

Abstract
NAD(P)H-dependent enzymes are ubiquitous in metabolism and cellular processes and are also of great interest for pharmaceutical and industrial applications. Here, we present a structure-guided enzyme engineering strategy for improving catalytic properties of NAD(P)H-dependent enzymes toward native or native-like reactions using mutations to the enzyme's adenine-binding pocket, distal to the site of catalysis. Screening single-site saturation mutagenesis libraries identified mutations that increased catalytic efficiency up to 10-fold in 7 out of 10 enzymes. The enzymes improved in this study represent three different cofactor-binding folds (Rossmann, DHQS-like, and FAD/NAD binding) and utilize both NADH and NADPH. Structural and biochemical analyses show that the improved activities are accompanied by minimal changes in other properties (cooperativity, thermostability, pH optimum, uncoupling), and initial tests on two enzymes (ScADH6 and EcFucO) show improved functionality in Escherichia coli.
Keywords: directed evolution, enzyme optimality, nicotinamide cofactors, protein engineering, site-saturation mutagenesis
Introduction
Engineering novel or improved metabolic pathways often changes the demands placed on enzymes evolved to carry out their natural functions in specific contexts. For instance, it has been proposed that enzymatic KM values have evolved to match physiological substrate concentrations (Bennett et al., 2009), which can change as a result of heterologous expression or pathway engineering that changes steady-state metabolite concentrations. Such changes in metabolic context might require alterations in enzyme kinetics through protein engineering for optimal metabolic flux and cell physiology. Beyond the room for improvement created by a novel physiological context, it has been hypothesized that the kinetics of many enzymes have not been maximized by evolution, particularly the ‘moderately efficient’ enzymes of secondary metabolism where kinetic enhancement is of minimal benefit to host fitness (Bar-Even et al., 2011; Bar-Even and Tawfik, 2013). Despite the suggestions that there is potential to improve native activities, engineering enzymes for the direct improvement of activities on their natural substrates under biologically relevant reaction conditions has proven challenging and, to our knowledge, broadly unsuccessful (Tcherkez et al., 2006; Savir et al., 2010; Bar-Even et al., 2011). Instead, enzyme engineering usually improves reactivity toward non-natural substrates, increases promiscuous reactivities or alters selectivity (Schmidt-Dannert and Arnold, 1999; Bornscheuer and Pohl, 2001; Brustad and Arnold, 2011; Li and Cirino, 2014). Unlike these goals, in which functional changes can often be ascribed to specific remodeling of the active site to accommodate or exclude certain substrates or transition states, the precise structural origins of an enzyme's kinetic properties are more enigmatic and to-date have been resistant to prediction. In this study, we empirically identify structural positions in NAD(P)H-dependent enzymes where mutations can provide significant boosts in enzyme activity and catalytic efficiency.
NAD(P)H-dependent enzymes are involved in a wide range of metabolic reactions, which makes them of interest for pharmaceutical and industrial applications. Protein engineering has been used to study and change cofactor binding to NAD(P)H-dependent enzymes (Hurley et al., 1996; Khoury et al., 2009; Bastian et al., 2011; Brinkmann-Chen et al., 2013). Previous work from this laboratory on engineering of cofactor specificity of ketol-acid reductoisomerase (KARI) enzymes revealed that mutation at a single position on a helix that runs parallel to the cofactor adenine moiety improved the catalytic activity of several KARIs, both wild type and cofactor-switched (Bastian et al., 2011; Brinkmann-Chen et al., 2013). This finding was replicated in another KARI by Reiße et al. (2015).
While screening a random mutant library of the Arabidopsis thaliana glyoxylate reductase (AtGR1) prepared for an unrelated study, we observed similar activating effects from a mutation adjacent to the position corresponding to that in the KARIs. AtGR1 with mutation C68R showed a significant improvement in activity in lysate, which we later established was due largely to a ∼5-fold decrease in KM for the substrate (this work). AtGR1 and KARIs possess similar overall folds, with highly similar Rossmann domains, and both are specific for NADPH over NADH. Because it is rare to find mutations that boost the activity of an enzyme for its native reaction, we set out to investigate whether modifications at similar positions with respect to the adenine could improve the activities of enzymes with more diverse folds and cofactor utilization profiles. These amino acids are situated in the internal lining of the adenine-binding pocket. More specifically, they contain atoms located within 5 Å of the N6 atom of the NAD(P)H adenine (see Fig. 1), but are not involved in determining cofactor preference through interaction with the phosphate or hydroxyl in the 2′-position of the ribose. Libraries of enzyme variants made by site-saturation mutagenesis at these positions can be screened rapidly for increased enzyme activity in lysate. We demonstrate that this simple structure-guided engineering strategy works to improve the activities of a surprising range of enzymes, opening the door to improving the catalytic properties of a broad array of industrially relevant enzymes and metabolic pathways.
Fig. 1.

(a) The standard numbering of atoms in the NAD(P)(H) adenine moiety. (b) An example of the amino acid positions around the adenine N6 mutated in this study (yellow spheres), in this case S. cerevisiae ADH6 (ScADH6), whose structure was reported in Valencia et al. (2004) (PDB 1PIW).
