

Abstract
BACKGROUND
Asthma exacerbations are a major cause of morbidity and medical cost.
OBJECTIVE
The objective of this study was to identify genetic predictors of exacerbations in subjects with asthma.
METHODS
We performed GWAS meta analysis of acute asthma exacerbation in two pediatric clinical trials: Childhood Asthma Management Program (CAMP, n=581) and Childhood Asthma Research and Education (CARE, n=205) network trials. Acute asthma exacerbations was defined as treatment with a five-day course of oral steroids. We obtained a replication cohort from BioVU (n=786), the Vanderbilt University electronic medical record-linked DNA biobank. We used CD4+ lymphocyte genome-wide mRNA expression profiling to identify associations of top SNPs with mRNA abundance of nearby genes.
RESULTS
A locus in CTNNA3 reached genome-wide significance (rs7915695, p=2.19*10-8, mean exacerbations 6.05 for minor alleles vs. 3.71 for homozygous major). Among four top SNPs replicated in BioVU, rs993312 in SEMA3D was significant (p=0.0083), and displayed stronger association among African Americans (p=0.0004 in BioVU, mean exacerbations 3.91 vs. 1.53; p=0.0089 in CAMP, mean exacerbations 6.0 vs. 3.25). CTNNA3 variants did not replicate in BioVU. A regulatory variant in the CTNNA3 locus was associated with CTNNA3 mRNA expression in CD4+ cells from asthmatics (p=0.00079). CTNNA3 appears to be active in immune response, and SEMA3D has a plausible role in airway remodeling. We also provide a replication of a previous association of P2RX7 with asthma exacerbation.
CONCLUSIONS
We identified two loci associated with exacerbations through GWAS. CTNNA3 met genome-wide significance thresholds and SEMA3D replicated in a clinical Biobank database.
Keywords: Asthma, Exacerbation, GWAS, eQTL, CTNNA3, SEMA3D, BioBank, Childhood Asthma
Introduction
Although many asthma patients maintain control of asthma symptoms, acute asthma exacerbations remain a dangerous and costly consequence of asthma. In 2008, asthma exacerbations resulted in 10.5 million school days lost and 14.2 million work days lost;[1] in 2007 asthma exacerbations resulted in 3,445 deaths, 1.75 million emergency-department visits, and 456 thousand hospitalizations.[1] The cost of these events is estimated at 50 billion US dollars in direct medical costs and another 6 billion in loss of productivity annually.[2] The investigation of exacerbation of asthma has the possibility to improve care and curtail an important public health burden.
An asthma exacerbation has been defined as “worsening of asthma requiring the use of systemic corticosteroids ... to prevent a serious outcome.”[3] Exacerbations have been associated with several clinical and demographic factors including respiratory infection,[4, 5] race,[6] sex,[7] treatment noncompliance[8], and smoke[9] and air pollution[10] exposures. A host of genetic variants have also been linked to asthma exacerbation rates (reviewed in Greenberg[11]), and also broader genomic features including gene methylation,[12] peripheral blood gene expression,[13] and gene-exposure interactions.[14] These studies have demonstrated the importance of genomic factors in influencing asthma exacerbation rates.
Previous genetic studies of asthma exacerbation include successful candidate gene investigations;[15-18] and genome-wide association studies (GWAS) that a) investigated variant-vitamin-D interactions,[14] b) resulted in no genome-wide significant results,[19] or c) identified a predictive model without identifying GWAS loci.[20] Our objective was to conduct a GWAS of asthma exacerbations in a pediatric cohort to identify additional genetic loci associated with increased or reduced exacerbation. We analyzed GWAS data from two clinical trials of childhood asthmatics, the Children's Asthma Management Program (CAMP)[21] and Childhood Asthma Research and Education (CARE)[22] network trials, in which measures of systemic oral steroid use were available, and performed replication in an electronic medical record (EMR)-linked biobank from Vanderbilt University (BioVU).[23] The combination of these trials provided an opportunity to identify genetic variants that may be associated with asthma exacerbation in children and uncover additional biology related to acute asthma episodes, while providing a replication in a more diverse outpatient sample.
Methods
CAMP
We performed a GWAS in the CAMP [21] cohort. CAMP enrolled 1,041 children ages 5 to 12 years with mild to moderate persistent asthma over a 23-month period, and followed them for an additional 4 years. Recruitment criteria and study characteristics have been described previously.[21, 24] CAMP participants were randomized into three treatment arms: budesonide, nedocromil, or placebo. Following the CAMP trial design, acute asthma exacerbation was treated with a prednisone dose of 2 mg/kg per day (maximum 60 mg) as a single morning dose for 2 days, followed by 1 mg/kg per day (maximum 30 mg) as a single morning dose for 2 days, with additional treatment extension up to the supervising physician's discretion, based on clinical response.[21] We considered the total number of these exacerbations (“bursts”) over the length of the CAMP trial (4 years) to be our primary outcome.
CARE
The CARE[22] network clinical trials enrolled children between the ages of 1-18 years at the time of their study participation who had a confirmed diagnosis of asthma or wheezing illness. In order to match the phenotype and clinical characteristics of CAMP, we studied the 205 participants from three CARE network trials: Pediatric Asthma Controller Trial (PACT, n = 130);[25] Characterizing the Response to a Leukotriene Receptor Antagonist and an Inhaled Corticosteroid (CLIC, n = 53)[26]; and Montelukast or Azithromycin for Reduction of Inhaled Corticosteroids in Childhood Asthma Study (MARS, n = 22).[27] These subjects were age 5 or more years at enrollment, had a diagnosis of asthma, and had detailed exacerbation data (supplemental prednisone courses prescribed) over the length of those studies (PACT 48 weeks; CLIC 16 weeks; MARS 6 weeks to 26 weeks). Total number of exacerbations was used as the primary outcome.
Vanderbilt BioVU
BioVU[23] accrues DNA samples during routine clinical care from Vanderbilt patients who have not opted out of participation, using blood that would have otherwise been discarded for clinical testing. Biological samples from BioVU are linked using research unique identifiers to the Synthetic Derivative, a de-identified version of Vanderbilt's EMR.
