

Abstract
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor activated by a range of oxidants and electrophiles. The transcriptional response to endogenous oxidative cues by Nrf2 plays an important role in mammalian redox physiology and oxidative pathology. Hyperglycemia induces oxidative stress in the heart where it leads to apoptosis and ultimately cardiomyopathy. Here we investigated the mechanism by which Nrf2 suppresses oxidative stress in diabetic mouse heart. Knockout (KO) of Nrf2 induced oxidative stress and apoptosis in KO heart; diabetes further increased oxidative damage. A pathway-focused gene array revealed that Nrf2 controls the expression of 24 genes in the heart, including the gene encoding thioredoxin-interacting protein (TXNIP). Nrf2 suppressed the basal expression of Txnip in the heart and blocked induction of Txnip by high glucose by binding to an antioxidant response element (ARE) (−1286 to −1276) of the Txnip promoter. Binding of Nrf2 to ARE also suppressed the binding of MondoA to the carbohydrate response element with or without high glucose. TXNIP promoted reactive oxygen species production and apoptosis by inhibiting thioredoxin. On the other hand, Nrf2 boosted thioredoxin activity by inhibiting Txnip. The findings revealed, for the first time, that Nrf2 is a key gatekeeper of Txnip transcription, suppressing both its basal expression and MondoA-driven induction to control the thioredoxin redox signaling in diabetes.
Introduction
Diabetes mellitus is a growing public health issue because of its fast-rising incidence worldwide and severe consequences from its complications (Zimmet et al., 2001). Diabetic cardiomyopathy, a complication causing heart failure, can occur independently of vascular complications, such as coronary artery disease and hypertension, thus representing a distinct disease process (Francis, 2001; Murarka and Movahed, 2010). The mechanism by which diabetes causes cardiomyopathy remains largely unclear. Diabetic hyperglycemia promotes the production of reactive oxygen species (ROS) to result in oxidative stress, apoptosis, and inflammation responsible for progressive deterioration of the structure and function of organ systems (Brownlee, 2001; Rösen et al., 2001). These findings imply that factors and pathways controlling ROS production and the cellular redox state hold a key to the development of diabetes and diabetes complications including cardiomyopathy (Giacco and Brownlee, 2010).
ROS is counterbalanced by a web of antioxidant systems in the body. The thioredoxin (Trx) system is a major intracellular thiol-reducing and ROS-scavenging component (Nordberg and Arnér, 2001; Ma, 2010). Trx reduces hydrogen peroxide via peroxiredoxin (Prx) and oxidized Trx is reduced by Trx reductase. Oxidation-reduction of Trx occurs at its two cysteine residues, Cys32 and Cys35. A unique feature of the Trx circuitry is that Trx is regulated by thioredoxin-interacting protein (TXNIP) (Chen and DeLuca, 1994; Nishiyama et al., 1999; Schulze et al., 2004). TXNIP binds reduced Trx and prevents it from reducing Prx, thereby blocking cyclic Trx redox reactions. TXNIP has been implicated in the development of type 2 diabetes through several mechanisms, including promoting β-cell apoptosis by regulating Trx2 and ASK1 in the mitochondria (Saxena et al., 2010), inhibiting glucose uptake in fat tissue and skeletal muscle to increase insulin resistance (Parikh et al., 2007), stimulating glucose production in the liver (Chutkow et al., 2008), and regulating adipogenesis (Chutkow et al., 2010) and fatty acid utilization (Oka et al., 2006). TXNIP promotes oxidative damage in vascular endothelia to cause vascular pathology (Schulze et al., 2004) and regulates Schwann cell reactions in peripheral nerve repair through a receptor for the advanced glycation end products-TXNIP axis to induce peripheral nerve injury (Sbai et al., 2010). TXNIP inhibits cell cycle progression and serves as a tumor suppressor in several tumor models (Han et al., 2003; Jeon et al., 2005; Sheth et al., 2006; Nishizawa et al., 2011). In addition, TXNIP is required for the development of natural killer cells, which is disrupted in TXNIP-deficient mice (Lee et al., 2005). As a member of the mammalian α-arrestin proteins, TXNIP contains an arrestin domain that exhibits some metabolic regulatory activities independently of Trx binding (Patwari et al., 2009).
