutes of Health (grant numbers R01-ES017250, R01-ES023260, T32-ES07026, T32-AI007285, T32-HL066988, K12-ES019852, and P30-ES01247) and funds from the University of Rochester.
ACKNOWLEDGMENTS
The authors are grateful to Dr Frances Lund for her advice on the initial study design, and for providing the initial stock of Gnaq+/− breeders. They also thank Dr Deborah Fowell for advice on CD4+ T-cell subset analyses, Dr Ravi Misra for his expertise and guidance in using the Gnaq+/− model, and Dr Scott Gerber for his assistance with microscopy. They thank the University of Rochester Medical Center Environmental Health Science Center Histology Core and Daria Krenitsky for help with the joint histology. They are also grateful to Dr Timothy Bushnell and the outstanding team at the URMC Flow Cytometry Core.
REFERENCES
- Bogdanos D. P., Smyk D. S., Rigopoulou E. I., Mytilinaiou M. G., Heneghan M. A., Selmi C., Gershwin M. E. (2012). Twin studies in autoimmune disease: Genetics, gender and environment. J. Autoimmun. 38, J156–J169. [DOI] [PubMed] [Google Scholar]
- Bommhardt U., Beyer M., Hunig T., Reichardt H. M. (2004). Molecular and cellular mechanisms of T cell development. Cell. Mol. Life Sci. 61, 263–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boule L. A., Winans B., Lambert K., Vorderstrasse B. A., Topham D. J., Pavelka M. S., Jr, Lawrence B. P. (2015). Activation of the aryl hydrocarbon receptor during development enhances the pulmonary CD4+ T cell response to viral infection. Am. J. Physiol. Lung Cell Mol. Physiol. 309, L305–L313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boule L. A., Winans B., Lawrence B. P. (2014). Effects of developmental activation of the AhR on CD4+ T-cell responses to influenza virus infection in adult mice. Environ. Health Perspect. 122, 1201–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouta E. M., Ju Y., Rahimi H., de Mesy-Bentley K. L., Wood R. W., Xing L., Schwarz E. M. (2013). Power Doppler ultrasound phenotyping of expanding versus collapsed popliteal lymph nodes in murine inflammatory arthritis. PLoS One 8, e73766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho I. A., Singh N., Hegde V. L., Nagarkatti M., Nagarkatti P. S. (2005). Treatment of mice with 2,3,7,8-tetrachlorodibenzo-p-dioxin leads to aryl hydrocarbon receptor-dependent nuclear translocation of NF-kappaB and expression of Fas ligand in thymic stromal cells and consequent apoptosis in T cells. J. Immunol. 175, 90–103. [DOI] [PubMed] [Google Scholar]
- Dailey M. O. (1998). Expression of T lymphocyte adhesion molecules: Regulation during antigen-induced T cell activation and differentiation. Crit. Rev. Immunol. 18, 153–184. [DOI] [PubMed] [Google Scholar]
- Debock I., Flamand V. (2014). Unbalanced neonatal CD4(+) T-cell immunity. Front. Immunol. 5, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinse G. E., Jusko T. A., Whitt I. Z., Co C. A., Parks C. G., Satoh M., Chan E. K., Rose K. M., Walker N. J., Birnbaum L. S., et al. (2015). Associations between selected xenobiotics and antinuclear antibodies in the national health and nutrition examination survey, 1999-2004. Environ. Health Perspect. DOI: 10.1289/ehp1409345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J. A., Kemp A. S., Ponsonby A. L. (2014). Gene-environment interaction in autoimmune disease. Expert. Rev. Mol. Med. 16, e4. [DOI] [PubMed] [Google Scholar]
- Esser C., Lai Z., Gleichmann E. (1994). Proliferation inhibition and CD4/CD8 thymocyte subset skewing by in vivo exposure of C57BL/6 mice to Ah receptor-binding 3,3',4,4'-tetrachlorobiphenyl. Exp. Clin. Immunogenet. 11, 75–85. [DOI] [PubMed] [Google Scholar]
- Fairweather D., Frisancho-Kiss S., Rose N. R. (2008). Sex differences in autoimmune disease from a pathological perspective. Am. J. Pathol. 173, 600–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith R. E., Moore J. A. (1977). Impairment of thymus-dependent immune functions by exposure of the developing immune system to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). J Toxicol. Environ. Health 3, 451–464. [DOI] [PubMed] [Google Scholar]
- Gilbert K. M., Woodruff W., Blossom S. J. (2014). Differential immunotoxicity induced by two different windows of developmental trichloroethylene exposure. Autoimmun. Dis. 2014, 982073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogal R. M., Jr, Holladay S. D. (2008). Perinatal TCDD exposure and the adult onset of autoimmune disease. J. Immunotoxicol. 5, 413–418. [DOI] [PubMed] [Google Scholar]