Materials and methods
Cloning and library construction
All genes were obtained from Integrated DNA Technologies (IDT) as gBlock linear fragments and were cloned into pET22b(+) in frame with the C-terminal his-tag for expression in Escherichia coli using Gibson cloning (Gibson et al., 2009) with overlap at the T7 promoter and terminator sequences. Mutagenic primers for site-saturation mutagenesis were obtained from IDT and treated as suggested by IDT protocols. Splicing by overlap extension polymerase chain reaction (SOE-PCR) was performed as described previously (Kunkel et al., 1987; Bastian et al., 2011). The quality of the library was assessed using DNA sequencing performed by Laragen (Los Angeles, CA, USA). Standard molecular biology methods were taken from Sambrook et al. (1989).
Heterologous gene expression for high-throughput screening and protein purification
All expression cultures were grown in Luria-Bertani broth, supplemented with ampicillin for selection (LB+Amp).
For high-throughput screening, pre-cultures of 300 µl LB+Amp were inoculated with single colony forming units (CFUs) in 96-deep-well plates (DWPs) using toothpicks. For each library, 84–88 CFUs were screened, corresponding to 93–95% theoretical library coverage (Patrick et al., 2003; Bosley and Ostermeier, 2005), along with the parent protein and the pET22b(+) vector as positive and negative controls. The pre-cultures were grown overnight at 37°C, 200 rpm and 80% humidity. The next day, expression cultures of 600 µl were inoculated with 50 µl of the overnight cultures in 96-DWPs. The pre-cultures were stored at 4°C until the screening was completed to serve as temporary stock from which positive hits were regrown. After incubation of the expression cultures for 4 h at 37°C, 200 rpm and 80% humidity, expression was induced by adding isopropyl thiogalactopyranoside (IPTG) to a final concentration of 0.25 mM. Expression occurred for 21 h at temperatures indicated in Supplementary Information S1 and 200 rpm without humidity control. The expression cultures were harvested through centrifugation, and the DWPs containing the cell pellets were stored at −20°C until screening.
For purified protein, pre-cultures grown overnight at 37°C and 200 rpm were used for inoculation of 200 ml expression cultures to an OD600 of 0.05–0.1 and incubated at 37°C and 210 rpm until an OD600 of ∼0.8 was reached. At this point, the expression cultures were cooled to their expression temperatures (Supplementary Information S1) before induction with IPTG to a final concentration of 0.5 mM and growth for an additional 21 h. The expression cultures were harvested by centrifugation, the supernatant was discarded and the pellets were frozen at −20°C until further use.
Enzyme assays and high-throughput screening
Escherichia coli cells were resuspended in the respective assay buffer (Supplementary Information S1) containing 750 mg/l lysozyme, 10 mg/l DNaseI and 2 mM MgCl2. Lysis was accomplished at 37°C for 1 h. Enzyme activities were then assayed by monitoring NAD(P)H consumption in the presence of the respective substrate (see Supplementary Information S1) and 250 μM NAD(P)H at 340 nm in a plate reader.
Thermostability was measured through determination of T50, the temperature where the enzyme activity is reduced to 50% of its initial activity after incubation for 10 min. The pH optimum was determined using a selection of four different buffer systems to cover the relevant pH scale from pH 3 to 10 (pH 3–6: 50 mM sodium citrate buffer, pH 6–8: 50 mM sodium phosphate buffer, pH 8–9: 50 mM Tris–HCl buffer, pH 9.2–10: 50 mM carbonate–bicarbonate buffer). All observations are averages of at least three replicates.
Protein purification and enzyme kinetics
Escherichia coli cell pellets were resuspended in 10 ml buffer A (25 mM Tris, 100 mM NaCl, 20 mM imidazole, pH 7.4) and lysed by sonication. The lysate was centrifuged, and the enzymes were purified via their C-terminal His6-tag using High Performance (HP) Ni-NTA Sepharose columns (GE Healthcare, Waukesha, WI, USA) on an Äkta Xpress FPLC (GE Healthcare). The concentration of purified protein was determined using the Bradford assay (Bio-Rad, Hercules, CA, USA).
For rate measurements, kcat values were determined using the same assay conditions as above with saturating cofactor and substrate, while Michaelis–Menten constants were determined by varying each individually. At least six cofactor concentrations and at least five substrate concentrations were used for these determinations, and all measurements were performed at least three times. MatLab (Mathworks, Natwick, MA, USA) was used for parameter fitting.
Protein crystallization and structure determination
Screening of crystallization conditions was conducted at the Beckman Molecular Observatory at the California Institute of Technology using commercially available crystal screens. Crystallization occurred with purified EcFucOM185C at a concentration of 15 mg/ml, 10 mM NAD+ and 10 mM isobutyraldehyde using the sitting drop method at ambient temperature. Crystals were obtained with 12% PEG 3350 and 200 mM NH4Cl as precipitant. The crystals were soaked with mother liquor containing 17% glycerol and 6.9 mM NAD+ before flash freezing in liquid nitrogen. Diffraction data were collected using a Dectris Pilatus 6 M detector on beamline 12–2 at the Stanford Synchrotron Radiation Laboratory at 100 K. Diffraction datasets were integrated with XDS (Kabsch, 2010) and scaled using SCALA (Evans, 2006).
The structure of EcFucO (pdb code 1RRM; Kumaran and Swaminathan, 2009) was used for molecular replacement. Refinement was conducted by iterating automatic refinement with Refmac5 (CCP4 suite) and manual refinement using Coot (Emsley and Cowtan, 2004). The structure was deposited in the RCSB Protein Data Bank with accession code 5BR4.
