

Summary
Autoimmune hemolytic anemia is characterized by shortened red blood cell survival and a positive Coombs test. The responsible autoantibodies may be either warm reactive or cold reactive. The rate of hemolysis and the severity of the anemia may vary from mild to severe and life-threatening. Diagnosis is made in the laboratory by the findings of anemia, reticulocytosis, a positive Coombs test, and specific serologic tests. The prognosis is generally good but renal failure and death sometimes occur, especially in cases mediated by drugs.
Keywords: Autoimmunity, Hemolysis, Agglutinin, Hemolysin, Direct antiglobulin test
Introduction and Classification
Autoimmune hemolytic anemia (AIHA) exhibits two important characteristics: shortened red blood cell (RBC) survival and presence of autoantibodies directed toward autologous RBCs, usually demonstrated by a positive direct antiglobulin (Coombs) test (DAT) [1]. AIHA is classified by the temperature at which autoantibodies bind optimally to RBCs. In warm antibody AIHA, which constitutes about 80-90% of adult cases, hemolysis is mediated by antibodies which bind to RBCs at 37 °C (98.6 °F) [2,3,4]. In cryopathic hemolytic syndromes, cold-reactive autoantibodies exhibit affinity for RBCs optimally at temperatures below body temperature. In adults, most of the cold-reactive antibodies are agglutinins of the IgM isotype. In children, cold hemolysins (generally IgG) are most common. Unusual patients with mixed AIHA exhibit both cold-reactive and warm-reactive autoantibodies [5,6].
Each of these types of AIHA may be sub-classified based on the presence or absence of underlying diseases. In the absence of an underlying disease, the AIHA is termed primary or idiopathic. When AIHA occurs as a manifestation or complication of another disease, the term secondary AIHA is used. The mere presence of another disease in a patient with AIHA does not permit classification of AIHA as secondary. Rather, AIHA should be considered secondary only when i) AIHA and the suspect disease occur together more frequently than by chance alone, ii) AIHA remits with correction of the suspect disease, or iii) the suspect disease causes immunologic aberration [3]. Chronic lymphocytic leukemia (CLL) and lymphomas account for about half of all secondary AIHA cases. Systemic lupus erythematosus (SLE) and other autoimmune diseases also account for a large number of secondary AIHA cases. Certain drugs also mediate immune injury to RBCs. Three mechanisms are recognized and two of these involve elements of autoimmunity, in which autoantibodies recognize and bind to specific RBC epitopes in concert with drugs or their metabolites or in which the autoantibody binds to RBC epitopes without the presence of drug [7]. The classification of AIHA is shown in table 1.
Table 1.
Classification of AIHA (modified from [1])
| I. Warm autoantibody type |
| A. Primary or idiopathic warm antibody AIHA |
| B. Secondary warm AIHA |
| 1. Associated with lymphoproliferative disorders (e.g., Hodgkin's lymphoma) |
| 2. Associated with the rheumatic disorders, (e.g., systemic lupus erythematosus (SLE) |
| 3. Associated with certain nonlymphoid neoplasms (e.g., ovarian tumors) or certain chronic inflammatory diseases (e.g., ulcerative colitis) |
| 4. Associated with ingestion of certain drugs (e.g., α-methyldopa) |
| II. Cold autoantibody type |
| A. Mediated by cold agglutinins |
| 1. Idiopathic (primary) chronic cold agglutinin disease (associated with clonal B-lymphocyte proliferation with or without symptomatic lymphoma) |
| 2. Secondary cold agglutinin hemolytic anemia Postinfectious (e.g., mycoplasma pneumonia, chickenpox or infectious mononucleosis) |
| B. Mediated by cold hemolysins |
| 1. Idiopathic (primary) paroxysmal cold hemoglobinuria |
| 2. Secondary |
| a. Donath-Landsteiner hemolytic anemia in children |
| b. Congenital or tertiary syphilis in adults |
| III. Mixed cold and warm autoantibodies |
| A. Primary or idiopathic mixed AIHA |
| B. Secondary mixed AIHA (e.g., associated with systemic lupus erythematosus) |
| IV. Drug-induced autoantibody type |
Clinical Features
Warm Antibody AIHA
Annual incidence of warm antibody AIHA is estimated at one per 75,000-80,000 population, occurring in people of all ages [3]. Primary warm antibody AIHA accounts for about half of the cases [3,4]. The presenting symptoms of warm antibody AIHA are commonly related to the anemia itself. Typically, onset of symptoms is insidious over months. Less often a patient may note sudden onset of symptoms of severe anemia and jaundice over a few days. In secondary AIHA, the symptoms and signs of the underlying disease may overshadow the hemolytic anemia and associated features.
In idiopathic warm antibody AIHA with only mild anemia, the physical examination is usually unremarkable. Even patients with severe hemolysis may have only mild splenomegaly. In very severe cases, including those with acute onset, patients may present with fever, pallor, jaundice, (fig. 1) hepatosplenomegaly, hyperpnea, tachycardia, angina, or heart failure.
Fig. 1.
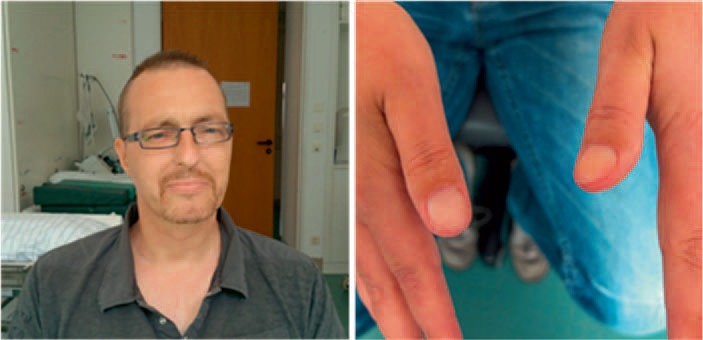
Physical examination in warm antibody AIHA. Skin pallor, icterus and nailbed pallor in a patient with treatment-refractory warm AIHA, hemoglobin 4.8 g/dl (photographs courtesy of Professor A. Salama, with kind permission of the patient).