Expression of TXNIP is highly induced by glucose but is inhibited by insulin (Schulze et al., 2004; Parikh et al., 2007). This tight regulation of TXNIP by the opposing key regulatory signals of metabolism makes TXNIP a fitting glucose-and insulin-sensitive homeostatic switch for the control of glucose uptake in the periphery and glucose production in the liver. In diabetes, TXNIP is persistently elevated and is implicated in β-cell apoptosis, peripheral insulin resistance, and defective glucose homeostasis (Parikh et al., 2007). The molecular mechanism that underlies the induction and inhibition of TXNIP expression remains largely unknown. Recent studies revealed that glucose- and adenosine-containing molecules induce TXNIP by activating MondoA and Max-like protein X, which form a heterodimer to bind to two carbohydrate responsive elements (ChoREs), each with two copies of E-box sequences, in the enhancer of Txnip to transactivate the gene (Minn et al., 2005; Stoltzman et al., 2008; Yu and Luo, 2009; Yu et al., 2009).
The nuclear factor erythroid 2-related factor 2 (Nrf2) has recently emerged as a critical regulator of resistance to ROS and electrophiles (Kensler et al., 2007; Ma, 2008; Taguchi et al., 2011; Ma and He, 2012). Nrf2 protects against glucose toxicity in cardiomyocytes by inhibiting ROS production (He et al., 2009). Here we report that Nrf2 is critical for defense against oxidative stress in diabetic cardiomyopathy. Nrf2 regulates Trx redox signaling by suppressing the basal and induced expression of Txnip in vivo and in vitro. Nrf2 binds to an ARE of the Txnip promoter; binding is increased by high glucose and binding inhibited the binding of MondoA to ChoRE. Therefore, Nrf2 functions as a key gatekeeper of Txnip transcription to keep Txnip basal expression at a low level and to suppress MondoA-driven induction of Txnip in diabetes.
Materials and Methods
Animal Treatment
Nrf2 knockout (KO) mice were originally provided by Dr. Y. W. Kan (University of California San Francisco, San Francisco, CA) and were rederived and backcrossed to a C57BL/6 genetic background as described previously (Chan et al., 1996; Ma et al., 2004). C57BL/6J mice [wild-type (WT)] from The Jackson Laboratory (Bar Harbor, ME) were used as control. Mice were maintained at an environmentally controlled animal facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. All animal studies were approved by the Animal Care and Use Committee at the National Institute for Occupational Safety and Health.
Male WT and KO mice (8 weeks old) were given streptozotocin (STZ) (150 mg/kg b.wt. i.p.; Sigma-Aldrich, St. Louis, MO) to induce diabetes (n = 8 for each genotype and treatment). Sodium citrate (0.1 M, pH 4.5) was used as the solvent control. Tail vein blood glucose was measured by using the OneTouch Ultra Blood Glucose Monitoring System (Lifescan, Milpitas, CA). Mice with blood glucose levels ≥250 mg/dl were considered diabetic. Mice with blood glucose levels ≥600 mg/dl were supplemented with insulin to keep blood glucose levels less than 600 mg/dl to avoid lethality. Mice were sacrificed 2 weeks after STZ injection.
Cell Culture and Treatment
H9C2 (rat cardiomyoblasts; American Type Culture Collection, Manassas, VA) were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum and 5% CO2 at 37°C. Adult mouse ventricular myocytes (AMVM) were isolated as described before (He et al., 2009). In brief, 8-week-old, male mice were euthanized with sodium pentobarbital. The heart was removed and perfused with Krebs-Henseleit bicarbonate buffer (KHB) using a peristaltic pump at a constant rate of 3 ml/min for 5 min, followed by a low Ca2+ KHB for 10 min. The heart was then immersed in recirculating KHB with low Ca2+-containing collagenase B for 30 min. The left ventricle were minced, placed in a 50-ml tube, adjusted to 25 ml with low Ca2+ KHB, and centrifuged at 50g for 2 min. The supernatant was aspirated. The KHB Ca2+ was increased in 3 increments (0.08, 0.6, and 1.2 mM). The mixture was filtered through a 225-μm nylon mesh and centrifuged at 50g for 2 min. Centrifugation was repeated until the preparation was composed of 80% viable left ventricular myocytes. Only myocytes with rod shape and clear striations but no blebs and spontaneous contraction were used for further analysis. AMVM were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum and 5% CO2 at 37°C, under which condition AMVM were typically viable and suitable for biochemical and molecular analyses for 24 to 48 h after isolation. Mouse embryonic fibroblasts were prepared from Nrf2 WT and KO embryos and were immortalized as described previously (He and Ma, 2009).