We identified subjects who had a diagnosis of asthma and were prescribed inhaled corticosteroids with an electronic algorithm developed and validated by physician chart review of 50 randomly-selected cases. We selected subjects aged 4-35 years who had ≥2 encounters with a diagnosis of asthma on 2 different days. We included subjects who had a prescription for an inhaled corticosteroid. The algorithm excludes subjects with confounding diseases such as cystic fibrosis, chronic obstructive pulmonary disease, pulmonary hypertension, pulmonary embolism, lung cancer, immunodeficiency, bronchiectasis, congestive heart failure, and other diseases. The algorithm identified asthma-related hospitalizations and emergency department visits. The positive predictive value of the algorithms was shown to be >92%. The algorithm is available on http://PheKB.org.
From the BioVU population, we identified a cohort of 786 asthmatic subjects. We defined exacerbation as an asthma-related hospitalization or emergency room visit for which there was an injected or oral steroid prescribed, occurring in 191 patients of the 786. We counted number of exacerbations occurring in a four-year window, a time period chosen to match the duration of observation in CAMP. This was used as our primary outcome for the BioVU cohort (Supplemental Figure S2).
GWAS
We used CAMP subjects passing genotype quality controls with self-designated European ancestry (n = 581). Genome-wide genotyping was previously performed at the Channing Division of Network Medicine (CDNM), using Illumina HumanHap 610 bead chips (Illumina, Inc., San Diego, CA), which was then imputed using MaCH[28] with reference to the 1000 Genomes Pilot data to obtain high-confidence imputations on over 6.5 million single-nucleotide polymorphisms (SNPs). We removed any SNPs that had an r2 MaCH imputation-quality score less than 0.5, as well as any SNPs with minor allele frequency less than 0.05. We used imputed SNP data and estimated minor alleles for each SNP (a continuous measure between 0 and 2) in order to use the most informative SNP calculation from the imputation. We performed a GWAS for association with number of bursts using a linear regression model with additive effects for each SNP using PLINK v1.07,[29] adjusting for age and gender as covariates. Similar results were obtained with MaCH r2 less than 0.3.
To verify that the top hits from the GWAS were not artifacts of a non-normally distributed phenotype, we performed permutation testing with the top GWAS SNPs (p < 10-5). This is a nonparametric method for adjusting for a non-normal phenotype distribution; we chose this method over other methods such as Poisson regression (unsuitable because phenotype distribution does not have variance = mean) and log-transformation of the phenotype (performs poorly, except under particular conditions)[30]. Permutation testing was done with PLINK, using 100 million permutations to estimate the null distribution for each of the top SNPs.
The top CAMP GWAS SNPs were tested for association in CARE using a linear regression test for association with number of steroid bursts. We obtained CARE data on genome-wide SNP measurements from the Asthma BioRepository for Integrative Genomic Exploration (ABRIDGE).[31] Genotyping was previously performed through the SNP Health Association Resource (SHARe) Asthma Resource Project (SHARP), by Affymetrix Inc, (Santa Clara, CA, USA) according to manufacturer's protocol using the Affymetrix Genome-Wide Human SNP Array 6.0. This genotype data is archived on the database of Genotypes and Phenotypes (dbGaP, www.ncbi.nlm.nih.gov/gap) for all CARE participants. Imputation was previously performed at CDNM as before (using MaCH, 6.5M SNPs imputed to 1000Genomes). Significance p-values obtained from the CAMP permutation testing and the CARE replication were combined using the Liptak-Stouffer method.
Four top loci were identified for replication in BioVU, and were assayed at single SNPs using Taqman (Applied Biosystems). To consider as similar a phenotype as possible to our CAMP bursts phenotype, we counted the number of exacerbations in a four-year period, choosing a 4-year period that maximized exacerbations for each patient. These SNPs were then tested for linear association with number of exacerbations, with adjustment for race, gender, and smoking status.
Expression analysis
We used all available CD4+ lymphocyte cells with genome-wide mRNA expression profiling from ABRIDGE to check our top SNPs for potential functional effects on gene expression. The ABRIDGE project contains a wide variety of blood and cell lines from various extant asthma trials, which includes CD4+ lymphocytes from the CARE and Genomic Research on Asthma in the African Diaspora (GRAAD)[32] cohorts of ABRIDGE. These were previously processed on Illumina Human HT-12 v4 arrays, according to manufacturers’ protocol (Illumina, San Diego CA). This array assays for 47036 different gene RNA products. These data were processed through quality control metrics at the CDNM for data missingness rates, arrays that failed across subjects, gene probes that displayed either outlying expression levels or limited changes in expression. This resulted in 154 samples from CARE and 195 samples from GRAAD passing quality control metrics that were available for analysis. Each SNP within 400kbp of rs7915695 was tested for association with CTNNA3 mRNA expression in the 349 CD4+ lymphocytes. Expression p-values were computed using regression and including the top 10 principal components of expression to account for stratification and batch effects. Bronchoalveolar cells from ABRIDGE (n = 43) were used in case / control expression analysis.
RNA-Seq analysis
We tested for differential expression of the SEMA3D gene in asthma airway smooth muscle (ASM) using RNA-Seq dataset GSE58434, which is available from the Gene Expression Omnibus Web site (http://www.ncbi.nlm.nih.gov/geo/). For that study, primary ASM cells were isolated from white donors, five with fatal asthma and ten age- and gender-matched controls with no chronic illness. ASM cell cultivation was described previously [33]. Total RNA was extracted, sequenced, and analyzed as described previously [34]. Briefly, TopHat-2.0.9 was used to align reads to the hg19 genome build, Cufflinks (v.2.1.1) was used to quantify hg19 transcripts based on reads that mapped to the provided hg19 reference file, and differential expression of genes was obtained using Cuffdiff (v.2.1.1) while applying bias correction for all samples.
Results
Baseline characteristics for our three cohorts (CAMP, CARE, and BioVU) are shown in Table 1. CAMP and CARE are similar in many regards, whereas subjects in BioVU are significantly older in age, have higher body-mass index (BMI), are more likely to be current smokers, less likely to be of European ancestry, and less likely to be male. Subjects in CAMP required more oral steroid bursts on average (4.12 +/- 4.84) compared to subjects in CARE (0.60 +/- 1.17) and BioVU (0.93 +/- 3.47, p < 0.001).
Table 1.