In vivo growth assays
For the in vivo growth assays, cells were grown in 24-well round-bottom plates (Invitrogen, Carlsbad, CA, USA). In each well, 3 ml of LB broth with 100 μg/ml ampicillin and 500 μM IPTG were inoculated with 10 μl of saturated overnight culture. After 3 h of growth, an additional 3 ml of LB (with ampicillin and IPTG at the same concentrations) were added, which contained 10 mM of furfural or trans-cinnamaldehyde. After 10 h, the OD600 of a 200-μl aliquot was measured on a plate reader (Tecan, Männedorf, Switzerland). All observations are averages of at least five replicates.
Results
Improvement of catalytic properties through mutations around adenine N6
In addition to the KARI enzymes previously studied, we selected eight distinct enzymes representing a range of cofactor preferences and cofactor-binding folds to test whether mutations at positions around the N6 nitrogen of adenine led to higher activity. The enzymes selected for this study include enzymes previously studied in our group and enzymes with potential for industrial applications: Saccharomyces cerevisiae cinnamyl alcohol dehydrogenase (ScADH6), AtGR1, Lactococcus lactis and Drosophila melanogaster alcohol dehydrogenases (LlAdhA and DmADH), Klebsiella pneumoniae propanediol dehydrogenase (KpDhaT), E. coli lactaldehyde reductase (EcFucO), Talaromyces emersonii xylose reductase (TeXR) and Lactobacillus sanfranciscensis NADH oxidase (LsNOX). Some details about these enzymes are provided in Table I, and more information can be found in Supplementary Information S2. Tables I and II include information on two KARIs, EcIlvC from Bastian et al. (2011) and MrKARI from Reiße et al. (2015), which come from two distinct structural classes. EcIlvC is also notable for undergoing a cofactor-induced conformational change opposite that observed in other KARIs studied (Cahn et al., 2015).
Table I.
The enzymes tested in this study (below the line) and previously reported (above the line), including the Structural Classification of Proteins (SCOP) classification of their cofactor-binding fold and a list of the positions mutated
| Enzyme | PDB accession | Cofactor-binding fold | Cofactor preference | Positions mutated |
|---|---|---|---|---|
| EcIlvC | 3ULK (Wong et al., 2012) | Rossmann (c.2.1.6) | NADPH | Q110 (Bastian et al., 2011) |
| MrKARI | None | Rossmann (c.2.1.6) | NADPH | T84 (Reiße et al., 2015) |
| AtGR1 | 3DOJ (cofactor from 3PEF) (G. Hoover et al., 2011; Zhang and Garavito, 2011) | Rossmann (c.2.1.0) | NADPH | C68, A69 |
| ScADH6 | 1PIW (Valencia et al., 2004) | Rossmann (c.2.1.1) | NADPH | S253, T255, D256 |
| LlAdhA | 4EEX (cofactor from 4GKV) (Liu et al., 2012; Thomas et al., 2013) | Rossmann (c.2.1.1) | NADH | A242, A245 |
| DmAdhA | 1MG5 (Benach et al., 2005) | Rossmann (c.2.1.2) | NADH | D65, V66, R104, V108 |
| KpDhaT | 3BFJ (cofactor from 3OX4) (Marcal et al., 2009; Moon et al., 2011) | DHQS-like (e.22.1.2) | NADH | K187 |
| EcFucO | 1RRM (Kumaran and Swaminathan, 2009) | DHQS-like (e.22.1.2) | NADH | T140, M185 |
| TeXR | 1K8C (Kavanagh et al., 2002) (homology) | TIM barrel (c.1.7.1) | NADPH | F217, A254, Q280, N281 |
| LsNOX | 2CDU (Lountos et al., 2006) | FAD/NAD-binding (c.3.1.5) | Bi-specific | I122, I155, V214, I243 |
Positions indicated in bold indicate those where one or more beneficial mutations were discovered.
A. thaliana glyoxylate reductase 1 (AtGR1), S. cerevisiae cinnamyl alcohol dehydrogenase (ScADH6), L. lactis alcohol dehydrogenase (LlAdhA), D. melanogaster alcohol dehydrogenase (DmADH), K. pneumoniae 1,3-propanediol dehydrogenase (KpDhaT), E. coli lactaldehyde reductase (EcFucO), T. emersonii xylose reductase (TeXR), L. sanfranciscensis NAD(P)H-oxidase (LsNOX), dihydroquinoate synthase (DHQS).
Table II.
