

ABSTRACT
Chronic hepatitis B (CHB), chronic hepatitis C (CHC), and associated hepatocellular carcinoma (HCC) are characterized by cholesterol imbalance and dyslipidemia; however, the key regulatory drivers of these phenotypes are incompletely understood. Using gene expression microarrays and high-throughput sequencing of small RNAs, we performed integrative analysis of microRNA (miRNA) and gene expression in nonmalignant and matched cancer tissue samples from human subjects with CHB or CHC and HCC. We also carried out follow-up functional studies of specific miRNAs in a cell-based system. These studies led to four major findings. First, pathways affecting cholesterol homeostasis were among the most significantly overrepresented among genes dysregulated in chronic viral hepatitis and especially in tumor tissue. Second, for each disease state, specific miRNA signatures that included miRNAs not previously associated with chronic viral hepatitis, such as miR-1307 in CHC, were identified. Notably, a few miRNAs, including miR-27 and miR-224, were components of the miRNA signatures of all four disease states: CHB, CHC, CHB-associated HCC, and CHC-associated HCC. Third, using a statistical simulation method (miRHub) applied to the gene expression data, we identified candidate master miRNA regulators of pathways controlling cholesterol homeostasis in chronic viral hepatitis and HCC, including miR-21, miR-27, and miR-33. Last, we validated in human hepatoma cells that both miR-21 and miR-27 significantly repress cholesterol synthesis and that miR-27 does so in part through regulation of the gene that codes for the rate-limiting enzyme 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase (HMGCR).
IMPORTANCE
Hepatitis B virus (HBV) and hepatitis C virus (HCV) are phylogenetically unrelated hepatotropic viruses that persistently infect hundreds of millions of people world-wide, often leading to chronic liver disease and hepatocellular carcinoma (HCC). Chronic hepatitis B (CHB), chronic hepatitis C (CHC), and associated HCC often lead to cholesterol imbalance and dyslipidemia. However, the regulatory mechanisms underlying the dysregulation of lipid pathways in these disease states are incompletely understood. MicroRNAs (miRNAs) have emerged as critical modulators of lipid homeostasis. Here we use a blend of genomic, molecular, and biochemical strategies to identify key miRNAs that drive the lipid phenotypes of chronic viral hepatitis and HCC. These findings provide a panoramic view of the miRNA landscape in chronic viral hepatitis, which could contribute to the development of novel and more-effective miRNA-based therapeutic strategies.
INTRODUCTION
Hepatitis B virus (HBV) and hepatitis C virus (HCV) are noncytopathic viruses that both exhibit marked tropism for liver parenchymal cells and persistently infect hundreds of millions of people world-wide, often leading to chronic liver disease and cancer (1). Together, these viral infections are responsible for ~80% of hepatocellular carcinoma (HCC), which is the third leading cause of cancer-related deaths. Though HBV and HCV are phylogenetically unrelated viruses, chronic infections with either virus can lead to dyslipidemia and cholesterol imbalance (2–5). Liver cancer is also associated with aberrant lipid and lipoprotein metabolism (6, 7). While some of the molecular features of this phenotype have been described, including altered transcriptional activity of sterol regulatory element-binding protein 1 (SREBP1) in chronic HCV infection (8), the pathogenic mechanisms, in particular the roles of microRNAs (miRNAs), underlying dysregulation of cholesterol pathways in chronic hepatitis C (CHC), and especially chronic hepatitis B (CHB) and HCC, remain poorly understood.
miRNAs, which are small RNAs that posttranscriptionally regulate gene expression, have emerged as critical modulators of lipid homeostasis (9–11). The first miRNA that was reported to control lipid pathways in the liver was miR-122 (12), which was also subsequently shown to promote HCV replication (13). Since the discovery of miR-122, several other miRNAs have been implicated in the control of lipid homeostasis. For example, miR-34a contributes to hepatic steatosis via regulation of sirtuins (14), miR-33 controls cholesterol flux through regulation of ATP-binding cassette transporters (15–17), miR-30 regulates lipoprotein secretion by repressing microsomal triglyceride transfer protein (18), and miR-29 fine-tunes the FOXA2-controlled gene network governing lipid homeostasis (19). We hypothesize that miRNAs play a key role in the lipid imbalance that has been observed in chronic viral hepatitis and associated HCC.
In this study, we performed an integrative analysis of miRNA and gene expression profiles in nonmalignant and matched cancer tissue samples from patients with CHB or CHC and associated HCC. This study represents only the second sequencing-based profiling of miRNAs in chronic viral hepatitis. We report four major findings. First, we established that cholesterol synthesis and metabolism pathways are among the most dominantly altered in each of the disease states, particularly HCC. Second, for each disease state, we defined distinct liver miRNA signatures that included miRNAs not previously associated with viral hepatitis. Third, we identified by integrative analysis of small-RNA and mRNA expression data a suite of miRNAs that represent candidate drivers of the altered cholesterol pathways in chronic viral hepatitis. Fourth, we validated through cell-based assays that miR-27, which is prominently dysregulated in both CHB and CHC as well as in HCC, significantly represses cholesterol synthesis in part through regulation of the gene that codes for the rate-limiting enzyme 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase (HMGCR).
RESULTS
Pathways mediating cholesterol homeostasis are prominently altered in chronic viral hepatitis and liver cancer.
We interrogated previously described gene expression microarray data sets from matched cancer and noncancer liver tissue samples collected from Japanese adults with chronic hepatitis B (CHB) (n = 10) or chronic hepatitis C (CHC) (n = 16) and chronic HCC (chronic hepatitis B-associated cancer [c-CHB] [n = 10] and chronic hepatitis C-associated cancer [c-CHC] [n = 16]) as well as uninfected controls (n = 9) (see Tables S1 and S2 in the supplemental material) (20, 21). Correlation analyses of gene expression profiles showed that all samples within each disease category were highly correlated with each other (Fig. 1A), although the cancer tissue samples were less strongly correlated (c-CHB median pairwise r2 = 0.86; c-CHC median pairwise r2 = 0.88) than the nonmalignant CHB and CHC samples (median pairwise r2 = 0.96 and r2 = 0.95, respectively).
FIG 1 .

Global gene expression in chronic viral hepatitis and cancer. (A and B) Pearson correlation coefficient heat maps. Each cell in the map represents the correlation in expression profiles between two samples. Cells along the diagonal line represent identical samples and are shown in white. Black lines divide each disease group. The midpoint for color bar r2 is 0.5. (A) Data from microarray analyses include only genes with average normalized expression of >1,000 (n = 8,162) in uninfected controls (n = 9), CHB and matched c-CHB (n = 10 each), and CHC and matched c-CHC (n = 16 each). (B) The Cancer Genome Atlas RNA-seq data include genes with an average normalized read count of >50 (n = 13,017) in each disease category: c-CHB (n = 12), CHB (n = 6), c-CHC (n = 18), and CHC (n = 5). (C and D) Analysis of differentially expressed (DE) genes in each disease category compared to the uninfected controls with a fold change of >2 and ANOVA P value of <0.05 after Benjamini-Hochberg step-up multiple testing correction. (C) Number of DE genes. (D) Proportional Venn diagrams comparing the DE genes of the different disease groups.
We then repeated this analysis using publically available RNA sequencing data from The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/) for 18 c-CHB tissue samples and 6 matched CHB tissue samples as well as 18 c-CHC tissue samples and 5 matched CHC tissue samples. We again found that the cancer tissue samples were more poorly correlated (c-CHB median pairwise r2 = 0.58; c-CHC median pairwise r2 = 0.70) with each other compared to the noncancer tissue samples (median pairwise r2 = 0.98) (Fig. 1B). The weaker pairwise correlations among c-CHB and c-CHC samples in the TCGA cohort compared to the microarray cohort may be due at least in part to the much higher dynamic range of expression provided by deep sequencing relative to microarray (22), as well as increased genetic heterogeneity (due to representation from several different ancestries) among individuals in the TCGA cohort relative to the Japanese cohort.
Next, using the microarray data from the Japanese cohort, we identified genes that were significantly differentially expressed (DE) in each of the four disease categories compared to the uninfected controls (see Table S2 in the supplemental material). We found that CHB and c-CHB had twice as many DE genes than CHC and c-CHC, respectively (Fig. 1C). Also, c-CHB and c-CHC had ~3-fold-more DE genes than their nonmalignant counterparts (Fig. 1C). For both CHB and CHC, 60% of DE genes were also significantly altered in the matched cancer tissue samples (Fig. 1D). While only 17% of DE genes in CHC were also shared with CHB, 82% of DE genes in c-CHC were shared with c-CHB (Fig. 1D).