Cryopathic Hemolytic Syndromes
Cold agglutinin disease accounts for about 10-20% of cases of AIHA [2,3]. The prevalence is estimated at about 14 per million [8]. It is more common in women than in men [2,3].
Idiopathic (primary) chronic cold agglutinin disease has its peak incidence after age 50 years. This disorder is characterized by monoclonal IgM cold agglutinins and is considered a special form of monoclonal gammopathy or low-grade lymphoproliferative malignancy. The majority of these patients exhibit clonal B-lymphocyte proliferation [8]. As with other ‘essential’ or idiopathic monoclonal gammopathies, some of these patients develop features of a B-cell lymphoproliferative disorder such as chronic lymphocytic leukemia, low-grade lymphoma or Waldenström macroglobulinemia. Other patients with established lymphoproliferative disorders may develop cold agglutinin disease as a manifestation of the lymphoma. Cases associated with symptomatic lymphoproliferative disorders are said to represent secondary cold agglutinin disease. However, since ‘idiopathic’ cases also generally have evidence of a clonal lymphoproliferative disorder, the distinction between these primary and secondary types of chronic cold agglutinin disease is not fixed.
Most patients with idiopathic cold agglutinin disease have chronic hemolytic anemia. These patients usually possess cold agglutinins of high thermal amplitude, i.e., agglutinins that can bind at or near body temperature. Other patients exhibit episodic, acute hemolysis with hemoglobinuria induced by chilling. In these patients, one tends to see antibodies with lower thermal amplitude requiring more severe chilling for the agglutinin to bind to RBCs. Secondary cold agglutinin disease is seen in adolescents or young adults with Mycoplasma pneumoniae infections, infectious mononucleosis and, sometimes in children with chickenpox. It is a self-limited process. Most patients with mycoplasma pneumonia have high cold agglutinin titers, but clinical hemolytic anemia is unusual [9]. Cold agglutinins occur in more than 60% of patients with infectious mononucleosis, usually unaccompanied by hemolysis [10]. Hemolysis in M. pneumoniae infections is acute in onset, and usually appears as the patient is recovering from pneumonia, coinciding with peak titers of cold agglutinins. The hemolysis is self-limited, lasting 1-3 weeks [3]. Hemolytic anemia in infectious mononucleosis develops within the first 3 weeks of illness [10].
Jaundice may be present. Livedo reticularis is occasionally seen (fig. 2a). Acrocyanosis involving the fingers, toes, nose, and ears is caused by sludging of RBCs in the cutaneous microvasculature (fig. 2b). Skin ulceration and necrosis are uncommon. Other physical findings are variable and depend upon the presence and nature of an underlying disease. Splenomegaly, a characteristic finding in lymphoproliferative diseases or infectious mononucleosis, may be observed in idiopathic cold agglutinin disease.
Fig. 2.

Physical examination in cold antibody AIHA. a Livedo reticularis (photograph courtesy of Professor A. Salama). b Acrocyanosis involving the toes in a patient with a high-titer IgM cold agglutinin (reprinted from Sinha A, Richardson G, Patel RT : Cold agglutinin related acrocyanosis and paroxysmal hemolysis. Eur J Vasc Endovasc Surg, 30:563-565, copyright 2005, with permission from Elsevier).
Paroxysmal cold hemoglobinuria is most common in children and accounts for about 2-5% of all cases of AIHA [2,3]. However, among children, Donath-Landsteiner hemolytic anemia accounts for almost one-third of cases [11]. Most childhood cases follow either specific viral infections or upper respiratory infections of undefined etiology [2,3,11,12]. In paroxysmal cold hemoglobinuria, constitutional symptoms are prominent during a paroxysm. A few minutes to hours after cold exposure, the patient develops aching pains in the back or legs, abdominal cramps, and headaches. Chills and fever typically follow. Hemoglobinuria is observed shortly after onset of symptoms. The constitutional symptoms and hemoglobinuria generally last a few hours. Raynaud phenomenon and cold urticaria sometimes occur during an attack; jaundice may follow.
Drug-Induced Immune Hemolytic Anemia
The incidence of drug-induced immune hemolytic anemia is about one per million. The great majority result from the second- and third-generation cephalosporins, cefotetan, and ceftriaxone [13]. A careful history of drug exposure should be elicited from all patients with hemolytic anemia or a positive DAT. The clinical picture is quite variable. As with other hemolytic anemias, the severity of symptoms depends upon the rate of hemolysis, which may be slow and mild or rapid and severe. Acute renal failure and death may accompany severe hemolysis [14,15,16].
Laboratory Features
General Features
Patients with warm antibody AIHA may present with hematocrit levels less than 10% or may have compensated hemolytic anemia and a near-normal hematocrit. Occasional patients have leukopenia and neutropenia [17]. Platelet counts are usually normal. Occasionally immune thrombocytopenia is seen in patients with warm antibody AIHA, a condition termed Evans syndrome [17].
