


Abstract
Ribosome biogenesis is one of the most essential pathways in eukaryotes although it is still not fully characterized. Given the importance of this process in proliferating cells, it is obvious that understanding the macromolecular details of the interactions that take place between the assembly factors, ribosomal proteins and nascent pre-rRNAs is essentially required for the development of new non-genotoxic treatments for cancer. Herein, we have studied the association between the WD40-repeat domains of Erb1 and Ytm1 proteins. These are essential factors for the biogenesis of 60S ribosomal subunits in eukaryotes that form a heterotrimeric complex together with the also essential Nop7 protein. We provide the crystal structure of a dimer formed by the C-terminal part of Erb1 and Ytm1 from Chaetomium thermophilum at 2.1 Å resolution. Using a multidisciplinary approach we show that the β-propeller domains of these proteins interact in a novel manner that leads to a high-affinity binding. We prove that a point mutation within the interface of the complex impairs the interaction between the two proteins and negatively affects growth and ribosome production in yeast. Our study suggests insights into the association of the Erb1-Ytm1 dimer with pre-ribosomal particles.
INTRODUCTION
Eukaryotic ribosome biogenesis is one of the most complex pathways in a cell and involves numerous steps of rRNA processing with concomitant association and dissociation of many non-ribosomal proteins (about 250 in yeast; more than 500 in human cells) that assure correct assembly of ribosomal proteins during maturation of the nascent 40S and 60S subunits (SSU and LSU, respectively) (1–6). Within these factors involved in ribosome maturation, many of them present enzymatic activities that include GTPases, ATPases, nucleases and RNA helicases, meanwhile other factors act as scaffolds that allow proper structural organization of the arising pre-ribosomal particles (7). Ribosome biogenesis has been studied in several eukaryotes, including human, but has been best characterized in the yeast Saccharomyces cerevisiae (4).
Erb1 (Bop1 in Mammals) is a conserved nucleolar factor involved in the biogenesis of LSUs in eukaryotes (8,9). It is essential for cell viability although its exact function in ribosome biogenesis remains unclear. Erb1 directly interacts with other two factors, Nop7 and Ytm1 (Pes1 and Wdr12 in Mammals, respectively) as part of a heterotrimeric Nop7-Erb1-Ytm1 sub-complex (PeBoW complex in mammals) that is present within pre-60S ribosomal particles (r-particles) but is also stable independently of those. The interaction of Erb1 with its partners is essentially required for the correct LSU maturation (10–12). Moreover, UV-cross-linking and cDNA analysis (CRAC) of the RNA binding sites of Erb1 and Nop7 in r-particles showed that both proteins cross-linked to specific but distinct sequences of the pre-rRNA in proximity to the region where the 5′ end of 25S rRNA forms a duplex with the 3′ end of 5.8S rRNA (13). In contrast, Ytm1 did not efficiently cross-link to RNA in CRAC analysis (13).
The binding of Erb1 to Ytm1 is essential for the release of the trimeric complex from pre-60S r-particles, which is triggered by the AAA-ATPase Rea1 (14,15). All, Erb1, Nop7 and Ytm1 belong to the group of proteins termed ‘A3 factors’, which is required for processing of 27SA3 pre-rRNA (13,16). Thus, depletion of Erb1, Nop7 or Ytm1 cause not only to accumulation of 27SA3 pre-rRNA but also of 35S and 27SA2 pre-rRNAs, and a reduction of 27SB pre-rRNAs, especially 27SBS pre-rRNA, and downstream 7S precursors; as a consequence, production of mature 5.8S and 25S rRNAs is decreased upon depletion of these factors (8,10,13,17–19).
From a structural point of view there is very little amount of information regarding the proteins that form the Nop7-Erb1-Ytm1 complex. Erb1 contains a conserved N-terminal BOP1NT domain (Pfam:PF08145) followed by seven WD40 repeats that fold into a β-propeller structure (20). The N-terminal region has been shown to bind Nop7 and is required for the nucleolar localization of the protein (11). We have previously solved the structure of the WD40 domain of Erb1 from S. cerevisiae at 1.6 Å resolution (20). This has allowed us to suggest that the conserved surface of the propeller could be a platform that establishes multiple interactions with other proteins and/or RNA (20). CRAC analyses have demonstrated that Erb1 binds rRNA and, indeed, additional studies have confirmed that the C-terminal region of the protein can bind nucleic acids in vitro (13,20). However, despite its apparent importance, it has been reported that the deletion of the WD40 motifs of Erb1 did neither cause a severe growth impairment nor defects in pre-rRNA processing and LSU assembly in yeast (11).
No structural data are available for Ytm1. Its primary sequence indicates that Ytm1 also contains a seven-bladed β-propeller domain on its C-terminus and a small ubiquitin-like (UBL) motif at the N-terminus (17,18). As Erb1, Ytm1 is an essential factor required for LSU maturation but, in contrast to Erb1, both its N- and C-terminal regions have been shown to be required for LSU assembly. It has been proven that the β-propeller domain of Ytm1 interacts directly with Erb1 and allows the association with pre-60S r-particles (10,11). The UBL domain of Ytm1 has been described to contact the MIDAS domain of Rea1 through a conserved aspartic acid. It has been postulated that the hydrolysis of ATP carried out by the catalytic domain of Rea1 induces a movement of MIDAS that releases the Nop7-Erb1-Ytm1 complex from pre-60S particles in a similar manner as described for Rsa4 protein in a subsequent step of LSU formation (7,15,21). Previous functional reports have suggested that Ytm1 binds to Erb1, in proximity to the Nop7 binding site (11), however no study has shown a direct interaction of Ytm1 with this region of Erb1.
In this work, we use proteins from the thermophilic ascomycete Chaetomium thermophilum to elucidate structural details of the interactions between Erb1 and Ytm1. Herein, we have determined the crystal structure of a dimer formed by Ytm1 bound to the β-propeller domain of Erb1 at a resolution of 2.1 Å. We show, for the first time, the structure of the UBL domain of Ytm1 and describe the particular features of its seven-bladed β-propeller domain. Moreover, we confirmed the in vitro interaction between Erb1 and Ytm1 proteins and proved that the binding occurs between their respective β-propellers. Functional analyses demonstrated that a mutation (R470E in yeast Erb1) that impairs binding of the β-propeller of Erb1 to Ytm1 results in apparent defects in growth and ribosome assembly in yeast. Most importantly, this mutation mildly affects the association of Erb1 to pre-60S r-particles and drastically prevents the recruitment of Ytm1. This is the first report regarding the biological significance of the C-terminal domain of Erb1 that provides an explanation for its function during ribosome assembly.
MATERIALS AND METHODS
Cloning, protein expression and purification
ERB1 and NOP7 genes were amplified from a C. thermophilum cDNA library (obtained as described in (20)) and cloned into the pET28-NKI/LIC 6His/3C vector (a gift from A. Perrakis). To obtain the C. thermophilum Erb1432–801 protein fragment (ChErb1432–801), the appropriate construct was generated by PCR using pET28-NKI-ChErb1 as a template. Site-directed mutagenesis of the alleles for ChErb1 and ChErb1432–801 was done by whole plasmid PCR amplification and resulting constructs were used for protein production. ChErb1, ChErb1432–801 and the mutated ChErb1 proteins were expressed in BL21 Codon Plus strain of E.coli using an autoinduction method (22) at 20°C for 16 h. ChNop7 was expressed in E. coli BL21 Codon Plus with 0.5 mM IPTG at 20°C for 16 h. Cells were collected by centrifugation, frozen in liquid nitrogen and stored at −80°C.
Purification of ChNop7 was performed as described for Nop7 from S. cerevisiae (20). In the case of ChErb1, ChErb1432–801 and the site-directed mutants, the lysis was done as for ChNop7. The purification was done in three steps. First, cleared lysates were loaded onto a HisTrap column (GE Healthcare) in Buffer A (20 mM Hepes pH 7.5; 0.5 M NaCl; 5% glycerol; 2 mM β-mercaptoethanol and 20 mM imidazole). Then, a low-salt containing Buffer ALS (20 mM Hepes pH 7.5; 0.15 M NaCl; 5% glycerol; 2 mM β-mercaptoethanol and 20 mM imidazole) was used to wash the column. The proteins were eluted with Buffer BLS (20 mM Hepes pH 7.5; 0.15 M NaCl; 5% glycerol; 2 mM β-mercaptoethanol and 500 mM imidazole). The fractions containing the corresponding ChErb1 protein were mixed and loaded onto a HiTrap Heparin HP column (GE Healthcare). After a 5 column volumes wash with Buffer SE (20 mM Hepes pH 7.5; 0.15 M NaCl; 5% glycerol; 2 mM β-mercaptoethanol), the protein was released by step elution using high-salt Buffer BHS (20 mM Hepes pH 7.5; 1.5 M NaCl; 5% glycerol; 2 mM β-mercaptoethanol). The fractions containing the protein were concentrated and injected into a Superdex200 16/60 column and eluted in Buffer SE. The fractions corresponding to the monomeric species of the protein were concentrated. Due to the low solubility of ChErb1432–801, the protein was concentrated only up to 1 mg/ml after size exclusion. The final concentrations of ChErb1 and ChNop7 were 50 mg/ml and 45 mg/ml, respectively.