To determine the pathways that are most affected in each disease category, we performed Ingenuity Pathway Analysis (IPA). We found that the DE genes in CHB and CHC were most significantly overrepresented in distinct immune response pathways (Table 1). For genes uniquely DE in CHB, the most enriched pathway was CD28 signaling, which is critical for T-cell activation and the production of interleukins (23). For genes uniquely DE in CHC, the most enriched pathway was interferon signaling, which is critical to innate immunity (24). For genes DE in both CHB and CHC, the most enriched pathways were related to lipid metabolism, including liver X receptor (LXR)/retinoid X receptor (RXR) activation, which plays a central role in cholesterol homeostasis (25–27). Lipid-related pathways were even more prevalent among the DE genes in c-CHB and c-CHC (Table 2). For both CHB and CHC, the top three most significantly affected pathways were related to the function of RXR, which together with LXR and FXR regulates cholesterol balance. For genes DE in both c-CHB and c-CHC, another significantly enriched pathway was aryl hydrocarbon receptor (AHR) signaling, which is involved in the suppression of hepatic cholesterol synthesis (28). Indeed, we found that many of the genes that code for enzymes in the cholesterol synthesis pathway were downregulated in all disease categories compared to uninfected controls (Fig. 2).
TABLE 1 .
Top 10 most significantly enriched pathways by pathway enrichment analysis of chronic viral hepatitis
| Most significantly enriched pathway (P value) using DE genes from the following disease group(s)a: | ||||
|---|---|---|---|---|
| CHB all | CHB only | CHB and CHC overlap | CHC all | CHC only |
| Signaling by Rho family GTPases (3.55E−04) | CD28 signaling in T helper cells (1.05E−04) | Phosphatidylcholine biosynthesis (1.02E−03) | Phosphatidylcholine biosynthesis (1.86E−06) | Interferon signaling (4.68E−07) |
| Natural killer cell signaling (3.63E−04) | Tec kinase signaling (1.7E−04) | Choline biosynthesis (3.72E−03) | Interferon signaling (1.66E−05) | Pathogenesis of multiple sclerosis (4.57E−05) |
| Role of PRRs (4.47E−04) | Signaling by Rho family GTPases (2.00E−04) | Polyamine regulation in colon cell proliferation/cancer (1.05E−02) | Pathogenesis of multiple sclerosis (2.82E−04) | iNOS signaling (4.79E−04) |
| CD28 signaling in T helper cells (7.08E−04) | Natural killer cell signaling (2.09E−04) | Epoxysqualene biosynthesis (1.41E−02) | Choline biosynthesis (9.2E−04) | Phosphatidylcholine biosynthesis (1.41E−03) |
| CXCR4 signaling (9.12E−04) | CTLA4 signaling in cytotoxic T lymphocytes (5.0E−04) | Toll-like receptor signaling (1.58E−02) | Type I diabetes mellitus signaling (1.58E−03) | Type I diabetes mellitus signaling (2.24E−03) |
| Integrin signaling (1.00E−03) | MSP-RON signaling pathway (6.04E−04) | TREM1 signaling (1.62E−02) | iNOS signaling (4.57E03) | Phosphatidylethanolamine biosynthesis (2.34E−03) |
| NF-κB signaling (1.26E−03) | Renin-angiotensin signaling (7.41E−04) | PRPP biosynthesis (2.14E−02) | Phosphatidylethanolamine biosynthesis (7.94E−03) | Prolactin signaling (3.16E−03) |
| Tec kinase signaling (1.35E−03) | PRRs of bacteria and viruses (7.76E−04) | Arsenate detoxification (2.82E−02) | LXR/RXR activation (1.12E−02) | B cell receptor signaling (3.55E−03) |
| Dendritic cell maturation (1.82E−03) | Integrin signaling (8.13E−04) | Tetrahydrofolate (3.47E−02) | Hepatic fibrosis/hepatic stellate cell activation (0.01) | Antigen presentationpathway (3.55E−03) |
| MSP-RON signaling pathway (1.91E−03) | CXCR4 signaling (1.23E−03) | LXR/RXR activation (5.5E−02) | Communication between innate and adaptive immune cells (0.013) | PDGF signaling (3.89E−03) |
Top ten most significantly enriched pathways from Ingenuity Pathway Analysis using either all differentially expressed (DE) genes of a disease group (“all”), DE genes of a disease group with no overlap with any other group (“only”), or DE genes shared between two disease groups (“overlap”). Abbreviations: PRRs, pattern recognition receptors; iNOS, inducible nitric oxide synthase; CXCR4, chemokine (C-X-C motif) receptor 4; CTLA4, cytotoxic T lymphocyte-associated protein 4; MSP, macrophage-stimulating protein; TREM1, triggering receptor expressed on myeloid cells 1; PRPP, phosphoribosyl pyrophosphate; PDGF, platelet-derived growth factor.
TABLE 2 .
Top 10 most significantly enriched pathways by pathway enrichment analysis of cancer
| Most significantly enriched pathway (P value) using DE genes from the following disease group(s)a: | ||||
|---|---|---|---|---|
| c-CHB all | c-CHB only | c-CHB and c-CHC overlap | c-CHC all | c-CHC only |
| FXR/RXR activation (1.00E−23) | FXR/RXR activation (2.00E−14) | LXR/RXR activation (1.1E−08) | LXR/RXR activation (4.27E−09) | Pathogenesis of multiple sclerosis (7.94E−05) |
| LPS/IL-1-mediated inhibition of RXR function (2.51E−18) | LPS/IL-1-mediated inhibition of RXR function (2.95E−10) | FXR/RXR activation (2.88E−08) | LPS/IL-1-mediated inhibition of RXR function (1.20E−07) | Cell cycle G1/S checkpoint regulation (3.89E−03) |
| LXR/RXR activation (2.51E−16) | Acute-phase response signaling (3.72E−10) | LPS/IL-1-mediated inhibition of RXR function (5.25E−08) | FXR/RXR activation (8.71E−07) | Protein kinase A signaling (5.50E−03) |
| Acute-phase response signaling (1.58E−12) | LXR/RXR activation (1.02E−07) | Chemokine signaling (1.17E−04) | Complement system (7.41E−05) | Small cell lung cancer signaling (5.62E−03) |
| Blood flow/coagulation system (3.31E−09) | Intrinsic prothrombin activation pathway (6.17E−07) | Aryl hydrocarbon receptor signaling (3.55E−04) | Phosphatidylcholine biosynthesis (3.31E−04) | Cyclins and cell cycle regulation (7.76E−03) |
| Intrinsic prothrombin activation pathway (4.27E−08) | Extrinsic prothrombin activation pathway (6.46E−07) | Fatty acid α-oxidation (6.76E−04) | Hepatic cholestasis (4.07E−04) | RAR activation (8.71E−03) |
| Complement system (6.31E−08) | Phenylalanine degradation (2.34E−06) | Dopamine degradation (7.24E−04) | Aryl hydrocarbon receptor signaling (4.27E−04) | Granulocyte adhesion and diapedesis (8.91E−03) |
| Estrogen biosynthesis (7.94E−08) | Blood flow/coagulation system (6.46E−06) | Atherosclerosis signaling (7.41E−04) | Bile acid biosynthesis, neutral pathway (5.50E−04) | Glioma signaling (1.55E−02) |
| Xenobiotic metabolism signaling (1.02E−07) | Xenobiotic metabolism signaling (2.29E−05) | Complement system (7.76E−04) | Chemokine signaling (6.76E−04) | Type I diabetes mellitus signaling (2.45E−02) |
| Valine degradation (1.2E−07) | Valine degradation (2.63E−05) | Blood flow/coagulation system (1.12E−03) | Blood flow/coagulation system (6.92E−04) | IL-17A signaling in gastric cells (2.57E−02) |
Top ten most significantly enriched pathways from Ingenuity Pathway Analysis using either all differentially expressed (DE) genes of a disease group (“all”), DE genes of a disease group with no overlap with any other group (“only”), or DE genes shared between two disease groups (“overlap”). Abbreviations: LPS, lipopolysaccharide; RAR, retinoid acid receptor.
FIG 2 .
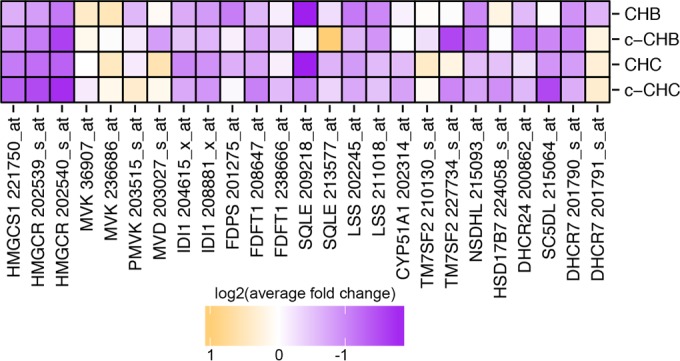
Expression of genes in the cholesterol synthesis pathway in chronic viral hepatitis and cancer. Log2 of average fold change in each disease group (CHB, c-CHB, CHC, and c-CHC) compared to uninfected controls for genes that code for major enzymes within the cholesterol biosynthesis pathway. Due to decreasing reliability from “_at,” “_s_at,” to “_x_at” for genes with multiple probe types, the most reliable annotations are displayed. Results are shown for “_s_at” only if “_at” were not available and for “_x_at” probes only if “_s_at” or “_at” probes were not available. HMGCS1, HMG-CoA synthase 1; HMGCR, HMG-CoA reductase; MVK, mevalonate kinase; PMVK, phosphomevalonate kinase; MVD, mevalonate decarboxylase; IDI1, isopentenyl-diphosphate delta isomerase 1; FDPS, farnesyl diphosphate synthase; FDFT1, farnesyl-diphosphate farnesyltransferase 1; SQLE, squalene epoxidase; LSS, lanosterol synthase; CYP51A1, cytochrome P450 family 41 subfamily A; TM7SF2, transmembrane 7 superfamily member 2; NSDHL, NAD(P)-dependent steroid dehydrogenase-like; HSD17B7, hydroxysteroid 17-beta dehydrogenase; DHCR24, 24-dehydrocholesterol reductase; SC5D, sterol-C5-desaturase; DHCR7, 7-dehydrocholesterol reductase.