Patients with chronic cold agglutinin disease exhibit mild to moderate anemia, hematocrit levels ranging sometimes as low as 15-20%. Patients with paroxysmal cold hemoglobinuria exhibit a rapid fall in hematocrit during a paroxysm. During a paroxysm, leukopenia is noted early, followed by leukocytosis. Consumption of complement proteins during hemolysis may lead to depression of complement levels.
In drug-induced immune hemolytic anemia, the hematologic findings are similar to those of warm antibody AIHA at one end of a spectrum, or may be as severe as those seen in paroxysmal cold hemoglobinuria at the other end, depending on the mechanism of the drug-induced immune process. Most patients exhibit anemia and reticulocytosis.
The blood film exhibits several features common to all types of AIHA. Polychromasia represents reticulocytosis, reflecting an increased production and egress of reticulocytes from the marrow. Spherocytes are frequently seen, a finding that suggests the presence of either hereditary spherocytosis (HS) or an immune hemolytic process. In more severe cases, RBC fragments, nucleated RBCs, and erythrophagocytosis by monocytes may be seen. Mild leukocytosis and neutrophilia are typical. RBC autoagglutination may be seen in the blood film and in chilled anticoagulated blood from patients with cold-antibody AIHA (fig. 3).
Fig. 3.

Blood films from patients with AIHA. Used with permission from Lichtman's Atlas of Hematology, www.accessmedicine.com. a Cold agglutinin disease, low power, slide made at room temperature. Note agglutination of RBCs. b Warm antibody AIHA. Note frequent small, round RBCs lacking central pallor, large bluish RBCs (polychromasia), Howell-Jolly body and nucleated RBCs.
The reticulocyte count usually is elevated. Nevertheless, early on, more than one-third of patients may exhibit transient reticulocytopenia [18,19]. Reticulocytopenia also may be seen in patients with compromised marrow function, parvovirus infection, toxic chemicals, or nutritional deficiency. Marrow examination is not necessary for the diagnosis, but usually reveals erythroid hyperplasia. Sometimes there is evidence of an underlying lymphoproliferative disorder.
Increased unconjugated bilirubin is characteristic of hemolytic anemia. Total bilirubin is only modestly increased and the conjugated (direct) fraction constitutes less than 15% of the total. Urinary urobilinogen is increased but bilirubin is not detected in the urine unless serum conjugated bilirubin is increased. Serum haptoglobin levels are low, and lactate dehydrogenase levels are increased. Hemoglobinuria is rare in warm antibody AIHA but is more common in patients with cold agglutinin disease, and especially in patients with paroxysmal cold hemoglobinuria and with some types of drug-induced immune hemolytic anemia.
Serologic Features
The DAT detects antibody and/or complement on RBCs. A positive DAT is an important feature of AIHA. A broad-spectrum antiglobulin reagent detects both immunoglobulin and complement components on patient RBCs. More specific reagents which react selectively with IgG or with C3 are used to determine which sensitizing agents are present on the RBCs. Monospecific antisera to IgM or IgA may be used in selected cases [1].
DAT may detect 3 principal patterns of RBC sensitization: IgG alone, complement alone, and IgG plus complement. Each pattern is associated with active hemolysis. The differential diagnostic significance of each of these major patterns of RBC sensitization is shown in table 2. IgA and IgM are encountered less frequently, usually in concert with bound IgG and/or complement [20,21,22,23].
Table 2.
Patterns of reactivity in the DAT and associated types of AIHA (modified from [1])
| Reaction components | Type of AIHA |
|---|---|
| IgG alone | warm antibody AIHA |
| drug immune hemolytic anemia | |
| Complement alone | warm antibody AIHA with subthreshold IgG deposition |
| cold agglutinin disease | |
| paroxysmal cold hemoglobinuria | |
| drug-immune hemolytic anemia | |
| IgG plus complement | warm-antibody AIHA |
| mixed warm and cold antibody AIHA | |
| drug-immune hemolytic anemia | |
In warm antibody AIHA, the autoantibodies exist in a reversible, dynamic equilibrium between RBCs and plasma [24]. Thus ‘free’ autoantibody may be found in the plasma or serum of patients by the indirect antiglobulin test (IAT). The occurrence of free autoantibody in plasma is an ‘overflow’ phenomenon, depending upon the amount of antibody present and the binding affinity of the antibody for RBC antigens. The intensity of the DAT reaction is dose-dependent on the number of IgG or complement molecules bound per RBC.
Occasionally, patients with warm antibody AIHA present with spherocytic hemolytic anemia but a negative DAT. There are 3 principle causes for the negative DAT: IgG or complement sensitization below the threshold of detection of antiglobulin reagents; low-affinity IgG causing loss of cell-bound antibody during the cell washing steps before the direct antiglobulin reaction; sensitization with IgA or IgM antibodies which many commercial DAT reagents cannot detect because they contain only anti-IgG and anti-C3. More sensitive methods for quantifying RBC-bound IgG allow identification of patients with so-called ‘Coombs negative’ AIHA [25]. Coombs-negative AIHA has been reviewed elsewhere [26].
The rate of hemolysis in patients with warm antibody AIHA patients is quite variable and depends only loosely on the strength of the antiglobulin reaction. The IgG subclass of warm autoantibodies appears to influence RBC survival. IgG1, the most commonly encountered subclass, and IgG3 autoantibodies appear to be more effective in decreasing RBC life span than do those of the IgG2 or IgG4 subclass [20,27,28].