Cohort characteristics.
| CAMP | CARE | BioVU | |
|---|---|---|---|
| N | 581 | 205 | 786 |
| Race, N white (%)* | 581 (100%) | 205 (100%) | 447 (56.7°%)** |
| Age, mean (std)* | 8.87 (+/− 2.14) | 10.59 (+/− 3.03) | 22.34 (+/− 10.76) |
| Gender, N males (%)* | 347 (59.72%) | 127 (61.95%) | 256 (32.57%) |
| Height (cm), mean (std)* | 133.17 (+/− 13.86) | 143.09 (+/− 17.37) | 156.71 (+/− 21.07) |
| BMI (kg/m2), mean (std)* | 17.91 (+/− 3.15) | 19.99 (+/− 4.87) | 26.97 (+/− 9.68) |
| Asthma Onset (age), mean (std)* | 3.12 (+/− 2.46) | 3.93 (+/− 3.26) | 17.04 (+/− 10.29) |
| Smoking, N smokers (%)* | 12 (2.36%) | 0 (0.00%) | 197 (25.06%) |
| ICS, N ICS (%)* | 172 (29.60%) | 198 (96.59%) | 766 (97.46%) |
| Bursts, mean count (std)* | 4.12 (+/− 4.84) | 0.60 (+/− 1.17) | 0.93 (+/− 3.47) |
All variables significantly different by one-sided ANOVA test, p < 0.001
Racial ancestry makeup in BioVU also contains 27% African Americans (n = 212) and 16% other/unknowns (n = 127).
GWAS results on 581 CAMP subjects as are shown in Supplemental Figure S4. The genomic inflation factor (GIF) of the total p-value distribution of the GWAS was 1.0048 (Supplemental Figure S3), indicating minimal stratification and the general suitability of the null hypothesis of no association for most assayed SNPs. Label-shuffling permutation tests (Supplemental Figure S5) on SNPs with raw p-value < 10-5 showed little variation in p-values for the indicated SNPs in the genes catenin (cadherin-associated protein), alpha 3 (CTNNA3), sema domain, immunoglobulin domain (Ig), short basic domain, secreted, (semaphorin) 3D (SEMA3D), titin (TTN), and purinergic receptor P2X, ligand-gated ion channel, 7 (P2RX7), providing further evidence the original (linear-regression) GWAS was a suitable test of the association of SNPs with exacerbation phenotype, despite the phenotype having a non-normal distribution. No SNPs reached statistical threshold for genome-wide significance in the permuted CAMP GWAS (p < 5 * 10-8).
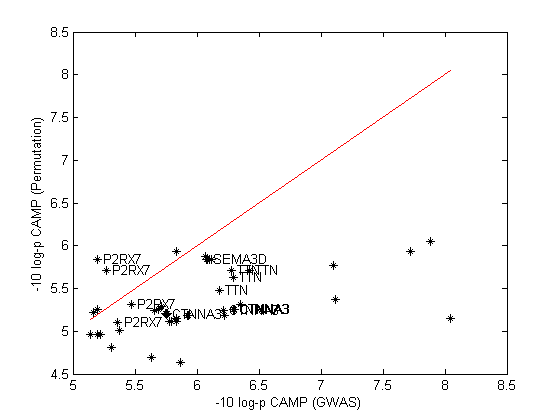
For these top SNPs, we combined analysis with a GWAS of the CARE cohort (n = 205). Figure 1 shows the combined (Liptak) CAMP-CARE p-values for the top SNPs vs. the original CAMP permutation p-values. This indicates that the strong signals in CTNNA3 and TTN were confirmed, while SEMA3D and P2RX7 were not significant in CARE. Table 2 shows the observed p-values (in CAMP and CARE) for the top genic SNPs (a full list of top SNPs appears in Supplemental Table S1). Considering the meta-analyses of these SNPs, the locus in CTNNA3 meets genome-wide threshold for statistical significance (p < 5 * 10-8), with SNPs rs7915695, rs7923078, rs7923279, and rs6480203 all reaching genome-wide significance. CAMP subjects with one or more C alleles for rs7915695 experienced more exacerbations (mean 6.05) than subjects homozygous for the reference T allele (mean 3.71) (Supplemental Figure S1). To test the generalizability of these variants to predict asthma exacerbations in the general population, we studied 786 asthmatic samples from BioVU.[23] The top SNPs in TTN represented 2 distinct linkage disequilibrium (LD) blocks, while the SNPs in CTNNA3 and SEMA3D each represented only one; accordingly four representative SNPs were chosen for genotyping in BioVU (Table 3). The SEMA3D SNP, rs993312, was significantly associated after Bonferonni correction for four independent tests, using covariates for race, sex, and smoking status. In stratified analysis, rs993312 was not significant in subjects of European ancestry in BioVU (p = 0.51), although it was more significant in African Americans (n = 212, p = 0.0004, mean exacerbations 3.91 vs. 1.53 for one or more A alleles, Supplemental Figure S2). We then surveyed 96 African Americans from the CAMP cohort, not included in the original GWAS analysis, and found an association of rs993312 with exacerbation (p = 0.0089, mean exacerbations 6.0 vs. 3.25, Supplemental Figure S1). The SNPs tested in TTN and CTNNA3 did not replicate in BioVU. The CTNNA3 SNP, rs7915695, is an intronic SNP within the CTNNA3 gene. It is in strong LD (r2 = 0.97) with a predicted regulatory variant, rs10997296, which is annotated as a strong enhancer (HaploReg v2)[34] in human mammary epithelial cells. Rs10997296 is further in a DNAse I hypersensitivity site in a large variety of epithelial and endothelial cells, including small airway epithelial cells.[34] This implies that rs10997296 may have a role in regulating expression of CTNNA3 in a cell type of relevance to asthma exacerbation. The risk allele of rs10997296 was associated with lower CTNNA3 expression in a sample of CD4+ lymphocyte cells drawn from 349 asthmatics in the ABRIDGE cohort (p = 7.9 * 10-4, Figure 2 and Supplemental Figure S6). We have included a list of top eQTL SNPs for CTNNA3 in Supplemental Table S3, which may be of interest to future investigators of this locus. This association was also significant in a smaller sample of 111 CD4+ lymphocytes from non-asthmatic controls in ABRIDGE (p = 0.0012).
Figure 1.

CAMP Permutation vs. joint CAMP-CARE p-values from meta-analysis for top GWAS SNPs. Stars indicate SNPs and are annotated with gene symbols in cases where the SNP is within a gene. Diagonal line indicates x = y; SNPs that did not change p-value from CAMP permutation testing to CAMP-CARE meta-analysis are close to this line.
Table 2.
