


Abstract
The family of ten-eleven translocation (Tet) dioxygenases is widely distributed across the eukaryotic tree of life, from mammals to the amoeboflagellate Naegleria gruberi. Like mammalian Tet proteins, the Naegleria Tet-like protein, NgTet1, acts on 5-methylcytosine (5mC) and generates 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) in three consecutive, Fe(II)- and α-ketoglutarate-dependent oxidation reactions. The two intermediates, 5hmC and 5fC, could be considered either as the reaction product of the previous enzymatic cycle or the substrate for the next cycle. Here we present a new crystal structure of NgTet1 in complex with DNA containing a 5hmC. Along with the previously solved NgTet1–5mC structure, the two complexes offer a detailed picture of the active site at individual stages of the reaction cycle. In the crystal, the hydroxymethyl (OH-CH2-) moiety of 5hmC points to the metal center, representing the reaction product of 5mC hydroxylation. The hydroxyl oxygen atom could be rotated away from the metal center, to a hydrophobic pocket formed by Ala212, Val293 and Phe295. Such rotation turns the hydroxyl oxygen atom away from the product conformation, and exposes the target CH2 towards the metal-ligand water molecule, where a dioxygen O2 molecule would occupy to initiate the next round of reaction by abstracting a hydrogen atom from the substrate. The Ala212-to-Val (A212V) mutant profoundly limits the product to 5hmC, probably because the reduced hydrophobic pocket size restricts the binding of 5hmC as a substrate.
INTRODUCTION
There is much interest in the effects of DNA cytosine modifications on epigenetic regulation, development and differentiation, neuron function, and diseases. DNA methyltransferases convert certain cytosines to 5-methylcytosine (5mC), usually within the sequence context CpG (1–3) or CpA (4). A subset of these 5mC residues is then converted to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) by the ten-eleven translocation (Tet) dioxygenases (5–7). The Tet dioxygenases are widely distributed across the eukaryotic tree of life, from mammals to the amoeboflagellate Naegleria gruberi (8), mushroom (Coprinopsis cinerea) (9), honey bee (Apis mellifera) (10) and Drosophila (11). High-throughput methods for characterizing DNA oxidation states at single base resolution are becoming available, so our understanding of Tet-mediated oxidation is expected to develop rapidly.
The Tet enzymes belong to a family of Fe(II)- and α-ketoglutarate-dependent dioxygenases that also includes the Jumonji-domain containing histone lysine demethylases, the N-methyl nucleic acid demethylase including E. coli AlkB and its mammalian homologs, and many others (12). The N-demethylation reaction catalyzed by most of these enzymes involves the transient formation of a N-hydroxymethyl intermediate followed by the spontaneous (non-enzymatic) release of formaldehyde (Supplemental Figure S1).
In contrast, formaldehyde is not released during Tet-mediated hydroxylation of 5mC (Figure 1A). Unlike methylation/demethylation of monoamines, the main mechanistic problem in methylation and demethylation of carbons is that the C5 atom of the cytosine ring is an inert carbon. DNA cytosine methyltransferases solve this problem by flipping the target base into a concave active site, where a transient covalent adduct forms at cytosine C6 (13,14). However, the 5-hydroxymethyl modification at C5 (5hmC) does not change the nature of the carbon-carbon bond (i.e., C5-CH3 versus C5-CH2OH) and thus either stays as a stable modification or is further converted to 5fC and 5caC in consecutive Tet-mediated oxidation reactions, generating higher oxidized modifications further away from 5mC (15,16). That Tet-mediated 5hmC remains a stable modification, rather than serving as an intermediate in direct demethylation, is supported by the observation that 5mC loss in the paternal genome immediately after fertilization during mouse development, is accompanied by a concurrent increase in 5hmC (17).
Figure 1.
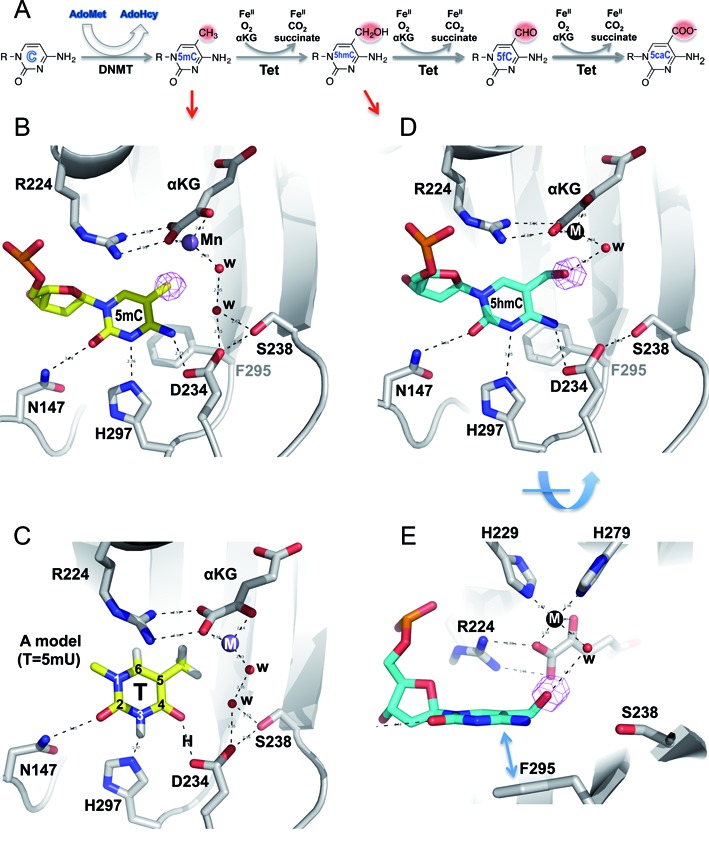
Structures of NgTet1 in complexes with 5mC and 5hmC DNA. (A) Schematic illustration of methylation and oxidation reactions. DNA methyltransferases convert a proportion of the cytosines (Cs) into 5mC in a S-adenosyl-l-methionine (AdoMet)-dependent reaction. The Tet dioxygenases then convert a fraction of 5mC to 5hmC, 5fC and 5caC in three consecutive, Fe(II)- and α-ketoglutarate-dependent oxidation reactions without releasing any formaldehyde (in contrast to demethylation of N-methylated substrates; Supplemental Figure S1). (B) The extrahelical 5mC in the active site forms planar π stacking contacts with F295 (away from the viewer in the background) and R224 (which forms an ion-pair interaction with α-ketoglutarate or αKG), as well as hydrogen bonds with three residues along the Watson–Crick polar edge. The simulated annealing omit electron density (in magenta) is shown for the methyl group of 5mC, contoured at 4.5σ above the mean. (C) A model of thymine (5mU) in the active site including hydrogen atoms. The side-chain imidazole ring of H297 could interact with the protonated N3 nitrogen. The distance between one of the carboxylate oxygen atoms of D234 and the O4 carbonyl oxygen would suggest the presence of a hydrogen bond and therefore the presence of a proton between them (labeled as H). The proton source may be the COOH group of D234, which itself might be protonated as a result of the water-mediated interaction. (D) The extrahelical 5hmC in the active site forms nearly identical interaction as that of 5mC (panel B). The simulated annealing omit electron density (in magenta) for the hydroxyl oxygen atom of 5hmC is shown, contoured at 4.5σ above the mean. (E) An orthogonal view from panel D shows the out-of-plane hydroxyl oxygen atom of 5hmC interacting with the metal-ligand water molecule.