The autoantibodies in warm antibody AIHA typically bind to all common types of human RBCs found in test panels used by blood banks. However, they are not ‘non-specific.’ Rather, the autoantibodies from any given patient recognize one or more antigens common to almost all human RBCs, so-called ‘public’ antigens. Nearly half of all warm antibody AIHA patients have autoantibodies specific for epitopes on Rh proteins [29,30,31,32]. These autoantibodies react with all human RBCs except those with the Rh-null phenotype. Autoantibodies with such specificity are designated collectively as Rh related [32,33]. The remaining patients with warm antibody AIHA have IgG autoantibodies that react with Rh-null RBCs [29,30,31,32]. Autoantibody specificity for these so-called non-Rh-related blood group antigens has been identified, including Wrb[29], Ena[34], LW [35], U [36], Ge [23,37], Sc1 [38], and K [39].
Cold agglutinins directly agglutinate RBCs at temperatures below body temperature, maximally at 0-5 °C (32-41 °F). Agglutination is reversible by warming. Healthy individuals often display low-titer cold agglutinins, but in chronic cold agglutinin disease, serum titers may range from 1:1,000 to 1:512,000 [3]. Cold agglutinins are typically IgM but IgA or IgG cold agglutinins have also been reported [3,40,41], sometimes with IgM [42]. In mixed warm and cold antibody AIHA, both warm-reactive IgG autoantibodies and IgM cold agglutinins are found concurrently [5].
The DAT is positive with anticomplement reagents. The cold agglutinin antibodies are not detected by the DAT because the cold agglutinins easily dissociate from the RBCs not only in vivo but also during the washing steps of the antiglobulin procedure. In contrast, C4b and C3b are covalently bound to target RBCs via thioester linkages.
The majority of cold agglutinins react with antigens of the I/i system, precursors of ABH and Lewis blood group substances [43]. I antigens are plentiful on adult RBCs but only weakly expressed on neonatal or cord RBCs. The opposite is true of i antigens. Anti-I is the principal specificity of cold agglutinins in idiopathic cold agglutinin disease, in patients with M. pneumoniae and in some cases of lymphoma. Anti-i specificity is predominant in patients with infectious mononucleosis and in some patients with lymphoma. Some cold agglutinins react equally well with adult and neonatal RBCs. These antibodies recognize antigens outside the I/i system, including Pr [40], M [44], or P [45] blood group antigens. Cold agglutinins associated with chickenpox often exhibit anti-Pr specificity. In hemolytic anemia of infectious mononucleosis, in addition to the usual IgM anti-i cold agglutinins, in some cases cold-reactive nonagglutinating IgG anti-i antibodies occur in concert with IgM cold-reactive anti-IgG antibodies (‘rheumatoid factors'). The latter may then cross-link the IgG-coated RBCs to produce agglutination [46].
In paroxysmal cold hemoglobinuria, surviving RBCs are generally coated with complement. The direct antiglobulin reaction may be positive for complement during and briefly following an acute attack. The non-agglutinating IgG Donath-Landsteiner antibody is responsible for complement deposition on the cells; however, it binds RBCs only in the cold and is not detected by the DAT. The DAT remains negative between attacks. The antibody is detected by the Donath-Landsteiner test, in which patient's fresh serum is first incubated with RBCs at 4 °C (39.2 °F) to allow binding of antibody. The mixture is then warmed to 37 °C (98.6 °F) to allow complement activation, resulting in intense hemolysis [3]. Antibody titers are generally low, rarely exceeding 1:16. The Donath-Landsteiner antibody exhibits specificity for the P blood group antigen, which is found on lymphocytes and skin fibroblasts [47]. The latter finding may account for episodes of cold urticaria in paroxysmal cold hemoglobinuria, a phenomenon that may be transferred passively by serum to normal skin [2].
The laboratory findings of drug-induced AIHA may resemble those of warm antibody AIHA or a cryopathic hemolytic syndrome, depending on the mechanism of the autoantibody induction and the target antigen [1]. A high index of suspicion on the part of the clinician and discussions with blood bank personnel can usually reveal the role of the drug.
Differential Diagnosis
HS, Zieve syndrome, clostridial sepsis, and the hemolytic anemia preceding Wilson disease resemble AIHA in several respects. HS may present in adulthood, and splenomegaly may be present in both HS and AIHA. The family history in HS, however, often identifies other affected individuals. Most importantly, among these diseases, the DAT is positive only in AIHA. Bites of the brown recluse spider, (Loxosceles reclusa) may cause severe hemolysis and a DAT positive for IgG and or complement. Spherocytes and RBC fragmentation are noted on the blood film [48,49].
The serologic nature of the autoantibody helps distinguish warm antibody AIHA from cryopathic syndromes. A careful drug history supported by matching serologic findings helps to distinguish drug-induced immune hemolytic anemia. A positive DAT reaction shortly following blood transfusion raises the specter of a delayed transfusion reaction, leading to an erroneous impression of an autoimmune process.
Other acquired types of hemolytic anemia are unlikely to mimic AIHA since spherocytosis is absent on the blood film and the DAT is negative. Patients with paroxysmal nocturnal hemoglobinuria (PNH) may note hemoglobinuria which can occur in patients with the cryopathic AIHA, but not usually in patients with warm autoantibodies. The DAT is negative in PNH. Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome are distinguished from AIHA by fragmentation of RBCs and relative absence of spherocytes on the blood film and the almost uniform presence of thrombocytopenia, which is lacking in most cases of AIHA.