Immunofluorescent Staining
Heart tissues were fixed, embedded in paraffin, and sectioned in 5-μm thickness. Slides were treated with sodium citrate (10 mM, pH 6.0) for 20 min in a microwave (output 1000 W, frequency 2450 mHz) to retrieve antigens. Slides were then blotted with mouse IgG for 1 h at room temperature and incubated with mouse monoclonal anti-TXNIP antibody (K02053; MBL International, Woburn, MA) at 1:1000 dilution or anti-8-hydroxydeoxyguanosine (8-OHdG) (Japan Institute for the Control of Age, Fukuro, Japan) at 1:100 dilution, in a mouse protein dilution buffer (Vector Laboratories, Burlingame, CA) overnight at 4°C, followed by Alexa Fluor 488 conjugated second antibody. The slides were mounted with a mounting solution with 4′,6-diamidino-2-phenylindole (Vectorshield, Vector Laboratories), which counterstained the nuclei. Fluorescence images were obtained with a Zeiss LSM 510 confocal microscope with a fluorescein isothiocyanate-DAPI setting and equal exposure time. Quantification of fluorescence intensity was performed by using Optimums software (version 6.51; Media Cybernetics, Silver Spring, MD). Quantification data represent means and S.D. from five separate fields for each treatment.
Detection of ROS
AMVM or H9C2 cells were cultured in four-well chamber slides and were treated as indicated. Thirty minutes before the end of treatment dihydroethium (DHE) (Invitrogen, Carlsbad, CA) was added at 5 μM as a fluorescent indicator of ROS production; DHE is oxidized by superoxide anion radical and intercalates into DNA to fluoresce. Cells were washed with ice-cold phosphate-buffered saline for 3 times, fixed with 4% paraformaldehyde, and mounted with the DAPI mounting solution. Fluorescent images were obtained with a Zeiss LSM510 confocal microscope using a rhodamine-DAPI setting, and DHE fluorescence intensity was quantified as described for immunofluorescent staining.
TUNEL Assay
Heart sections in paraffin were examined for apoptosis by using the DeadEnd Fluorometric TUNEL system (Promega, Madison, WI). Slides were deparaffinized, rehydrated, fixed with 4% formaldehyde, and permeated with protease K. Slides were then labeled with a terminal deoxynucleotidyl transferase reaction mixture for 90 min and were mounted with the DAPI mounting solution. Fluorescent images of apoptotic cells (green) and nuclei (blue) were obtained and fluorescence intensity was quantified as described for immunofluorescent staining.
Caspase-Glo Assay
H9C2 cells grown in six-well plates were transfected with control siRNA or Txnip siRNA. Twenty-four hours later, the cells were detached, and 100 μl of cell suspension were transferred to a 96-well plate. The cells were then treated with glucose (40 mM) for 24 h. Caspase-Glo 3/7 reagent (100 μl each) (Promega) was added to each well, briefly mixed on an orbital shaker for 30 s, and incubated at room temperature for 30 min. Caspase 3/7 activities were measured as luminescence intensity with a luminescence plate reader.
Multiparameter Flow Cytometry
Apoptosis of H9C2 cells were assayed with the multiparameter flow cytometry as described previously (Hu et al., 2006).
Northern Blotting
Total RNA was prepared from tissue homogenate or cultured cells with the RNeasy kit (QIAGEN, Valencia, CA). Total RNA (3 μg/lane) was fractionated in a 1% agarose-formaldehyde gel, transferred to a Nytran membrane, and probed with a digoxigenin-labeled riboprobe specific for mouse Txnip or actin. mRNA was visualized by chemiluminescence using CDP-Star as substrate (Roche Molecular Biochemicals, Indianapolis, IN).
Immunoblotting
Fresh tissues were snap-frozen in liquid nitrogen and stored at −80°C until use. Tissues were homogenated with the Zirconia beads. Proteins were extracted from tissue homogenate with the T-PER tissue protein extraction reagent containing Halt protease inhibitors (Thermo Fisher Scientific, Waltham, MA). For cultured cells, cells were lysed on ice with radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitors for 30 min. Cell homogenate was sonicated briefly and centrifuged at 14,000g for 20 min. Proteins (40–60 μg each sample) were separated on a 4 to 20% SDS-polyacrylamide gel electrophoresis gradient gel (Bio-Rad Laboratories, Hercules, CA), transferred to polyvinylidene difluoride membranes, and blocked with 5% nonfat milk for 1 h at room temperature. The membrane was blotted with anti-TXNIP, Nrf2, and actin antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) overnight at 4°C with gentle shaking, followed by a horse-radish peroxidase-conjugated secondary antibody at room temperature for 1 h and visualization by chemiluminescence using ECL (Thermo Fisher Scientific).
Real-Time PCR
Total RNA was reverse-transcribed into single-strand cDNAs and analyzed by real-time PCR using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) following standard procedures. Primer sequences used for real-time PCR are available upon request. Representative data were from three separate experiments.