Small-RNA transcriptome analysis identifies novel differentially expressed microRNAs in chronic viral hepatitis and liver cancer.
Next we performed small-RNA sequencing in four liver samples from each of the disease categories in the Japanese cohort (uninfected controls, CHB, c-CHB, CHC, and c-CHC). Seven out of the 8 c-CHB and c-CHC samples were matched with their corresponding nonmalignant samples. We obtained an average of ~18 million reads from each, of which ~70% mapped to the genome (see Table S3 in the supplemental material). miRNAs and their isomiRs (sequence variants of canonical miRNAs with a different seed sequence) were annotated and quantified by our previously described small-RNA transcriptome sequencing (small-RNA-seq) analysis pipeline (Materials and Methods). Within-category correlation analyses of miRNA expression profiles showed that uninfected controls were highly correlated (median pairwise r2 = 0.98), and as in the case of gene expression (Fig. 1A and B), the c-CHB samples were the most poorly correlated (median pairwise r2 = 0.70) (Fig. 3A).
FIG 3 .

Global miRNA expression in chronic viral hepatitis and cancer. (A and B) Pearson correlation coefficient heat maps. Each cell in the map represents the correlation in expression profiles between two samples. Cells along the diagonal represent identical samples and are shown in white. Black lines divide each disease group. The midpoint for color bar r2 is 0.5. (A) miRNAs included are those from small-RNA sequencing with average reads per million mapped (RPMM) of >100 (n = 208) in uninfected controls (n = 4), CHB (n = 4), matched c-CHB (n = 4), CHC (n = 4), and matched c-CHC (n = 4) tissue samples. (B) miRNAs from The Cancer Genome Atlas small-RNA-seq with an overall average RPMM of >100 (n = 180) in each disease category: CHB (n = 6), c-CHB (n = 12), CHC (n = 5), and c-CHC (n = 15). (C and D) Analysis of differentially expressed (DE) miRNAs in each disease category compared to the uninfected controls, including only miRNAs that have the following characteristics: (i) RPMM of >50, (ii) a fold change of >1.5, and (iii) changes in the same direction for at least 3 out of 4 samples from each disease group. 5′-shifted isomiRs (sequence variants of canonical miRNAs with a different seed sequence) are included in this analysis. (C) Number of DE miRNAs. (D) Proportional Venn diagrams comparing the DE miRNAs of different disease groups.
We repeated this analysis using publically available small-RNA sequencing data from TCGA (http://cancergenome.nih.gov/) for 12 c-CHB tissue samples and 6 matched CHB tissue samples as well as 18 c-CHC tissue samples and 5 matched CHC tissue samples. These patients represent a subset of the individuals for whom RNA-seq data were available and analyzed (Fig. 1B). We quantified miRNAs according to our previously described small-RNA RNA-seq analysis pipeline (29) and confirmed that the cancer tissue samples were more poorly correlated (c-CHB median pairwise r2 = 0.83; c-CHC median pairwise r2 = 0.79) with each other compared to the noncancer tissue samples (median pairwise r2 of >0.95) (Fig. 3B).
Next, using the small-RNA-seq data from the Japanese cohort, we identified DE miRNAs in each of the disease categories compared to the uninfected controls (Materials and Methods). The cancer tissue samples (c-CHB and c-CHC) had a greater number of DE miRNAs than the noncancer tissue samples (CHB and CHC) (Fig. 3C). Among the cancer tissue samples, c-CHB had the greatest number of DE miRNAs (Fig. 3C). For each of CHB and CHC, ~60% of DE miRNAs were also altered in the matched cancer tissue samples (Fig. 3D). While only 35% of DE miRNAs in CHC were also shared with CHB, 67% of the DE miRNAs in c-CHC were shared with those in c-CHB (Fig. 3D).
Figure 4 shows all 134 miRNAs that were DE in at least one disease category compared to uninfected controls. Of these 134 miRNAs, 10 (miR-378a-3p, miR-486-5p, miR-148a-3p, miR-148a-5p, miR-27a-3p, miR-146b-5p, miR-21-3p, miR-99b, miR-221, and miR-224) were DE in all four disease categories compared to the uninfected controls. Several of these, including miR-378a-3p and miR-27a-3p, have been associated previously with cholesterol homeostasis (9). Using an automated PubMed literature search (Materials and Methods), we determined that among the miRNAs that were DE in at least one disease category, the one most studied in hepatitis and associated liver disease is miR-122 (Fig. 4), which has also previously been shown to influence lipid metabolism (12). Although miR-122-5p, which promotes HCV replication (13), did not satisfy our criteria for differential expression, miR-122-3p was substantially decreased in CHB, c-CHB, and CHC (Fig. 4). While miR-122-3p is relatively understudied compared to miR-122-5p, we found that it was robustly expressed in all of the liver tissue samples (see Fig. S1 in the supplemental material). We also found that miR-122-3p coimmunopurifies with Argonaute 2 (AGO2) in extracts from human hepatoma cells (FT3-7) (20) and that its abundance in AGO2 immunoprecipitates is proportional to total intracellular levels, indicating efficient loading (Fig. S1). These data suggest that despite the extensive focus in the literature on miR-122-5p, miR-122-3p is likely also a functional miRNA and merits further detailed experimental investigation.
FIG 4 .

Published literature on miRNAs that are differentially expressed in chronic viral hepatitis and liver cancer. (Left of each panel shown) Heat map of significantly differentially expressed miRNAs in each disease group (CHB, c-CHB, CHC, and c-CHC) compared to the uninfected controls that have the following characteristics: (i) an average fold change of >1.5 in one disease state, (ii) a change in the same direction in at least 3 out of 4 samples within each disease group, and (iii) reads per million mapped reads (RPMM) of >50 in at least one disease group. The color represents log2 (average fold change) compared to the uninfected controls. Black boxes indicate that the miRNA for a specific disease state did not fit the criteria of DE (center and right of each panel shown). Results of automated searches of PubMed on 5 December 2014 using the following search terms for the miRNA: e.g., “miRNA-X,” “miR-X,” or “microRNA-X” with or without the miRNA family letter (e.g., for miR-148a, “miRNA-148a,” “miR-148a,” “microRNA-148a,” “miRNA-148,” “miR-148,” or “microRNA-148”]) and ignoring the “-3p” or “-5p.” (Center) Number of total published papers indexed on PubMed based on searches of just the miRNA name. The color bar midpoint at 100 is shown on the right. Proportion of total published papers related to each miRNA that was associated with liver (search term “liver”), HBV (search term “hepatitis B” or “HBV”), HCV (search term “hepatitis C” or “HCV”), or hepatocellular carcinoma (search term “HCC,” “hepatocellular,” or “liver cancer”).
We identified several previously reported DE miRNAs, including miR-21 (30), miR-27 (31), and miR-181 (32) in CHC, miR-199 (33), miR-148 (34), and miR-125 (35) in CHB, and miR-221/222 (36), miR-101 (37), and miR-224 (38) in chronic viral hepatitis-associated liver cancer. Many of the other DE miRNAs that we identified, such as miR-215 and miR-340 in CHB, miR-1307 and miR-484 in CHC, and miR-136-5p and miR-3591-5p in c-CHC, have not been well studied in the liver. All of these miRNAs represent candidate regulators of the pathways altered in chronic viral hepatitis and liver cancer, including those governing cholesterol synthesis and metabolism.
Several microRNAs are identified as candidate master regulators of pathways mediating cholesterol homeostasis in chronic viral hepatitis and liver cancer.
We next sought to identify DE miRNAs that represent potential key control points (master regulators) in the networks that regulate gene expression in chronic viral hepatitis and cancer. Using our previously developed tool miRHub (Materials and Methods), we identified 21 candidate master miRNA regulators of genes downregulated in at least one disease group: CHB, c-CHB, CHC, or c-CHC (Fig. 5A). Among these 21, six were predicted to be master regulators in at least two different disease categories: miR-33, miR-199-3p, miR-194, miR-450b, miR-23, and miR-27-3p (Fig. 5A). miR-33, miR-199-3p, miR-450b, and miR-27-3p were upregulated in c-CHB and c-CHC (Fig. 4) and identified as potential master regulators of downregulated genes in the same disease categories (Fig. 5A). Although hepatic miR-33 is a known regulator of cholesterol homeostasis, it has only recently been studied in the context of chronic viral hepatitis and HCC (39). Both miR-199-3p and miR-27-3p are among the few miRNAs that have been reported as regulators of phenotypes associated with chronic viral hepatitis (cancer [40] and dyslipidemia [41, 42], respectively). miR-194 and miR-23 were downregulated in the same disease categories for which they were predicted as master regulators of downregulated genes. This finding is suggestive of transcriptional coregulation of miR-194/miR-23 and their target genes, consistent with the participation of these miRNAs in complex regulatory motifs (43).