Two X-ray structures are currently available for Tet enzymes in complex with 5mC: the catalytic domain of human TET2 (18) (Supplemental Figure S2a) and NgTet1 from Naegleria gruberi (8) (Supplemental Figure S2b). Like DNA methyltransferases, Tet enzymes use a base-flipping mechanism to access 5mC. This is a process that involves rotation of backbone bonds in double-stranded DNA to expose an out-of-stack base, which can then be a substrate for an enzyme-catalyzed chemical reaction or for a specific protein binding interaction (19). Structurally, NgTet1 contains the core structure of the catalytic domain of the mammalian Tet enzymes (Supplemental Figure S2c), including conserved residues involved in structural integrity and functional significance (8). Like other structurally characterized α-ketoglutarate-dependent dioxygenases (such as AlkB), NgTet1 has a core double-stranded β-helix fold that binds Fe(II) and α-ketoglutarate (Supplemental Figure S2b). Two twisted β-sheets (a four-stranded minor sheet and an eight-stranded major sheet) pack together with five helices on the outer surface of the major sheet to form a three-layered structure (Supplemental Figure S2b). The unequal number of strands of the two sheets creates the active site located asymmetrically on the side of the molecule where the extra strands of the major sheet are located. Here, we focus on the NgTet1 active site and its ability to bind 5hmC as a reaction product as well as a reaction substrate in two different conformations. The binding of the hydroxyl oxygen atom of 5hmC in a hydrophobic pocket away from the metal center enables NgTet1 to pursue the next reaction cycle. Substitution of Ala212 to Val (A212V) in the pocket robustly limits the product to 5hmC.
MATERIALS AND METHODS
Crystallography
In our previous determined NgTet1–5mC structure, the first 56 residues were not modeled due to lack of continuous electron density (8). We thus generated a hexahistidine–SUMO-tagged construct deleting the first 56 residues of NgTet1 (pXC1336). The protein was expressed in E. coli BL21 (DE3)-Gold cells with the RIL-Codon plus plasmid (Stratagene). Cultures were grown at 37°C until the OD at 600 nm reached 0.5; the temperature was then shifted to 16°C, and isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to 0.4 mM to induce expression. Cell pellets were re-suspended with 4 volumes of 500 mM NaCl, 20 mM sodium phosphate, pH 7.4, 20 mM imidazole, 1 mM dithiothreitol (DTT) and 0.25 mM phenylmethyl-sulphonyl fluoride (PMSF) and sonicated for 5 min (1 s on and 2 s off). The lysate was clarified by centrifugation at 38 000g for 60 min. The hexahistidine fusion protein was isolated on a nickel-charged chelating column (GE Healthcare). The His-SUMO tag was removed by incubating with Ulp1 (purified in-house) for 16 h at 4°C. The cleaved protein was further purified by a tandem HiTrap Q and SP column (GE-Healthcare), eluted from SP column and concentrated. The protein was then loaded onto a Superdex 75 (16/60) column (equilibrated with 150 mM NaCl, 20 mM HEPES, pH 8.0, 1 mM DTT) where it eluted as a single peak corresponding to a monomeric protein.
For co-crystallization, we used oligonucleotides (either 14 or 12bp + one overhang) containing 5hmC or 5mC (synthesized by New England Biolabs) (Table 1). An equimolar mixture of protein and DNA (0.5 mM) was incubated in 2 mM α-ketoglutarate (αKG), 2 mM MnCl2, 100 mM NaCl, and 20 mM HEPES-NaOH, pH 8.0, for 30 min at 4°C. Crystallization was carried out in a 2 μl sitting drop with equal volume of the complex solution and well solution. Crystals appeared within 2 days at 16°C under the conditions of 25% (w/v) polyethylene glycol monomethyl ether 550, 10 mM ZnSO4, and 100 mM MES, pH 6.5, for the 5hmC–NgTet1 complex, and 20% (w/v) polyethylene glycol 8000, 200 mM Mg oxaloacetate, 100 mM Na cacodylate, pH 6.5 for the 5mC–NgTet1complex.