Fold enhancement of catalytic properties of selected variants compared with the respective wild-type enzyme
|
kcat (fold increase) |
KM (fold decrease) |
Catalytic efficiency (fold increase) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Enzyme | Mutation | NADH | NADPH | NADH | NADPH | Substrate | kcatNADH/KMNADH | kcatNADPH/KMNADPH | kcatNADH/KMsubstrate | kcatNADPH/KMsubstrate |
| EcIlvC | Q110V | 11.0 | 2.0 | 7.7 | 3.1 | – | 82.7 | 6.5 | – | – |
| EcIlvC | Q110A | 10.3 | 2.0 | 3.9 | 1.7 | – | 36.7 | 3.5 | – | – |
| MrKARI | T84S | 2.6 | 4.9 | 13.8 | 0.5 | – | 37.9 | 2.5 | – | – |
| AtGR1 | C68E | 0.4 | 0.5 | 1.1 | 2.0 | 11.5 | 0.4 | 1.0 | 4.2 | 5.5 |
| AtGR1 | C68R | 1.8 | 0.3 | 1.4 | 0.8 | 5.7 | 2.6 | 0.2 | 10.1 | 1.6 |
| ScADH6 | T255K | 2.4 | 0.6 | 0.5 | 3.8 | 0.7 | 1.3 | 2.2 | 1.7 | 0.4 |
| DmADH | V108I | 1.2 | – | 1.4 | – | 1.2 | 1.6 | – | 1.5 | – |
| EcFucO | M185A | 3.4 | – | 1.8* | – | 3.5 | 6.1 | – | 11.8 | – |
| EcFucO | M185C | 3.6 | – | 1.3* | – | 1.5 | 4.6 | – | 5.4 | – |
| LsNOX | I122V | 3.3 | 1.5 | 4.2 | 0.7 | – | 13.7 | 1.0 | – | – |
| LsNOX | I155L | 2.2 | 3.8 | 0.9 | 2.3 | – | 1.9 | 8.8 | – | – |
| LsNOX | I243M | 2.2 | 3.3 | 0.6 | 1.3 | – | 1.2 | 4.3 | – | – |
| LsNOX | I122V-I155L | 2.3 | 5.6 | 1.2 | 0.6 | – | 2.8 | 3.3 | – | – |
| LsNOX | I22V-I243M | 4.6 | 5.3 | 1.5 | 2.3 | – | 6.6 | 12.3 | – | – |
| LsNOX | I155L-I243M | 2.1 | 2.7 | 1.0 | 0.6 | – | 2.1 | 1.7 | – | – |
| LsNOX | I122V-I155L-I243M | 6.6 | 12.1 | 0.9 | 0.5 | – | 6.0 | 6.4 | – | – |
EcIlvC results are quoted from Bastian et al. (2011), and MrKARI results are from Reiße et al. (2015). Due to low enzymatic activity with NADPH as cofactor, kcatNADPH and KMNADPH could not be determined for the wild-type and variant enzymes of EcFucO and DmADH. KMsubstrate could not be determined for LsNOX due to experimental constraints. Asterisks indicate binding constants that showed cooperative behavior. For numerical values including errors, please refer to Supplementary Information S4.
For each enzyme, we identified residues within 5 Å of the N6 atom of the NAD(P)H adenine, based on published crystal structures or homology models produced using the SWISS-MODEL server (Arnold et al., 2006). A site-saturation mutagenesis library was generated at each position listed in Table I, and the enzyme variants were screened for activity on both NADH and NADPH using the substrates indicated in Supplementary Information S1. Mutants with significant activity enhancements toward either cofactor were purified, and Michaelis–Menten kinetic parameters were determined for the cofactors and the substrate. Improved kinetics were found in variants of five out of the eight new enzymes tested, in addition to the KARIs previously studied by Brinkmann-Chen et al. (2013) and Reiße et al. (2015). This number is notable given that these mutations are (i) boosting kinetic properties of native or native-like reactions, (ii) distal to the active sites and (iii) obtained by screening small saturation libraries made at only a very few positions (between 1 and 4; see Table I).
The combined results are summarized in Table II, where improvements in kcat (1.2–11.0-fold increase) as well as in the KM for substrate (1.2–11.5-fold decrease) and cofactor (1.1–7.7-fold decrease) are reported for 7 out of 10 enzymes. (Here, and elsewhere, we use KM to refer to both the Michaelis constant and the dissociation constant KH for enzymes displaying cooperativity. Such enzymes are marked in Table II. No significant changes in the Hill coefficient were observed for these cooperative enzymes.) Accordingly, the catalytic efficiencies were increased from 1.2-fold to as much as 83-fold with respect to cofactor or substrate (Table II). The improved enzymes contained Rossmann (KARIs, AtGR1, DmADH, ScADH6), FAD/NAD-binding (LsNOX) and DHQS-like (EcFucO) cofactor-binding folds and included enzymes with both cofactor preferences. We were unable to find beneficial mutations at the targeted positions in three enzymes: LlAdhA (Rossmann fold), KpDhaT (DHQS-like fold) and TeXR (TIM barrel fold).
Because we identified beneficial mutations at multiple residues in the LsNOX enzymes, we also tested the double and triple combinatorial mutations. All three double mutants showed activity enhancements with respect to wild type, although not necessarily above the single mutants (Table II). The triple mutant had considerably elevated kcat values but expressed quite poorly compared with the wild-type enzyme and the other single and combinatorial mutants (data not shown). Losses in expression or stability upon the accumulation of mutations are to be expected; stability can often be recovered by further mutagenesis, without compromising activity (Bloom et al., 2007; Fasan et al., 2008; Gong et al., 2013). Although expression levels were not quantified here, no other large changes in purified protein yields were observed for the other mutants of LsNOX or any other protein tested.
The evolutionary fitness of an enzyme, that is how it contributes to the survival and fitness of its source, is determined by factors beyond catalytic efficiency, and thus catalytic efficiency may be sacrificed to achieve other properties. In this case, improving activity might be expected to come at the expense of those other properties. To test whether other enzyme properties may have been perturbed by the mutations we identified, we analyzed selected enzymes for changes in pH optimum (DmADHV108I) and thermostability (DmADHV108I, EcFucOM185A and EcFucOM185C). We found no significant changes in the improved variants compared with the wild-type enzymes (Supplementary Information S3). Additionally, none of the enzymes characterized were shown to be uncoupled—that is, none consumed cofactor in the absence of substrate—although we did not test LsNOX (its molecular oxygen substrate requires a controlled atmosphere for kinetic measurements). We also examined the in vivo activities of two of the enzymes (ScADH6 and EcFucO) whose activities could be directly tied to cell survival (Larroy et al., 2002; Wang et al., 2011). Under standard E. coli expression conditions, no growth defect was observed as a result of the mutations (Fig. 2, top row). Furthermore, upon the addition of a toxic aldehyde (trans-cinnamaldehyde or furfural, respectively), growth was enhanced with the mutant ScADH6 and EcFucO enzymes providing improved reductive detoxification ability compared with the wild-type enzymes (Fig. 2, bottom row).