FIG 5 .

Candidate miRNA master regulators of gene expression profiles in chronic viral hepatitis and associated liver cancer. (A and B) Heat map of the –log10(P value) of the predicted targeting score for each miRNA (determined using miRHub) among genes deregulated in each disease category (CHB, c-CHB, CHC, and c-CHC) compared to uninfected controls. Only miRNAs with an empirical P value of <0.05 (based on miRHub Monte Carlo simulation) in at least one disease group are shown. Black boxes indicate that the miRNA for a specific disease state did not fit the criteria of DE. (A) Candidate miRNA master regulators of significantly downregulated genes in each disease category. (B) Candidate miRNA master regulators of significantly upregulated genes in each disease category.
We also identified eight candidate master miRNA regulators of upregulated genes in at least one of the disease categories (Fig. 5B). Interestingly, all eight miRNAs were upregulated in the same disease categories for which they were predicted to be master regulators of upregulated genes. As with miR-194 and miR-23, one possible explanation for this finding is the widespread use of incoherent feed-forward miRNA loops in liver gene regulatory networks (43).
The strongest candidate master miRNA regulators in any disease category were miR-21 and miR-33, which were both upregulated in c-CHB (Fig. 4) and predicted to exert greater control of downregulated genes in c-CHB than any other miRNA (Fig. 5A). miR-21 has been reported previously as a critical mediator of c-CHB and suppressor of the host immune system in CHC (30). miR-33 is a key regulator of cholesterol homeostasis and has recently been associated with hepatic steatosis and fibrosis in patients with CHC and CHB, respectively (39).
For each miRNA predicted as a candidate master regulator of up- or downregulated genes in each disease category, we identified biological pathways significantly enriched among its predicted target genes (see Table S4 in the supplemental material). We found that most candidate master regulator miRNAs were predicted to target genes overrepresented in pathways regulating lipid homeostasis. For example, among downregulated genes in c-CHB, 12 of the 13 candidate master miRNA regulators were predicted to target genes significantly overrepresented in the peroxisome proliferator-activated receptor α (PPARα)/RXRα activation pathway. Several of these 12 candidate master miRNA regulators, including miR-21 and miR-33, have been recently reported as direct modulators of PPARα expression (44), and miR-27-3p has been shown to target and repress both PPARα (44) and RXRα (45). Transcriptional regulation by PPARα/RXRα contributes to hepatic control of lipid biosynthesis (46, 47). These findings lead to the hypothesis that several key miRNAs mediate the robust suppression of cholesterol synthesis pathways in chronic viral hepatitis and associated liver cancer.
miR-27 suppresses cholesterol synthesis via regulation of the rate-limiting enzyme HMG-CoA reductase.
The evaluation of this hypothesis is hindered by a lack of cell culture models that accurately recapitulate virus-host interactions in vivo during HBV or HCV infection. Nonetheless, to determine whether the DE miRNAs are capable of regulating cholesterol synthesis in liver cancer cells, we selected miR-21, miR-27, and miR-224 for functional follow-up analyses in human hepatoma cells (Huh7). As a control for these experiments, we similarly evaluated miR-151, which our computational analyses did not identify as a master regulator. Specifically, we overexpressed each miRNA at a standard effective concentration (see Fig. S2 in the supplemental material) separately in Huh7 cells grown in lipoprotein-deficient serum and evaluated the effect on de novo cholesterol synthesis (Materials and Methods). We found that all three DE miRNAs, but not the control miR-151, suppressed cholesterol synthesis to various degrees (Fig. 6A). Interestingly, all three of these miRNAs have at least one predicted seed sequence target site in the 3′ untranslated region (UTR) of the HMG-CoA reductase (HMGCR) gene (Fig. 6B), which encodes the rate-limiting enzyme in the cholesterol synthesis pathway. The predicted target sites for miR-27 and miR-224 are conserved across several mammalian species, whereas the predicted site for miR-21 is found only in humans (Fig. 6B).
FIG 6 .

Candidate miRNA master regulators miR-27 and miR-21 control cholesterol synthesis in vitro. (A) Human hepatoma (Huh7) cells transfected with a 10 nM concentration of either miRNA mimic or transfection reagent alone (mock transfected) for the different RNAs: for miR-151, mock (n = 6) and mimic (n = 3); for miR-27, mock (n = 6) and mimic (n = 6); for miR-21, mock (n = 6) and mimic (n = 6); for miR-224, mock (n = 5) and mimic (n = 5). De novo cholesterol synthesis was measured by radiolabeled acetate incorporation assay (Materials and Methods). Relative change compared to mock transfection is shown. (B) Predicted target sites in the HMGCR 3′ UTR for miR-27, miR-21, and miR-224 as determined by the TargetScan algorithm. Gaps introduced to maximize alignment are indicated by dashes. (C) Relative levels of HMGCR mRNA, miR-21, miR-27, and miR-224 across the disease categories. Fold change values for HMGCR are based on comparison to the values for uninfected controls. RPMM, reads per million mapped reads. (D) Effects of miR-27 mimic (10 nM) and miR-375 (10 nM; negative control) on the activity of Renilla (RL) luciferase containing either wild-type (WT) or mutated (Mut) HMGCR 3′ UTR normalized to firefly luciferase (FL) in transfected Huh7 cells. Each condition was tested twice with three technical replicates each (total n = 6). The mutation was targeted to the predicted miR-27 target site. P values were determined using two-tailed unpaired Student’s t test and are indicated as follows: *, P < 0.05; ***, P < 0.005.
HMGCR mRNA was significantly decreased in all four disease categories, whereas miR-27 and miR-224 were significantly elevated in all four disease categories, and miR-21 was increased in c-CHB and CHC (Fig. 6C). We have shown previously that miR-27 represses HMGCR expression in hepatoma cells (41), and another study using mouse brain tissue suggested that miR-27 may regulate HMGCR directly by targeting its 3′ UTR (48). To test this hypothesis, we performed a 3′ UTR reporter gene assay. Specifically, the entire HMGCR 3′ UTR was PCR amplified from human genomic DNA and cloned into the PsiCheck2 expression vector, downstream of the Renilla luciferase (RL) coding region (Materials and Methods). The recombinant PsiCheck2 vector was then transfected into HEK293T cells, and relative RL activity was measured in the presence and absence of oligonucleotide mimics of miR-27 or a negative-control miRNA (miR-375, which is not predicted to have a target site in the HMGCR 3′ UTR). The miR-27 mimic induced significant repression (Fig. 6D), whereas miR-375 had no effect. We then introduced a mutation in the miR-27 target site via site-directed mutagenesis and demonstrated a partial but still significant (P < 0.01) rescue of relative RL activity.
Model of miRNA-mediated regulation of cholesterol metabolism in chronic viral hepatitis and associated HCC.
At least nine genes in the cholesterol synthesis pathway were significantly downregulated in at least one disease category. Of these nine genes, five (HMGCS1, HMGCR, NSDHL, DHCR24, and SC5DL) have evolutionarily conserved predicted target sites for ≥2 miRNAs that were upregulated more than 5-fold in at least one disease category and/or identified as master regulators of downregulated genes in at least one disease category (Fig. 7; see Fig. S3 in the supplemental material). Predicted target sites for miR-499 and miR-224 are shared among two of the genes (HMGCS1/HMGCR and HMGCR/SC5DL, respectively), and three genes (HMGCS1, HMGCR, and SC5DL) harbor putative target sites for miR-27. These three miRNAs represent key direct regulators of the cholesterol synthesis pathway in chronic viral hepatitis and associated liver cancer.
FIG 7 .
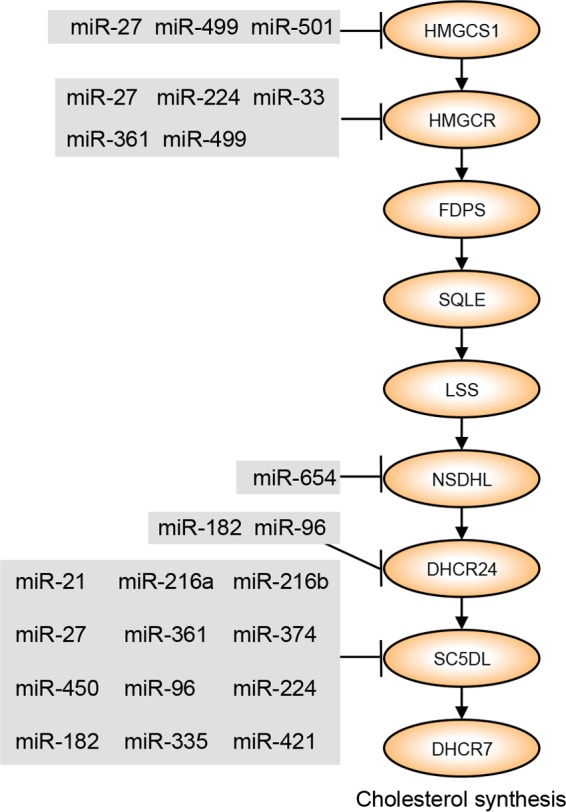
Model of miRNA regulation of cholesterol synthesis in chronic viral hepatitis and associated liver cancer. Cholesterol synthesis genes that are significantly downregulated in at least one disease state are shown. Each miRNA is significantly upregulated in at least one disease state relative to uninfected controls and is either a predicted miRNA master regulator of gene expression in at least one disease state based on miRHub P value of <0.05 or has a fold change of >5 compared to uninfected controls. See Fig. S2 and S3 for greater detail. HMGCS1, HMG-CoA synthase 1; HMGCR, HMG-CoA reductase; FDPS, farnesyl diphosphate synthase; SQLE, squalene epoxidase; LSS, lanosterol synthase; NSDHL, NAD(P)-dependent steroid dehydrogenase-like; DHCR24, 24-dehydrocholesterol reductase; SC5D, sterol-C5-desaturase; DHCR7, 7-dehydrocholesterol reductase.