Pathway Gene Expression Array
Mouse heart RNA was extracted with TRIzol reagent (Invitrogen) followed by purification with a QIAGEN RNeasy Mini Kit (QIAGEN). Five micrograms of RNA was reverse-transcribed into cDNA with the RT2 First Strand Kit (SABiosciences, Frederick, MD). Synthesized cDNA was diluted to appropriate concentrations and a PCR array was performed with the Mouse Oxidative Stress and Antioxidant Defense PCR Array system (SABiosciences) using an ABI-7900 HT real-time PCR machine. PCR array data were analyzed by using SABiosciences software. The PCR array profiles the expression of 84 genes related to oxidative stress. The genes include peroxidases, such as glutathione peroxidases and peroxiredoxins, and antioxidants; genes involved in ROS metabolism, such as superoxide dismutases, and oxidative stress responsive genes; and oxygen transporters.
Cloning and Mutagenesis of Txnip Promoter and Luciferase Reporter Assay
The promoter region (−1 to −2000) of mouse Txnip gene was amplified by PCR and subcloned into the pGL3-Luc basic vector at KpnI and XhoI sites (designated pTxnip-P). The promoter sequence was confirmed by DNA sequencing. Deletion mutant pTxnip-P-ΔARE lacks the ARE between −1286 and −1276 base pairs. Deletion mutant pTxnip-P-ΔTGAC lacks the ARE core sequence TGAC. The reporter plasmids were each transfected into H9C2 cells using Lipofectamine Plus reagent (Invitrogen). Twenty-four hours after transfection, the cells were treated with glucose for 16 h and lysed with a passive reporter lysis buffer (Promega). Cell lysate was vortexed and centrifuged briefly to remove cell debris. Then 20 μl of supernatant was mixed with 100 μl of luciferase reagent (Promega). Luciferase activity was detected using a TD-20/20 luminometer and was normalized to protein concentrations.
ChIP Assay
ChIP was performed as described previously with modifications (He et al., 2006). Immunoprecipitation was performed with rabbit anti-Nrf2, anti-MondoA antibody, or normal rabbit IgG (as a control) (Santa Cruz Biotechnology, Inc.). Primer sequences for amplifying ARE or ChoRE-a of Txnip were available upon request.
siRNA Knockdown
H9C2 cells were transfected with either control siRNA (Santa Cruz Biotechnology, Inc.), TXNIP siRNA (SiGENOME SMARTpool, Thermo Fisher Scientific; Dharmacon Products, Lafayette, CO), or rNrf2 siRNA (Invitrogen) with the Lipofectamine Plus reagents. Knockdown of TXNIP or Nrf2 was confirmed by immunoblotting of TXNIP or Nrf2 protein.
Trx Activity Assay
Trx activity was measured using the endpoint insulin assay described elsewhere (Holmgren, 1985). In brief, cell extracts of 60 μg were incubated at 37°C for 15 min with activation buffer to reduce thioredoxin. After addition of reaction buffer, the reaction was started with bovine thioredoxin reductase, and incubation continued for 1 h at 37°C. The reaction was terminated by addition of stopping buffer, followed by absorption measurement at 412 nm.
Statistical Analysis
Statistic analysis was performed with one-way analysis of variance followed by a t test using Microsoft Excel. p < 0.05 was considered statistically significant.
Results
Nrf2 Suppresses Oxidative Stress and Apoptosis in Normal and Diabetic Mouse Hearts
We used Nrf2 KO mice and a STZ-induced diabetes model to analyze Nrf2 function in diabetic heart in vivo. STZ-induced diabetes developed more rapidly and severely in KO than WT mice. The mean whole blood glucose level is 560 mg/dl for KO and 380 mg/dl for WT mice, respectively (Fig. 1A). Some KO mice had blood glucose levels exceeding 600 mg/dl and were supplemented with insulin to keep the level at less than 600 mg/dl to avoid lethality.
Fig. 1.