Results of top SNPs after meta-analysis in CAMP and CARE cohorts.
| CHR | SNP | Effect Allele | MAF (hapmap CEU) | Gene | SNP role | CAMP p : Permutation | CARE p | Effect Dirs | Combined p |
|---|---|---|---|---|---|---|---|---|---|
| 10 | rs7915695 | C | 9.3% | CTNNA3 | Intronic | 6.45 * 10−6 | 2.14 * 10−5 | +/+ | 2.19 * 10−8* |
| 10 | rs7923078 | C | 11.5% | CTNNA3 | Intronic | 5.54 * 10−6 | 6.52 * 10−5 | +/+ | 3.02 * 10−8* |
| 10 | rs7923279 | T | 11.7% | CTNNA3 | Intronic | 5.54 * 10−6 | 8.09 * 10−5 | +/+ | 3.34 * 10−8* |
| 10 | rs6480203 | C | 11% | CTNNA3 | Intronic | 5.71 * 10−6 | 9.88 * 10−5 | +/+ | 3.79 * 10−8* |
| 10 | rs10997296 | A | 8.3% | CTNNA3 | Intronic | 6.09 * 10−5 | 6.34 *10−5 | +/+ | 4.9 * 10−7 |
| 2 | rs1484118 | A | 7.6% | TTN | Intronic | 1.95 * 10−6 | 0.08122 | +/+ | 7.24 * 10−7 |
| 2 | rs11893580 | G | 8.2% | TTN | Intronic | 2.36 * 10−6 | 0.09024 | +/+ | 9.60 * 10−7 |
| 2 | rs746578 | A | 7.5% | TTN | Coding | 1.93 * 10−6 | 0.09668 | -/- | 8.41 * 10−7 |
| 2 | rs4894042 | G | 8.3% | TTN | Intronic | 3.32 * 10−6 | 0.08119 | +/+ | 1.22 * 10−6 |
| 12 | rs7978398 | T | ?* | P2RX7 | Intronic | 1.45 * 10−6 | 0.2357 | +/− | 1.51 * 10−5 |
| 12 | rs11065464 | A | 19.5% | P2RX7 | Intronic | 1.95 * 10−6 | 0.2032 | −/+ | 2.28 * 10−5 |
| 12 | rs7958311 | A | 20% | P2RX7 | Coding | 4.89 * 10−6 | 0.08952 | −/+ | 9.85 * 10−5 |
| 7 | rs993312 | A | 27% | SEMA3D | Intronic | 1.45 * 10−6 | 0.9734 | +/− | 8.23 * 10−5 |
Indicates SNPs meeting significance thresholds for genome-wide significance (p < 5 * 10−8). Combined p-value computed using Liptak-Stouffer method. Effect directions refer to the sign of CAMP beta values (leading the slash) and CARE beta values (trailing the slash), which are interpreted as the direction of exacerbations for each additional copy of the effect allele; ie, ‘+’ means each additional copy of the allele resulted in an increase of exacerbations. Effect Allele shown is the minor allele, and was used for meta analysis.
Table 3.
SNP replication results in BioVU.
| Gene | Chr | SNP | Position | Allele | P |
|---|---|---|---|---|---|
| TTN | 2 | rs13390491 | 179582327 | T | 0.7638 |
| TTN | 2 | rs4894044 | 179632133 | A | 0.1982 |
| SEMA3D | 7 | rs993312 | 84747844 | A | 0.00825* |
| CTNNA3 | 10 | rs7915695 | 68444734 | C | 0.8236 |
SNPs assayed in BioVU using Taqman (Applied Biosystems). These represent four distinct loci in different linkage disequilibrium blocks.
Indicates rs993312 was significant after multiple testing correction for four tests.
Figure 2.

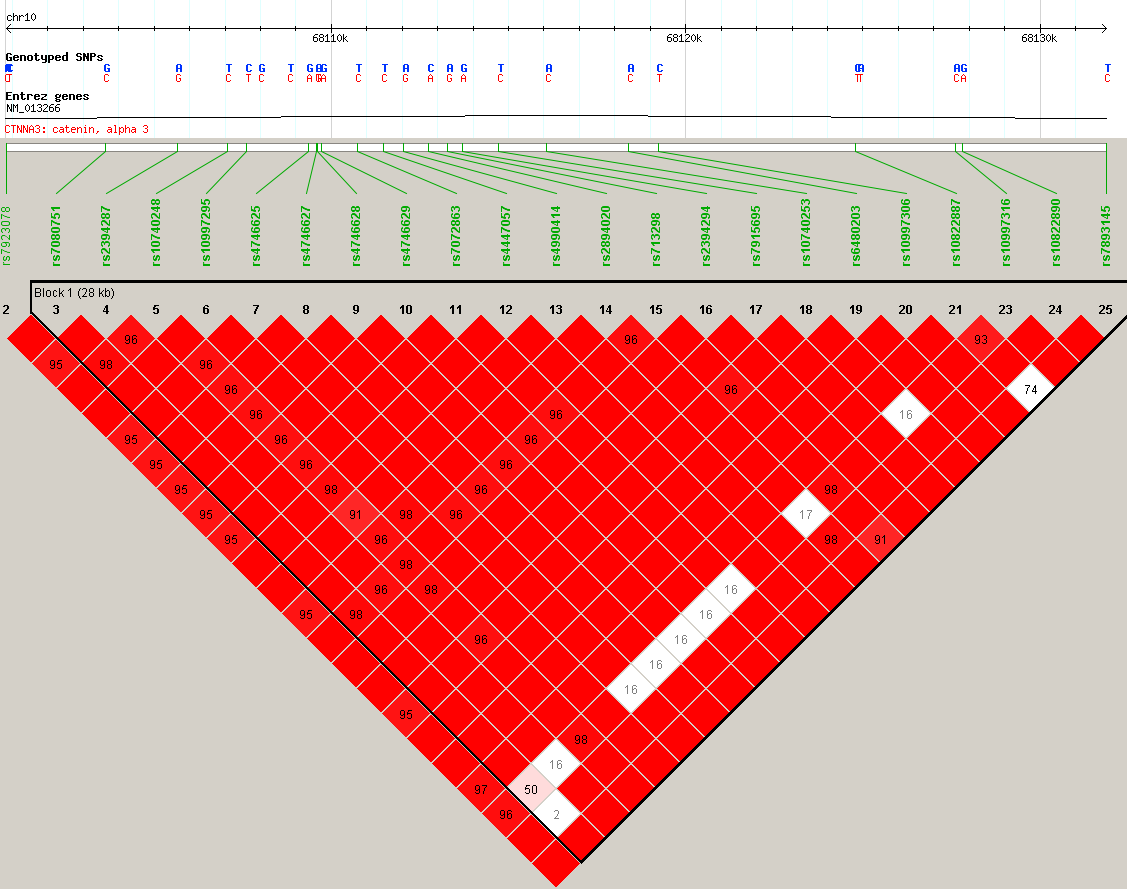
CTNNA3 expression quantitative trait locus (eQTL) plot for rs10997296. The suspected regulatory SNP, rs10997269, is labeled and highlighted with a purple diamond. Rs7915695 is indicated with a black arrow. Other SNPs are colored with their linkage disequilibrium to rs10997269 (r2), indicating a locus of SNPs nearby that may have action on CTNNA3 expression. Figure generated with LocusZoom.