The LXR/RXR regulatory pathway is significantly enriched among dysregulated genes in many of the disease groups (Table 2). This pathway is involved in the control of several aspects of cholesterol metabolism, most notably cholesterol efflux (see Fig. S4 in the supplemental material). Numerous genes in the cholesterol efflux and transport pathway are significantly downregulated in at least one disease state, including ABCA1, also known as cholesterol efflux regulatory protein (CERP), which is a direct target of LXR/RXR and is significantly decreased in expression in c-CHB and c-CHC. ABCA1 is predicted to be a target gene of 15 miRNAs that are upregulated in at least one disease state (c-CHB or c-CHC), including miR-33 and miR-27, which are established regulators of cholesterol efflux through suppression of ABCA1 (42, 49).
DISCUSSION
On the basis of gene expression microarray data, we found pathways relating to cholesterol synthesis and metabolism to be the most significantly enriched among genes dysregulated in the various disease states we studied, especially c-CHB and c-CHC. Interestingly, in the noncancer tissue samples, although immune pathways were the most significantly enriched, the specific immune pathways were different for CHB and CHC, which is consistent with distinct inflammatory features and innate immune responses in each disease. The top enriched immune pathway for CHC is interferon signaling, and for CHB, the top pathways are associated with both the innate and adaptive immune responses, although not interferon signaling. Some pathways, such as those controlling blood flow and coagulation (c-CHB and c-CHC), as well as amino acid degradation (c-CHB), merit future investigation.
Studies of miRNAs in hepatitis C have been dominated by miR-122-5p, and only a few other miRNAs have been investigated. miR-122-5p was previously found to be reduced in patients with chronic hepatitis C, but the reduction was dependent on the interleukin 28B (IL-28B) genotype (20). In this study, we performed small-RNA-seq on two subjects from each IL-28B genotype class (T/T and T/G, based on SNP rs8099917). Similar to what was reported previously (20), miR-122 levels were reduced only in the individuals with the T/G genotypes and not the T/T genotype. Because our criteria for DE stipulated that the levels of a miRNA must change significantly and in the same direction in three of the four samples in a disease group, miR-122-5p was not deemed to be DE in our study.
Our study points to the potential functional relevance of miR-122-3p, which has received substantially less attention due to its lower expression than miR-122-5p, but we find that its expression is robust and that it is effectively loaded and abundantly present on AGO2. miR-122-3p was not a predicted miRNA master regulator of gene expression in any of the disease states, but it does have several key predicted mRNA targets, including the interferon-stimulated gene, interferon-induced protein with tetratricopeptide repeats 3 (IFIT3). We also show that a suite of other miRNAs, including miR-27, miR-33, miR-21, and miR-199, are differentially expressed in one or more of the disease states and also are predicted to be master regulators of the disease gene expression profiles.
We showed that several of these “master regulator” miRNAs, notably miR-33, miR-21, and miR-27, are strongly associated with liver programs that control cholesterol homeostasis. Since miR-33 is already well established as a modulator of cholesterol metabolism, we focused our functional studies on miR-27 and miR-21, as well as miR-224, which were among the most highly upregulated miRNAs in all disease states. While miR-21 and miR-27 have been linked to lipid homeostasis previously (42, 50, 51), we have shown here that miR-27 and miR-21 both significantly suppress de novo cholesterol synthesis in Huh7 cells by ~30% and ~70%, respectively. Furthermore, we demonstrated that the effect of miR-27 is mediated in part through direct regulation of the HMGCR gene, which encodes the rate-limiting enzyme in cholesterol biosynthesis. Specifically, in a reporter gene assay in which the HMGCR 3′ UTR was cloned downstream of a luciferase gene, we showed that overexpression of miR-27 markedly reduced luciferase activity and that a mutation in the target site of miR-27 within the HMGCR 3′ UTR significantly rescued luciferase activity. We note that the rescue was not complete, and there are several possible explanations for this observation. For example, miR-27 may have additional noncanonical target sites within HMGCR (even within its open reading frame) that are not predicted by current bioinformatics methods; such sites would, of course, remain functional even when a mutation is introduced in the canonical target site. Very few studies have linked miRNAs to the direct regulation of HMGCR. Both miR-27 and miR-21 may be candidate therapeutic targets for hypercholesterolemia.
We found that the LXR/RXR pathway was prominently dysregulated in all disease states. This nuclear receptor pathway has known functions in the regulation of both lipid metabolism and immune and inflammatory responses. For example, recent in vivo studies have shown that LXR activation promotes cholesterol efflux and inhibits inflammation in part by suppressing NF-κB signaling (52–54). For most of the disease states we studied, well-established LXR target genes, such as CYP7A1 and ABCA1, were downregulated, whereas genes promoted by NF-κB signaling were upregulated (Table 1). More-detailed analysis is required to determine the roles of miRNAs in controlling LXR/RXR in vivo during chronic viral hepatitis and thereby integrating lipid metabolic and inflammatory signaling.
CHB, CHC, and HCC have all been linked to cholesterol and lipid imbalance; however, much remains unknown about the underlying molecular mechanisms, particularly with regard to miRNAs. Through integrative analysis of small-RNA-seq and microarray data and follow-up validation experiments in a cell-based system, our study provides important clues about the candidate miRNA drivers of this phenotype. In terms of CHC, the results of our studies are most applicable to HCV genotype 1, because the tissue samples we analyzed were primarily from patients infected with this strain. Similar studies are warranted in CHC resulting from infections by other HCV genotypes, in particular genotype 3, which has been associated with fatty liver disease and which may have unique, genotype-specific interactions with host genes (55, 56). It is important to note that we did not study liver tissue from individuals with nonviral liver diseases, such as alcoholic hepatitis or nonalcoholic fatty liver disease. Future studies with such samples will help determine whether the miRNAs that we found to be associated with hepatitis and HCC in this study are specific to viral hepatitis or shared across other types of hepatic disease. Also, future investigations should focus on more-detailed functional characterization of specific miRNAs of interest and their roles in regulating cholesterol homeostasis, particularly in the background of cirrhosis and the chronic inflammation present in viral hepatitis. As the use of locked nucleic acid (LNA) inhibitors expand, such studies may lead to the development of novel and effective miRNA-based therapeutic strategies.
MATERIALS AND METHODS
Human subjects.
Written informed consent was obtained from all human subjects. Ethics approval was obtained from the Ethics Committee for Human Genome/Gene Analysis Research at Kanazawa University Graduate School of Medical Science.
Gene expression analysis.
Gene expression microarray data (20, 21) for CHB, c-CHB, CHC, and c-CHC were contrasted to the uninfected tissue samples to determine differentially expressed (DE) genes (analysis of variance [ANOVA] P value of <0.05 after Benjamini-Hochberg step-up multiple testing correction and fold change of >2 compared to the uninfected tissue samples) using Partek Genomics Suite (Partek Inc., St. Louis, MO). Pathway enrichment was determined with Ingenuity Pathway Analysis software (Qiagen, Hilden, Germany).
Small-RNA sequencing.
RNA was isolated as described previously (20). RNA purity was assessed with Nanodrop 2000 (Thermo Scientific, Rockford, IL), and integrity was determined with an Agilent 2100 bioanalyzer (Agilent, Santa Clara, CA). RNA integrity and sequencing quality were comparable for all specimens. Small-RNA libraries were generated using Illumina TruSeq small-RNA sample preparation kit (Illumina, San Diego, CA). Sequencing was performed on the Illumina HiSeq 2000 platform. Sequencing reads were trimmed using Cutadapt (parameters O −10 e 0.1). Trimmed reads were mapped to genomic regions spanning annotated miRNAs (±20 nucleotides [nt]) using Bowtie 0.12.7, allowing for no mismatches. Next, reads that did not map without mismatches were aligned to the same regions using SHRiMP2.2.2. SHRiMP2.2.2 seeds were set based on the length of the read allowing one mismatch anywhere in the body and up to three mismatches at the 3′ end of the read (based on the length of the read). Small-RNA sequencing data were deposited in GEO (accession no. GSE57381). TCGA liver cancer small RNA-seq BAM files annotated as having a risk factor of either “hepatitis B” or “hepatitis C” and no other annotated risk were downloaded and processed using the bioinformatics method described above.
Automated PubMed searches.