The YTM1 gene from C. thermophilum was amplified from a cDNA library and cloned into pOPINF or pOPINJ vector using the In-Fusion system (Clontech). Sf9 insect cells were grown in Sf900 II SFM medium and were transfected with ChYtm1-pOPINF or ChYtm1-pOPINJ and linearized Ian Jones bacmid (PMCID: PMC140531). Resulting baculovirus were amplified for 5 days and used to induce protein expression. Large scale cultures (300 ml each) were incubated for 72 h at 27°C upon infection with the virus. Cells pellets were frozen in liquid nitrogen and stored at −80°C. The 6xHis tagged ChYtm1 protein was extracted and purified with a HisTrap column (GE Healthcare) followed by size exclusion chromatography as described for ChNop7. GST-tagged ChYtm1 purification was done as for 6xHis-ChYtm1 but the cellular lysate was loaded onto a GSTrap column (GE Healthcare) previously equilibrated with 5 column volumes of Buffer SE. Next, the column was washed with 5 column volumes of Buffer SE and the protein was eluted with Buffer GSH (20 mM Hepes pH 7.5; 150 mM NaCl; 5% glycerol; 2 mM β-mercaptoethanol and 20 mM reduced glutathione). The protein-containing fractions were concentrated and injected into a Superdex200 16/60 column (GE Healthcare) equilibrated with Buffer SE. The eluted protein was concentrated up to 5 mg/ml and flash-frozen in liquid nitrogen until use.
Crystallization and data collection
ChErb1427–801-ChYtm1 dimer in the P65 2 2 space group
Hexagonal crystals were obtained upon co-crystallization trials of ChErb1 full-length mixed with ChYtm1 (1:1 ratio, final concentration 38 mg/ml). Proteins were purified separately, stored in SE buffer and mixed prior to the crystallization trials. The best resolution was achieved for crystals that grew in 15% PEG 4000; 0.1 M sodium citrate pH 5.6; 0.2 M ammonium sulfate that diffracted up to 3.1 Å in XALOC beamline of ALBA Synchrotron (Spain). Data were processed using iMosflm (23) and CCP4 (24) and the phasing was done using a combined Molecular Replacement approach. In the first step Balbes server was used to place the β-propeller of ChYtm1 (PDB model:3PSL) (25). The resulting model, together with the PDB of the previously solved Erb1416–807 from yeast (PDB:4U7A), were used as an input for Phaser-MR as implemented in Phenix (26). Next, AutoBuild from Phenix was used to partially place residues corresponding to both propellers. The final model was completed by manual fitting of missing loops and regions of the UBL domain of ChYtm1. Cycles of phenix.refine and manual refinement in Coot (27) were performed to obtain the final structure with R and Rfree factors of 19.4% and 23.8%, respectively. Ramachandran plot was: 94% favored and 0.13% outliers.
ChErb1432–801-ChYtm1 dimer in the P21 21 2 space group
Orthorhombic crystals appeared in sitting drops that contained ChNop7/ChErb1/ChYtm1 trimer (35 mg/ml) that co-eluted from gel filtration in SE buffer. The trimer was purified by size exclusion chromatography. The best diffracting crystals were obtained in 20% PEG 8000 and 0.1 M Hepes pH 7.5 at 294 K. The crystals were cryo-protected by adding 10% ethylene glycol to the crystallization condition and flash-cooled in liquid nitrogen. The data were collected at 100 K at I04 beamline at DLS facility (UK) where automated data processing was performed by xia2 pipeline (28). The highest resolution data set (2.1 Å) was used for phasing with Phaser-MR as implemented in Phenix and the refined 3.1 Å structure served as the input model. The resulting model was refined and the position of UBL domain of ChYtm1 was manually corrected in Coot (27). The final structure was refined manually with Coot and using phenix.refine, until reaching the values of R and Rfree of 16.5% and 21.5%, respectively. Ramachandran statistics were: 96% favored and 0.25% outliers.
ChErb1[R486E]435–801-ChYtm1 dimer in the P65 2 2 space group
ChErb1[R486E]432–801 and ChYtm1 were purified separately and stored in SE Buffer. The proteins were mixed prior to the crystallization trials. Crystals grew in a drop containing 0.3 μl of ChErb1[R486E]432–801-ChYtm1 dimer at 6 mg/ml with 0.3 μl of 0.1 M Hepes pH 7.5 and 2 M ammonium sulfate. They were flash-cooled in liquid nitrogen and diffracted at 100 K at I02 Beamilne of DLS Synchrotron (UK). Acquired data at 3.0 Å were automatically processed and scaled with xia2. Phenix.refine was used for phasing and the P65 2 2 structure of ChErb1435–801/ChYtm1 was used as input. The resulting model was manually inspected with Coot and refined using phenix.refine. Final R and Rfree values were 20.2% and 26.2%, respectively. Ramachandran plot showed 94% of favored residues and 0.13% of outliers.
Computational analysis
Dali Server and iPBA server were used for similarity searches in the PDB (29,30). Sequence multi-alignments were done with JalView (31). Dimer stability and assembly analysis were performed by PISA server (32). Structure representations were generated by Chimera (33), which was also used for structure superimposition.
In vitro binding assays
Pull-down assays
100 μg of GST-ChYtm1 in Buffer SE were mixed with 50 μl Glutathione Sepharose 4B beads (GE Healthcare) equilibrated with the same buffer. After 15 min of incubation on ice, the beads were extensively washed with Buffer SE and equimolar amounts of ChErb1 (full-length, truncated or mutant) were added to the mixture and left on ice for additional 15 min. Free protein was removed in three wash steps (1 ml of Buffer SE each) and the bound fraction was eluted with reduced glutathione (Buffer GSH). Fractions corresponding to the input, final wash and elution steps were analyzed by SDS-PAGE.
Gel filtration
Equimolar amounts of 6xHis-ChYtm1 and 6xHis-ChErb1432–801 were mixed, incubated 30 min on ice and injected into a Superdex200 16/60 column equilibrated with Buffer SE. The fractions corresponding to the peaks were analyzed by SDS-PAGE. Analytical gel filtration was performed using a Superdex200 10/30 (GE Healthcare) column and Buffer SE for all the runs. 6xHis-ChYtm1 and His-tagged full-length mutants of ChErb1 were analyzed. The proteins were mixed in 1:1 ratio and incubated on ice for 15 min, then they were injected together into the column; collected fractions were analyzed by SDS-PAGE.
Interferometry
Biolayer interferometry system (BLItz, PALL) was used to calculate KD values for the interaction between ChYtm1 and ChErb1 or ChErb1432–801 versus ChErb1 mutants. A sample containing 40 ng/μl of GST-ChYtm1 in SE Buffer was loaded onto an Anti-GST Biosensor (ForteBio) during 180 s which was followed by 60 s of equilibration in Buffer SE. Next, association and dissociation steps (180 s each) were carried out using different concentrations of binding partners also diluted in SE Buffer. Curve fitting and KD calculation were done using the BLItz Pro 1.2 Software.
Isothermal titration calorimetry (ITC)
Nano ITC from TA Instruments was used to perform the measurements. The experiments were done at 20°C and consisted of 24 serial injections of 1.5 μl of 280 μM 6xHis-ChYtm1 into the cell compartment containing 170 μl of 23 μM 6xHis-ChErb1432–801. Both samples were diluted in Buffer SE. Generated heat was corrected by subtracting heat of dilution measured by injecting 280μM 6xHis-ChYtm1 into the cell compartment containing only 170μl of SE Buffer with the same experimental setup. Base-line correction was done with Nitpic; data fitting and binding kinetics calculation was performed with Sedphat (34,35). The graphs were generated with GUSSI.
Strains, plasmids and microbiological methods
BY4741 was used as the parental strain (36). Strain JDY1232 (BY4741 but erb1::kanMX4) is a haploid segregant of Y26184 (Euroscarf) that requires a plasmid-borne copy of ERB1 for cell viability. Growth and handling of yeast and preparation of standard media were prepared by established procedures (37). To generate YCplac111-ERB1 and YCplac33-ERB1, a 4150 kb PstI/BamHI fragment containing the ERB1 ORF and 1000 bp up- and downstream the ATG and stop codons, respectively, was amplified from genomic DNA of the BY4741 strain and cloned into the PstI/BamHI-restricted vectors YCplac111 and YCplac33 (38). Both constructs apparently complemented the erb1 null allele to the wild-type extent. The mutant erb1[R470E] was generated by site-directed mutagenesis in YCplac111-ERB1 with appropriate primers and confirmed by sequencing. This construct and YCplac111-ERB1 were transformed into the shuffle strain JDY1232 YCplac33-ERB1 and subsequently the YCplac33-ERB1 was counter-selected on plates containing 5-fluoroorotic acid (5-FOA).