Table 1. Summary of Statistics of X-ray diffraction and refinement*.
| Protein | NgTet1Δ57 | |
|---|---|---|
| DNA | 5′-TGGAAHGCAATTCT-3′ | 5′-TGTCAGMGCATGG-3′ |
| (M = 5mC; H = 5hmC) | 3′-ACCTTGCGTTAAGA-5′ | 3′-CAGTCGCGTACCT-5′ |
| Cofactor / Metal | αKG/Mn(II) | αKG/Mn(II) |
| PDB | 5CG8 | 5CG9 |
| Beamline/wavelength | SER-CAT 22-BM/1.0 Å | SER-CAT 22-ID/1.0 Å |
| Space group | I212121 | P3221 |
| Unit cell (a, b, c (Å)) | 83.8, 107.4, 167.7 | 191.2, 191.2, 51.3 |
| (α, β, γ (°)) | 90, 90, 90 | 90, 90, 120 |
| Resolution (Å) | 27.6–2.69 (2.79–2.69) | 29.7–2.69 (2.79–2.69) |
| aRmerge | 0.066 (0.977) | 0.154 (0.894) |
| b <I/σI> | 29.2 (2.9) | 13.7 (2.2) |
| Completeness (%) | 99.7 (100.0) | 98.7 (92.8) |
| Redundancy | 9.9 (10.0) | 9.3 (8.9) |
| CC 1/2, CC | (0.908/0.976) | (0.796/0.942) |
| Reflections (observed) | 208 582 | 274 840 |
| (Unique) | 20 985 | 29 462 |
| Refinement | (1 complex in asymmetric unit) | (Two complexes in asymmetric unit) |
| Resolution (Å) | 2.70 | 2.69 |
| No. of reflections | 20 958 | 29 452 |
| cRwork/dRfree | 0.189/0.228 | 0.217/0.238 |
| No. of atoms | ||
| Protein | 2094 | 4211 |
| DNA | 570 | 953 |
| αKG | 10 | 20 |
| Mn(II) | 1 | 2 |
| Solvent | 11 | 42 |
| B-factors (Å2) | ||
| Protein | 86.2 | 70.9 |
| DNA | 111.1 | 98.8 |
| αKG | 80.5 | 69.6 |
| Mn(II) | 65.9 | 68.2 |
| Solvent | 97.5 | 73.7 |
| R.M.S. deviations | ||
| Bond length (Å) | 0.008 | 0.006 |
| Bond angles (°) | 1.0 | 0.8 |
| All atom clash score | 0.8 | 2.1 |
| Ramachandran plot (%) | ||
| Favored | 98.5 | 99.0 |
| Allowed | 1.5 | 1.0 |
| Rotamer outliers (%) | 0 | 0.2 |
| Cβ deviation | 0 | 0 |
*Values in parenthesis correspond to highest resolution shell.
aRmerge = Σ|I – <I>| /ΣI, where I is the observed intensity and <I> is the averaged intensity from multiple observations.
b <I/σI> = averaged ratio of the intensity (I) to the error of the intensity (σI).
cRwork = Σ|Fobs – Fcal |/Σ| Fobs |, where Fobs and Fcal are the observed and calculated structure factors, respectively.
dRfree was calculated using a randomly chosen subset (5%) of the reflections not used in refinement.
Crystals were cryoprotected by soaking in mother liquor supplemented with 20% (v/v) glycerol or ethylene glycol and by plunging into liquid nitrogen. X-ray diffraction data sets were collected at the SER-CAT beamline (22-ID-D or 22-BM-D) at the Advanced Photon Source, Argonne National Laboratory and processed using HKL2000 (20). Initial crystallographic phases were determined by molecular replacement using the coordinates of the NgTet1–5mC complex structure (PDB 4LT5) as a search model. Phasing, molecular replacement, map production, and model refinement were performed using PHENIX (21,22). The two structures were solved, built, and refined independently. The statistics were calculated for the entire resolution range. The Rfree and Rwork values were calculated for 5% (randomly selected) and 95%, respectively, of the observed reflections. Molecular graphics were generated using PyMol (DeLano Scientific, LLC).
Site-directed mutagenesis
Mutagenesis of NgTet1 was performed using the Q5 Site-Directed Mutagenesis Kit and confirmed by sequencing. Wild-type and variant proteins containing an N-terminal 6X histidine tag (in pTXB1 constructs) were expressed in E. coli T7 Express competent cells (NEB) and purified as previously described (8), using a HiTrap Heparin HP column followed by a HisTrap HP column (GE Healthcare). Purified proteins were stored at -20°C in 20 mM Tris pH 7.5, 300 mM NaCl, and 50% glycerol (Supplemental Figure S3a). The protein concentrations were estimated by Bradford assay and equal amount of proteins (WT and mutants) were used in each reaction.
NgTet1 activity assay using liquid chromatography–mass spectrometry (LC–MS/MS)
The activities of NgTet1 wild type and variants were measured using a LC–MS/MS-based assay (8,23) [for LC–MS traces of a sample reaction carried out by NgTet1, see Supplemental Figure S3b]. A 20 μL NgTet1 reaction in 50 mM MOPS, pH 6.75, 50 mM NaCl, 1 mM DTT, 2 mM ascorbic acid, 1 mM αKG, and 100 μM FeSO4 contained 8 μM NgTet1 and 4 μM 56-bp, hemi-modified dsDNA (5′-CGG CGT TTC CGG GTT CCA TAG GCT CCG CCC XGG ACT CTG ATG ACC AGG GCA TCA CA-3′ where X = 5mC, 5hmC or 5fC) and its complementary strand with no modification.
All reactions were incubated at 34°C for the specified amount of time. For time courses, reactions were quenched at the specified time by heating at 95°C for 3 min, and subsequent chilling on ice for 5 min. All samples were digested with 0.8 units proteinase K (NEB) for 1 h at 50°C, and the DNA was purified using the DNA Clean & Concentrator Kit (Zymo Research). DNA was then digested to nucleosides as described previously (8), and analyzed by Agilent 1290 UHPLC and 6490 Triple Quad Mass detector on a Waters XSelect HSS T3 column (2.1 × 100 mm, 2.5 μm).