Diabetes induced marked oxidative stress and apoptosis in Nrf2 KO cardiomyocytes. A, B, and D, WT and Nrf2 KO mice (8 weeks old, male) were given STZ (150 mg/kg b.wt.) or 0.1 M citrate buffer (vehicle control) intraperitoneally. Mice were sacrificed, and heart tissues were examined 2 weeks after STZ injection. A, blood glucose level. Tail blood glucose levels were measured at the end of the experiment (n = 8). B, 8-OHdG formation. Oxidative DNA damage was assayed by immunofluorescent staining of 8-OHdG in heart sections. C, ROS production. AMVM were isolated from WT and KO mouse hearts, treated with glucose (30 mM, 18 h), and stained with DHE for production of ROS. Fluorescence intensities were quantified and are shown as means and S.D. from four to six samples. D, apoptosis. Apoptosis was measured by using the DeadEnd Fluorometric TUNEL assay of heart sections. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Examination of the heart from normal and diabetic mice revealed a significantly higher level of oxidative stress in KO than in WT heart with or without diabetes. The amount of 8-OHdG, a marker of oxidative damage to DNA, was significantly higher in control KO than in control WT heart, indicating that Nrf2 is required for suppression of the basal level oxidative damage to DNA (Fig. 1B). Diabetes increased the levels of 8-OHdG significantly as expected, but the increase was higher in KO than in WT hearts, suggesting a protective role of Nrf2 against diabetes-induced oxidative damage to DNA.
ROS production was directly measured in isolated WT and KO ventricular cardiomyocytes (AMVM) with the use of ROS fluorophore DHE (Fig. 1C). In agreement with the 8-OHdG result, ROS level was significantly higher in control KO than in WT AMVM (3-fold). High glucose significantly increased ROS production in both WT and KO than in control AMVM (2.5- and 2-fold, respectively). The ROS level was significantly higher in diabetic KO than in diabetic WT heart (2.4-fold).
Heightened oxidative stress in diabetic KO heart was accompanied with marked pathological changes including apoptosis of cardiomyocytes compared with normal KO and diabetic WT heart, which is indicative of cardiomyopathy (Fig. 1D). Apoptosis was significantly higher in control KO than in control WT heart (2.5-fold). Diabetes increased apoptosis in WT heart by 3.5- and in KO heart by 3.8-fold compared with that in WT and KO controls, respectively. Apoptosis in diabetic KO heart was significantly higher than that in diabetic WT heart (2.6-fold). These and previous data obtained with ventricular cardiomyocytes (He et al., 2009) demonstrated that Nrf2 is required for suppressing both the basal and high glucose-induced oxidative stress and apoptosis in cardiomyocytes. The above observed pathology was not due to toxicity of STZ on the heart or organs other than islet β-cells but was attributed to hyperglycemia induced by STZ, because mice treated with STZ followed by insulin had normal levels of blood glucose and did not develop oxidative damage in the heart (data not shown).
Nrf2 Regulates the Expression of Multiple Genes in the Oxidative Stress and Antioxidant Defense Pathways in Diabetic Mouse Heart
Because Nrf2 is a transcription factor, we posit that Nrf2 directly regulates pathways that control ROS homeostasis and antioxidant defense in the heart by modulating the transcription of critical genes of the pathways to influence the onset and severity of diabetic heart pathology. To test this notion, we performed an oxidative stress and antioxidant defense pathway-focused gene expression array using WT and KO mouse heart RNA. Total RNA pooled from four hearts of each genotype was analyzed by PCR array.
Among the 84 genes examined, 6 genes were up-regulated (Table 1), and 18 genes were down-regulated (Table 2), all with fold changes ≥1.8 in KO heart compared with WT heart. The genes are involved in a range of cellular functions that center on ROS homeostasis and antioxidant defense. These include production and detoxification of superoxide and peroxides, mitochondrial respiration and ROS production, detoxification of ROS-promoting chemicals, antioxidants, oxygen transporters, and oxidative stress responsive genes. For example, Scd1, which encodes stearoyl-CoA-desaturase 1, and Txnip, which encodes Trx-interacting protein, have been associated with insulin resistance and oxidative stress in diabetes; both genes were up-regulated in Nrf2 KO hearts by 8-and 4-fold, respectively. On the other hand, a number of cellular protective genes were down-regulated in the absence of Nrf2, including Mpp4 (membrane palmitoylated protein 4, 4.9-fold), Lpo (lactoperoxidase, 2.5-fold), Gpx8 (glutathione peroxidase 8, 2.3-fold), Srxn1 (sulfiredoxin 1, 2.1-fold), Noxo1 (NADPH oxidase organizer 1, 2.0-fold), Mpo (myeloperoxidase, 1.8-fold), and Nqo1 (NADPH:quinone oxidoreductase 1, 1.8-fold). The findings support the notion that Nrf2 protects against oxidative stress and other lesions in the heart under physiological conditions by regulating the transcription of oxidative and antioxidative genes.
TABLE 1.