Sucrose gradient centrifugation
Cell extracts for polysome and r-subunit studies were prepared and analyzed as previously described using an ISCO UA-6 system equipped to continuously monitor A254 (39). For r-subunit quantifications, low-Mg2+ gradients were used and the profiles were recorded on chart paper. Peaks corresponding to the 40S and 60S ribosomal subunits were carefully cut out and weighed. Then, the 60S/40S ratio was calculated for each sample. At least three independent replicas were done for each strain or condition. When needed, fractions of 0.5 ml were collected from the gradients and proteins were extracted as exactly described (40), and analyzed by western blot analyses. Polyclonal antibodies against Erb1, Nop7 and Ytm1 were used as primary antibodies. These antibodies were raised by immunizing rabbits with purified proteins at the facilities of the Service for Animal Production (University of Seville, Spain). Polyclonal antibodies against L1 (a gift from F. Lacroute (41)) were used to define the position of LSU in the gradients. As a loading control for whole cell extracts, monoclonal anti-Pgk1 antibodies (Invitrogen) were used.
RNA analyses
RNA extraction, northern blot hybridization and primer extension analyses were carried out according to standard procedures (42). In all experiments, RNA was extracted from samples corresponding to 10 OD600 units of exponentially grown cells. Equal amounts of total RNA (5 μg) were loaded on gels or used for primer extension reactions. Specific probes, whose sequences are listed in Supplementary Table S1, were 5′-end labelled with [γ32-P] ATP. Signal intensities were quantified using a FLA-5100 imaging system and Image Gauge (Fujifilm).
RESULTS
Crystal structure of ChYtm1-ChErb1432–801 dimer
To study the structure of the Nop7-Erb1-Ytm1 sub-complex from C. thermophilum, we first purified ChYtm1 and ChErb1 and performed crystallization trials with both proteins and after a successful expression and purification of ChNop7, it was included in the in vitro reconstitution assays and crystallization trials. Crystals appeared for both, dimer- and trimer-containing, crystallization screenings and the highest resolution achieved was of 3.1 Å and 2.1 Å, respectively (Supplementary Table S2). Solved structures revealed that the asymmetric unit content was identical in both cases although the space group and unit cell parameters differed. Crystals contained a dimer formed by ChYtm1 and the C-terminal β-propeller domain of ChErb1 (Figure 1A) but the relative orientation of some elements was different among the space groups. The C-terminal domain of ChErb1 superposed well with the previously solved β-propeller of Erb1 from yeast (RMSD 0.98 Å). Interestingly, the large positively charged area described for the C-terminal domain of S. cerevisiae Erb1, which had been shown to bind RNA in vitro, is well preserved in the C-terminal domain of ChErb1 (20).
Figure 1.
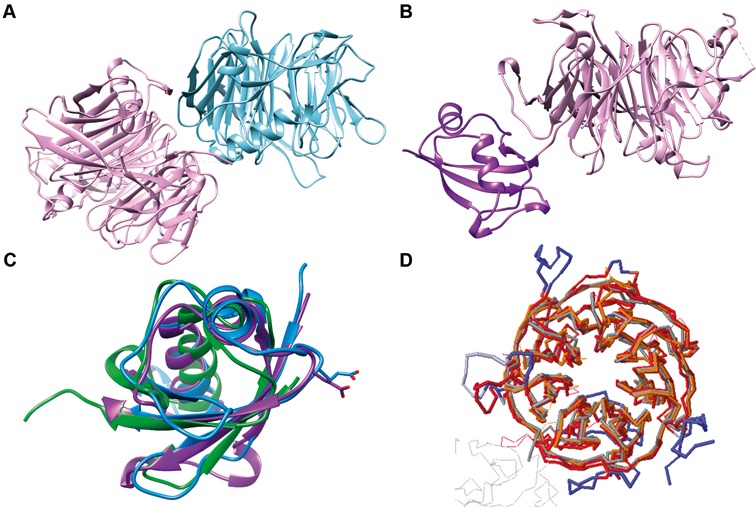
Structural overview of the dimer formed between ChYtm1 and the β-propeller of ChErb1. (A) Global view of the asymmetric unit content. The β-propeller of ChErb1 is shown in blue and ChYtm1 protein is depicted in pink. (B) ChYtm1 protein folds into two separate domains. The amino-terminal region contains 97 residues that form an ubiquitin-like (UBL) domain (in purple), which is attached to the base of a large seven-bladed β-propeller domain (pink). (C) Superimposition of the structures of UBL of ChYtm1 (purple) with ubiquitin (PDB:UBQ1, green) and UBL of ChRsa4 (PDB:4WJS, blue) shows that the ubiquitin-like fold is well preserved. Side chains of the glutamic acid involved in binding to the MIDAS domain of Rea1 are shown for ChYtm1 and ChRsa4. (D) The three most similar structures from PDB (as calculated by Dali Server) were superimposed to the one of ChYtm1100–487. (Chains were colored as follows: 4d6v:A-dark red, 2ymu:A-gray, 4esg:A-orange, ChYtm1-red). Additional segments of ChYtm1 that do not appear in canonical β-propeller fold are represented in blue.
ChYtm1 contains an N-terminal ubiquitin-like domain followed by a seven-bladed β-propeller
Crystals contained full-length ChYtm1; we could trace nearly the entire polypeptide (residues 12–487) and only two disordered loops were not visible in the electron density (274–282 and 379–381) (Supplementary Figure S1). The first 97 residues folded into an UBL domain and the rest of the protein into a seven-bladed β-propeller domain (Figure 1B). The UBL is formed by two short α-helices and four antiparallel β-strands. It superimposes well with ubiquitin (RMSD: 1.2 Å) and with the N-terminal domain of C. thermophilum Rsa4 (ChRsa4) (RMSD: 1.8 Å), a protein than in yeast has been shown to participate in subsequent steps during LSU maturation (Figure 1C) (43). Strikingly, only few residues are fully conserved between the ChYtm1 and ChRsa4 proteins, mainly those that maintain the hydrophobic core of UBL. Similarly to ChRsa4, the N-terminal part of ChYtm1 contains an exposed loop with a conserved glutamic acid (E88) that mediates interaction with the MIDAS domain of the Rea1 ATPase. This structural similarity between both UBLs confirms previous functional studies that proposed a similar role of the N-terminal region from Ytm1 and Rsa4 in yeast ribosome biogenesis. Strikingly, the N-terminal domain of ChYtm1 (Figure 1B) is found in a different orientation between both crystal forms we could solve, indicating a certain degree of flexibility for this domain (Supplementary Figure S2A). A similar behavior had been previously observed for the UBL of ChRsa4 (43).
The C-terminal domain of ChYtm1 is formed by seven WD repeats that arrange in a tight propeller structure. Although the core of the domain highly resembles other 7-bladed β-propellers, the main difference can be attributed to the loops between β-strands that, in several regions, fold into additional secondary structures (Figure 1D). Noticeably, some of these extended loops seem to be specific for the C. thermophilum protein and might be correlated with the thermal stability of the protein. In the bottom part of the domain, arising from the first blade appears a short α-helix (residues 125–131) that partially occludes the central aperture. A long non-conserved loop between WD repeats 6 and 7 contains a β-strand that becomes the fifth strand of blade 5. Blade 2 is also built by 5 and not 4 β-strands, but in this case the strand ‘e’ is conserved in higher eukaryotes. The last extended loop connects blade 6 and 7 (residues 444–461) and forms a protrusion on the upper face of the propeller forming a knob-like structure that appears in the same position as previously described for the RACK1 protein (44). When we analyzed the conserved patches on the surface of the propeller, it was clearly noticeable that the most invariable area corresponded to the ChErb1-interacting face.
ChYtm1 interacts with the β-propeller domain of ChErb1
The unexpected disposition of the full-length ChYtm1 bound to the C-terminal domain of ChErb1 in the asymmetric unit led us to further investigate the possibility of a direct interaction between the β-propellers of both proteins. Since previously published data suggested that the binding of Ytm1 to Erb1 in yeast did not involve the β-propeller of Erb1, but instead its N-terminal conserved region (11), we first computationally checked whether the dimer we have solved was stable in solution. PISA server confirmed that the complex formation was energetically favorable, being the buried area between both monomers 2458 Å2 (32). Next, we performed in vitro binding assays using ChYtm1 and truncated ChErb1 containing the residues 432–801 (ChErb1432–801). As a result, both proteins co-eluted in gel filtration and 6xHis-tagged ChErb1432–801 co-purified with GST-tagged ChYtm1 on Glutathione Sepharose beads, thus clearly indicating that the β-propeller of ChErb1 was stably associating with ChYtm1 (Figure 2A). In order to quantify the strength of this interaction, we calculated the KD using biolayer interferometry. Both, ChErb1 and ChErb1432–801 strongly associated with GST-ChYtm1 immobilized on an Anti-GST biosensor (Figure 2B). Comparable KD of 4 × 10−9 M and 2.7 × 10−9 M were obtained for ChErb1 and ChErb1432–801, respectively, indicating that the N-terminal portion of ChErb1 was dispensable for the stable association to occur. The affinity of the interaction between ChYtm1 and the β-propeller of ChErb1 was also confirmed by ITC (calculated KD∼9 nM) (Figure 2C). Altogether, these results indicate that ChYtm1 and ChErb1 interact with each other through their respective β-propeller domains.
Figure 2.