Fig. 2.

In vivo growth of cells containing mutant enzymes. Top row shows cells under normal growth conditions. None of the characterized mutations cause a growth defect relative to overexpression of the wild-type enzyme. In the presence of toxic aldehydes (bottom row), the respective wild-type enzymes improved growth rate compared with no enzyme, and the mutant enzymes led to higher growth rates than the wild-type enzymes. Error bars indicate the standard deviation of the measurements.
Structural alterations in EcFucO
The structure of EcFucOM185C was solved at a resolution of 0.91 Å (Table III), which allowed us to investigate whether the mutation at position M185 caused structural changes in the enzyme or the bound cofactor. In EcFucOM185C, activity was enhanced when M185 was substituted by a smaller and slightly more polar cysteine. The side chain of this cysteine lies almost perfectly along the β and γ carbons of the wild-type methionine, and aligning the structure of EcFucOM185C to the previously published wild-type structure (Kumaran and Swaminathan, 2009) reveals no major changes in the protein structure or the cofactor-binding pocket. However, the axis of the cofactor is tilted slightly, and the adenine is slightly shifted (0.3 Å) in the direction of the active site (Fig. 3). This leads to a more significant shift of 1.1 Å at the N1 of the nicotinamide at the other end of the cofactor (Fig. 3). As the nicotinamide is the catalytically active part of the cofactor, we assume that this change in position in the active site enhances catalysis in the EcFucOM185C variant, mainly via a 3.6-fold increase in kcat.
Table III.
Data collection and refinement statistics for the crystal structure of EcFucOM185C (PDB ID 5BR4)
| Data collection | |
|---|---|
| Space group | P 21 |
| Cell dimensions | |
| a, b, c (Å) | 69.7, 63.8, 91.7 |
| α, β, γ (°) | 90, 111.2, 90 |
| Resolution (Å) | 85.50–0.91 (0.96–0.91) |
| Rp.i.m (%) | 0.044 (1.669) |
| Mn(I)/sd | 8.8 (0.4) |
| Completeness (%) | 91.8 (74.6) |
| Redundancy | 2.7 (2.5) |
| Refinement | |
| No. of reflections | 466 646 (22 931) |
| Rwork/Rfree (%) | 12.9/14.7 (35.9/37.1) |
| No. of atoms | |
| Protein | 5818 |
| Ligand/ion | 112 |
| Water | 1167 |
| Root Mean Square Deviation | |
| Bond lengths (Å) | 0.021 |
| Bond angles (°) | 2.035 |
| Ramachandran map analysis | |
| Favored | 774 |
| Allowed | 11 |
| Outliers | 0 |
Values in parentheses refer to the highest-resolution shell.
Fig. 3.
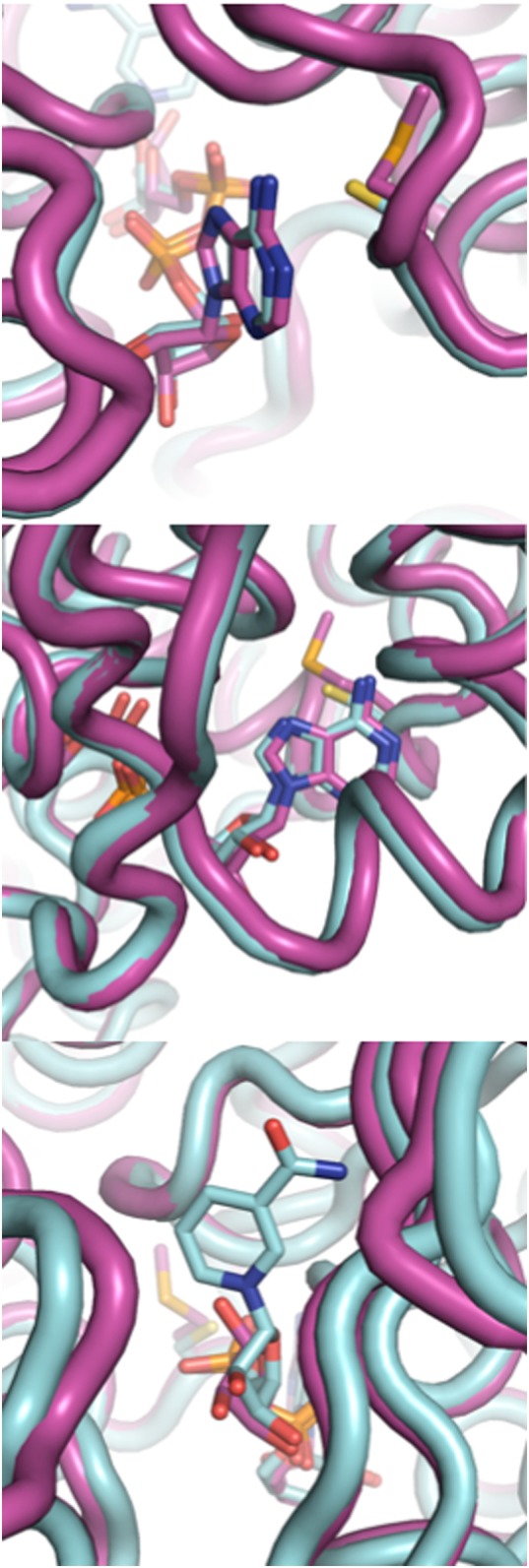
Structures of wild-type EcFucO (purple) and the M185C mutant (cyan), showing two angles on the adenine and one of the nicotinamide. The nicotinamide moiety is not resolved in the wild-type structure.