Chronic cold agglutinin disease is marked by the presence of a cold agglutinin in high titer and a DAT positive for complement and not IgG. Some cases of drug-induced immune hemolytic anemia also exhibit a positive DAT only with an anti-complement reagent. A history of recent drug ingestion and absence of a significant cold agglutinin titer help to differentiate drug-induced immune hemolytic anemia from cold agglutinin disease. A patient with high titer cold agglutinins and a positive DAT for both IgG and complement may have a mixed-type AIHA. When hemolysis is chronic, warm antibody AIHA, hereditary hemolytic anemia, and PNH should be excluded. The family history, serology, and flow analysis of CD55/CD59 on blood cells offer added support in such cases. Episodic hemolysis raises the possibility of paroxysmal cold hemoglobinuria, march hemoglobinuria, and PNH. Cryopathic peripheral vasoocclusive symptoms call to mind cryoglobulinemia and Raynaud phenomenon, possibly related to a rheumatic disorder. In the appropriate clinical setting, infectious mononucleosis, M. pneumoniae infection, or lymphoma should be considered.
Unusual clinical presentations of AIHA may cause initial confusion in the diagnosis. For example, patients may present with a normal reticulocyte count [18] or a negative DAT [26]. Other evidence of hemolysis such as elevated indirect bilirubin and lactate dehydrogenase, decreased serum haptoglobin, and the presence of spherocytes on the blood film along with a high index of suspicion will lead to the correct diagnosis.
In other cases, hemolysis is less prominent, and blood cell injury is mediated primarily by agglutination. A recent case report describes a patient with livedo reticularis, minimal hemolysis, and a warm IgA non-complement-fixing autoagglutinin, who died of complications of hemagglutination [50]. In another example, a patient presented with acrocyanosis and a high-titer, high-thermal-amplitude, complement-fixing IgM cold agglutinin, without evidence for hemolysis. [51] In atypical cases, serologic examination for mixed warm and cold autoantibodies [5], warm IgM autoantibodies [52], or IgA autoantibodies [53] may prove useful since many of these patients exhibit more aggressive disease. Discussion between the clinician and the blood bank physician is helpful to solve difficult cases and has the potential to affect therapy.
Paroxysmal cold hemoglobinuria with its characteristic episodic hemolysis and hemoglobinuria is generally distinguished from chronic cold agglutinin disease, warm antibody AIHA, march hemoglobinuria, myoglobinuria, and PNH by the DAT and Donath-Landsteiner test.
Drug-induced immune hemolysis may mimic warm or cold antibody AIHA, hereditary hemolytic anemias such as HS, and other drug-mediated hemolytic anemias such as glucose-6-phosphate dehydrogenase deficiency. Patients with drug-induced immune hemolytic anemia have a positive DAT that distinguishes this group from patients with inherited RBC defects.
Course and Prognosis
Relapses and remissions characterize the unpredictable clinical course of idiopathic warm antibody AIHA. An older study reports survival of 73% at 10 years [51] but it is likely higher nowadays. Thromboembolic episodes are relatively common during active phases of the disease, frequently associated with anti-phospholipid antibodies [55,56,57]. The prognosis in secondary warm antibody AHA largely depends upon the course of the underlying disease.
In children, warm antibody AIHA frequently exhibits a self-limited course following a rapid response to glucocorticoids. Most of these cases occur after an acute infection or immunization [58,59]. Those who recover from the initial hemolytic episode have a good prognosis and are unlikely to relapse, although exceptions occur. Children with chronic AIHA tend to be older [60]. The mortality rate ranges from 10 to 30% [58,60,61], with higher mortality rates in those with chronic AIHA and in cases with simultaneous immune thrombocytopenia (Evans syndrome) [62].
Patients with idiopathic cold agglutinin disease generally follow a benign direction, often surviving for many years [3]. Death may result from infection or severe anemia or sometimes from an underlying lymphoma.
Postinfectious cold agglutinin disease is usually self-limited, with recovery occurring in a few weeks. Postinfectious forms of paroxysmal cold hemoglobinuria cease spontaneously within days to weeks after onset [11,12], although the Donath-Landsteiner antibody may persist in low titer for several years [2]. Patients with chronic idiopathic paroxysmal cold hemoglobinuria survive for years despite occasional paroxysms of hemolysis.
Drug-induced immune hemolytic anemia is usually mild, and the prognosis is good. Severe hemolysis leading to renal failure or death has been reported [14,15,16]. In most cases, hemolysis ceases, and the DAT becomes negative shortly after the drug is discontinued and clears from the circulation. However, in those cases mediated by drug-induced autoantibodies, the DAT may remain positive for weeks to months after discontinuation of the offending drug and cessation of hemolysis.
Disclosure Statement
The author declares no conflicts of interest with respect to this manuscript.
References
- 1.Packman CH. Hemolytic anemia resulting from immune injury; in Kaushansky K, Lichtman MA, Prchal JT, Levi MM, Press OW, Burns LJ, Caligiuri MA (eds): Williams Hematology, 9th ed. New York, McGraw-Hill, chapter 54, 2016 (in press).
- 2.Dacie JV. The Haemolytic Anaemias, vol 3, The Autoimmune Haemolytic Anaemias, 3rd ed. New York, Churchill Livingstone, 1992.
- 3.Petz LD, Garratty G. Acquired Immune Hemolytic Anemias. Philadelphia, Churchill Livingstone, 2004.