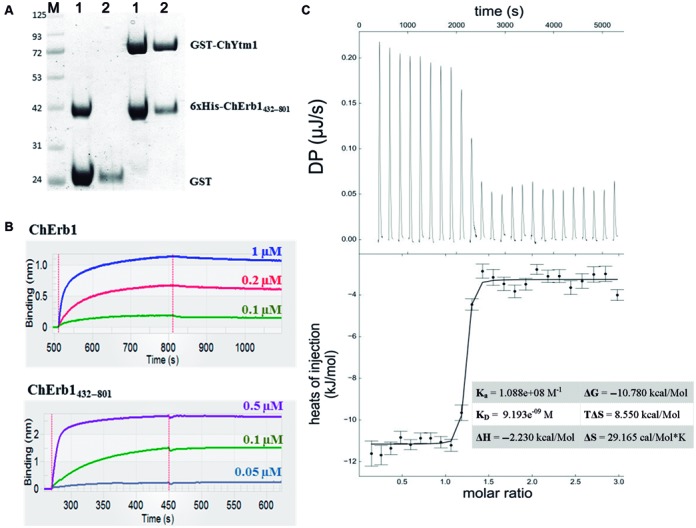
Analysis of the interaction between ChYtm1 and the β-propeller of ChErb1. (A) Pull-down experiment showed that 6xHis-tagged ChErb1432–801 co-purified with GST-tagged ChYtm1 but not with GST alone on Glutathione Sepharose beads. M: Molecular weight marker, 1: Input, 2: Elution. (B) Biolayer Interferometry graphs show association and dissociation steps using different concentrations of full-length ChErb1 (top) or ChErb1432–801 (bottom) to GST-ChYtm1 immobilized on the Anti-GST biosensors. (C) ITC was done to validate high-affinity binding between ChYtm1 and ChErb1432–801. Raw heat of each injection is shown in the upper panel. Curve fitting confirmed that the binding affinity was in low nanomolar range (KD ∼9 nM). Calculated thermodynamic parameters for the ITC experiment are shown in the table.
Structural insights into the dimer formation of ChYtm1-ChErb1432–801
As calculated by PISA Server, there are 18 hydrogen bonds and 11 salt bridges across the ChYtm1–ChErb1 dimer interface. Manual inspection of the model confirmed that the dimer was mainly maintained by electrostatic forces although several hydrophobic regions were also involved in the interaction. ChYtm1 uses the extensive top face of its β-propeller to establish contacts with different areas at the bottom part and the circumference of the C-terminal β-propeller of ChErb1 (Figure 3A). The manner in which both propellers interact has not been previously observed for a WD40–WD40 assembly as the axis of one of the domains is tilted 55° with respect to the other (Figure 3B).
Figure 3.
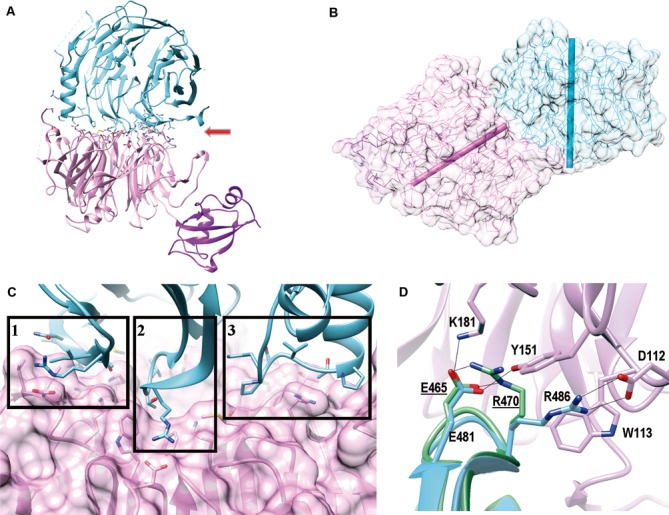
The β-propellers of ChErb1 and ChYtm1 interact in a novel manner. (A) Ribbon representation of the dimer shows that ChYtm1 (pink) binds to the C-terminal domain of ChErb1 (blue) through the top surface of the β-propeller (red arrow). UBL domain of ChYtm1 (purple) does not participate in the interaction. Side chains of interacting residues are shown. (B) The central axes of both β-propellers (shown as bars, ChYtm1:pink; ChErb1: blue) form an angle of 55°. (C) Three areas of ChErb1432–801 (blue) contact the β-propeller of ChYtm1 (pink): (i) Strand 1d from the blade 7 establishes non-conserved interactions with the knob that appears between blades 6 and 7 of ChYtm1. (ii) A well conserved loop ‘c–d’ from blade 1 of ChErb1 binds to the central channel of ChYtm1. (iii) The insertion from blade 2 of ChErb1 mediates binding to an extended loop from ChYtm1 on one side of the propeller. (D) Superimposition of the β-propeller of Erb1 alone (PDB: 4U7A, in green) shows that upon binding to ChYtm1 (pink) the interacting residues from ChErb1 (blue) reorganize and force R486 of ChErb1 toward salt-bridge formation with D112 of ChYtm1. Electrostatic interactions are shown as black lines. Labels corresponding to the residues of Erb1 from yeast are underlined.
The first interacting region includes the last β-strand (‘1d’) of blade 7 in ChErb1432–801 that contacts loop ‘6d-6a’ and a long extension that appears between strands ‘7d’ and ‘7a’ of ChYtm1 (the knob formed by residues 444–460) (1 in Figure 3C). Since this extended loop is not conserved in other Fungi or higher eukaryotes, it is possible that the way both proteins interact at this point is specific for the dimer present in C. thermophilum. Blade 7 of ChErb1 is also engaged in another contact through its ‘7a-7b’ loop, which contains E785 that forms salt bridge with fully conserved H320 from ChYtm1. Secondly, the entrance of the central tunnel of ChYtm1 serves as place of docking for a loop between strands ‘1c-2d’ from the first blade of ChErb1432–801 (2 in Figure 3C). The loop contains three well conserved residues, E481, T484 and R486, which establish a network of electrostatic interactions with also conserved amino acids from blades 1, 2, 3 and 7 of ChYtm1. A salt bridge is formed between R486 of ChErb1 and D112 from ChYtm1. Two additional residues of ChYtm1, W113 and Y151, ensure the proper orientation of R486 side chain thus promoting electrostatic contacts. Interaction with ChYtm1 actually induces a conformational reorganization that allows the salt bridge formation because in the structure of the C-terminal part of yeast Erb1 alone, the residue corresponding to R486 in ChErb1 (R470 in Erb1 from S. cerevisiae) is coordinated by E481 (E465 in Erb1 from S. cerevisiae). Upon binding, E481 forms hydrogen bonds with K181 and Y151 from ChYtm1 and the side chain of R486 is displaced toward D112 (Figure 3D).
At last, the crystal structure we have solved showed the importance of the insertion that appeared within the second blade of the β-propeller of Erb1 (20). We observed that in the ChYtm1-ChErb1432–801 dimer, this region provided additional surface of interaction because it made important contacts with the loops from blade 2 and 3 of ChYtm1 and with another long extension, between strands ‘3c’ and ‘4d’ that projects out of the plane formed by the top surface of ChYtm1, thus increasing the dimerization area (3 in Figure 3C). In conclusion, the central part of the propeller of ChYtm1 provides a large docking surface for the bottom face of blades 1, 2 and 7 from the β-propeller of ChErb1 that is additionally held in place by two lateral extensions from ChYtm1.
A salt bridge between R486 of ChErb1 and D112 of ChYtm1 stabilizes the dimer
In order to validate the structural information provided by our model, we address whether the β-propeller of ChErb1 plays a crucial role in the binding to ChYtm1. We first analyzed the conservation of the interfaces that were engaged in dimer formation and we found an area that was fully conserved in both proteins (Figure 4A). We then designed a set of ChErb1 point mutations that altered the most conserved residues from the central area of interaction (Figure 4B). Those variants that expressed in a soluble form were purified and assayed for their binding to ChYtm1 (Figure 4C). To this extend, we checked whether any mutant of ChErb1 would lose its ability to co-elute with ChYtm1 in gel filtration indicating lack of binding or, at least, an important decrease in its affinity (column SE in Figure 4C). As a result, only one of the tested mutant proteins, ChErb1[R486E], did not elute together with ChYtm1 in size exclusion column. Next, we carried out a pull-down experiment to assess if this point mutation was completely disrupting the binding, however, we observed that there was still a detectable amount of 6xHis-ChErb1[R486E] co-purifying with GST-ChYtm1 on Sepharose Glutathione beads. This result suggested that both proteins were still associating but less stably as in the case of wild-type ChErb1 and ChYtm1. Indeed, when we quantified the decrease of affinity produced by the mutant, the KD measured by interferometry using ChYtm1 with full-length ChErb1[R486E] or ChErb1[R486E]432–801 resulted to be two orders of magnitude lower than that previously calculated for the ChErb1-ChYtm1 interaction (Figure 4C). Interestingly, disruption of the salt bridge by the R486A mutation was not sufficient to weaken the binding (KD∼7 nM) and only the electrostatic repulsion induced by the R486E change had an observable effect on the in vitro interaction between ChErb1 and ChYtm1 (Figure 4C).
Figure 4.