RESULTS
Overall structures
Previously, we determined the crystal structure of NgTet1 with a 14-base-pair (bp) oligonucleotide containing a single, fully methylated CpG site (8). Only one of the 5mC nucleotides flips out and is positioned in the active site. Here, we used the same, hemi-modified oligonucleotide with a 5hmC in the position of the flipped nucleotide (Table 1). We used Mn(II), instead of Fe(II), to generate catalytically inert complexes. The 5hmC structure was solved by molecular replacement and refined to a resolution of 2.7 Å (Table 1). The two structures are highly similar, with a root mean squared deviation of less than 0.3 Å when comparing protein components of 265 pairs of Cα atoms. During the screen for crystallization, we also crystallized a second NgTet1–5mC complex using a 12-bp DNA plus a 5′-overhanging thymine in a different space group (Table 1). The crystallographic asymmetric unit contains two NgTet1-DNA complexes (discussed in Supplemental Figure S4) and the structure was determined to the same resolution of 2.7 Å.
The active site (5mC versus thymine)
The extrahelical nucleotide, 5mC, is bound in a cage-like active site via stacking of the flipped base in between the phenyl ring of F295 and the guanidino group of R224 (Figure 1B). The polar groups of the (modified) cytosine ring that normally form the Watson–Crick pairings with guanine now form hydrogen bonds with the side-chain amide group of N147 (interacting with the O2 oxygen), the side-chain imidazole ring of H297 (interacting with the deprotonated N3 nitrogen), and the side chain carboxylate oxygen atoms of D234 (interacting with the N4 amino group NH2) (Figure 1B). Interactions between the carboxylate oxygen atom of D234 and the exocyclic amino group N4 define the binding pocket specificity, resulting in the strong preference for C5 modified cytosines as substrates by NgTet1 (8,23).
However, like mammalian Tet1 (24), NgTet1 has a minor activity on thymine (23). Like 5mC, thymine (5-methyluracil) contains a methyl group at C5, but the hydrogen bonding potentials at N3 and O3 are reversed compared with cytosine's N3 and N4 atoms. We modeled a thymine in the same active site configuration (Figure 1C). The N3-H297 interaction would remain, as the imidazole ring could serve as a proton donor/acceptor depending on the N3 protonation status. However, a protonated D234 is needed to accommodate the O4 carbonyl oxygen, which could occur via a water molecule (Figure 1C). We note that mammalian Tet enzymes have asparagines at the corresponding position of D234 (N1387 in human TET2) (18), and can oxidize thymine to generate 5-hydroxymethyluracil (5hmU) in vivo (24). Indeed, the aspartate-to-asparagine (D234N) mutant of NgTet1 has a ∼2-fold increase of the activity on thymine while the activity on 5mC was decreased by ∼2-fold (23). Asparagine can donate one H-bond to the O4 atom of thymine via its side chain amide nitrogen (NH2). The equivalent residue in the catalytic site of thymidylate synthase, whose substrate is dUMP, is also asparagine (25).
5hmC in the active site
The flipped 5hmC in the active site has almost identical interactions as those of 5mC, in terms of base-stacking and polar edge hydrogen bonding interactions (Figure 1D). The hydroxymethyl moiety of 5hmC points to the metal center, and the out-of-planar hydroxyl oxygen atom is only 3.3 Å away from the metal-ligand water molecule (where a dioxygen O2 molecule would occupy to initiate the reaction) (Figure 1E). This observation suggests that the observed conformation of the hydroxymethyl moiety almost certainly represents the reaction product of 5mC hydroxylation, rather than the posture of a substrate ready for the next round of reaction. However, we note that the substitution of Mn(II) for Fe(II), which generated an inert enzyme-cofactor complex, might induce 5hmC into the product conformation.
The hydroxyl oxygen atom of 5hmC could rotate freely along the C5-CH2 bond in the absence of spatial constraint. Starting from the observed conformation 1 (the product conformation), rotating the C5-CH2 bond 120° generates conformations 2 and 3 (Figure 2A–C). We note that all three conformations have been observed previously in our study of 5hmC-containing DNA bound by transcription factors WT1 and Egr1 (26). Both conformations 2 and 3 expose the target CH2 towards the metal-ligand water molecule (or the dioxygen molecule during the reaction), which would allow the next round of reaction to occur (Figure 2B and C). However, the hydroxyl oxygen atom in conformation 2 would be too close to the cofactor α-ketoglutarate (Figure 2B) (an O…O distance of ∼2 Å), resulting in repulsion and/or interference with the binding of α-ketoglutarate and the metal ion. In contrast, conformation 3 would place the hydroxyl oxygen atom in the vicinity of A212, V293 and F295 (Figure 2C and D), without interfering with cofactor binding. The oxygen and carbon distances in the range of 3–3.2 Å would allow the hydroxyl oxygen atom to make a number of interactions, including a potential C-H…O hydrogen bond - a common interaction found in bio-molecular recognition (27,28), and a potential O–H…π interaction (29) between the hydroxyl oxygen and the aromatic ring of residue F295. We note that the observation of hydrogen atoms will require other techniques such as neutron protein crystallography (29).
Figure 2.
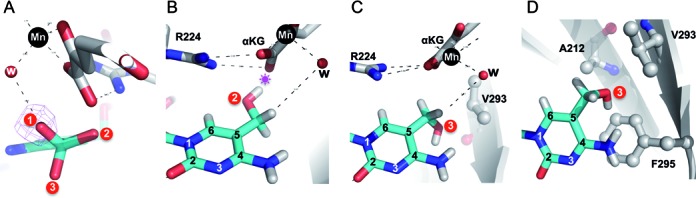
Alternative conformation of 5hmC as a reaction product or substrate. (A) The hydroxyl oxygen atom of 5hmC could adopt three alternative conformations by rotating the C5–CH2 bond every 120°. Conformation 1 represents the experimentally observed product state of 5mC hydroxylation. The metal–ligand water molecule suggests the position where the dioxygen molecule would occupy during the reaction. (B) Conformation 2 would place the hydroxyl oxygen atom of 5hmC in the vicinity of the carboxylate group of α-ketoglutarate (αKG), potentially resulting in repulsion (indicated by a star). (C and D) Two views of conformation 3 with the hydroxyl oxygen atom of 5hmC in close contact with the hydrophobic side chains of A212, V293 and F295.