A similar repositioning of the cofactor was also observed in the crystal structure of Se_KARIDDV with mutation I95V close to the adenine of the cofactor (Brinkmann-Chen et al., 2013). Mutations in Se_KARIDDV caused a 1-Å shift of the adenine compared with the parent structure, and Brinkmann-Chen et al. proposed that this readjustment placed the cofactor in a more favorable position for catalysis, although the presence of two additional mutations and a reversal of the cofactor specificity made it impossible to attribute the shift to I95V alone.
Discussion
It is often assumed in protein and metabolic engineering that enzymes have already been optimized toward their native functions and that their native catalytic efficiencies cannot be improved (Tokuriki et al., 2012). Despite evidence that metabolically crucial enzymes display, on average, faster kinetics than those involved in secondary metabolism and the corresponding prediction that the kinetics of secondary metabolic enzymes could be improved (Bar-Even et al., 2011), the engineering of more active enzymes has remained elusive (Bar-Even and Tawfik, 2013). Even when modest successes have been described (e.g. as in Bastian et al., 2005), no method for finding activating distal mutations (other than random mutagenesis) has been reported. In this study, we have empirically identified structural positions across a broad category of enzymes where mutations can improve kinetics of native or native-like reactions. Our results with ScADH6 and EcFucO also indicate improved overall functionality in an in vivo context where higher activity promotes better growth in the presence of toxic substrates.
We propose a few factors that may contribute to this unusual finding. The mutations identified in this study are remote from the catalytically active centers in the proteins (the average distance between the Cα of residues targeted in this study and the hydride-carrying C4N atom of the cofactor is 16.9 Å), lowering the chance of disrupting the active sites. Nevertheless, the extended NAD(P)(H) cofactor can transmit perturbations to the active site and may even magnify them (Mesecar et al., 1997). This is particularly relevant given the role of adenosine as a common energetic and recognition ‘handle’ in the binding of enzyme cofactors (Moodie et al., 1996). Furthermore, despite (or perhaps because of) its ubiquity, structurally diverse binding pockets for adenine have evolved (Chakrabarti and Samanta, 1995; Moodie et al., 1996; Nobeli et al., 2001; Pyrkov et al., 2007). In contrast to moieties with more specialized, conserved binding motifs, adenine binding is governed by a ‘fuzzy recognition template’ consisting of hydrophobic residues above and below adenine rings and polar residues around its rim (Moodie et al., 1996; Nobeli et al., 2001). This might suggest that the adenine-binding pocket can tolerate mutations that fine-tune the kinetics or energetics of binding as long as these general structural elements are present. Although no pattern in the beneficial mutations described here is readily apparent, such as an increase or decrease in steric bulk, side chain polarity or conformational entropy, a number of structural factors can be linked to the catalytic efficiency of enzymes. We offer two possible mechanisms for the enhancements observed and discuss them below in the specific context of nicotinamide cofactor binding.
In a 1997 study of isocitrate dehydrogenase, Mesecar et al. observed that subtle chemical modification of the adenine ring of NADPH significantly reduced catalytic activity. High-resolution crystal structures showed how the change to the adenine binding resulted in a slight shift in the position of the nicotinamide with respect to the substrate, and the large decrease in catalytic activity was attributed to this subtle misalignment (Mesecar et al., 1997). In a study of aldo–keto reductases, Campbell et al. (2013) similarly concluded that the relative positioning of the cofactor and substrate in the active site was the major factor contributing to efficient turnover. Although the structure of wild-type EcFucO does not have the nicotinamide resolved and the structure of EcFucOM185C lacks the substrate, it is possible that the beneficial mutations identified here realign the cofactor in a way that improves catalytic preorganization with respect to the substrate and active site.
Alternately, the mutations may affect the binding and unbinding kinetics of the cofactor. In a comprehensive study of adenine-binding pockets, Nobeli et al. (2001) found that protein-bound adenine moieties form, on average, only 67% of theoretically possible hydrogen bonds, suggesting that modulating the binding energy to fine-tune kon and koff is more important than achieving the tightest possible binding. Furthermore, for several kinds of NAD(P)(H)-dependent enzymes, it has been shown that long-range conformational changes occur during binding of cofactor and/or substrate, resulting in allosteric cooperativity (Kavanagh et al., 2002; Plapp, 2010; Liu et al., 2012; Campbell et al., 2013), which could explain why, in some of the enzymes tested, mutations affect the substrate KM, even though they are far from the substrate-binding portion of the enzyme.