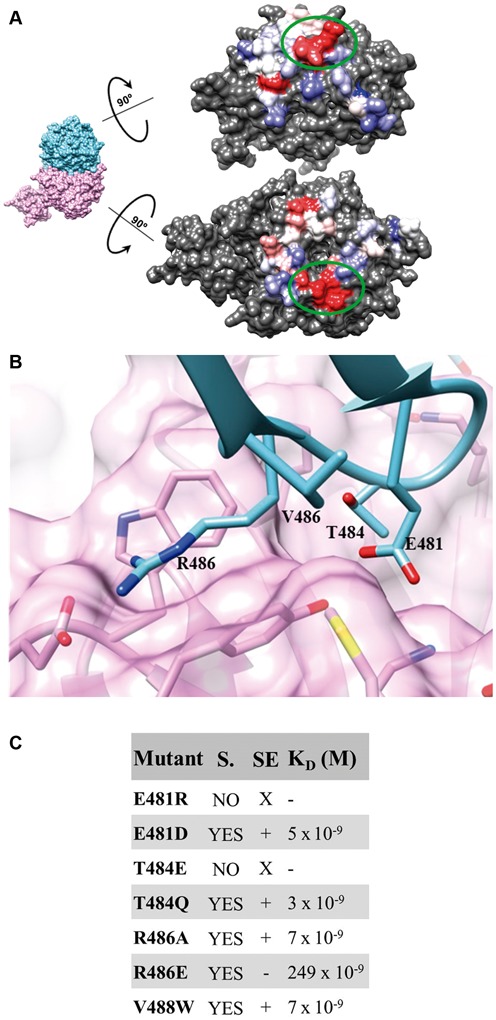
Contacts between the β-propellers from ChErb1 and ChYtm1. (A) The colored surfaces of the C-terminal part of ChErb1 (top) and ChYtm1 (bottom) show the area that mediates the binding between both molecules. The most variable residues are shown in blue and the most conserved ones are depicted in red. Green circles mark the region that was chosen for mutant generation (corresponding to Box 1 in Figure 3C). (B) Detailed view of the most conserved interface of interaction that has been modified in order to alter the binding. The amino acids that have been mutated are labeled. (C) Table showing mutations done in ChErb1. Soluble variants (YES in column S.) were injected with ChYtm1 in gel filtration (SE column: X: not checked, +: bound, −: no binding) and interaction affinity was measured by interferometry (column KD).
The β-propeller architecture is preserved in the ChErb1[R486E] mutant protein
To further investigate the impact of the R486E mutation on the binding between ChErb1 and ChYtm1, we address whether the decreased affinity was not a side effect of an aberrant β-propeller folding or a reduced stability in vitro of the ChErb1[R486E] protein but the result of the direct repulsion between E486 residue from ChErb1 and D112 from ChYtm1. Using Thermofluor, we confirmed that the thermal stability of the truncated mutated ChErb1[R486E]432–801 protein was not compromised in vitro and its melting temperature was comparable to the one of wild-type ChErb1432–801 (data not shown). Next, we performed crystallization trials with ChErb1[R486E]432–801 and ChYtm1 that yielded crystals, which let us solve a 3.0 Å structure of the dimer. The analysis of this structure confirmed that the overall architecture of the C-terminal domain of Erb1 was not altered in the context of the mutation (Supplementary Table S2). The β-propeller domains of wild-type ChErb1 and ChErb1[R486E] superposed with RMSD of 0.48 Å. As expected, both proteins still interacted in vitro at concentrations used in the crystallization drop and the electron density for E486 from ChErb1 clearly showed lack of coordination with D112 from ChYtm1 (Supplementary Figure S2B).
Altered binding of Ytm1 and Erb1 is of functional importance in vivo
In S. cerevisiae, it has been shown that the in vivo interaction of Ytm1 and Erb1 is essential for ribosome biogenesis (10). Therefore, to study the biological relevance of the salt bridge between R486 from ChErb1 and D112 from ChYtm1, we wonder whether the weakened binding produced by R486E mutation in the ChErb1 protein would have an effect in its S. cerevisiae counterpart. Since the residues forming the above mentioned salt bridge are fully conserved in all species, we mutated the equivalent arginine in the Erb1 protein from yeast (R470) and assessed its impact on growth and ribosome production.
Given the fact that Erb1 is essential (8), we made use of an erb1 null strain containing a plasmid-borne functional copy of ERB1. Then, to determine whether the R470 plays a role in the in vivo function of Erb1, we generated a YCplac111 plasmid-borne erb1[R470E] mutant allele by site-directed mutagenesis. This plasmid was introduced into the JDY12323 [YCplac33–ERB1] strain, which apparently grows as a wild-type strain (data not shown), and several transformants were subjected to 5-FOA plasmid shuffling. As a control, a wild-type ERB1 gene was also cloned in YCplac111 and used. As shown in Figure 5A, the erb1[R470E] allele affected significantly growth in a temperature-dependent manner. Thus, while growth rate is mildly reduced at 30°C, it is severely affected at 37°C.
Figure 5.
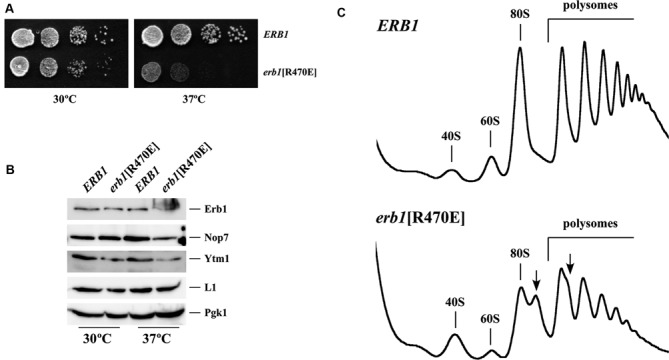
(A) The erb1[R470E] mutation confers slow growth and temperature sensitivity. A null erb1 strain containing YCplac111 plasmid-borne wild-type ERB1 or the erb1[R470E] allele were grown in liquid YPD, diluted to an OD600 of 0.05 and spotted in 10-fold serial dilutions onto YPD plates. Plates were incubated at 30°C and 37°C for 3 days. (B) The amounts of Erb1[R470E], Nop7 and Ytm1 proteins are reduced in the erb1[R470E] mutant. Whole cell extracts were prepared from the indicated strains, growing in exponential phase at 30°C and 37°C in YPD. Equivalent amounts of extracts were analyzed by western blotting using antibodies against Erb1, Nop7, Ytm1, L1 and Pgk1. (C) The erb1[R470E] mutation results in a deficit in 60S r-subunits. The indicated strains were grown in YPD at 37°C to exponential phase. Cell extracts were prepared and 10 A260 of each extract was resolved on 7–50% sucrose gradients. The A254 was continuously measured. Sedimentation is from left to right. The peaks of free 40S, 60S r-subunits, 80S vacant ribosomes/monosomes and polysomes are indicated. Half-mers are labeled by arrows.
To test whether this growth defect was due to altered expression or decreased stability of the Erb1[R470E] protein, we determined the steady-state levels of Erb1 in the mutant strain and its wild-type counterpart by western blot analysis. As shown in Figure 5B, reduced levels of Erb1[R470E] protein compared to those of wild-type Erb1 were detected when mutant and wild-type cells were grown at 37°C. A diffuse smear was reproducibly migrating at the top of the full Erb1[R470E] protein. The significance of this smear is still unclear but it could be presumably due to a modification or aggregation of the protein. Interestingly, the levels of Nop7 and Ytm1 proteins were also reduced in the erb1[R470E] mutant strain. We conclude that the erb1[R470E] mutation leads to decreased levels of the heterotrimer Erb1-Nop7-Ytm1 complex at 37°C.
The erb1[R470E] mutation leads to a deficiency in 60S ribosomal subunits
To determine the effect of reduced affinity between Ytm1 and Erb1 on ribosome biogenesis, we assayed steady-state levels of r-subunits and polysomes in erb1[R470E] cells grown at either 30°C or 37°C. Compared to wild-type ERB1 cells, erb1[R470E] mutant cells showed a strong decrease in the levels of free LSUs, an increase in the levels of free SSUs, an overall decrease in the 80S peak and in polysomes. Most importantly, there was an accumulation of half-mer polysomes only in erb1[R470E] cells. These defects were more pronounced when cells were grown at 37°C (Figure 5C). Taking into account the role of Erb1 in ribosome biogenesis, these results strongly suggest that the erb1[R470E] mutation severely impairs LSU biogenesis at high temperatures. Consistently, quantification of total r-subunits showed a 35% reduction in the overall quantity of LSUs in the erb1[R470E] mutant relative to the wild-type strain at 37°C (A254 60S/40S ratio of about 2.2 in the wild-type strain versus of 1.4 in the erb1[R470E] mutant).