- 4.Sokol RJ, Hewitt S, Stamps BK. Autoimmune haemolysis: an 18 year study of 865 cases referred to a regional transfusion centre. Br Med J. 1981;282:2023–2027. doi: 10.1136/bmj.282.6281.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mayer B, Yürek S, Kiesewetter H, Salama A. Mixed‐type autoimmune hemolytic anemia: differential diagnosis and a critical review of reported cases. Transfusion. 2008;48:2229–2234. doi: 10.1111/j.1537-2995.2008.01805.x. [DOI] [PubMed] [Google Scholar]
- 6.Shulman IA, Branch DR, Nelson JM, Thompson JC, Saxena S, Petz LD. Autoimmune hemolytic anemias with both cold and warm autoantibodies. JAMA. 1985;253:1746–1748. [PubMed] [Google Scholar]
- 7.Salama A, Mueller-Eckhardt C. On the mechanisms of sensitization and attachment of antibodies to RBCs in drug-induced immune hemolytic anemia. Blood. 1987;69:1006–1010. [PubMed] [Google Scholar]
- 8.Berentsen S, Bo K, Shammas F, Myking A, Ulvestad D. Chronic cold agglutinin disease of the ‘idiopathic’ type is a premalignant or low-grade malignant lymphoproliferative disease. APMIS. 1997;105:354–362. doi: 10.1111/j.1699-0463.1997.tb00581.x. [DOI] [PubMed] [Google Scholar]
- 9.Jacobson LB, Longstreth GF, Edington TS. Clinical and immunologic features of transient cold agglutinin hemolytic anemia. Am J Med. 1973;54:514–521. doi: 10.1016/0002-9343(73)90047-8. [DOI] [PubMed] [Google Scholar]
- 10.Worlledge SM, Dacie JV. Haemolytic and other anaemias in infectious mononucleosis; in Carter RL, Penman HG (eds): Infectious Mononucleosis. Oxford, Blackwell Science, 1969, p 82.
- 11.Gottsche B, Salama A, Mueller-Eckhardt C. Donath-Landsteiner autoimmune hemolytic anemia in children: a study of 22 cases. Vox Sang. 1990;58:281–286. doi: 10.1111/j.1423-0410.1990.tb05000.x. [DOI] [PubMed] [Google Scholar]
- 12.Nordhagen R, Stensvold K, Winsnes A, Skyberg D, Storen A. Paroxysmal cold hemoglobinuria. The most frequent autoimmune hemolytic anemia in children? Acta Paediatr Scand. 1984;73:258–262. doi: 10.1111/j.1651-2227.1984.tb09939.x. [DOI] [PubMed] [Google Scholar]
- 13.Arndt PA, Garratty G. Cross-reactivity of cefotetan and ceftriaxone antibodies, associated with hemolytic anemia, with other cephalosporins and penicillin. Coagul Transfusion Med. 2002;118:256–262. doi: 10.1309/JFJE-VUKN-221T-G6EV. [DOI] [PubMed] [Google Scholar]
- 14.Garratty G, Nance S, Lloyd M, Domen R. Fatal immune hemolytic anemia due to cefotetan. Transfusion. 1992;32:269–271. doi: 10.1046/j.1537-2995.1992.32392213813.x. [DOI] [PubMed] [Google Scholar]
- 15.Habibi B, Basty R, Chodez S, Prunat A. Thiopental- related immune hemolytic anemia and renal failure. N Engl J Med. 1985;312:353–355. doi: 10.1056/NEJM198502073120606. [DOI] [PubMed] [Google Scholar]
- 16.Garratty G, Postoway N, Schwellenbach J, McMahill PC. A fatal case of ceftriaxone (Rocephin)-induced hemolytic anemia associated with intravascular immune hemolysis. Transfusion. 1991;31:176–179. doi: 10.1046/j.1537-2995.1991.31291142951.x. [DOI] [PubMed] [Google Scholar]
- 17.Evans RS, Duane RT. Acquired hemolytic anemia: I. The relation of erythrocyte antibody production to activity of the disease: II. The significance of thrombocytopenia and leukopenia. Blood. 1949;4:1196–1213. [PubMed] [Google Scholar]
- 18.Liesveld JL, Rowe JM, Lichtman MA. Variability of the erythropoietic response in autoimmune hemolytic anemia: analysis of 109 cases. Blood. 1987;69:820–826. [PubMed] [Google Scholar]
- 19.Conley CL, Lippman SM, Ness P. Autoimmune hemolytic anemia with reticulocytopenia: a medical emergency. JAMA. 1980;244:1688–1690. [PubMed] [Google Scholar]
- 20.Engelfriet CP, von dem Borne AE, Beckers D, van Loghem JJ. Autoimmune haemolytic anaemia: serological and immunochemical characteristics of the autoantibodies: mechanisms of cell destruction. Ser Haematol. 1974;7:328–347. [PubMed] [Google Scholar]
- 21.Wolf CF, Wolf DJ, Peterson P, Brandstetter RD, Hansen DE. Autoimmune hemolytic anemia with predominance of IgA autoantibody. Transfusion. 1982;22:238–240. doi: 10.1046/j.1537-2995.1982.22382224949.x. [DOI] [PubMed] [Google Scholar]
- 22.Szymanski IO, Teno R, Rybak ME. Hemolytic anemia due to a mixture of low-titer IgG lambda and IgM lambda agglutinins reacting optimally at 22 degrees C. Vox Sang. 1986;51:112–116. doi: 10.1111/j.1423-0410.1986.tb00225.x. [DOI] [PubMed] [Google Scholar]
- 23.Göttsche B, Salama A, Mueller-Eckhardt C. Autoimmune hemolytic anemia associated with an IgA autoanti-Gerbich. Vox Sang. 1990;58:211–214. doi: 10.1111/j.1423-0410.1990.tb02093.x. [DOI] [PubMed] [Google Scholar]