An automated search of PubMed was performed using a Ruby program with the HTML parser Nokogiri on 5 December 2014. Search terms for the miRNAs were, for example, “miRNA-X,” “miR-X,” or “microRNA-X” with or without the miRNA family letter (e.g., miR-148a [“miRNA-148a,” “miR-148a,” “microRNA-148a,” “miRNA-148,” “miR-148,” “microRNA-148”]). Other search terms (search term “liver”), HBV (search term “hepatitis B” or “HBV”), HCV (search term “hepatitis C” or “HCV”), or hepatocellular carcinoma (search term “HCC” or “hepatocellular” or “liver cancer”).
Identification of miRNA candidate master regulators using miRHub.
To identify miRNA candidate master regulators, we used a tool we developed previously (29), which we subsequently named “miRHub.” We have since used miRHub successfully in several follow-up studies (57–59), including one in which we provided a more detailed description of the methods (57). Briefly, miRHub uses the TargetScan algorithm to predicted target sites for miRNAs of interest in the 3′ UTRs of DE genes and then determines by Monte Carlo simulation for each miRNA whether the number and strength of predicted targets is significantly greater than expected by chance. Such miRNAs are designated “master regulators.” Using miRHub, we predicted the miRNA master regulators for eight different gene lists (significantly DE upregulated genes in each disease group and significantly DE downregulated genes in each disease group), using four different miRNA lists (DE miRNAs in each disease category irrespective of their fold change direction).
Measuring cholesterol synthesis.
Human hepatoma cells (Huh7) were seeded into six-well plates at a density of 1 × 105 cells/ml. After overnight growth, cells were transfected with miR-151 mimic, miR-21 mimic, miR-27 mimic, or miR-224 mimic at 10 nM each (Exiqon, Woburn, MA), or Lipofectamine 2000 transfection reagent only (mock transfected) (Life Technologies, Grand Island, NY). Forty-eight hours after transfection, the medium was removed and replaced with Dulbecco modified Eagle medium (DMEM) with low-glucose lipoprotein-deficient serum (LPDS). Seventeen hours later, low-glucose medium was removed and replaced with fresh serum-free low-glucose medium (cholesterol free) with 1 μCi [3H]acetic acid per well (0.5 µCi/ml medium) and incubated for 6 h. The cells were washed twice with 1× phosphate-buffered saline. Two milliliters of 3:2 hexane-isopropanol were added to each well; the cells were allowed to sit for 2 h and then placed in glass vials. Samples were dried by using nitrogen, and 30 µl of cold 0.1 µg cholesterol-cholesteryloleate was added. Thirty microliters of resuspended lipids was placed on a plate, and thin-layer chromatography was performed (230:60:3 petroleum ether-diethyl ether-acetic acid). Spots were resolved with iodine, and cholesteryl ester, cholesterol, and triglyceride spots were cut out and placed in 10 ml scintillation fluid. Total protein for each well was quantified using the bicinchoninic acid (BCA) assay. Disintegrations per minute (dpm) values were normalized to the values for total protein.
miRNA overexpression and reporter gene assays.
HEK293T cells were maintained in DMEM with 25 mM glucose (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS) and 2 mM l-glutamine (Invitrogen, Grand Island, NY) and cultured in a humidified incubator at 37°C and 5% CO2. HEK293T cells were seeded into 24-well plates and allowed to grow overnight. Once the cells were approximately 70% confluent, they were transfected with 200 ng of pEZX-MT01 empty vector, vector containing the 3′ UTR of HMGCR (GeneCopoeia,; Rockville, MD) and 10 nM miRIDIAN microRNA hsa-miR-27b mimic (5′ UAGCACCAUCUGAAAUCGGUUA 3′, Dharmacon, Lafayette, CO) or 10 nM miRIDIAN microRNA hsa-miR-375 mimic (5′ UUUGUUCGUUCGGCUCGCGUGA 3′). Transfection was performed using Lipofectamine 2000 (Life Technologies, Grand Island, NY). After 48 h, the cells were lysed, and luciferase activity was measured using the Luc-Pair luciferase assay kit (Agilent, Santa Clara, CA) on a GloMax 96 microplate luminometer (Promega, Madison, WI). Site-directed mutagenesis was performed with the QuikChange II XL site-directed mutagenesis kit (Agilent, Santa Clara, CA).
Statistics and graphics.
P values were calculated and plots were generated using R and Prism. Proportional Venn diagrams were generated by the R package “VennDiagram.”
SUPPLEMENTAL MATERIAL
miR-122-3p and miR-122-5p comparison. (A) Small-RNA sequencing results from AGO2 immunoprecipitation and total RNA from FT3-7 cells for miR-122-3p and miR-122-5p. (B) Small-RNA sequencing results for miR-122-3p and miR-122-5p in liver tissue from each of the disease states (uninfected control, CHB, c-CHB, CHC, and c-CHC). Download
Transfection efficiency of miRNA mimics. In experiments separate from those described in Fig. 6, mimics formiR-21 (n = 4) (A) and miR-27b (n = 4) (B) were transfected into human hepatoma (Huh7) cells at a concentration of 10 nM, and miRNA levels were measured by quantitative PCR (qPCR) and compared to transfection reagent alone. Download
Detailed model of miRNA regulation of cholesterol synthesis in chronic viral hepatitis and associated liver cancer. Cholesterol synthesis genes that are significantly altered in at least one disease state are shown. The color of the pie quadrant to the right of each gene indicates the disease state in which each gene is significantly altered as defined previously (light pink, c-CHB; dark pink, CHB; light blue, c-CHC; dark blue, CHC). Green outline indicates that the gene is downregulated. Each of the miRNAs displayed has at least one predicted target site in at least one of the genes listed, which is conserved in humans and at least one other species. Each miRNA is significantly upregulated in at least one disease state relative to uninfected controls and is either a predicted miRNA master regulator of gene expression in at least one disease state based on miRHub P value of <0.05 (pie piece outlined in blue) or has a fold change of >5 compared to uninfected controls (outlined in orange) or both (outlined in black). Download
Detailed model of miRNA regulation of cholesterol transport and efflux in chronic viral hepatitis and associated cancer. Significantly altered cholesterol transport and efflux genes in at least one disease state are shown. The color of the pie quadrant to the right of each gene indicates the disease state in which each gene is significantly altered as defined previously (light pink, c-CHB; dark pink, CHB; light blue, c-CHC; dark blue, CHC). Green or red outline indicates that the gene is downregulated or upregulated, respectively. Each of the miRNAs displayed has at least one predicted target site, in at least one of the genes listed, which is conserved in humans and at least one other species. Each miRNA is significantly upregulated in at least one disease state relative to uninfected controls and is either a predicted miRNA master regulator of gene expression in at least one disease state based on miRHub P value of <0.05 (pie piece outlined in blue) or has a fold change of >5 compared to uninfected controls (outlined in orange) or both (outlined in black). Download
Basic clinical patient data. Patient identifier (ID), sex (M, male; F, female), age (in years), tumor grade (p, poor; m, moderate; w, well-differentiated), and METAVIR score are shown.
Significantly altered genes in each disease state. Any probe significantly DE in one or more of the four disease groups listed below, with the significantly changed disease group’s fold change listed.
Small-RNA-seq mapping statistics. Patient ID, number of total reads for each patient, number of reads successfully trimmed, percentage of reads trimmed of total reads, number of mapped reads, percentage of reads mapped of trimmed reads are shown.
Enriched pathways of miRNA master regulator targets. For each miRNA predicted as a candidate master regulator of up- and downregulated genes in each disease category, we identified biological pathways significantly enriched among its predicted target genes. Each biological pathway is significant (P < 0.01) for the miRNAs listed.
ACKNOWLEDGMENTS
We thank members of the Sethupathy and Lemon laboratories for helpful comments on the study.
This work was supported by grants from the National Institutes of Health: grants R00-DK091318 (P.S.), R01-AI095690 and R01-CA164029 (S.M.L), and T32-GM067553 and T32-AI007419 (S.R.S)
Footnotes
Citation Selitsky SR, Dinh TA, Toth CL, Kurtz CL, Honda M, Struck BR, Kaneko S, Vickers KC, Lemon SM, Sethupathy P. 2015. Transcriptomic analysis of chronic hepatitis B and C and liver cancer reveals microRNA-mediated control of cholesterol synthesis programs. mBio 6(6):e01500-15. doi:10.1128/mBio.01500-15.