- Hogaboam J. P., Moore A. J., Lawrence B. P. (2008). The aryl hydrocarbon receptor affects distinct tissue compartments during ontogeny of the immune system. Toxicol. Sci. 102, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holladay S. D., Mustafa A., Gogal R. M., Jr (2011). Prenatal TCDD in mice increases adult autoimmunity. Reprod. Toxicol. 31, 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimaru N., Takagi A., Kohashi M., Yamada A., Arakaki R., Kanno J., Hayashi Y. (2009). Neonatal exposure to low-dose 2,3,7,8-tetrachlorodibenzo-p-dioxin causes autoimmunity due to the disruption of T cell tolerance. J. Immunol. 182, 6576–6586. [DOI] [PubMed] [Google Scholar]
- Jancar S., Sanchez Crespo M. (2005). Immune complex-mediated tissue injury: A multistep paradigm. Trends Immunol. 26, 48–55. [DOI] [PubMed] [Google Scholar]
- Ju Y., Rahimi H., Li J., Wood R. W., Xing L., Schwarz E. M. (2012). Validation of 3-dimensional ultrasound versus magnetic resonance imaging quantification of popliteal lymph node volume as a biomarker of erosive inflammatory arthritis in mice. Arthritis. Rheum. 64, 2048–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jusko T. A., Sonneborn D., Palkovicova L., Kocan A., Drobna B., Trnovec T., Hertz-Picciotto I. (2012). Pre- and postnatal polychlorinated biphenyl concentrations and longitudinal measures of thymus volume in infants. Environ. Health Perspect. 120, 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlson E. W., Deane K. (2012). Environmental and gene-environment interactions and risk of rheumatoid arthritis. Rheum. Dis. Clin. North Am. 38, 405–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkvliet N. I., Steppan L. B., Vorachek W., Oda S., Farrer D., Wong C. P., Pham D., Mourich D. V. (2009). Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy 1, 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein L., Hinterberger M., Wirnsberger G., Kyewski B. (2009). Antigen presentation in the thymus for positive selection and central tolerance induction. Nat. Rev. Immunol. 9, 833–844. [DOI] [PubMed] [Google Scholar]
- Lai Z. W., Kremer J., Gleichmann E., Esser C. (1994). 3,3',4,4'-Tetrachlorobiphenyl inhibits proliferation of immature thymocytes in fetal thymus organ culture. Scand. J. Immunol. 39, 480–488. [DOI] [PubMed] [Google Scholar]
- Laiosa M. D., Mills J. H., Lai Z. W., Singh K. P., Middleton F. A., Gasiewicz T. A., Silverstone A. E. (2010). Identification of stage-specific gene modulation during early thymocyte development by whole-genome profiling analysis after aryl hydrocarbon receptor activation. Mol. Pharmacol. 77, 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Wang D., Li F., Shi G. (2015). Galphaq controls rheumatoid arthritis via regulation of Th17 differentiation. Immunol. Cell Biol. 93, 616–624. [DOI] [PubMed] [Google Scholar]
- Mayadas T. N., Tsokos G. C., Tsuboi N. (2009). Mechanisms of immune complex-mediated neutrophil recruitment and tissue injury. Circulation 120, 2012–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra R. S., Shi G., Moreno-Garcia M. E., Thankappan A., Tighe M., Mousseau B., Kusser K., Becker-Herman S., Hudkins K. L., Dunn R., et al. (2010). G alpha q-containing G proteins regulate B cell selection and survival and are required to prevent B cell-dependent autoimmunity. J. Exp. Med. 207, 1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni L., Bianchi I., Lleo A. (2012). Geoepidemiology, gender and autoimmune disease. Autoimmun. Rev. 11, A386–A392. [DOI] [PubMed] [Google Scholar]
- Mustafa A., Holladay S., Witonsky S., Zimmerman K., Manari A., Countermarsh S., Karpuzoglu E., Gogal R. (2011a). Prenatal TCDD causes persistent modulation of the postnatal immune response, and exacerbates inflammatory disease, in 36-week-old lupus-like autoimmune SNF1 mice. Birth Defects Res. B Dev. Reprod. Toxicol. 92, 82–94. [DOI] [PubMed] [Google Scholar]
- Mustafa A., Holladay S. D., Goff M., Witonsky S. G., Kerr R., Reilly C. M., Sponenberg D. P., Gogal R. M., Jr (2008). An enhanced postnatal autoimmune profile in 24 week-old C57BL/6 mice developmentally exposed to TCDD. Toxicol. Appl. Pharmacol. 232, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa A., Holladay S. D., Witonsky S., Sponenberg D. P., Karpuzoglu E., Gogal R. M., Jr (2011b). A single mid-gestation exposure to TCDD yields a postnatal autoimmune signature, differing by sex, in early geriatric C57BL/6 mice. Toxicology 290, 156–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai H., Kubo M., Abe R., Yamamoto M., Nohara K. (2006). Constitutive activation of the aryl hydrocarbon receptor in T-lineage cells induces thymus involution independently of the Fas/Fas ligand signaling pathway. Int. Immunopharmacol. 6, 279–286. [DOI] [PubMed] [Google Scholar]