Pre-rRNA processing is impaired in the erb1[R470E] mutant
To further study the role of the erb1[R470E] mutation in the biogenesis of LSUs, we assessed the steady-state levels of pre- and mature rRNA by northern blot hybridization and primer extension analyses in the erb1[R470E] strain and compared them to those in the isogenic wild-type strain. Both strains were grown at either 30°C or 37°C in liquid YPD medium to mid-log phase, harvested and total RNA was extracted and analyzed. As shown in Figure 6A, Northern blot analysis revealed mild differences in the levels of pre- and mature rRNAs in wild-type and erb1[R470E] cells grown at either 30°C or 37°C. A decrease in levels of mature 25S rRNA is appreciable and consistent with the role of Erb1 in LSU biogenesis (Figure 6A). More interestingly, primer extension analyses showed a clear accumulation of the 27SA3 pre-rRNA and a reduction of the immediate downstream 27SBS pre-rRNA in the erb1[R470E] cells grown at 37°C (Figure 6B). Taken together, these results indicated that the erb1[R470E] mutation had a major impact on processing of 27SA3 pre-rRNA, which had been previously shown as the most prominent defect upon the loss-of-function of any member of the so-called group of A3 factors (11,13,16) and/or upon a mutation in the YTM1 gene that negatively affects the interaction of Ytm1 with Erb1 (10).
Figure 6.
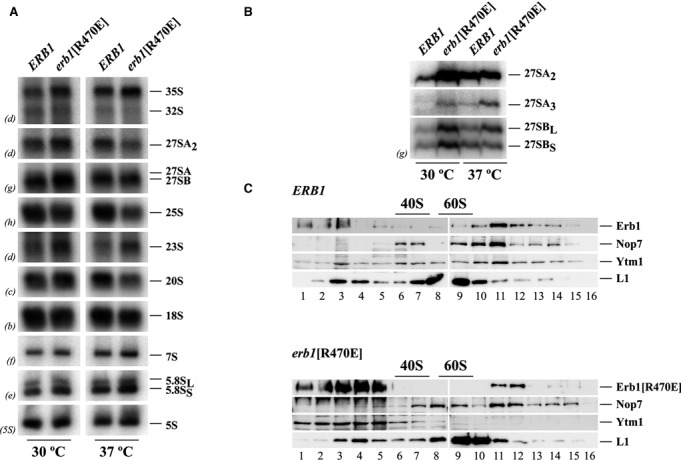
The erb1[R470E] mutation impairs 27SA3 pre-rRNA processing. The indicated strains were grown in YPD at 30°C and 37°C to exponential phase. Total RNA was extracted and subjected to northern hybridization or primer extension. Probes (in parenthesis) are described in Supplementary Table S1. (A) Northern analysis of high- and low-molecular pre- and mature rRNAs (B) Primer extension analysis of 27S pre-rRNAs, Probe g within ITS2 was used. (C) Interaction of Ytm1 with pre-60S ribosomal particles is significantly impaired in the erb1[R470E] mutant. Total cell extracts were resolved on 7–50% sucrose gradients containing a low concentration of Mg2+ to dissociate ribosomes into subunits. Sedimentation is shown from left to right. The sedimentation positions of 40S and 60S ribosomal subunits are indicated. Fractions (numbered 1–16 under the panels) were collected from the gradients; proteins were extracted from the same volume of each fraction and subjected to western blot analysis with specific antibodies detecting Erb1, Nop7, Ytm1 and L1 proteins.
Association of Ytm1 with pre-60S ribosomal particles is drastically reduced in the erb1[R470E] mutant
The erb1[R470E] mutation is expected to impair binding of the β-propeller of Erb1 to Ytm1, we therefore analyzed the sedimentation behavior of wild-type and mutant Erb1 proteins in sucrose gradients. Since both Ytm1 and Nop7 bind directly to Erb1, we also tested the distribution of these two proteins, and thus the integrity of the Erb1-Nop7-Ytm1 in the gradients. For this purpose, total extracts were prepared from wild-type and erb1[R470E] cells grown in YPD at 37°C and subjected to low Mg2+ sucrose gradient ultracentrifugation. Then, fractions were collected and analyzed by western blot. As shown in Figure 6C, a peak of wild-type Erb1 was found associated with high-molecular-mass complexes that sediment slightly lower than mature 60S r-subunits. These complexes most likely represent pre-60S r-particles. Consistent with the association of the Erb1, Nop7 and Ytm1 together as a heterotrimer within pre-60S r-particles, we found that Nop7 and Ytm1 also co-sediment with these particles in wild-type cells (Figure 6C, top, lanes 10–11). Moreover, practically no signal could be detected sedimenting at the top of the gradient for none of the proteins, suggesting that the bulk of the Erb1-Nop7-Ytm1 heterotrimer is stably associated with pre-60S r-particles while only few is present as a free sub-complex (Figure 6C, top, lanes 1–5). In contrast, when we examined the distribution of Erb1[R470E] in sucrose gradients, less protein was detected in high-molecular-mass fractions compared to wild-type Erb1, however, a large amount of Erb1[R470E], mainly as an smeared signal, was present at the top of the gradient (Figure 6C, bottom, lane 1–5). The distribution of Nop7 did apparently not change in the mutant cells but, remarkably, practically no Ytm1 was found associated with pre-60S r-particles.
Altogether, these findings strongly suggest that while Nop7 and, to a lesser extent, Erb1[R470E] are still able to bind pre-60S r-particles in the erb1[R470E] cells, Ytm1 has lost its ability to stably associate with these particles, likely as the consequence of its decrease affinity for the Erb1[R470E] protein. We conclude that Erb1 employs its β-propeller to stably recruit Ytm1 into pre-60S r-particles.
DISCUSSION
In yeast and mammals, a nucleolar sub-complex (called the PeBoW complex in mammals) formed by the conserved Nop7 (Pes1), Erb1 (Bop1) and Ytm1 (WDR12) proteins is essential for cell proliferation, pre-rRNA processing and LSU biogenesis (8,10,17–19). Additional roles for the proteins of the trimer have been described in DNA replication (45) and chromosome stability (46), however, the precise function of the complex in all these processes remains unknown. It has been shown that it is associated with pre-ribosomal particles and it can be stably separated from the particles by different means (10,17,45,47).
We are interested in obtaining structural insights into the architecture of the Nop7-Erb1-Ytm1 sub-complex. We now have been able to crystallize a dimer formed by practically full-length ChYtm1 and the C-terminal part of ChErb1 from C. thermophilum. In both crystal forms we have obtained, Ytm1 consists of an N-terminal UBL domain followed by a C-terminal β-propeller, which interacts with the β-propeller of ChErb1. Similarly to the structure we have reported for the yeast Erb1 (20), ChErb1 also suffered a proteolytic cleavage in its central part, thus the obtained crystals lacked the N-terminal domain of the protein. Additionally, we could observe that the relative orientation of the N-terminal UBL domain of ChYtm1 varies from one type of crystal to another. This conformational shift could be a side effect induced by crystal formation but, in any case, it demonstrates that this domain presents certain flexibility that could be important for its function. A similar observation has been made by Bassler et al. (43) for the crystal structure of Rsa4. As Ytm1, Rsa4 also consists of an N-terminal UBL domain followed by a C-terminal β-propeller. These authors have shown that the UBL domain of both, Rsa4 and Ytm1, contacts the MIDAS domain of Rea1 ATPase and suggested that the energy from hydrolyzed ATP could be transformed into mechanical force through those flexible UBL domains.
Our structure demonstrates that the C-terminal β-propeller of ChYtm1 stably binds to an extensive conserved surface in the C-terminal β-propeller region of ChErb1. There are few known structures that describe interacting WD40 domains, like the one of the τ60–τ91 complex where two propellers bind perpendicularly to each other (48). In the dimer we have solved, both β-propellers orientate in a manner that had not been previously described because the central axes of the domains form an angle of 55°. Whereas the top face area of ChYtm1 is known as a ‘supersite’ of the β-propellers since it is commonly employed in binding to other molecules (49), the portion of ChErb1 involved in the interaction is more unusual. It includes residues from the bottom face of the propeller and others that belong to the circumference. This way, the edge of the domain is inserted into the central aperture of ChYtm1. Additional loops on the sides of ChYtm1 further strengthen the dimer by providing supplementary clamp-like interactions. It has been postulated that interacting β-propellers can act as scaffold platform indispensable for the assembly of larger macromolecular complexes (50). The dimer ChYtm1-ChErb1432–801 is a good example of such a platform since it would elegantly connect Nop7 bound to the N-terminal region of Erb1 with the mechanochemical force produced by Rea1 within the context of pre-60S r-particles. In contrast to our structural results, previous studies have suggested that the β-propeller domain of Erb1 is not required for the formation of the Nop7-Erb1-Ytm1 sub-complex, thus, the N-terminal part of Erb1 interacts with both Nop7 and Ytm1 (11). However, the high-affinity binding that leads to ChErb1-ChYtm1 crystal formation does not seem to depend on the N-terminal portion of ChErb1 although it is still possible that there is an additional area of interaction that cannot be seen in the crystal structure. Still, the fact that we used the full-length ChErb1 in the study but only the C-terminal part of ChErb1 crystallized with ChYtm1 strongly supports the idea that any other kind of interaction between both proteins would be very weak and/or transient. Our model is supported by different binding assays techniques such as co-elution by gel filtration, pull-down experiments, interferometry and ITC. This is further sustained by mutational analysis of the interface involved in the interaction between the β-propellers of the two proteins. Our results proved that a point mutation on the surface of the β-propeller of ChErb1 results in a two-orders of magnitude decrease in affinity with ChYtm1, even in the context of full-length ChErb1. This mutation, R486E for ChErb1 and R470E for yeast Erb1, disrupts a salt bridge that is formed between the D112 residue of ChYtm1 and R486 of ChErb1 (E104 and R470 in S. cerevisiae, respectively). We have further demonstrated that the weaker binding between ChYtm1 and ChErb1[R486E] is not the consequence of an aberrant folding or diminished stability of the β-propeller domain in the ChErb1[R486E] protein since the crystal structure of the ChYtm1-ChErb1[R486E]432–801 dimer clearly shows that the seven-bladed architecture is perfectly preserved in both, Erb1 and Ytm1. This is a significant observation because the central area of the interaction includes the conserved WE di-peptide from the first WD repeat which may be relevant for the folding of the β-propellers. Its importance has been confirmed by the fact that E481R mutant failed to express in a soluble form so any modification of ‘1c-2d’ loop (residues 481–486) of ChErb1 could yield unfolded and insoluble protein (as also observed for the T484E mutant). The ChErb1-ChYtm1 dimer structure also sheds light on the role of evolutionary conserved insertion that had been previously described in the β-propeller of Erb1 from yeast (20). In many WD40 domains, these extra segments or extended loops have been shown to bind different ligands (51). This is the case of Erb1 because the fragment between H528 and A534 of ChErb1 maintains a network of electrostatic interactions with ChYtm1 and provides additional area of association between both proteins thus increasing binding specificity and affinity.