- 24.Evans RS, Bingham M, Boehni P. Autoimmune hemolytic disease: antibody dissociation and activity. Arch Intern Med. 1961;108:338–352. doi: 10.1001/archinte.1961.03620090010003. [DOI] [PubMed] [Google Scholar]
- 25.Gilliland BC, Leddy JP, Vaughan JH. The detection of cell-bound antibody on complement-coated human red cells. J Clin Invest. 1970;49:898–906. doi: 10.1172/JCI106309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Segel GB, Lichtman MA. Direct antiglobulin (‘Coombs') test-negative autoimmune hemolytic anemia: A review. Blood Cells Mol Dis. 2014;52:152–160. doi: 10.1016/j.bcmd.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Sokol RJ, Hewitt S, Booker DJ, Bailey A. Erythrocyte autoantibodies, subclasses of IgG and autoimmune haemolysis. Autoimmunity. 1990;6:99–104. doi: 10.3109/08916939008993374. [DOI] [PubMed] [Google Scholar]
- 28.von dem Borne AE, Beckers D, van der Meulen W, Engelfriet CP. IgG4 autoantibodies against erythrocytes, without increased hemolysis: a case report. Br J Haematol. 1977;37:137–144. [PubMed] [Google Scholar]
- 29.Issitt PD, Pavone BG, Goldfinger D, Zwixker H, Issitt CH, Tessel JA, Kroovand SW, Bell CA. Anti-Wrb and other autoantibodies responsible for positive direct antiglobulin test in 150 individuals. Br J Haematol. 1976;34:5–17. doi: 10.1111/j.1365-2141.1976.tb00168.x. [DOI] [PubMed] [Google Scholar]
- 30.Weiner W, Vos GH. Serology of acquired hemolytic anemia. Blood. 1963;22:606–613. [PubMed] [Google Scholar]
- 31.Vos GH, Petz L, Funenberg HH. Specificity of acquired haemolytic anaemia autoantibodies and their serological characteristics. Br J Haematol. 1970;19:57–66. doi: 10.1111/j.1365-2141.1970.tb01601.x. [DOI] [PubMed] [Google Scholar]
- 32.Leddy JP, Peterson P, Yeaw MA, Bakemeier RF. Patterns of serologic specificity of human γ G erythrocyte autoantibodies. J Immunol. 1970;105:677–686. [PubMed] [Google Scholar]
- 33.Leddy JP, Falany JL, Kissel GE, Passodor, ST, Rosenfeld SI. Erythrocyte membrane proteins reactive with human (warm-reacting) anti-red cell autoantibodies. J Clin Invest. 1993;91:1672–1680. doi: 10.1172/JCI116376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bell CA, Zwicker H. Further studies on the relationship of anti-Ena and anti-Wrb in warm autoimmune hemolytic anemia. Transfusion. 1978;18:572–575. doi: 10.1046/j.1537-2995.1978.18579036389.x. [DOI] [PubMed] [Google Scholar]
- 35.Celano MJ, Levine P. Anti-LW specificity in autoimmune acquired hemolytic anemia. Transfusion. 1967;7:265–268. doi: 10.1111/j.1537-2995.1967.tb05515.x. [DOI] [PubMed] [Google Scholar]
- 36.Marsh WL, Reid ME, Scott EP. Autoantibodies of U blood group specificity in autoimmune haemolytic anaemia. Br J Haematol. 1972;22:625–629. doi: 10.1111/j.1365-2141.1972.tb05709.x. [DOI] [PubMed] [Google Scholar]
- 37.Shulman IA, Vengelen-Tyler V, Thompson JC, Nelson JM, Chen DC. Autoanti-Ge associated with severe autoimmune hemolytic anemia. Vox Sang. 1990;59:232–234. doi: 10.1111/j.1423-0410.1990.tb00243.x. [DOI] [PubMed] [Google Scholar]
- 38.Owen I, Chowdhury V, Reid ME, Poole J, Marsh JC, Hows JM. Autoimmune hemolytic anemia associated with anti-Sc 1. Transfusion. 1992;32:173–176. doi: 10.1046/j.1537-2995.1992.32292180150.x. [DOI] [PubMed] [Google Scholar]
- 39.Marsh WL, Oyen R, Alicea E, Linter M, Horton S. Autoimmune hemolytic anemia and the Kell blood groups. Am J Hematol 1979;155-162. [DOI] [PubMed]
- 40.Roelcke D. Cold agglutination: antibodies and antigens. Clin Immunol Immunopathol. 1974;2:266–280. doi: 10.1016/0090-1229(74)90044-0. [DOI] [PubMed] [Google Scholar]
- 41.Curtis BR, Lamon J, Roelcke D, Chaplin H. Life-threatening, antiglobulin test-negative, acute autoimmune hemolytic anemia due to a non-complement-activating IgG 1k cold antibody with Pra specificity. Transfusion. 1990;30:838–843. doi: 10.1046/j.1537-2995.1990.30991048792.x. [DOI] [PubMed] [Google Scholar]
- 42.Silberstein LE, Berkman EM, Schreiber AD. Cold hemagglutinin disease associated with IgG cold reactive antibody. Ann Intern Med. 1987;106:238–242. doi: 10.7326/0003-4819-106-2-238. [DOI] [PubMed] [Google Scholar]
- 43.Feizi T, Kabat EA, Vicari G, Anderson B, Marsh WL. Immunochemical studies on blood groups: XLVII. The I antigen complex precursors in the A, B, H, Lea and Leb blood group system: hemagglutination inhibition studies. J Exp Med. 1971;133:39–52. doi: 10.1084/jem.133.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chapman J, Murphy MF, Waters AH. Chronic cold hemagglutinin disease due to an anti-M-like autoantibody. Vox Sang. 1982;42:272–277. doi: 10.1111/j.1423-0410.1982.tb00756.x. [DOI] [PubMed] [Google Scholar]