- Nguyen L. P., Bradfield C. A. (2008). The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 21, 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermanns S., Hashimoto K., Watanabe M., Sun W., Kurihara H., Thompson R. F., Inoue Y., Kano M., Simon M. I. (1997). Impaired motor coordination and persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking Galphaq. Proc. Natl. Acad. Sci. U.S.A. 94, 14089–14094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H. Y., Hertz-Picciotto I., Petrik J., Palkovicova L., Kocan A., Trnovec T. (2008). Prenatal PCB exposure and thymus size at birth in neonates in Eastern Slovakia. Environ. Health Perspect. 116, 104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks C. G., Miller F. W., Pollard K. M., Selmi C., Germolec D., Joyce K., Rose N. R., Humble M. C. (2014). Expert panel workshop consensus statement on the role of the environment in the development of autoimmune disease. Int. J. Mol. Sci. 15, 14269–14297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana F. J., Basso A. S., Iglesias A. H., Korn T., Farez M. F., Bettelli E., Caccamo M., Oukka M., Weiner H. L. (2008). Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71. [DOI] [PubMed] [Google Scholar]
- Quintana F. J., Sherr D. H. (2013). Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol. Rev. 65, 1148–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael I., Nalawade S., Eagar T. N., Forsthuber T. G. (2014). T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. 74, 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S., Ono M., Setoguchi R., Yagi H., Hori S., Fehervari Z., Shimizu J., Takahashi T., Nomura T. (2006). Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 212, 8–27. [DOI] [PubMed] [Google Scholar]
- Satoh M., Chan E. K., Sobel E. S., Kimpel D. L., Yamasaki Y., Narain S., Mansoor R., Reeves W. H. (2007). Clinical implication of autoantibodies in patients with systemic rheumatic diseases. Expert. Rev. Clin. Immunol. 3, 721–738. [DOI] [PubMed] [Google Scholar]
- Sauce D., Appay V. (2011). Altered thymic activity in early life: How does it affect the immune system in young adults? Curr. Opin. Immunol. 23, 543–548. [DOI] [PubMed] [Google Scholar]
- Somers E. C., Richardson B. C. (2014). Environmental exposures, epigenetic changes and the risk of lupus. Lupus 23, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan E. M. (1989). Antinuclear antibodies: Diagnostic markers for autoimmune diseases and probes for cell biology. Adv. Immunol. 44, 93–151. [DOI] [PubMed] [Google Scholar]
- Thomas P. T., Hinsdill R. D. (1979). The effect of perinatal exposure to tetrachlorodibenzo-p-dioxin on the immune response of young mice. Drug Chem. Toxicol. 2, 77–98. [DOI] [PubMed] [Google Scholar]
- Vorderstrasse B. A., Cundiff J. A., Lawrence B. P. (2004). Developmental exposure to the potent aryl hydrocarbon receptor agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin Impairs the cell-mediated immune response to infection with influenza a virus, but enhances elements of innate immunity. J. Immunotoxicol. 1, 103–112. [DOI] [PubMed] [Google Scholar]
- Vos J. G., Moore J. A. (1974). Suppression of cellular immunity in rats and mice by maternal treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Int. Arch. Allergy Appl. Immunol. 47, 777–794. [DOI] [PubMed] [Google Scholar]
- Wang D., Zhang Y., He Y., Li Y., Lund F. E., Shi G. (2014). The deficiency of Galphaq leads to enhanced T-cell survival. Immunol. Cell. Biol. 92, 781–790. [DOI] [PubMed] [Google Scholar]
- Wang G., Miyahara Y., Guo Z., Khattar M., Stepkowski S. M., Chen W. (2010). “Default” generation of neonatal regulatory T cells. J. Immunol. 185, 71–78. [DOI] [PubMed] [Google Scholar]
- Weiss N., Koepsell T. (2014). Epidemiologic Methods: Studying the Occurence of Illness, 2th ed. Oxford University Press, New York, NY. [Google Scholar]
- Winans B., Humble M. C., Lawrence B. P. (2011). Environmental toxicants and the developing immune system: A missing link in the global battle against infectious disease? Reprod. Toxicol. 31, 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winans B., Nagari A., Chae M., Post C. M., Ko C. I., Puga A., Kraus W. L., Lawrence B. P. (2015). Linking the aryl hydrocarbon receptor with altered DNA methylation patterns and developmentally induced aberrant antiviral CD8+ T cell responses. J. Immunol. 194, 4446–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane H., Paul W. E. (2013). Early signaling events that underlie fate decisions of naive CD4(+) T cells toward distinct T-helper cell subsets. Immunol. Rev. 252, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]