We have studied whether the observations derived from the ChErb1-ChYtm1 structure have in vivo relevance in S. cerevisiae. Expression of the orthologous protein Erb1[R470E] as the sole source of Erb1 impaired cell growth and ribosome biogenesis in yeast, especially at high temperatures. Most importantly, the mutant protein associates slightly worse with pre-60S r-particles and drastically impedes the interaction of Ytm1 with these particles. However, we cannot rigorously exclude that the weakened binding of Ytm1 and Erb1 as a consequence of the R486E mutation in vivo, enhances their mutual dissociation as soon as whole cell extracts are performed. Similar results have been reported for the temperature-sensitive yeast ytm1–1 mutant by the Woolford lab (10). The ytm1–1 mutations consists in the G398D and S442N substitutions (10). These residues correspond to G411 and S475 in C. thermophilum, respectively. As discussed, the Ytm1–1 protein did not stably associate with Erb1 in vitro and affected the stability of the Nop7-Erb1-Ytm1 sub-complex in vivo (10). In the dimer structure we have solved, the G411 and S475 residues from ChYtm1 are not directly involved in the binding to the C-terminal β-propellers of ChErb1 but they are located close to the interface between blades 6 and 7 of ChYtm1 that interacts with blade 7 of ChErb1. Moreover, S475 contributes to the blade stability through a network of hydrogen bonds with neighboring residues. Thus, it is conceivable that the mutations G398D and S442N in yeast Ytm1 affect the proper folding of blades 6 and 7 of Ytm1, thus perturbing the area of interaction with Erb1. Altogether, these results indicate that the binding of Erb1 with Ytm1 is not strictly required for the interaction of either Nop7 or Erb1 with pre-60S r-particles and that Ytm1 could be recruited in a second step to the pre-ribosome. Supporting this model, the study of the electrostatic surface of the β-propeller of ChErb1 shows a large positively charged area, which we have previously suggested for the yeast Erb1 to be involved in RNA binding (20). This region maps on the side of the β-propeller that is opposite to the interface of interaction with ChYtm1. Thus, these findings suggest that the C-terminal domain of Erb1 could actually be a platform for binding of other proteins and RNA (Supplementary Figure S3).
In conclusion, our data describe a novel way of a high affinity protein–protein recognition that employs two β-propellers. The structural characterization of such an interaction and the consequences of its disruption provide a good outset for the study of the effects of directed ribosome biogenesis impairment. It is of special interest to emphasize that rapid proliferation depends on high rates of ribosome production (52). In this context the PeBoW complex is required for cell proliferation (17). Moreover, Pes1, Bop1 and/or WDR12 has been found to be upregulated by c-Myc (53), Bop1 is over-expressed in certain cancers and its up-regulation has been correlated to the tumorigenesis of colorectal and hepatic cancers (46,54). Therefore, the interaction of Erb1 with its binding partners may be a good target to inhibit cell growth and proliferation.
ACCESION CODES
Coordinates and structure factors for ChErb1432–801-ChYtm1 were deposited in the PDB under 5CXB (P 21 21 2 space group) and 5CXC (P 65 2 2) codes. Coordinates and structure factors of ChErb1[R486E]432–801-ChYtm1 were deposited under 5CYK code.
Supplementary Material
Acknowledgments
Part of the X-ray data collection experiments was performed at XALOC beamline at ALBA Synchrotron with the collaboration of ALBA staff. The authors would like to thank Diamond Light Source for beamtime (proposals mx8035 and mx10130), and the staff of beamlines I02, I04 for assistance with crystal testing and data collection.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Spanish Ministry of Economy and Competitiveness [SAF2012–31405]; Generalitat Valenciana [PROMETEO/2012/061 to J.B.]; Spanish Ministry of Economy and Competitiveness and ERDF [BFU2013–42958-P]; Andalusian Government [BIO-271 to J.d.l.C.]; European Community's Seventh Framework Programme [FP7/2007–2013] under BioStruct-X (grant agreement N°283570). M.W. received a JAE-Predoc from the Spanish National Research Council. Funding for open access charge: Ministerio de Economía y Competitividad, Madrid [SAF2012-31405].
Conflict of interest statement. None declared.
REFERENCES
- 1.Harnpicharnchai P., Jakovljevic J., Horsey E., Miles T., Roman J., Rout M., Meagher D., Imai B., Guo Y., Brame C.J., et al. Composition and functional characterization of yeast 66S ribosome assembly intermediates. Mol. Cell. 2001;8:505–515. doi: 10.1016/s1097-2765(01)00344-6. [DOI] [PubMed] [Google Scholar]
- 2.Henras A.K., Plisson-Chastang C., O'Donohue M.-F., Chakraborty A., Gleizes P.-E. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip. Rev. RNA. 2014;6:225–242. doi: 10.1002/wrna.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henras A.K., Soudet J., Gérus M., Lebaron S., Caizergues-Ferrer M., Mougin A., Henry Y. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell. Mol. Life Sci. 2008;65:2334–2359. doi: 10.1007/s00018-008-8027-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woolford J.L., Baserga S.J. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics. 2013;195:643–681. doi: 10.1534/genetics.113.153197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernández-Pevida A., Kressler D., de la Cruz J. Processing of preribosomal RNA in Saccharomyces cerevisiae. Wiley Interdiscip. Rev. RNA. 2015;6:191–209. doi: 10.1002/wrna.1267. [DOI] [PubMed] [Google Scholar]
- 6.De la Cruz J., Karbstein K., Woolford J.L. Functions of ribosomal proteins in assembly of eukaryotic ribosomes in vivo. Annu. Rev. Biochem. 2015;84:93–129. doi: 10.1146/annurev-biochem-060614-033917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kressler D., Hurt E., Bassler J. Driving ribosome assembly. Biochim. Biophys. Acta. 2010;1803:673–683. doi: 10.1016/j.bbamcr.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 8.Pestov D.G., Stockelman M.G., Strezoska Z., Lau L.F. ERB1, the yeast homolog of mammalian Bop1, is an essential gene required for maturation of the 25S and 5.8S ribosomal RNAs. Nucleic Acids Res. 2001;29:3621–3630. doi: 10.1093/nar/29.17.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strezoska Z., Pestov D.G., Lau L.F. Bop1 is a mouse WD40 repeat nucleolar protein involved in 28S and 5. 8S RRNA processing and 60S ribosome biogenesis. Mol. Cell. Biol. 2000;20:5516–5528. doi: 10.1128/mcb.20.15.5516-5528.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miles T.D., Jakovljevic J., Horsey E.W., Harnpicharnchai P., Tang L., Woolford J.L. Ytm1, Nop7, and Erb1 form a complex necessary for maturation of yeast 66S preribosomes. Mol. Cell. Biol. 2005;25:10419–10432. doi: 10.1128/MCB.25.23.10419-10432.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang L., Sahasranaman A., Jakovljevic J., Schleifman E., Woolford J.L. Interactions among Ytm1, Erb1, and Nop7 required for assembly of the Nop7-subcomplex in yeast preribosomes. Mol. Biol. Cell. 2008;19:2844–2856. doi: 10.1091/mbc.E07-12-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rohrmoser M., Ho M., Grimm T., Malamoussi A., Harasim T., Orban M., Pfisterer I., Gruber-Eber A., Kremmer E., Eick D., et al. Interdependence of Pes1, Bop1, and WDR12 controls nucleolar localization and assembly of the PeBoW complex required for maturation of the 60S ribosomal subunit. Mol. Cell. Biol. 2007;27:3682–3694. doi: 10.1128/MCB.00172-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Granneman S., Petfalski E., Tollervey D. A cluster of ribosome synthesis factors regulate pre-rRNA folding and 5.8S rRNA maturation by the Rat1 exonuclease. EMBO J. 2011;30:4006–4019. doi: 10.1038/emboj.2011.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kressler D., Hurt E., Bergler H., Bassler J. The power of AAA-ATPases on the road of pre-60S ribosome maturation–molecular machines that strip pre-ribosomal particles. Biochim. Biophys. Acta. 2012;1823:92–100. doi: 10.1016/j.bbamcr.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bassler J., Kallas M., Pertschy B., Ulbrich C., Thoms M., Hurt E. The AAA-ATPase Rea1 drives removal of biogenesis factors during multiple stages of 60S ribosome assembly. Mol. Cell. 2010;38:712–721. doi: 10.1016/j.molcel.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sahasranaman A., Dembowski J., Strahler J., Andrews P., Maddock J., Woolford J.L. Assembly of Saccharomyces cerevisiae 60S ribosomal subunits: role of factors required for 27S pre-rRNA processing. EMBO J. 2011;30:4020–4032. doi: 10.1038/emboj.2011.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hölzel M., Rohrmoser M., Schlee M., Grimm T., Harasim T., Malamoussi A., Gruber-Eber A., Kremmer E., Hiddemann W., Bornkamm G.W., et al. Mammalian WDR12 is a novel member of the Pes1-Bop1 complex and is required for ribosome biogenesis and cell proliferation. J. Cell Biol. 2005;170:367–378. doi: 10.1083/jcb.200501141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nal B., Mohr E., Silva M.-I., Tagett R., Navarro C., Carroll P., Depetris D., Verthuy C., Jordan B.R., Ferrier P. Wdr12, a mouse gene encoding a novel WD-Repeat Protein with a notchless-like amino-terminal domain. Genomics. 2002;79:77–86. doi: 10.1006/geno.2001.6682. [DOI] [PubMed] [Google Scholar]