Up-regulated genes of oxidative stress/antioxidant defense pathways in Nrf2 KO heart
| Gene Symbol | Fold Change | Function | Oxidative Stress Pathway |
|---|---|---|---|
| Scd1 | 8.1 | Stearoyl-CoA desturase-1; fatty acid and cholesterol metabolism, insulin resistance, oxidative stress | Superoxide production |
| Txnip | 4.2 | Thioredoxin-interacting protein; energy metabolism, ROS metabolism, insulin resistance, β-cell function | Inhibiting reduced thioredoxin |
| Ccs | 2.6 | Copper chaperone for superoxide dismutase; controls copper incorporation into SOD1 | Copper chaperone for SOD1 |
| Hbq1 | 2.5 | Hemoglobin subunit theta 1; early erythroid function | Oxygen transport |
| Noxa1 | 2.3 | NADPH oxidase activator 1; regulates superoxide anion production and redox signaling through NOX | NOX activator |
| Vim | 1.9 | Vimentin; cell attachment, migration, and signaling | Oxygen transport |
NOX, NADPH oxidase.
TABLE 2.
Down-regulated genes of oxidative stress/antioxidant defense pathways in Nrf2 KO heart
| Gene Symbol | Fold Change | Function | Oxidative Stress Pathway |
|---|---|---|---|
| Mpp4 | 4.9 | Membrane palmitoylated protein 4; calcium homeostasis, ROS production, photoreceptor signaling | Oxidative stress responsive gene |
| Ift172 | 3.0 | Intraflagellar transport 172; intraflagellar transporter, oxygen transporter | Oxygen transport |
| Rag2 | 3.0 | Recombination activating gene 2; immune, inflammation, ROS response | Peroxidase function |
| IL22 | 2.6 | Interleukin 22; inflammation, ROS production, natural killer and T-cell functions | Regulates ROS metabolism |
| Lpo | 2.5 | Lactoperoxidase; innate immune function, bacterial killing | Peroxidase function |
| Recql4 | 2.4 | ATP-dependent DNA helicase Q4; skeleton and skin development, ROS production | Superoxide metabolism |
| Gpx8 | 2.3 | Glutathione peroxidase 8; peroxide metabolism, antioxidant function | Glutathione peroxidase |
| Srxn1 | 2.1 | Sulfiredoxin 1; reduction of peroxiredoxin-sulfinic acid (Prx-SO2H) to sulfenic acid (Prx-SOH) | Reduce Prx for peroxide metabolism |
| Ngb | 2.1 | Neuroglobin; protection against hypoxia and ROS injuries | Oxygen transport |
| Noxo1 | 2.0 | NADPH oxidase organizer 1; regulates superoxide production | Regulates NOX to produce superoxide |
| Prnp | 2.0 | Major prion protein; prion protein, glucose uptake, transport of Cu2+ | Oxidative stress responsive gene |
| Gpx2 | 2.0 | Glutathione peroxidase 2; detoxification of hydrogen peroxide | Glutathione peroxidase |
| Gpx6 | 1.9 | Glutathione peroxidase 6; detoxification of hydrogen peroxide | Glutathione peroxidase |
| Ercc2 | 1.9 | Excision repair cross-complementing rodent repair deficiency, complementation group 2; transcription-coupled excision repair | Oxidative stress responsive gene |
| Ptgs2 | 1.8 | Prostaglandin-endoperoxide synthase 2; prostaglandin biosynthesis | Prostaglandin metabolism |
| Mpo | 1.8 | Myeloperoxidase; inflammation, innate immune function, oxidant production | Myeloperoxidase function |
| Nqo1 | 1.8 | NAD(P)H:quinone oxidoreductase 1 | Two electron reduction of quinones |
| Cygb | 1.8 | Cytoglobin; oxygen transport, scavenging NO and ROS | Oxygen transport, antioxidant |
NOX, NADPH oxidase; NO, nitric oxide.
Nrf2 Negatively Controls the Basal and Diabetes-Induced Expression of Txnip
Given the prominent role of TXNIP in hyperglycemia-induced oxidative pathology (Nishiyama et al., 1999; Wang et al., 2002; Schulze et al., 2004; Minn et al., 2005; Muoio, 2007; Parikh et al., 2007; Chen et al., 2008) and a strong inhibition of Txnip expression by Nrf2 (Table 1), we further analyzed the regulation of Txnip by Nrf2. Real-time PCR revealed that the Txnip mRNA level was low in WT heart but was significantly elevated in KO heart (6-fold), confirming a critical role of Nrf2 in suppressing the basal expression of Txnip mRNA (Fig. 2A). The protein level of TXNIP was barely detectable in normal WT heart but was markedly increased in KO heart (Fig. 2B). The results indicate that suppression by Nrf2 is a major mechanism by which the expression of Txnip mRNA and protein is kept at low levels in normal heart.