While we have analyzed these variants for association with an asthma severity phenotype: exacerbations; for comparison, we also checked the top two associations in SEMA3D and CTNNA3 for association with asthma affection status. Additionally the rs993312 risk allele A, in SEMA3D, was also associated with asthma occurrence in a large asthma meta-analysis (p = 0.047, GABRIEL)[35]. The CTNNA3 risk allele, C, for rs7915695 showed suggestive evidence in the same (p = 0.14, GABRIEL)[35]. Compared with our own results, the same alleles both increase occurrence of asthma and occurrence of asthma exacerbations. We further checked SEMA3D gene products for expression differences in asthmatic vs. control samples from ABRIDGE, finding a significant effect in bronchoalveolar samples (p = 0.0295, n = 43, regression test, Supplemental Figure 10), where SEMA3D expression was somewhat increased in cases relative to controls. In a small sample of human airway smooth muscle cells (n = 15), SEMA3D expression was found to differ significantly between asthma cases and age/sex matched controls (q = 0.011, transcriptome-wide adjusted value, Cuffdiff v.2.1.1), with greater expression in asthmatic samples. CTNNA3 was not expressed in ASM cells, nor was it significantly differentially expressed in bronchoalveolar samples.
Discussion
We found evidence for two loci of association with asthma exacerbation: one in CTNNA3 reached genome-wide significance thresholds in the meta-analysis of two pediatric cohorts; the other in SEMA3D replicated in an adult, diverse, real-life population, and in particular in African Americans.
SEMA3D has been previously implicated in both schizophrenia[36] and anomalous pulmonary venous connections[37]. The SEMA3D SNP, rs993312 is intronic to the SEMA3D gene, and in strong LD (r2 = 0.99) with a regulatory intronic SNP, rs55834466. The ENCODE project identifies rs55834466 as a strong enhancer in epidermal keratinocytes and human mammary epithelial cells (HaploReg v2[34]). In addition to brain and nervous tissue, SEMA3D is relatively overexpressed in the lung, in comparison to other tissue types (Illumina Human BodyMap 2.0[38]).
SEMA3D is a member of the semaphorin class 3 signaling molecules; of which SEMA3A and SEMA3E are secreted transmembrane proteins involved in immune response and the recruitment of CD4+ and CD8+ T-cells.[39] SEMA3A and SEMA3E are adjacent to SEMA3D on chromosome 7. SEMA3D is a signaling protein responsible for endothelial cell migration,[40] and has been shown to be essential to healthy angiogenesis during development.[41] Angiogenesis is also a feature of airway remodeling,[42] the process of tissue changes in the airways that occurs in some asthmatics and leads to worsening asthma symptoms and development of chronic airway obstruction.[43] Since SEMA3D is known to direct angiogenesis in development and can repress angiogenesis in cancer,[44] it is possible that SEMA3D plays a role in airway remodeling. In fact, there are three plausible mechanisms of SEMA3D's involvement in airway remodeling and thus asthma exacerbation[43]: 1) that it directs angiogenesis, 2) that it directs airway epithelium migration resulting in a reduction of epithelial cells, and 3) that possibly, like other semaphorins, it has effects on immune cell recruitment during inflammatory response, which leads to remodeling. Examination of SEMA3D expression in ABRIDGE samples did not reveal a significant eQTL in linkage disequilibrium with rs993312.
While the CTNNA3 SNP did not replicate in BioVU, the associations were very strong in both the CAMP and CARE cohorts, and both effects were in the same direction, with the minor allele decreasing the probability of asthma exacerbation. This is similar to our previous work where strong associations in pediatric asthma were not replicated in adult cohorts.[45] We verified that rs10997296, which was in strong LD with the top CTNNA3 GWAS hit rs7915695, was associated with CTNNA3 expression in CD4+ lymphocytes, immune cells that represent an important cell type in asthma exacerbation. CTNNA3 has previously been associated with diisocyanide toluene induced asthma, in at least two separate cohorts, one Korean[46] and one of European ancestry.[47] A SNP in the promoter region of CTNNA3 was also associated with response to asthma treatment (glucocorticoid therapy) in a population of childhood asthmatics.[48] The rs7915695 variant is itself in a strong enhancer (HaploReg v2)[34] and has been observed to bind the transcription factor CEBP-Beta (CEBP-B), a member of the CCAAT/Enhancer Binding Proteins (CEBPs) family of transcription factors that help regulate a wide variety of processes, including inflammatory response.[49] Two of these factors, CEBP-B and CEBP-D, have been shown to be induced by glucocorticoids in lung epithelium[50] and skeletal muscle cells.[51] Additionally, the transcription of CEBP-B has been shown to be induced by Interleukin 1 (IL1), Interleukin 6 (IL6), and lipopolysaccharides (LPS).[49] CEBP-B's broad influence on inflammatory processes and genes, and the known binding of CEBP-B with CTNNA3 provide evidence for CTNNA3's likely role in immune response. Additionally the CTNNA3 protein has been shown to play a role in muscle cell coherence, which may also provide insight to its possible action on asthma exacerbation.[52]
While not carried forward for replication in BioVU, the P2RX7 gene has recently been associated with asthma exacerbation.[16] A coding SNP in P2RX7 was suggestively significant in the CAMP cohort (p < 5 * 10-6) after permutation, and this CAMP result can thus be seen as a replication of the earlier finding that P2RX7 is associated with asthma exacerbation. We also checked our top hits for correspondence with a previous GWAS, and did not find any overlap with results reported by Anderson et al. [19] A recent GWAS of early-childhood asthma and wheeze identified CDHR3 as a risk gene[53]; the top SNP identified in that study was rs6967330, which was non-significant in CAMP (p = 0.9). Of ~270 SNPs tested in CDHR3 in CAMP, rs28605757 was the most significant (p = 0.0026), however this is not significant after a multiple-testing correction for 270 tested SNPs in this gene.