- 19.Adams C.C., Jakovljevic J., Roman J., Harnpicharnchai P., Woolford J.L. Saccharomyces cerevisiae nucleolar protein Nop7p is necessary for biogenesis of 60S ribosomal subunits. RNA. 2002;8:150–165. doi: 10.1017/s1355838202010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wegrecki M., Neira J.L., Bravo J. The Carboxy-Terminal Domain of Erb1 Is a Seven-Bladed ß-Propeller that Binds RNA. PLoS One. 2015;10:e0123463. doi: 10.1371/journal.pone.0123463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ulbrich C., Diepholz M., Bassler J., Kressler D., Pertschy B., Galani K., Böttcher B., Hurt E. Mechanochemical removal of ribosome biogenesis factors from nascent 60S ribosomal subunits. Cell. 2009;138:911–22. doi: 10.1016/j.cell.2009.06.045. [DOI] [PubMed] [Google Scholar]
- 22.Studier F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 23.Leslie A.W., Powell H. Processing diffraction data with mosflm. In: Read R, Sussman J, editors. Evolving Methods for Macromolecular Crystallography SE - 4, NATO Science Series. Vol. 245. Houten: Springer; 2007. pp. 41–51. [Google Scholar]
- 24.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A., et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Long F., Vagin A.A., Young P., Murshudov G.N. BALBES: a molecular-replacement pipeline. Acta Crystallogr. D. Biol. Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams P.D., Afonine P.V, Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winter G. xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2009;43:186–190. [Google Scholar]
- 29.Holm L., Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gelly J.-C., Joseph A.P., Srinivasan N., de Brevern A.G. iPBA: a tool for protein structure comparison using sequence alignment strategies. Nucleic Acids Res. 2011;39:W18–W23. doi: 10.1093/nar/gkr333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waterhouse A.M., Procter J.B., Martin D.M.A., Clamp M., Barton G.J. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 33.Meng E.C., Pettersen E.F., Couch G.S., Huang C.C., Ferrin T.E. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics. 2006;7:339. doi: 10.1186/1471-2105-7-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keller S., Vargas C., Zhao H., Piszczek G., Brautigam C.A., Schuck P. High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 2012;84:5066–5073. doi: 10.1021/ac3007522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Houtman J.C.D., Brown P.H., Bowden B., Yamaguchi H., Appella E., Samelson L.E., Schuck P. Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Protein Sci. 2007;16:30–42. doi: 10.1110/ps.062558507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brachmann C.B., Davies A., Cost G.J., Caputo E., Li J., Hieter P., Boeke J.D. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 37.Kaiser C., Michaelis S., Mitchell A., Cold Spring Harbor Laboratory . Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual. NY: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- 38.Gietz R.D., Akio S. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- 39.Kressler D., Rojo M., Linder P., Cruz J. Spb1p is a putative methyltransferase required for 60S ribosomal subunit biogenesis in Saccharomyces cerevisiae. Nucleic Acids Res. 1999;27:4598–4608. doi: 10.1093/nar/27.23.4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De la Cruz J., Kressler D., Rojo M., Tollervey D., Linder P. Spb4p, an essential putative RNA helicase, is required for a late step in the assembly of 60S ribosomal subunits in Saccharomyces cerevisiae. RNA. 1998;4:1268–1281. doi: 10.1017/s1355838298981158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petitjean A., Bonneaud N., Lacroute F. The duplicated Saccharomyces cerevisiae gene SSM1 encodes a eucaryotic homolog of the eubacterial and archaebacterial L1 ribosomal proteins. Mol. Cell. Biol. 1995;15:5071–5081. doi: 10.1128/mcb.15.9.5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Venema J., Planta R., Raué H. In Vivo Mutational Analysis of Ribosomal RNA in Saccharomyces cerevisiae. In: Martin R, editor. Protein Synthesis SE - 19, Methods in Molecular Biology. Vol. 77. NY: Springer; 1998. pp. 257–270. [DOI] [PubMed] [Google Scholar]
- 43.Bassler J., Paternoga H., Holdermann I., Thoms M., Granneman S., Barrio-Garcia C., Nyarko A., Stier G., Clark S.a, Schraivogel D., et al. A network of assembly factors is involved in remodeling rRNA elements during preribosome maturation. J. Cell Biol. 2014;207:481–498. doi: 10.1083/jcb.201408111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coyle S.M., Gilbert W.V., Doudna J.A., Berkeley L., Bibliotecas D. Direct link between RACK1 function and localization at the ribosome in vivo. Mol. Cell. Biol. 2009;29:1626–1634. doi: 10.1128/MCB.01718-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Du Y.N., Stillman B. Yph1p, an ORC-Interacting Protein: Potential Links between Cell Proliferation Control, DNA Replication, and Ribosome Biogenesis. Cell. 2002;109:835–848. doi: 10.1016/s0092-8674(02)00773-0. [DOI] [PubMed] [Google Scholar]
- 46.Killian A., Le Meur N., Sesboüé R., Bourguignon J., Bougeard G., Gautherot J., Bastard C., Frébourg T., Flaman J.-M. Inactivation of the RRB1-Pescadillo pathway involved in ribosome biogenesis induces chromosomal instability. Oncogene. 2004;23:8597–8602. doi: 10.1038/sj.onc.1207845. [DOI] [PubMed] [Google Scholar]
- 47.Krogan N.J., Peng W., Cagney G., Robinson M.D., Haw R., Zhong G., Guo X., Zhang X., Canadien V., Richards D.P., et al. High-definition macromolecular composition of yeast RNA-processing complexes. Mol. Cell. 2004;13:225–239. doi: 10.1016/s1097-2765(04)00003-6. [DOI] [PubMed] [Google Scholar]
- 48.Mylona A., Fernández-Tornero C., Legrand P., Haupt M., Sentenac A., Acker J., Müller C.W. Structure of the tau60/Delta tau91 subcomplex of yeast transcription factor IIIC: insights into preinitiation complex assembly. Mol. Cell. 2006;24:221–232. doi: 10.1016/j.molcel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 49.Russell R.B., Sasieni P.D., Sternberg M.J. Supersites within superfolds. Binding site similarity in the absence of homology. J. Mol. Biol. 1998;282:903–918. doi: 10.1006/jmbi.1998.2043. [DOI] [PubMed] [Google Scholar]
- 50.Stirnimann C.U., Petsalaki E., Russell R.B., Müller C.W. WD40 proteins propel cellular networks. Trends Biochem. Sci. 2010;35:565–574. doi: 10.1016/j.tibs.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 51.Xu C., Min J. Structure and function of WD40 domain proteins. Protein Cell. 2011;2:202–214. doi: 10.1007/s13238-011-1018-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Sluis M., McStay B. Ribosome biogenesis: Achilles heel of cancer? Genes Cancer. 2014;5:152–153. doi: 10.18632/genesandcancer.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schlosser I., Hölzel M., Mürnseer M., Burtscher H., Weidle U.H., Eick D. A role for c-Myc in the regulation of ribosomal RNA processing. Nucleic Acids Res. 2003;31:6148–6156. doi: 10.1093/nar/gkg794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Killian A., Sarafan-Vasseur N., Sesboüé R., Le Pessot F., Blanchard F., Lamy A., Laurent M., Flaman J.-M., Frébourg T. Contribution of the BOP1 gene, located on 8q24, to colorectal tumorigenesis. Genes Chromosomes Cancer. 2006;45:874–881. doi: 10.1002/gcc.20351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.