Fig. 2.
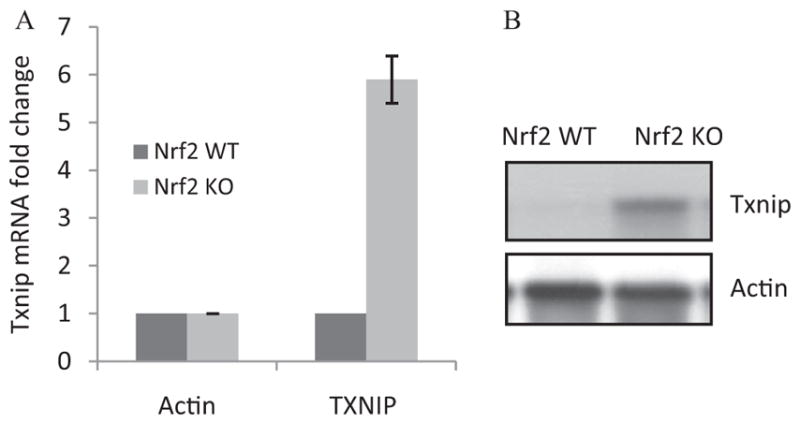
Nrf2 negatively controls the basal expression of Txnip in mouse heart. Heart tissues from adult male WT and KO mice were collected. A, Txnip mRNA expression. Total RNA was analyzed for Txnip mRNA expression by real-time PCR (n = 4 for each genotype). B, TXNIP protein expression. Heart homogenate pooled from four mice was examined for TXNIP protein levels by immunoblotting.
Nrf2 protein was detected in WT but not KO heart as expected. Nrf2 protein was significantly increased by STZ in WT heart, indicating activation of Nrf2 in diabetes (Fig. 3A). TXNIP protein was significantly induced by STZ in WT heart (Fig. 3B). TXNIP protein in control KO heart was markedly higher than that in control WT heart and was further increased by STZ to a level even higher than that of diabetic WT heart. Immunofluorescent staining of TXNIP protein in heart tissue showed that expression of TXNIP protein was very low in control WT heart (Fig. 3, C and D). Diabetes significantly increased TXNIP level in WT heart (2.3-fold). Expression of TXNIP in control KO heart was significantly induced (2.5-fold, compared with that in control WT heart). Diabetes further increased TXNIP expression in diabetic KO heart (2.9-fold, compared with that in control WT heart). Analysis of Txnip mRNA expression in diabetic heart revealed results similar to those with TXNIP protein. These findings demonstrated that Nrf2 suppresses TXNIP expression in diabetic heart.
Fig. 3.

Nrf2 suppresses the induction of TXNIP by diabetes in mouse heart. Adult male WT and KO mice were treated with STZ to induce diabetes. Heart tissues were collected as described for Fig. 1, immunoblotting. Heart homogenate was immunoblotted for Nrf2 (A) or TXNIP (B) and quantified. Actin was blotted for loading variation. C, immunofluorescent staining. Heart sections were stained with anti-TXNIP and green fluorescence. DAPI was used to stain for nuclei. D, quantification of TXNIP immunofluorescence intensity from C. Data represent means and S.D. from four to six samples. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To analyze the mechanism of TXNIP regulation by Nrf2, we examined TXNIP expression in cultured H9C2 cells. Both Txnip mRNA (Fig. 4A) and protein (Fig. 4B) were expressed in H9C2 at low levels. High glucose significantly induced Txnip mRNA and protein expression. Overexpression of Nrf2 in H9C2 cells reduced Txnip mRNA and protein expression in the absence of high glucose and blocked induction of Txnip mRNA and protein in the presence of high glucose. On the other hand, knocking down of Nrf2 in H9C2 cells by transfecting Nrf2-specific siRNA increased both the basal and high glucose-induced expression of TXNIP protein (Fig. 4C, top). Expression of Nrf2 protein was increased by high glucose in H9C2 cells transfected with control siRNA but was significantly down-regulated by Nrf2 siRNA as expected (Fig. 4C, middle). Taken together, both in vivo and in vitro data revealed that Nrf2 is activated in diabetic heart and that Nrf2 suppresses both the basal and high glucose-induced expression of Txnip mRNA and protein in vivo and in vitro.
Fig. 4.