A212V variant has altered product specificity
The intimate fitting of the hydroxyl oxygen atom of 5hmC into the space between the hydrophobic side chains of A212, V293 and F295 is consistent with 5hmC being a substrate of NgTet1, allowing the target CH2 to be exposed to the activated dioxygen which can abstract a hydrogen atom to eventually yield a formylated product (Figure 2C and D). We reasoned that the residues in the hydrophobic binding pocket are important for further oxidation beyond 5hmC by NgTet1. Several outcomes could be anticipated. First, increasing the pocket size should allow the binding of 5hmC without affecting the generation of 5fC and 5caC. Second, decreasing the pocket size would sterically exclude the binding of 5hmC and thus limit NgTet1's ability to generate 5fC and 5caC. Third, considerable alteration of the pocket shape and size might interfere with the binding of the cytosine ring and thus affect overall catalytic activity. We reasoned that the aromatic ring of F295 is important for stacking with the flipped cytosine ring and thus focused mutagenesis on the two smaller aliphatic residues (A212 and V293) and analyzed the reaction products of the mutant proteins.
We substituted alanine 212 with the smaller glycine (A212G), larger side chains with increasing sizes (A212-to-V, L, I and F), or a polar side chain (A212-to-N). All mutants behaved similarly to the wild type (WT) enzyme during purification with comparable final protein yield (Figure 3A, bottom panel, and Supplemental Figure S3a). As expected, A212G, which presumably has increased pocket size, had similar activity as that of WT on a hemi-methylated single 5mC-containing, 56-bp oligonucleotide DNA substrate (Figure 3A, top panel). When the alanine is changed to valine (A212V), slightly less 5mC was converted but a significant buildup of 5hmC was observed compared to WT, while only a very small amount of 5caC was formed after the 10-min reaction. Further increasing the size of residue 212 to leucine (A212L), isoleucine (A212I), or phenylalanine (A212F), almost abolished the activity forming only a small amount of 5hmC (Figure 3A), indicating the larger size chains at residue 212 prevented the initial 5mC binding. Surprisingly, A212-to-asparagine (A212N), which has a similar size to that of leucine, formed 5hmC as the major reaction product, similar to A212V.
Figure 3.
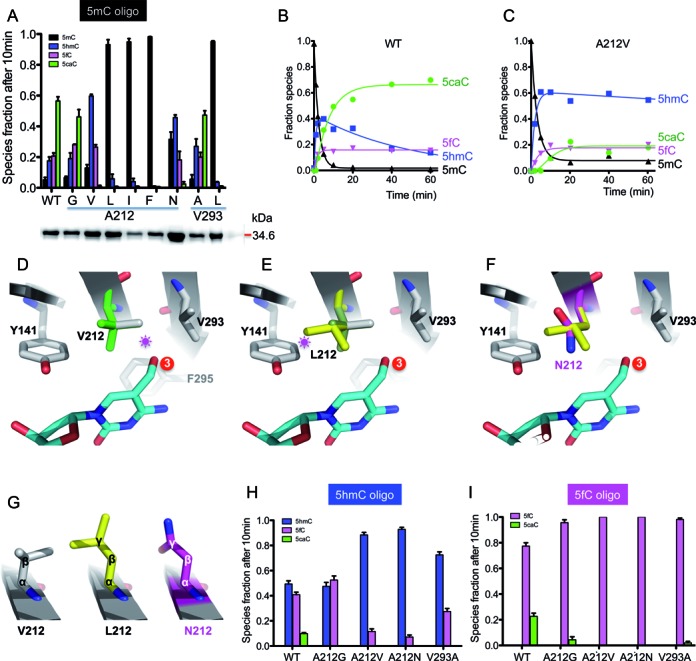
Oxidation activities of A212 and V293 mutants. (A) LC–MS/MS quantification of 5mC and its oxidized derivatives after a 10-min reaction of NgTet1 WT and variant proteins. Inserted is a SDS-PAGE gel (bottom) of the proteins (WT and 8 mutants) used for activity (see Supplemental Figure S3a). The protein concentrations were adjusted and equal amount of enzymes (WT and mutants) were used in each reaction. Error bars indicate standard error (s.e.) of the mean value from three independent experiments. (B and C) The time courses of quantitative LC–MS/MS measurements of 5mC (black) disappearance and formation of 5hmC (blue), 5fC (magenta) and 5caC (green) by WT (panel b) or 5hmC by A212V (panel c). (D) A model of V212 with two alternative conformations (in green or grey). The conformation in grey would clash with 5hmC (indicated by a star). (E) A model of L212. Only one of the three possible conformations was shown (in yellow), which would clash with Y141. The other two conformations would clash with V293 or F295 (in the background away from the reader). (F) A model of N212 (magenta) superimposed with L212 (yellow). The planar side chain conformation allowed N212 to be accommodated near Y141. (G) Comparison of V212, L212 and N212. (H and I) LC–MS/MS quantification of oxidation of a 5hmC- (panel G) or 5fC-containing oligo (panel h) after 10-min reactions of NgTet1 WT and mutant variants. Error bars indicate standard error (s.e.) of the mean value from three independent experiments.