REFERENCES
- 1.Arzumanyan A, Reis HMGPV, Feitelson MA. 2013. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer 13:123–135. doi: 10.1038/nrc3449. [DOI] [PubMed] [Google Scholar]
- 2.Su TC, Lee YT, Cheng TJ, Chien HP, Wang JD. 2004. Chronic hepatitis B virus infection and dyslipidemia. J Formos Med Assoc 103:286–291. [PubMed] [Google Scholar]
- 3.Corey KE, Kane E, Munroe C, Barlow LL, Zheng H, Chung RT. 2009. Hepatitis C virus infection and its clearance alter circulating lipids: implications for long-term follow-up. Hepatology 50:1030–1037. doi: 10.1002/hep.23219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Z, Keech A, Collins R, Slavin B, Chen J, Campbell TC, Peto R. 1993. Prolonged infection with hepatitis B virus and association between low blood cholesterol concentration and liver cancer. BMJ 306:890–894. doi: 10.1136/bmj.306.6882.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Felmlee DJ, Hafirassou ML, Lefevre M, Baumert TF, Schuster C. 2013. Hepatitis C virus, cholesterol and lipoproteins—impact for the viral life cycle and pathogenesis of liver disease. Viruses 5:1292–1324. doi: 10.3390/v5051292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang J, Nilsson-Ehle P, Xu N. 2006. Influence of liver cancer on lipid and lipoprotein metabolism. Lipids Health Dis 5:4. doi: 10.1186/1476-511X-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang JT, Xu N, Zhang XY, Wu CP. 2007. Lipids changes in liver cancer. J Zhejiang Univ Sci B 8:398–409. doi: 10.1631/jzus.2007.B0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaefer EA, Chung RT. 2013. HCV and host lipids: an intimate connection. Semin Liver Dis 33:358–368. doi: 10.1055/s-0033-1358524. [DOI] [PubMed] [Google Scholar]
- 9.Fernández-Hernando C, Suárez Y, Rayner KJ, Moore KJ. 2011. MicroRNAs in lipid metabolism. Curr Opin Lipidol 22:86–92. doi: 10.1097/MOL.0b013e3283428d9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flowers E, Froelicher ES, Aouizerat BE. 2013. MicroRNA regulation of lipid metabolism. Metab Clin Exp 62:12–20. doi: 10.1016/j.metabol.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vickers KC, Sethupathy P, Baran-Gale J, Remaley AT. 2013. Complexity of microRNA function and the role of isomiRs in lipid homeostasis. J Lipids Res 54:1182–1191. doi: 10.1194/jlr.R034801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, Subramaniam A, Propp S, Lollo BA, Freier S, Bennett CF, Bhanot S, Monia BP. 2006. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab 3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 14.Choi SE, Fu T, Seok S, Kim DH, Yu E, Lee KW, Kang Y, Li X, Kemper B, Kemper JK. 2013. Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMPT. Aging Cell 12:1062–1072. doi: 10.1111/acel.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernandez-Hernando C. 2010. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marquart TJ, Allen RM, Ory DS, Baldan A. 2010. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci U S A 107:12228–12232. doi: 10.1073/pnas.1005191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Naar AM. 2010. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soh J, Iqbal J, Queiroz J, Fernandez-Hernando C, Hussain MM. 2013. MicroRNA-30c reduces hyperlipidemia and atherosclerosis in mice by decreasing lipid synthesis and lipoprotein secretion. Nat Med 19:892–900. doi: 10.1038/nm.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurtz CL, Peck BCE, Fannin EE, Beysen C, Miao J, Landstreet SR, Ding S, Turaga V, Lund PK, Turner S, Biddinger SB, Vickers KC, Sethupathy P. 2014. MicroRNA-29 fine-tunes the expression of key FOXA2-activated lipid metabolism genes and is dysregulated in animal models of insulin resistance and diabetes. Diabetes 63:3141–3148. doi: 10.2337/db13-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selitsky SR, Baran-Gale J, Honda M, Yamane D, Masaki T, Fannin EE, Guerra B, Shirasaki T, Shimakami T, Kaneko S, Lanford RE, Lemon SM, Sethupathy P. 2015. Small tRNA-derived RNAs are increased and more abundant than microRNAs in chronic hepatitis B and C. Sci Rep 5:7675. doi: 10.1038/srep07675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spaniel C, Honda M, Selitsky SR, Yamane D, Shimakami T, Kaneko S, Lanford RE, Lemon SM. 2013. MicroRNA-122 abundance in hepatocellular carcinoma and non-tumor liver tissue from Japanese patients with persistent HCV versus HBV infection. PLoS One 8:e76867. doi: 10.1371/journal.pone.0076867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, Gerstein M, Snyder M. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson CB, Lindsten T, Ledbetter JA, Kunkel SL, Young HA, Emerson SG, Leiden JM, June CH. 1989. CD28 activation pathway regulates the production of multiple T-cell-derived lymphokines/cytokines. Proc Natl Acad Sci U S A 86:1333–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le Page C, Genin P, Baines MG, Hiscott J. 2000. Interferon activation and innate immunity. Rev Immunogenet 2:374–386. [PubMed] [Google Scholar]
- 25.Zhao C, Dahlman-Wright K. 2010. Liver X receptor in cholesterol metabolism. J Endocrinol 204:233–240. doi: 10.1677/JOE-09-0271. [DOI] [PubMed] [Google Scholar]
- 26.Edwards PA, Kennedy MA, Mak PA. 2002. LXRs; oxysterol-activated nuclear receptors that regulate genes controlling lipid homeostasis. Vasc Pharmacol 38:249–256. doi: 10.1016/S1537-1891(02)00175-1. [DOI] [PubMed] [Google Scholar]
- 27.Lalloyer F, Fievet C, Lestavel S, Torpier G, van der Veen J, Touche V, Bultel S, Yous S, Kuipers F, Paumelle R, Fruchart JC, Staels B, Tailleux A. 2006. The RXR agonist bexarotene improves cholesterol homeostasis and inhibits atherosclerosis progression in a mouse model of mixed dyslipidemia. Arterioscler Thromb Vasc Biol 26:2731–2737. doi: 10.1161/01.ATV.0000248101.93488.84. [DOI] [PubMed] [Google Scholar]
- 28.Tanos R, Patel RD, Murray IA, Smith PB, Patterson AD, Perdew GH. 2012. Aryl hydrocarbon receptor regulates the cholesterol biosynthetic pathway in a dioxin response element-independent manner. Hepatology 55:1994–2004. doi: 10.1002/hep.25571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baran-Gale J, Fannin EE, Kurtz CL, Sethupathy P. 2013. Beta cell 5′-shifted isomiRs are candidate regulatory hubs in type 2 diabetes. PLoS One 8:e73240. doi: 10.1371/journal.pone.0073240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Chen J, Wang H, Shi J, Wu K, Liu S, Liu Y, Wu J. 2013. HCV-induced miR-21 contributes to evasion of host immune system by targeting MyD88 and IRAK1. PLoS Pathog 9:e1003248. doi: 10.1371/journal.ppat.1003248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singaravelu R, Chen R, Lyn RK, Jones DM, O’Hara S, Rouleau Y, Cheng J, Srinivasan P, Nasheri N, Russell RS, Tyrrell DL, Pezacki JP. 2014. Hepatitis C virus induced up-regulation of microRNA-27: a novel mechanism for hepatic steatosis. Hepatology 59:98–108. doi: 10.1002/hep.26634. [DOI] [PubMed] [Google Scholar]
- 32.Li GY, Zhou Y, Ying RS, Shi L, Cheng YQ, Ren JP, Griffin JWD, Jia ZS, Li CF, Moorman JP, Yao ZQ. 2015. Hepatitis C virus-induced reduction in miR-181a impairs CD4 T-cell responses through overexpression of DUSP6. Hepatology 61:1163–1173. doi: 10.1002/hep.27634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang GL, Li YX, Zheng SQ, Liu M, Li X, Tang H. 2010. Suppression of hepatitis B virus replication by microRNA-199a-3p and microRNA-210. Antiviral Res 88:169–175. doi: 10.1016/j.antiviral.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 34.Xu X, Fan Z, Kang L, Han J, Jiang C, Zheng X, Zhu Z, Jiao H, Lin J, Jiang K, Ding L, Zhang H, Cheng L, Fu H, Song Y, Jiang Y, Liu J, Wang R, Du N, Ye Q. 2013. Hepatitis B virus X protein represses miRNA-148a to enhance tumorigenesis. J Clin Invest 123:630–645. doi: 10.1172/JCI64265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coppola N, Potenza N, Pisaturo M, Mosca N, Tonziello G, Signoriello G, Messina V, Sagnelli C, Russo A, Sagnelli E. 2013. Liver microRNA hsa-miR-125a-5p in HBV chronic infection: correlation with HBV replication and disease progression. PLoS One 8:e65336. doi: 10.1371/journal.pone.0065336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B, Mazzaferro V, Lowe SW, Croce CM, Dejean A. 2010. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A 107:264–269. doi: 10.1073/pnas.0907904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheng Y, Ding S, Chen K, Chen J, Wang S, Zou C, Zhang J, Cao Y, Huang A, Tang H. 2014. Functional analysis of miR-101-3p and Rap1b involved in hepatitis B virus-related hepatocellular carcinoma pathogenesis. Biochem Cell Biol 92:152–162. doi: 10.1139/bcb-2013-0128. [DOI] [PubMed] [Google Scholar]