Although asthma exacerbation rates are dependent upon the medication prescribed for asthma maintenance, and in particular inhaled corticosteroids can be efficacious in avoiding exacerbations,[24] these were not included in the CAMP analysis. Our results are presented without regard to ICS regimen, but rerunning the main CAMP GWAS with presence or absence of ICS as a covariate did not appreciably alter the results or p-values obtained; similarly, no interaction was detected with ICS and either rs993312 or rs7915695 (p = 0.4 and p = 0.7, respectively). Furthermore, stratified analysis in CAMP showed both rs993312 and rs7915695have consistent directions of effect and effect sizes. In the non-ICS arm of CAMP each minor allele, A, of the SEMA3D SNP rs993312 increased the number of exacerbations by 1.75 (ci [0.94, 2.55], n = 343, p = 2.73 * 10^-5) and in the ICS arm effects were similar 1.2 (ci [0.19, 2.21], n = 238, p = 0.021). Similarly, the CTNNA3 SNP, rs7915695, has consistent strength of effect in both the placebo (1.81 ci [0.68, 2.95], p = 0.0019) and ICS arms (2.25 ci [0.74, 3.76], p = 0.0038). In CARE and BioVU, the majority of participants were receiving ICS therapy (Table 1). That CTNNA3 and SEMA3 appear to influence exacerbation regardless of ICS therapy implies that their effect is not pharmacogenetic; although it is interesting that CTNNA3 has previously been associated with ICS response.
While both rs993312 and rs7915695 were found to be associated with asthma exacerbation, there was some evidence of an effect on asthma status in the GABRIEL asthma meta-analysis. Both SNPs show stronger association with asthma exacerbation than with asthma affection status; SEMA3D showed expression was associated with case/control status in both a small bronchoalveolar sample and an airway smooth muscle sample; and rs7915695 was identified as a regulator of CTNNA3 expression in both asthmatic CD4+ lymphocytes in ABRIDGE and in case/control analysis. Thus, SEMA3D may influence exacerbations by being an asthma susceptibility gene, while CTNNA3 may have a different mechanism of effect. Further work will be needed to identify precise biological mechanisms that lead to poorly-controlled asthma rather than merely to persistent asthma.
Failure to replicate of CTNNA3 may be due to some differences between the CAMP and CARE studies and the BioVU cohort. CAMP and CARE were clinical trials with detailed phenotyping and close attention from care providers, while BioVU, a real-life population, provides snapshots of a patient's interactions with the hospital and emergency department. Additionally, CAMP and CARE are pediatric cohorts, while BioVU contained a higher proportion of adults, and the exacerbation rates have been shown to be different in these groups.[7] Examining a rarely-occurring phenotype like exacerbation leads to small sample sizes, and this may also have contributed to difficultly in replicating the CTNNA3 locus. However, as more asthma cohorts are genotyped and collected, it would be important to perform additional GWAS of asthma exacerbation with larger sample sizes. As always, there is a tension between the quality of the phenotyping and the feasible size of the study, and our results here highlight the strengths of both approaches. We have provided complete lists of our top SNPs from the CAMP permutation association tests and from the CTNNA3 expression association investigation to aide future researchers that may reconsider these phenotypes with larger populations (tables S1, S3).
Our study has presented two promising loci that have several possible biochemical mechanisms for action in asthma exacerbation to augment their statistical associations in multiple cohorts. One of our variants replicated from childhood clinical trials of European ancestry to adult, biobank cohort of mixed races; this SEMA3D variant was associated in African Americans and further replicated in a sample of pediatric African Americans. This analysis demonstrates the utility of using disparate populations for identifying genetic associations. We also leverage public ABRIDGE expression data to identify DNAse I sensitivity SNPs in high LD with our top associations, and demonstrate that this in turn is associated with expression of CTNNA3. Additionally, we have demonstrated the use of a simple label-shuffling permutation-testing scheme for assessing genetic associations with non-normally distributed phenotypes. That SEMA3D seems to affect airway epithelium and CTNNA3 plays a role in adhesion of smooth muscle cells reinforces our results and the role of these cells in asthma pathogenesis and exacerbation.
In conclusion, we identified two regulatory SNPs, within CTNNA3 and SEMA3D, associated with asthma exacerbations. SEMA3D has strong evidence of a role in airway remodeling and CTNNA3 is believed to play a role in immune response; both are important processes in the causes and frequency of asthma exacerbation.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Capsule Summary.
Variants in two genes are associated with asthma exacerbation: SEMA3D that is implicated in airway remodeling; and CTNNA3 that plays a role in immune response.
Key Messages.
An eQTL in CTNNA3 is identified that is genome-wide significant for association with asthma exacerbation in children.
Genetic variants in SEMA3D are associated with asthma exacerbation and replicated in an adult BioBank population.
Acknowledgments
We thank all CAMP subjects for their ongoing participation in this study. We acknowledge the CAMP investigators and research team, supported by the National Heart, Lung and Blood Institute (NHLBI), for collection of CAMP Genetic Ancillary Study data. All work on data collected from the CAMP Genetic Ancillary Study was conducted at the Channing Division of Network Medicine of the Brigham and Women's Hospital under appropriate CAMP policies and human subject's protections.
Funding Sources
MJM is supported by a grant from the Parker B. Francis Foundation (PI: McGeachie). ACW is supported by K08 HL088046 (PI: Wu). BEH was funded by K99 HL105663 (PI: Himes). PW was funded by an unrestricted research grant from the Tryg Foundation (J.nr. 7343-09, TrygFonden, Denmark). This work was further supported by NIH grants U01 HL065899 (PIs: Weiss and Tantisira), U19 HL065962 (PI: Roden), R01 NR013391 (PI: Tantisira), and R01 HL086601 (PI: Raby).
Abbreviations
- ABRIDGE
Asthma BioRepository for Integrative Genomic Exploration
- BioVU
Biobank of Vanderbilt University Medical Center.
- BMI
body-mass index.
- CAMP
Childhood Asthma Management Program.
- CARE
Childhood Asthma Research and Education network.
- CDNM
Channing Division of Network Medicine.