Because A212V had the most pronounced effect on product composition while only minimally reducing the rate of 5mC conversion, we further analyzed this variant by performing a time course to track the relative levels of reaction species. During the WT reaction, 5caC was the major product after ∼10 min while 5hmC initially increased rapidly to 40% and then gradually decreased, concomitant with a further increase of 5caC (Figure 3B). The A212V reaction had an even faster initial increase of 5hmC to 60% but then stalled, corresponding to a very slow increase in 5caC (Figure 3C). This result indicates that A212V can form 5hmC normally, but the subsequent oxidation of 5hmC to 5fC and 5caC is drastically reduced, probably due to the abnormal binding of 5hmC as substrate. We modeled valine, leucine and asparagine at the residue 212, respectively, for conformation 3 containing 5hmC in the active site (Figure 3D–F). The side chain of valine could have two alternative rotomer conformations (Figure 3D). One of them would clash with the hydroxymethyl moiety of 5hmC, thus limiting the enzyme's ability to use 5hmC as a substrate. The second conformation would allow the binding of 5hmC and permit further oxidation. This is in agreement with the observation that A212V has 5hmC as the major product and 5fC and 5caC as the minor products. The side chain of leucine could be modeled in three conformations; however, each of them would clash with the side chain of F295, V293, and Y141, respectively, consistent with the nearly abolished activity (Figure 3E). One of the major differences between leucine and asparagine lies in the Cγ atom, which is an sp3 carbon in leucine and an sp2 carbon in asparagine, with the amide nitrogen and carbonyl oxygen staying in the same plane of the Cγ atom (Figure 3G). This difference allowed asparagine to be placed near Y141 without serious clash (Figure 3F), accounting for the ability for A212N enzyme to carry out further oxidation reactions. Like A212V, the other possible rotomer conformations of A212N would result in a clash with neighboring residues, consistent with a majority product of 5hmC and reduced formation of 5fC and 5caC.
To further understand the defect of A212V and A212N in generating 5fC and 5caC, we compared activity of NgTet1 variants on oligonucleotide substrates bearing the same sequence but with a 5hmC or 5fC in place of the 5mC. We found that WT NgTet1 can catalyze the oxidation of both the 5hmC- and 5fC-containing DNA as a starting substrate, although not as efficiently as the 5mC counterpart. After a 10-min reaction with the 5hmC oligo, approximately 50% of 5hmC has been converted to products (∼40% 5fC and ∼10% 5caC) (Figure 3H), whereas for the 5fC oligo, only ∼20% of 5fC is converted to the 5caC product under the conditions tested (Figure 3I). For A212V and A212N the extent of reaction with both 5hmC and 5fC is drastically reduced, with ∼10% 5hmC being oxidized or nearly no 5fC being oxidized (Figure 3H and I). Overall, these results are consistent with the critical size and positioning of residue 212 in 5hmC recognition and catalysis.
We also substituted valine 293 with smaller alanine (V293A) or larger leucine (V293L). An increase in size at residue 293 (V293L) resulted in negligible activity on 5mC substrate (Figure 3A), demonstrating that a larger side chain at this position is not well tolerated. On the other hand, two variants with smaller side chains (therefore larger pocket sizes), A212G and V293A, behave similar to WT for the 5mC-containing oligo (Figure 3A), but have somewhat decreased activity on 5hmC- and very limited activity on 5fC-containing oligos, particularly in generating the final product of 5caC (Figure 3H and I). Clearly the pocket size alone is insufficient to account for these results. We speculate these may be related to the fact that 5hmC and 5fC are intrinsically poor substrates, as discussed below.
One interesting, but unexplained, observation is that 5hmC- and 5fC-containing oligos were poor substrates for NgTet1 (Figure 3G and H) compared to 5mC, for WT and all active variants characterized. When a 5hmC or 5fC oligo was used as the initial substrate for WT, the amount of substrate remained at 50% for 5hmC or 80% for 5fC after a 10 min reaction (Figure 3H–I), compared to ∼20–30% 5hmC and ∼20% 5fC remaining when a 5mC oligo was used as the initial substrate (Figure 3A and B). One potential explanation is that NgTet1 strongly prefers 5mC DNA as a substrate and is able to processively carry out the three consecutive, oxidation reactions without releasing the bound DNA. The observed hydroxymethyl moiety of 5hmC in the product conformation (conformation 1 in Figure 2A) might suggest that the enzyme prefers to bind 5hmC as a product rather than substrate, in the presence of α-ketoglutarate (used in the crystallization). The conversion of product to substrate requires the rotation of the hydroxymethyl moiety (Figure 2B and C), as well as the exchange of the co-product succinate with another α-ketoglutarate between the successive reactions. One can speculate that in the presence of succinate, which is smaller (the product of the decarboxylation of α-ketoglutarate), 5hmC might have more freedom to rotate to assume the substrate conformation. We do not know how the cofactor exchange and the rotation of product-to-substrate conformation are coordinated in the active site, and how these processes could occur without releasing DNA. Additional experiments will be required to settle this point.
DISCUSSION
We have determined the crystal structures of NgTet1 from Naegleria gruberi, in complexes with a DNA containing either 5mC or 5hmC and carried out mutagenesis and biochemical studies to elucidate the catalytic mechanism and the product specificity of this enzyme. We use the term ‘product specificity’ of NgTet1 to describe the consecutive oxidation events: 5hmC (mono-oxidation), 5fC (di-oxidation), and 5caC (tri-oxidation). The concept is analogous to the product specificity of histone lysine methyltransferases (Supplemental Figure S1a) that transfer one, two, or three methyl groups to the target lysines (30–32). In a number of histone lysine methyltransferases, a key residue (Phe/Tyr) in the active site determines how many methyl groups the enzyme can add. Here we show that a small change (A212V) in the binding pocket of the hydroxymethyl moiety of 5hmC limits the major product of NgTet1 to be 5hmC. It will be interesting to see whether equivalent mammalian TET mutants also generate 5hmC primarily; if so, these mutants can be interesting tools for dissecting the roles of 5hmC in signaling.