- 38.Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ, Tsai TF, Lin YJ, Wu CT, Liu HS. 2014. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology 59:505–517. doi: 10.1002/hep.26659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lendvai G, Jarmay K, Karacsony G, Halasz T, Kovalszky I, Baghy K, Wittmann T, Schaff Z, Kiss A. 2014. Elevated miR-33a and miR-224 in steatotic chronic hepatitis C liver biopsies. World J Gastroenterol 20:15343–15350. doi: 10.3748/wjg.v20.i41.15343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hou J, Lin L, Zhou W, Wang Z, Ding G, Dong Q, Qin L, Wu X, Zheng Y, Yang Y, Tian W, Zhang Q, Wang C, Zhang Q, Zhuang SM, Zheng L, Liang A, Tao W, Cao X. 2011. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell 19:232–243. doi: 10.1016/j.ccr.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 41.Vickers KC, Shoucri BM, Levin MG, Wu H, Pearson DS, Osei-Hwedieh D, Collins FS, Remaley AT, Sethupathy P. 2013. MicroRNA-27b is a regulatory hub in lipid metabolism and is altered in dyslipidemia. Hepatology 57:533–542. doi: 10.1002/hep.25846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shirasaki T, Honda M, Shimakami T, Horii R, Yamashita T, Sakai Y, Sakai A, Okada H, Watanabe R, Murakami S, Yi M, Lemon SM, Kaneko S. 2013. MicroRNA-27a regulates lipid metabolism and inhibits hepatitis C virus replication in human hepatoma cells. J Virol 87:5270–5286. doi: 10.1128/JVI.03022-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsang J, Zhu J, van Oudenaarden A. 2007. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol Cell 26:753–767. doi: 10.1016/j.molcel.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kida K, Nakajima M, Mohri T, Oda Y, Takagi S, Fukami T, Yokoi T. 2011. PPARalpha is regulated by miR-21 and miR-27b in human liver. Pharm Res 28:2467–2476. doi: 10.1007/s11095-011-0473-y. [DOI] [PubMed] [Google Scholar]
- 45.Ji J, Zhang J, Huang G, Qian J, Wang X, Mei S. 2009. Over-expressed microRNA-27a and 27b influence fat accumulation and cell proliferation during rat hepatic stellate cell activation. FEBS Lett 583:759–766. doi: 10.1016/j.febslet.2009.01.034. [DOI] [PubMed] [Google Scholar]
- 46.van der Meer DLM, Degenhardt T, Vaisanen S, de Groot PJ, Heinaniemi M, de Vries SC, Muller M, Carlberg C, Kersten S. 2010. Profiling of promoter occupancy by PPARalpha in human hepatoma cells via ChIP-chip analysis. Nucleic Acids Res 38:2839–2850. doi: 10.1093/nar/gkq012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernandez-Alvarez A, Alvarez MS, Gonzalez R, Cucarella C, Muntane J, Casado M. 2011. Human SREBP1c expression in liver is directly regulated by peroxisome proliferator-activated receptor alpha (PPARalpha). J Biol Chem 286:21466–21477. doi: 10.1074/jbc.M110.209973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chi SW, Zang JB, Mele A, Darnell RB. 2009. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fernández-Hernando C. 2013. Emerging role of microRNAs in the regulation of lipid metabolism. Hepatology 57:432–434. doi: 10.1002/hep.25960. [DOI] [PubMed] [Google Scholar]
- 50.Zhang M, Wu JF, Chen WJ, Tang SL, Mo ZC, Tang YY, Li Y, Wang JL, Liu XY, Peng J, Chen K, He PP, Lv YC, Ouyang XP, Yao F, Tang DP, Cayabyab FS, Zhang DW, Zheng XL, Tian GP, Tang CK. 2014. MicroRNA-27a/b regulates cellular cholesterol efflux, influx and esterification/hydrolysis in THP-1 macrophages. Atherosclerosis 234:54–64. doi: 10.1016/j.atherosclerosis.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 51.Sun C, Huang F, Liu X, Xiao X, Yang M, Hu G, Liu H, Liao L. 2015. miR-21 regulates triglyceride and cholesterol metabolism in non-alcoholic fatty liver disease by targeting HMGCR. Int J Mol Med 35:847–853. doi: 10.3892/ijmm.2015.2076. [DOI] [PubMed] [Google Scholar]
- 52.Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. 2003. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med 9:213–219. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- 53.Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, Rosenfeld MG, Glass CK. 2009. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev 23:681–693. doi: 10.1101/gad.1773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Castrillo A, Joseph SB, Marathe C, Mangelsdorf DJ, Tontonoz P. 2003. Liver X receptor-dependent repression of matrix metalloproteinase-9 expression in macrophages. J Biol Chem 278:10443–10449. doi: 10.1074/jbc.M213071200. [DOI] [PubMed] [Google Scholar]
- 55.Cai T, Dufour JF, Muellhaupt B, Gerlach T, Heim M, Moradpour D, Cerny A, Malinverni R, Kaddai V, Bochud M, Negro F, Bochud PY, Swiss Hepatitis C Cohort Study Group . 2011. Viral genotype-specific role of PNPLA3, PPARG, MTTP, and IL28B in hepatitis C virus-associated steatosis. J Hepatol 55:529–535. doi: 10.1016/j.jhep.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 56.Goossens N, Negro F. 2014. Is genotype 3 of the hepatitis C virus the new villain? Hepatology 59:2403–2412. doi: 10.1002/hep.26905. [DOI] [PubMed] [Google Scholar]
- 57.Rutledge H, Baran-Gale J, de Villena FP, Chesler EJ, Churchill GA, Sethupathy P, Kelada SNP. 2015. Identification of microRNAs associated with allergic airway disease using a genetically diverse mouse population. BMC Genomics 16:633. doi: 10.1186/s12864-015-1732-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kurtz CL, Fannin EE, Toth CL, Pearson DS, Vickers KC, Sethupathy P. 2015. Inhibition of miR-29 has a significant lipid-lowering benefit through suppression of lipogenic programs in liver. Sci Rep 5:12911. doi: 10.1038/srep12911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peck BCE, Weiser M, Lee SE, Gipson GR, Iyer VB, Sartor RB, Herfarth HH, Long MD, Hansen JJ, Isaacs KL, Trembath DG, Rahbar R, Sadiq TS, Furey TS, Sethupathy P, Sheikh SZ. 2015. MicroRNAs classify different disease behavior phenotypes of Crohn’s disease and may have prognostic utility. Inflamm Bowel Dis 21:2178–2187. doi: 10.1097/MIB.0000000000000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miR-122-3p and miR-122-5p comparison. (A) Small-RNA sequencing results from AGO2 immunoprecipitation and total RNA from FT3-7 cells for miR-122-3p and miR-122-5p. (B) Small-RNA sequencing results for miR-122-3p and miR-122-5p in liver tissue from each of the disease states (uninfected control, CHB, c-CHB, CHC, and c-CHC). Download
Transfection efficiency of miRNA mimics. In experiments separate from those described in Fig. 6, mimics formiR-21 (n = 4) (A) and miR-27b (n = 4) (B) were transfected into human hepatoma (Huh7) cells at a concentration of 10 nM, and miRNA levels were measured by quantitative PCR (qPCR) and compared to transfection reagent alone. Download
Detailed model of miRNA regulation of cholesterol synthesis in chronic viral hepatitis and associated liver cancer. Cholesterol synthesis genes that are significantly altered in at least one disease state are shown. The color of the pie quadrant to the right of each gene indicates the disease state in which each gene is significantly altered as defined previously (light pink, c-CHB; dark pink, CHB; light blue, c-CHC; dark blue, CHC). Green outline indicates that the gene is downregulated. Each of the miRNAs displayed has at least one predicted target site in at least one of the genes listed, which is conserved in humans and at least one other species. Each miRNA is significantly upregulated in at least one disease state relative to uninfected controls and is either a predicted miRNA master regulator of gene expression in at least one disease state based on miRHub P value of <0.05 (pie piece outlined in blue) or has a fold change of >5 compared to uninfected controls (outlined in orange) or both (outlined in black). Download
Detailed model of miRNA regulation of cholesterol transport and efflux in chronic viral hepatitis and associated cancer. Significantly altered cholesterol transport and efflux genes in at least one disease state are shown. The color of the pie quadrant to the right of each gene indicates the disease state in which each gene is significantly altered as defined previously (light pink, c-CHB; dark pink, CHB; light blue, c-CHC; dark blue, CHC). Green or red outline indicates that the gene is downregulated or upregulated, respectively. Each of the miRNAs displayed has at least one predicted target site, in at least one of the genes listed, which is conserved in humans and at least one other species. Each miRNA is significantly upregulated in at least one disease state relative to uninfected controls and is either a predicted miRNA master regulator of gene expression in at least one disease state based on miRHub P value of <0.05 (pie piece outlined in blue) or has a fold change of >5 compared to uninfected controls (outlined in orange) or both (outlined in black). Download
Basic clinical patient data. Patient identifier (ID), sex (M, male; F, female), age (in years), tumor grade (p, poor; m, moderate; w, well-differentiated), and METAVIR score are shown.
Significantly altered genes in each disease state. Any probe significantly DE in one or more of the four disease groups listed below, with the significantly changed disease group’s fold change listed.
Small-RNA-seq mapping statistics. Patient ID, number of total reads for each patient, number of reads successfully trimmed, percentage of reads trimmed of total reads, number of mapped reads, percentage of reads mapped of trimmed reads are shown.
Enriched pathways of miRNA master regulator targets. For each miRNA predicted as a candidate master regulator of up- and downregulated genes in each disease category, we identified biological pathways significantly enriched among its predicted target genes. Each biological pathway is significant (P < 0.01) for the miRNAs listed.