- CLIC
Characterizing the Response to a Leukotriene Receptor Antagonist and an Inhaled Corticosteroid.
- CTNNA3
catenin (cadherin-associated protein), alpha 3.
- EMR
electronic medical record.
- eQTL
expression quantitative trait locus.
- GIF
genomic inflation factor
- GRAAD
Genomic Research on Asthma in the African Diaspora
- GWAS
genome-wide association study.
- ICS
inhaled corticosteroid
- LD
linkage disequilibrium
- MARS
Montelukast or Azithromycin for Reduction of Inhaled Corticosteroids in Childhood Asthma Study.
- PACT
Pediatric Asthma Controller Trial.
- P2RX7
purinergic receptor P2X, ligand-gated ion channel, 7.
- NHLBI
National Heart, Lung, and Blood Institute.
- SEMA3D
sema domain, immunoglobulin domain (Ig), short basic domain, secreted, (semaphorin) 3D.
- SHARe
SNP Health Association Resource
- SHARP
SNP Health Asthma Resource Project
- SNP
single-nucleotide polymorphism.
- TTN
titin.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Akinbami LJ, Moorman JE, Liu X. Asthma prevalence, health care use, and mortality: United States, 2005-2009. National health statistics reports. 2011(32):1–14. [PubMed] [Google Scholar]
- 2.Barnett SB, Nurmagambetov TA. Costs of asthma in the United States: 2002-2007. The Journal of allergy and clinical immunology. 2011;127(1):145–52. doi: 10.1016/j.jaci.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 3.Fuhlbrigge A, et al. Asthma outcomes: exacerbations. The Journal of allergy and clinical immunology. 2012;129(3 Suppl):S34–48. doi: 10.1016/j.jaci.2011.12.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busse WW, Lemanske RF, Jr., Gern JE. Role of viral respiratory infections in asthma and asthma exacerbations. Lancet. 2010;376(9743):826–34. doi: 10.1016/S0140-6736(10)61380-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dulek DE, Peebles RS., Jr. Viruses and asthma. Biochimica et biophysica acta. 2011;1810(11):1080–90. doi: 10.1016/j.bbagen.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rumpel JA, et al. Genetic ancestry and its association with asthma exacerbations among African American subjects with asthma. The Journal of allergy and clinical immunology. 2012;130(6):1302–6. doi: 10.1016/j.jaci.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moorman JE, et al. Vital & health statistics. Series 3, Analytical and epidemiological studies. 35. Vol. 2012. U.S. Dept. of Health and Human Services, Public Health Service, National Center for Health Statistics; National surveillance of asthma: United States, 2001-2010. pp. 1–67. [PubMed] [Google Scholar]
- 8.Williams LK, et al. Quantifying the proportion of severe asthma exacerbations attributable to inhaled corticosteroid nonadherence. The Journal of allergy and clinical immunology. 2011;128(6):1185–1191 e2. doi: 10.1016/j.jaci.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chilmonczyk BA, et al. Association between exposure to environmental tobacco smoke and exacerbations of asthma in children. The New England journal of medicine. 1993;328(23):1665–9. doi: 10.1056/NEJM199306103282303. [DOI] [PubMed] [Google Scholar]
- 10.Samoli E, et al. Acute effects of air pollution on pediatric asthma exacerbation: evidence of association and effect modification. Environmental research. 2011;111(3):418–24. doi: 10.1016/j.envres.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 11.Greenberg S. Asthma exacerbations: predisposing factors and prediction rules. Current opinion in allergy and clinical immunology. 2013;13(3):225–36. doi: 10.1097/ACI.0b013e32836096de. [DOI] [PubMed] [Google Scholar]
- 12.Curtin JA, et al. Methylation of IL-2 promoter at birth alters the risk of asthma exacerbations during childhood. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2013;43(3):304–11. doi: 10.1111/cea.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bjornsdottir US, et al. Pathways activated during human asthma exacerbation as revealed by gene expression patterns in blood. PLoS One. 2011;6(7):e21902. doi: 10.1371/journal.pone.0021902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du R, et al. Genome-wide association study reveals class I MHC-restricted T cell-associated molecule gene (CRTAM) variants interact with vitamin D levels to affect asthma exacerbations. The Journal of allergy and clinical immunology. 2012;129(2):368–73. 373, e1–5. doi: 10.1016/j.jaci.2011.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tantisira KG, et al. FCER2: a pharmacogenetic basis for severe exacerbations in children with asthma. The Journal of allergy and clinical immunology. 2007;120(6):1285–91. doi: 10.1016/j.jaci.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 16.Denlinger LC, et al. P2X7-regulated protection from exacerbations and loss of control is independent of asthma maintenance therapy. American journal of respiratory and critical care medicine. 2013;187(1):28–33. doi: 10.1164/rccm.201204-0750OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cunningham J, et al. The CHI3L1 rs4950928 polymorphism is associated with asthma-related hospital admissions in children and young adults. Annals of allergy, asthma & immunology : official publication of the American College of Allergy, Asthma, & Immunology. 2011;106(5):381–6. doi: 10.1016/j.anai.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 18.Tavendale R, et al. A polymorphism controlling ORMDL3 expression is associated with asthma that is poorly controlled by current medications. The Journal of allergy and clinical immunology. 2008;121(4):860–3. doi: 10.1016/j.jaci.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 19.Anderson WH, et al. Genetic analysis of asthma exacerbations. Annals of allergy, asthma & immunology : official publication of the American College of Allergy, Asthma, & Immunology. 2013;110(6):416–422 e2. doi: 10.1016/j.anai.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 20.Xu M, et al. Genome Wide Association Study to predict severe asthma exacerbations in children using random forests classifiers. BMC medical genetics. 2011;12:90. doi: 10.1186/1471-2350-12-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The Childhood Asthma Management Program (CAMP): design, rationale, and methods. Childhood Asthma Management Program Research Group. Control Clin Trials. 1999;20(1):91–120. [PubMed] [Google Scholar]
- 22.Guilbert TW, et al. The Prevention of Early Asthma in Kids study: design, rationale and methods for the Childhood Asthma Research and Education network. Controlled clinical trials. 2004;25(3):286–310. doi: 10.1016/j.cct.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Roden DM, et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clinical pharmacology and therapeutics. 2008;84(3):362–9. doi: 10.1038/clpt.2008.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long-term effects of budesonide or nedocromil in children with asthma. The Childhood Asthma Management Program Research Group. N Engl J Med. 2000;343(15):1054–63. doi: 10.1056/NEJM200010123431501. [DOI] [PubMed] [Google Scholar]