Unlike the histone lysine methylation and demethylation that conduct the exact opposite reactions (Supplemental Figure S1), the Tet-mediated oxidation is a one-way reaction, generating oxidation products rather than reversing to unmodified cytosine (Figure 1A). These modifications protrude into the major groove of DNA, the primary recognition surface for proteins, and change its atomic shape and pattern of electrostatic charge. In principle, such changes can alter the interaction with DNA binding proteins by strengthening, weakening, or abolishing the interaction altogether. This, in turn, can modulate gene expression and control cellular metabolism and is believed to be one of the principal mechanisms underlying epigenetic processes such as differentiation, development, aging, and disease. Recent work suggests that the Rett syndrome protein MeCP2 binds methylated and hydroxymethylated CpA/TpG sites with similar affinity to that of fully methylated CpG/CpG (33,34). The SRA domain of UHRF2 has a slightly stronger affinity for 5hmC DNA than 5mC DNA (35). Wilms tumor protein 1 (WT1) physically interacts with Tet2 (36,37), either recruiting Tet2 to its target genes and/or binding to the ultimate product of the Tet2 enzyme, 5caC DNA (26). Additional examples of cytosine modification-specific effects on DNA binding factors include the stem cell factor Tcf3 (38). Furthermore, 5fC and 5caC in DNA retarded Pol II elongation on gene bodies and the polymerase formed specific hydrogen bonds with the 5-carboxyl group of 5caC (39). These observations suggest that the oxidized derivatives of 5mC by Tet-mediated enzymatic reactions, 5hmC, 5fC, and 5caC act as distinct epigenetic signals. Variants that differ in production of 5hmC, 5fC, and 5caC provide a resource to investigate the possibility that different oxidation states on a given 5mCpG (or 5mCpA) may signal differently.
ACCESSION NUMBERS
The X-ray structures (coordinates and structure factor files) of NgTet1–5hmC (14 bp) and NgTet1–5mC (12+1 bp) have been submitted to Protein Data Bank (PDB) under accession number 5CG8 and 5CG9, respectively.
Supplementary Material
Acknowledgments
We thank B. Baker at the organic synthesis unit of New England Biolabs for synthesizing the oligonucleotides used in the crystallization.
Author Contributions: H.H. performed crystallographic experiments; J.E.P. performed mutagenesis and characterized the mutants; N.D. and I.R.C performed LC-MS/MS analysis. X.Z., Y.Z. and X.C. organized and designed the scope of the study, and all were involved in analyzing data and preparing the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
U.S. National Institutes of Health (NIH) [GM049245-22 to X.C. and GM105132-02 to Y.Z.]; Department of Biochemistry of Emory University School of Medicine supported the use of the Southeast Regional Collaborative Access Team (SERCAT) synchrotron beamlines at the Advanced Photon Source of Argonne National Laboratory; Georgia Research Alliance Eminent Scholar (to X.C.). Funding for open access charge: New England Biolabs.
Conflict of interest statement. None declared.
REFERENCES
- 1.Bestor T., Laudano A., Mattaliano R., Ingram V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol. 1988;203:971–983. doi: 10.1016/0022-2836(88)90122-2. [DOI] [PubMed] [Google Scholar]
- 2.Okano M., Xie S., Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 3.Okano M., Bell D.W., Haber D.A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 4.Gowher H., Jeltsch A. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J. Mol. Biol. 2001;309:1201–1208. doi: 10.1006/jmbi.2001.4710. [DOI] [PubMed] [Google Scholar]
- 5.Tahiliani M., Koh K.P., Shen Y., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R., Aravind L., et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C., Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He Y.F., Li B.Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L., et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hashimoto H., Pais J.E., Zhang X., Saleh L., Fu Z.Q., Dai N., Correa I.R., Jr, Zheng Y., Cheng X. Structure of a Naegleria Tet-like dioxygenase in complex with 5-methylcytosine DNA. Nature. 2014;506:391–395. doi: 10.1038/nature12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang L., Chen W., Iyer L.M., Hu J., Wang G., Fu Y., Yu M., Dai Q., Aravind L., He C. A TET homologue protein from Coprinopsis cinerea (CcTET) that biochemically converts 5-methylcytosine to 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxylcytosine. J. Am. Chem. Soc. 2014;136:4801–4804. doi: 10.1021/ja500979k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wojciechowski M., Rafalski D., Kucharski R., Misztal K., Maleszka J., Bochtler M., Maleszka R. Insights into DNA hydroxymethylation in the honeybee from in-depth analyses of TET dioxygenase. Open biology. 2014:4. doi: 10.1098/rsob.140110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang G., Huang H., Liu D., Cheng Y., Liu X., Zhang W., Yin R., Zhang D., Zhang P., Liu J., et al. N(6)-methyladenine DNA modification in Drosophila. Cell. 2015;161:893–906. doi: 10.1016/j.cell.2015.04.018. [DOI] [PubMed] [Google Scholar]
- 12.Hausinger R.P., Schofield C.J. 2-Oxoglutarate-dependent oxygenases. RSC metallobiology series no. 3. Roy. Soc. Chem. 2015 [Google Scholar]
- 13.Klimasauskas S., Kumar S., Roberts R.J., Cheng X. HhaI methyltransferase flips its target base out of the DNA helix. Cell. 1994;76:357–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 14.Wu J.C., Santi D.V. Kinetic and catalytic mechanism of HhaI methyltransferase. J. Biol. Chem. 1987;262:4778–4786. [PubMed] [Google Scholar]