Bar-Even et al. (2015) postulate in a recent paper that the sub-optimality of moderately efficient enzymes (enzymes whose second-order rate constants lie well below the diffusion limit) reflects, at least in part, a high proportion of ‘futile encounters’ between enzyme and substrate before a productive complex forms. In this context, the mutations we observed could increase the likelihood of formation of productive enzyme–substrate complexes either by improving the energetics of proper cofactor alignment or by increasing the rate at which futile complexes dissociate. The observation that enhancements in all three kinetic parameters (kcat, cofactor KM and substrate KM) arise from mutations distal to the site of catalysis or substrate binding may seem counter-intuitive. In this context, it is useful to remember that kcat and KM values are indirect ‘black-box’ measures of the formation and dissociation of a catalytically productive enzyme–cofactor–substrate complex (Bar-Even et al., 2015). Therefore, cofactor binding that is better suited to the formation of a productive complex can be manifested in the turnover rate (kcat) or may promote substrate binding that is more likely to be catalytically productive. Measurement of the microscopic kinetic parameters governing these reactions may be able to de-convolute these effects and shed more light on how the mutations described here promote activity.
In 3 out of 10 enzymes tested thus far (LlAdhA, KpDHAT and TeXR), no single mutation at these positions created variants with improved kinetics, indicating that these enzymes already lie at a local fitness optimum with respect to the targeted residues. No clear factor unites these three enzymes as distinct from the others studied, suggesting that the optimality of these positions is determined stochastically by the balance of genetic drift and natural selection (Lynch, 2012; Sung et al., 2012).
In this study, we have empirically identified structural sites that have a strong effect on activity without themselves being catalytically crucial and demonstrated that we can find mutations that boost the catalytic efficiency of the enzyme through subtle structural and/or energetic changes that cannot be rationally designed or predicted. We propose targeting mutations near the adenine for site-saturation mutagenesis and screening for improved kinetics as a fast and simple way to improve or tune the catalytic properties of NAD(P)H-dependent enzymes. With a demonstrated success rate of 7 in 10, including the 2 previously published KARIs, this is the first general strategy that has been proposed for improving a broad category of enzymes for their natural functions. Furthermore, different kinetic properties of the enzymes were changed, including substrate and cofactor affinity as well as turnover rate, allowing for fine-tuning of enzymatic properties for specific applications. Finally, these results demonstrate that many enzymes have room for improvement of catalytic properties in vitro and likely also in vivo, which holds promise for the engineering of improved biocatalysts and metabolic pathways.
Supplementary data
Funding
This work was supported by the Gordon and Betty Moore Foundation through grant number GBMF2809 to the Caltech Programmable Molecular Technology Initiative and by American Recovery and Reinvestment Act (ARRA) funds through the National Institutes of Health Shared Instrumentation Grant Program, grant number S10RR027203, to F.H.A. J.K.B.C. acknowledges the support of the Resnick Sustainability Institute (Caltech).
Supplementary Material
Acknowledgements
The authors thank Nelson Chou and Drs Sheel Dodani, Tillmann Heinisch and Devin Trudeau for their experimental assistance; Drs Devin Trudeau and John McIntosh for their helpful discussions; and Prof. Dan Tawfik, Prof. Pat Cirino and Prof. Monica Gerth for their helpful comments on this manuscript. They also thank Dr Jens Kaiser and Pavle Nicolovski of the Caltech Molecular Observatory for their continued support.
References
- Arnold K., Bordoli L., Kopp J., Schwede T. (2006) Bioinformatics, 22, 195–201. [DOI] [PubMed] [Google Scholar]
- Bar-Even A., Tawfik D.S. (2013) Curr Opin Biotech, 24, 310–319. [DOI] [PubMed] [Google Scholar]
- Bar-Even A., Noor E., Savir Y., Liebermeister W., Davidi D., Tawfik D.S., Milo R. (2011) Biochemistry, 50, 4402–4410. [DOI] [PubMed] [Google Scholar]
- Bar-Even A., Milo R., Noor E., Tawfik D.S. (2015) Biochemistry, Article ASAP. [DOI] [PubMed]
- Bastian S., Rekowski M.J., Witte K., Heckmann-Pohl D.M., Giffhorn F. (2005) Appl Microbiol Biot, 67, 654–663. [DOI] [PubMed] [Google Scholar]
- Bastian S., Liu X., Meyerowitz J.T., Snow C.D., Chen M.M.Y., Arnold F.H. (2011) Metab Eng, 13, 345–352. [DOI] [PubMed] [Google Scholar]
- Benach J., Winberg J.O., Svendsen J.S., Atrian S., Gonzalez-Duarte R., Ladenstein R. (2005) J Mol Biol, 345, 579–598. [DOI] [PubMed] [Google Scholar]
- Bennett B.D., Kimball E.H., Gao M., Osterhout R., Van Dien S.J., Rabinowitz J.D. (2009) Nat Chem Biol, 5, 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom J.D., Arnold F.H., Wilke C.O. (2007) Mol Syst Biol, 3, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornscheuer U.T., Pohl M. (2001) Curr Opin Chem Biol, 5, 137–143. [DOI] [PubMed] [Google Scholar]
- Bosley A.D., Ostermeier M. (2005) Biomol Eng, 22, 57–61. [DOI] [PubMed] [Google Scholar]