- 25.Sorkness CA, et al. Long-term comparison of 3 controller regimens for mild-moderate persistent childhood asthma: the Pediatric Asthma Controller Trial. The Journal of allergy and clinical immunology. 2007;119(1):64–72. doi: 10.1016/j.jaci.2006.09.042. [DOI] [PubMed] [Google Scholar]
- 26.Strunk RC, et al. Relationship of exhaled nitric oxide to clinical and inflammatory markers of persistent asthma in children. The Journal of allergy and clinical immunology. 2003;112(5):883–92. doi: 10.1016/j.jaci.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 27.Strunk RC, et al. Azithromycin or montelukast as inhaled corticosteroid-sparing agents in moderate-to-severe childhood asthma study. The Journal of allergy and clinical immunology. 2008;122(6):1138–1144. e4. doi: 10.1016/j.jaci.2008.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, et al. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34(8):816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O'Hara RB, Kotze DJ. Do not log-transform count data. Methods in Ecology and Evolution. 2010;1(2):118–122. [Google Scholar]
- 31.Raby B, et al. D101. ASTHMA GENETICS. American Thoracic Society; 2011. Asthma Bridge: The Asthma Biorepository For Integrative Genomic Exploration. [Google Scholar]
- 32.Mathias RA, et al. A genome-wide association study on African-ancestry populations for asthma. The Journal of allergy and clinical immunology. 2010;125(2):336–346. e4. doi: 10.1016/j.jaci.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Himes BE, et al. RNA-Seq transcriptome profiling identifies CRISPLD2 as a glucocorticoid responsive gene that modulates cytokine function in airway smooth muscle cells. PLoS One. 2014;9(6):e99625. doi: 10.1371/journal.pone.0099625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic acids research. 2012;40(Database issue):D930–4. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moffatt MF, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363(13):1211–21. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujii T, et al. Possible association of the semaphorin 3D gene (SEMA3D) with schizophrenia. Journal of psychiatric research. 2011;45(1):47–53. doi: 10.1016/j.jpsychires.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 37.Degenhardt K, et al. Semaphorin 3d signaling defects are associated with anomalous pulmonary venous connections. Nature medicine. 2013;19(6):760–5. doi: 10.1038/nm.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petryszak R, et al. Expression Atlas update--a database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic acids research. 2014;42(Database issue):D926–32. doi: 10.1093/nar/gkt1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takamatsu H, Kumanogoh A. Diverse roles for semaphorin-plexin signaling in the immune system. Trends in immunology. 2012;33(3):127–35. doi: 10.1016/j.it.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 40.Kruger RP, Aurandt J, Guan KL. Semaphorins command cells to move. Nature reviews. Molecular cell biology. 2005;6(10):789–800. doi: 10.1038/nrm1740. [DOI] [PubMed] [Google Scholar]
- 41.Gitler AD, Lu MM, Epstein JA. PlexinD1 and semaphorin signaling are required in endothelial cells for cardiovascular development. Developmental cell. 2004;7(1):107–16. doi: 10.1016/j.devcel.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 42.Barbato A, et al. Epithelial damage and angiogenesis in the airways of children with asthma. American journal of respiratory and critical care medicine. 2006;174(9):975–81. doi: 10.1164/rccm.200602-189OC. [DOI] [PubMed] [Google Scholar]
- 43.James AL, Wenzel S. Clinical relevance of airway remodelling in airway diseases. The European respiratory journal. 2007;30(1):134–55. doi: 10.1183/09031936.00146905. [DOI] [PubMed] [Google Scholar]
- 44.Kigel B, et al. Successful inhibition of tumor development by specific class-3 semaphorins is associated with expression of appropriate semaphorin receptors by tumor cells. PLoS One. 2008;3(9):e3287. doi: 10.1371/journal.pone.0003287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park HW, et al. Genetic predictors associated with improvement of asthma symptoms in response to inhaled corticosteroids. The Journal of allergy and clinical immunology. 2014;133(3):664–9. doi: 10.1016/j.jaci.2013.12.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim SH, et al. Alpha-T-catenin (CTNNA3) gene was identified as a risk variant for toluene diisocyanate-induced asthma by genome-wide association analysis. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2009;39(2):203–12. doi: 10.1111/j.1365-2222.2008.03117.x. [DOI] [PubMed] [Google Scholar]
- 47.Bernstein DI, et al. CTNNA3 (alpha-catenin) gene variants are associated with diisocyanate asthma: a replication study in a Caucasian worker population. Toxicological sciences : an official journal of the Society of Toxicology. 2013;131(1):242–6. doi: 10.1093/toxsci/kfs272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perin P, Potocnik U. Polymorphisms in recent GWA identified asthma genes CA10, SGK493, and CTNNA3 are associated with disease severity and treatment response in childhood asthma. Immunogenetics. 2014;66(3):143–51. doi: 10.1007/s00251-013-0755-0. [DOI] [PubMed] [Google Scholar]
- 49.Lekstrom-Himes J, Xanthopoulos KG. Biological role of the CCAAT/enhancer-binding protein family of transcription factors. The Journal of biological chemistry. 1998;273(44):28545–8. doi: 10.1074/jbc.273.44.28545. [DOI] [PubMed] [Google Scholar]
- 50.Berg T, et al. Glucocorticoids regulate the CCSP and CYP2B1 promoters via C/EBPbeta and delta in lung cells. Biochemical and biophysical research communications. 2002;293(3):907–12. doi: 10.1016/S0006-291X(02)00319-4. [DOI] [PubMed] [Google Scholar]
- 51.Yang H, et al. Expression and activity of C/EBPbeta and delta are upregulated by dexamethasone in skeletal muscle. Journal of cellular physiology. 2005;204(1):219–26. doi: 10.1002/jcp.20278. [DOI] [PubMed] [Google Scholar]
- 52.Janssens B, et al. alphaT-catenin: a novel tissue-specific beta-catenin-binding protein mediating strong cell-cell adhesion. J Cell Sci. 2001;114(Pt 17):3177–88. doi: 10.1242/jcs.114.17.3177. [DOI] [PubMed] [Google Scholar]
- 53.Bonnelykke K, et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. 2014;46(1):51–5. doi: 10.1038/ng.2830. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.