- 15.Bachman M., Uribe-Lewis S., Yang X., Williams M., Murrell A., Balasubramanian S. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem. 2014;6:1049–1055. doi: 10.1038/nchem.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bachman M., Uribe-Lewis S., Yang X., Burgess H.E., Iurlaro M., Reik W., Murrell A., Balasubramanian S. 5-Formylcytosine can be a stable DNA modification in mammals. Nat. Chem. Biol. 2015;11:555–557. doi: 10.1038/nchembio.1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li E., Zhang Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014;6:a019133. doi: 10.1101/cshperspect.a019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu L., Li Z., Cheng J., Rao Q., Gong W., Liu M., Shi Y.G., Zhu J., Wang P., Xu Y. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell. 2013;155:1545–1555. doi: 10.1016/j.cell.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 19.Roberts R.J., Cheng X. Base flipping. Annu Rev Biochem. 1998;67:181–198. doi: 10.1146/annurev.biochem.67.1.181. [DOI] [PubMed] [Google Scholar]
- 20.Otwinowski Z., Borek D., Majewski W., Minor W. Multiparametric scaling of diffraction intensities. Acta Crystallogr. A. 2003;59:228–234. doi: 10.1107/s0108767303005488. [DOI] [PubMed] [Google Scholar]
- 21.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams P.D., Grosse-Kunstleve R.W., Hung L.W., Ioerger T.R., McCoy A.J., Moriarty N.W., Read R.J., Sacchettini J.C., Sauter N.K., Terwilliger T.C. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 23.Pais J.E., Dai N., Tamanaha E., Vaisvila R., Fomenkov A.I., Bitinaite J., Sun Z., Guan S., Correa I.R., Jr, Noren C.J., et al. Biochemical characterization of a Naegleria TET-like oxygenase and its application in single molecule sequencing of 5-methylcytosine. Proc. Natl. Acad. Sci. U.S.A. 2015;112:4316–4321. doi: 10.1073/pnas.1417939112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pfaffeneder T., Spada F., Wagner M., Brandmayr C., Laube S.K., Eisen D., Truss M., Steinbacher J., Hackner B., Kotljarova O., et al. Tet oxidizes thymine to 5-hydroxymethyluracil in mouse embryonic stem cell DNA. Nat. Chem. Biol. 2014;10:574–581. doi: 10.1038/nchembio.1532. [DOI] [PubMed] [Google Scholar]
- 25.Liu L., Santi D.V. Mutation of asparagine 229 to aspartate in thymidylate synthase converts the enzyme to a deoxycytidylate methylase. Biochemistry. 1992;31:5100–5104. doi: 10.1021/bi00137a002. [DOI] [PubMed] [Google Scholar]
- 26.Hashimoto H., Olanrewaju Y.O., Zheng Y., Wilson G.G., Zhang X., Cheng X. Wilms tumor protein recognizes 5-carboxylcytosine within a specific DNA sequence. Genes Dev. 2014;28:2304–2313. doi: 10.1101/gad.250746.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horowitz S., Trievel R.C. Carbon-oxygen hydrogen bonding in biological structure and function. J. Biol. Chem. 2012;287:41576–41582. doi: 10.1074/jbc.R112.418574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yesselman J.D., Horowitz S., Brooks C.L. 3rd, Trievel R.C. Frequent side chain methyl carbon-oxygen hydrogen bonding in proteins revealed by computational and stereochemical analysis of neutron structures. Proteins. 2015;83:403–410. doi: 10.1002/prot.24724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen J.C., Hanson B.L., Fisher S.Z., Langan P., Kovalevsky A.Y. Direct observation of hydrogen atom dynamics and interactions by ultrahigh resolution neutron protein crystallography. Proc. Natl. Acad. Sci. U.S.A. 2012;109:15301–15306. doi: 10.1073/pnas.1208341109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X., Yang Z., Khan S.I., Horton J.R., Tamaru H., Selker E.U., Cheng X. Structural basis for the product specificity of histone lysine methyltransferases. Mol. Cell. 2003;12:177–185. doi: 10.1016/s1097-2765(03)00224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collins R.E., Tachibana M., Tamaru H., Smith K.M., Jia D., Zhang X., Selker E.U., Shinkai Y., Cheng X. In vitro and in vivo analyses of a Phe/Tyr switch controlling product specificity of histone lysine methyltransferases. J. Biol. Chem. 2005;280:5563–5570. doi: 10.1074/jbc.M410483200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Couture J.F., Dirk L.M., Brunzelle J.S., Houtz R.L., Trievel R.C. Structural origins for the product specificity of SET domain protein methyltransferases. Proc. Natl. Acad. Sci. U.S.A. 2008;105:20659–20664. doi: 10.1073/pnas.0806712105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo J.U., Su Y., Shin J.H., Shin J., Li H., Xie B., Zhong C., Hu S., Le T., Fan G., et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014;17:215–222. doi: 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gabel H.W., Kinde B., Stroud H., Gilbert C.S., Harmin D.A., Kastan N.R., Hemberg M., Ebert D.H., Greenberg M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature. 2015;522:89–93. doi: 10.1038/nature14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou T., Xiong J., Wang M., Yang N., Wong J., Zhu B., Xu R.M. Structural basis for hydroxymethylcytosine recognition by the SRA domain of UHRF2. Mol. Cell. 2014;54:879–886. doi: 10.1016/j.molcel.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 36.Rampal R., Alkalin A., Madzo J., Vasanthakumar A., Pronier E., Patel J., Li Y., Ahn J., Abdel-Wahab O., Shih A., et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014;9:1841–1855. doi: 10.1016/j.celrep.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y., Xiao M., Chen X., Chen L., Xu Y., Lv L., Wang P., Yang H., Ma S., Lin H., et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol. Cell. 2015;57:662–673. doi: 10.1016/j.molcel.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Golla J.P., Zhao J., Mann I.K., Sayeed S.K., Mandal A., Rose R.B., Vinson C. Carboxylation of cytosine (5caC) in the CG dinucleotide in the E-box motif (CGCAG|GTG) increases binding of the Tcf3|Ascl1 helix-loop-helix heterodimer 10-fold. Biochem. Biophys. Res. Commun. 2014;449:248–255. doi: 10.1016/j.bbrc.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L., Zhou Y., Xu L., Xiao R., Lu X., Chen L., Chong J., Li H., He C., Fu X.D., et al. Molecular basis for 5-carboxycytosine recognition by RNA polymerase II elongation complex. Nature. 2015;523:621–625. doi: 10.1038/nature14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.