- Brinkmann-Chen S., Flock T., Cahn J.K.B., Snow C.D., Brustad E.M., McIntosh J.A., Meinhold P., Zhang L., Arnold F.H. (2013) Proc Natl Acad Sci USA, 110, 10946–10951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustad E.M., Arnold F.H. (2011) Curr Opin Chem Biol, 15, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahn J.K.B., Brinkmann-Chen S., Spatzal T., Wiig J.A., Buller A.R., Einsle O., Hu Y., Ribbe M.W., Arnold F.H. (2015) Biochem J, 468, 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell E., Chuang S., Banta S. (2013) Protein Eng Des Sel, 26, 181–186. [DOI] [PubMed] [Google Scholar]
- Chakrabarti P., Samanta U. (1995) J Mol Biol, 251, 9–14. [DOI] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. (2004) Acta Crystallogr D, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Evans P. (2006) Acta Crystallogr D, 62, 72–82. [DOI] [PubMed] [Google Scholar]
- Fasan R., Meharenna Y.T., Snow C.D., Poulos T.L., Arnold F.H. (2008) J Mol Biol, 383, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D.G., Young L., Chuang R.-Y., Venter J.C., Hutchison C.A., Smith H.O. (2009) Nat Meth, 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Gong L.I., Suchard M.A., Bloom J.D. (2013) Elife, 2, e00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover G.J., Jorgensen R., Merrill A.R., Shelp B.J. (2011) Structure of glyoxylate reductase 1 from Arabidopsis (AtGLYR1). RCSB Protein Data Bank. [Google Scholar]
- Hurley J.H., Chen R.D., Dean A.M. (1996) Biochemistry, 35, 5670–5678. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) Acta Crystallogr D, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh K.L., Klimacek M., Nidetzky B., Wilson D.K. (2002) Biochemistry, 41, 8785–8795. [DOI] [PubMed] [Google Scholar]
- Khoury G.A., Fazelina H., Chin J.W., Pantazes R.J., Cirino P.C., Maranas C.D. (2009) Protein Sci, 18, 2125–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran D., Swaminathan S. (2009) Crystal structure of lactaldehyde reductase. RCSB Protein Data Bank. [Google Scholar]
- Kunkel T.A., Roberts J.D., Zakour R.A. (1987) Method Enzymol, 154, 367–382. [DOI] [PubMed] [Google Scholar]
- Larroy C., Fernández M.R., González E., Parés X., Biosca J.A. (2002) Biochem J, 361(Pt 1), 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Cirino P.C. (2014) Biotechnol Bioeng, 111, 1273–1287. [DOI] [PubMed] [Google Scholar]
- Liu X., Bastian S., Snow C.D., Brustad E.M., Saleski T.E., Xu J.-H., Meinhold P., Arnold F.H. (2012) J Biotechnol, 164, 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lountos G.T., Jiang R., Wellborn W.B., Thaler T.L., Bommarius A.S., Orville A.M. (2006) Biochemistry, 45, 9648–9659. [DOI] [PubMed] [Google Scholar]
- Lynch M. (2012) Proc Natl Acad Sci USA, 109, 18851–18856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcal D., Rego A.T., Carrondo M.A., Enguita F.J. (2009) J Bacteriol, 191, 1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesecar A.D., Stoddard B.L., Koshland D.E Jr (1997) Science, 277, 202–206. [DOI] [PubMed] [Google Scholar]
- Moodie S.L., Mitchell J.B., Thornton J.M. (1996) J Mol Biol, 263, 486–500. [DOI] [PubMed] [Google Scholar]
- Moon J.-H., Lee H.-J., Park S.-Y. et al. , (2011) J Mol Biol, 407, 413–424. [DOI] [PubMed] [Google Scholar]
- Nobeli I., Laskowski R.A., Valdar W.S.J., Thornton J.M. (2001) Nucleic Acids Res, 29, 4294–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick W.M., Firth A.E., Blackburn J.M. (2003) Protein Eng, 16, 451–457. [DOI] [PubMed] [Google Scholar]
- Plapp B.V. (2010) Arch Biochem Biophys, 493, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrkov T.V., Kosinsky Y.A., Arseniev A.S., Priestle J.P., Jacoby E., Efremov R.G. (2007) Proteins, 66, 388–398. [DOI] [PubMed] [Google Scholar]
- Reiße S., Garbe D., Brück T. (2015) Biochimie, 108, 76–84. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Frisch E.F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, New York. [Google Scholar]
- Savir Y., Noor E., Milo R., Tlustly T. (2010) Proc Natl Acad Sci USA, 107, 3475–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Dannert C., Arnold F.H. (1999) Trends Biotechnol, 17, 135–136. [DOI] [PubMed] [Google Scholar]
- Sung W., Ackerman M.S., Miller S.F., Doak T.G., Lynch M. (2012) Proc Natl Acad Sci USA, 109, 18488–18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcherkez G.G., Farquhar G.D., Andrews T.J. (2006) Proc Natl Acad Sci USA, 103, 7246–7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas L.M., Harper A.R., Miner W.A., Ajufo H.O., Branscum K.M., Kao L., Sims P.A. (2013) Acta Crystallogr F, 69, 730–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki N., Jackson C.J., Afriat-Jurnou L., Wyganowski K.T., Tang R., Tawfik D.S. (2012) Nat Commun, 3, 1257. [DOI] [PubMed] [Google Scholar]
- Valencia E., Larroy C., Ochoa W.F., Parés X., Fita I., Biosca J.A. (2004) J Mol Biol, 341, 1049–1062. [DOI] [PubMed] [Google Scholar]
- Wang X., Miller E.N., Yomano L.P., Zhang X., Shanmugam K.T., Ingram L.O. (2011) Appl Environ Microb, 77, 5132–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S.-H., Lonhienne T.G.A., Winzor D.J., Schenk G., Guddat L.W. (2012) J Mol Biol, 424, 168–179. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Garavito R.M. (2011) Crystal structure of gamma-hydroxybutyrate dehydrogenase from Geobacter metallireducens in complex with NADP+. RCSB Protein Data Bank. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.