- 45.von dem Borne AE, Mol JJ, Joustra-Maas N, Pegels JG, Langenjuijsen MMAC, Engelfriet CP. Autoimmune hemolytic anemia with monoclonal IgM (K) anti-P cold autohemolysins. Br J Haematol. 1982;50:345–350. doi: 10.1111/j.1365-2141.1982.tb01925.x. [DOI] [PubMed] [Google Scholar]
- 46.Capra JD, Dowling P, Cook S, Kunkel HG. An incomplete cold-reactive λ-G antibody with i specificity in infectious mononucleosis. Vox Sang. 1969;16:10–17. doi: 10.1111/j.1423-0410.1969.tb04711.x. [DOI] [PubMed] [Google Scholar]
- 47.Fellous M, Gerbal A, Tessier C, Frezal J, Dausset J, Salmon C. Studies on the biosynthetic pathway of human P erythrocyte antigens using somatic cells in culture. Vox Sang. 1974;26:518–536. doi: 10.1111/j.1423-0410.1974.tb02729.x. [DOI] [PubMed] [Google Scholar]
- 48.Gehrie EA, Nian H, Young PP. Brown recluse spider bite mediated hemolysis: clinical features, a possible role for complement inhibitor therapy and reduced RBC surface glycophorin A as a potential biomarker of venom exposure. Plos One. 2013;8:e76558. doi: 10.1371/journal.pone.0076558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDade J, Aygun B, Ware RE. Brown recluse spider (Loxosceles reclusa) envenomation leading to acute hemolytic anemia in six adolescents. J Pediatr. 2010;156:155–157. doi: 10.1016/j.jpeds.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salama A, Janvier D, Mayer B, Saison C, Moscatelli H, Aucouturier F, Yilmaz P, Arnaud L, Wild V, Knop S, Cartron J-P. Lethal autoimmune hemagglutination due to an immunoglobulin A autoagglutinin with Band 3 specificity. Transfusion. 2014;54:1988–1995. doi: 10.1111/trf.12638. [DOI] [PubMed] [Google Scholar]
- 51.Arndt P, Do JB, Garratty G, Kuriyan MA, Strair RK, Baranwal A. A high-titer, high-thermal amplitude autoanti-B associated with acrocyanosis but no obvious hemolytic anemia. Transfusion. 2003;43:1133–1137. doi: 10.1046/j.1537-2995.2003.00453.x. [DOI] [PubMed] [Google Scholar]
- 52.Arndt P, Leger RM, Garratty G. Serologic findings in in autoimmune hemolytic anemia associated with immunoglobulin M warm autoantibodies. Transfusion. 2009;49:235–242. doi: 10.1111/j.1537-2995.2008.01957.x. [DOI] [PubMed] [Google Scholar]
- 53.Sokol RJ, Booker DJ, Stamps R, Booth J R, Hook V. IgA red cell autoantibodies and autoimmune hemolysis. Transfusion. 1997;37:175–181. doi: 10.1046/j.1537-2995.1997.37297203520.x. [DOI] [PubMed] [Google Scholar]
- 54.Silverstein MN, Gomes MR, Elveback LR, ReMine WH, Linman JD. Idiopathic acquired hemolytic anemia: survival in 117 cases. Arch Intern Med. 1972;129:85–87. [PubMed] [Google Scholar]
- 55.Dausset J, Colombani J. The serology and the prognosis of 128 cases of autoimmune hemolytic anemia. Blood. 1959;14:1280–1301. [PubMed] [Google Scholar]
- 56.Pullarkat V, Ngo M, Iqbal S, Espina B, Liebman HA. Detection of lupus anticoagulant identifies patients with autoimmune haemolytic anaemia at increased risk of venous thromboembolism. Br J Haematol. 2002;118:1166–1169. doi: 10.1046/j.1365-2141.2002.03729.x. [DOI] [PubMed] [Google Scholar]
- 57.Hendrick AM. Auto-immune haemolytic anaemia - a high-risk disorder for thromboembolism? Hematology. 2003;8:53–56. doi: 10.1080/1024533021000059474. [DOI] [PubMed] [Google Scholar]
- 58.Habibi B, Homberg JC, Schaison G, Salmon C. Autoimmune hemolytic anemia in children: a review of 80 cases. Am J Med. 1974;56:61–69. doi: 10.1016/0002-9343(74)90751-7. [DOI] [PubMed] [Google Scholar]
- 59.Buchanan GR, Boxer LA, Nathan DG. The acute and transient nature of idiopathic immune hemolytic anemia in childhood. J Pediatr. 1976;88:780–783. doi: 10.1016/s0022-3476(76)81114-6. [DOI] [PubMed] [Google Scholar]
- 60.Heisel MA, Ortega JA. Factors influencing prognosis in childhood autoimmune hemolytic anemia. Am J Pediatr Hematol Oncol. 1983;5:147–152. [PubMed] [Google Scholar]
- 61.Sokol RJ, Hewitt S, Stamps BK, Hitchen PA. Autoimmune haemolysis in childhood and adolescence. Acta Haematol. 1984;72:245–257. doi: 10.1159/000206397. [DOI] [PubMed] [Google Scholar]
- 62.Wang WC. Evans syndrome in childhood: pathophysiology, clinical course, and treatment. Am J Pediatr Hematol Oncol. 1988;10:330–338. doi: 10.1097/00043426-198824000-00013. [DOI] [PubMed] [Google Scholar]