Nrf2 suppresses the basal and glucose-induced expression of Txnip in H9C2. Cells were transfected with Nrf2-expressing vector (pCMV-Nrf2) (A and B) or Nrf2 siRNA (C). Control vector (pCMV) (A and B) or control siRNA (C) was used as a control. The cells were then treated with glucose (40 mM, 16 h). A, Northern blotting of Txnip mRNA. B, immunoblotting of TXNIP protein. C, immunoblotting of TXNIP and Nrf2 protein expression. Actin was blotted as the loading control.
Nrf2 Inhibits Txnip Transcription
To examine whether Nrf2 controls Txnip transcription, the regulatory region of the Txnip promoter from −1 to −2000 upstream of the transcription start site was cloned into a luciferase reporter vector (designated pTxnip-P) (Fig. 5A, top). Luciferase was induced by high glucose in H9C2 cells transfected with pTxnip-P by 3-fold (Fig. 5A). Cotransfection of the reporter with an Nrf2 expression plasmid (pCMV-Nrf2) reduced high glucose-induced expression of the luciferase reporter by 47% (Fig. 5B). On the other hand, cotransfection of the reporter vector with Nrf2 siRNA increased luciferase expression by 3-fold, and high glucose further induced the expression of the reporter in Nrf2 knockdown cells (5-fold) (Fig. 5C). The findings demonstrate that Nrf2 suppresses transcription of the Txnip promoter under both basal and high-glucose conditions.
Fig. 5.

Nrf2 inhibits the basal and induced transcription by Txnip promoter. A, induction of Txnip promoter-driven luciferase expression. The Txnip promoter sequence from −1 to −2000 was sub-cloned into the pGL3-basic/Luc vector to generate the promoter-reporter plasmid pTxnip-P. H9C2 cells were transfected with the plasmid, followed by treatment with glucose. Luciferase activity was measured. B, suppression of Txnip promoter transcription by overexpressing Nrf2. H9C2 cells were cotransfected with pTxnip-P and pCMV-Nrf2, followed by treatment with glucose. C, increase in Tx-nip promoter transcription by knocking down Nrf2. H9C2 cells were cotransfected with pTxnip-P and Nrf2 siRNA, followed by treatment with glucose. Luciferase activities were measured and are expressed as means and S.D. from 3 samples. *, p < 0.05; **, p < 0.01.
Inspection of the promoter sequence of Txnip revealed a putative ARE element from −1286 to −1276 that is identical to the known ARE core sequence and highly similar to an ARE-like consensus sequence (Fig. 6A) (Nguyen et al., 2003; Hayes et al., 2010). The role of ARE in Txnip suppression was examined with Txnip promoter mutation; pTxnip-P-ΔARE lacks ARE from −1286 to −1276 base pairs and pTxnip-P-ΔTGAC lacks the ARE core sequence (Fig. 6B). Wild-type pTxnip-P supported induction of reporter expression by glucose and suppression of the basal and glucose-stimulated expression by Nrf2 as expected (Fig. 6B, lanes 1–4). Deletion of ARE increased basal expression (Fig. 6B, compare lane 5 with 1) and further increased induction by glucose (Fig. 6B, compare lane 6 with lane 2) but did not support suppression by Nrf2 on basal expression (Fig. 6B, compare lane 7 with 5) and induction by glucose (Fig. 6B, compare lane 8 with 6). Deletion of the ARE core sequence resulted in similar results (Fig. 6B, lanes 9–12). Therefore, ARE is required for suppression of basal and glucose-induced transcription activities of the Txnip promoter.
Fig. 6.

Interaction between Nrf2 and Txnip ARE. A, sequences of ARE and ChoRE-a of Txnip and alignment with ARE core and E-box consensus. A putative ARE sequence was identified in Txnip promoter (−1286 to −1276) that matches the ARE core sequence and is highly similar to the ARE-like sequence consensus. Shown also is the Txnip ChoRE-a sequence and its alignment with E-box consensus. N or n = G, C, T, or A; M = A or C; R = A or G; and Y = C or T. Positions represent base pairs upstream of the transcription start site of mouse Txnip. References are shown in brackets: [41] = Nguyen et al. (2003); [42] = Hayes et al. (2010); and [31,33] = Minn et al. (2005). B, mutational analysis of the Txnip promoter. pTxnip-P or a deletion mutant of Txnip promoter, pTxnip-P-ΔARE or pTxnip-P-ΔTGAC, was transfected into H9C2 cells with or without pCMV-Nrf2 and glucose treatment as described for Fig. 5. Luciferase activities were measured and are expressed as means and STD from three samples. Statistical significance was shown for some comparisons; statistical significance for the pTxnip-P-ΔTGAC group (lanes 9–12) (not shown) is similar to that for the pTxnip-P- ΔARE group (lanes 5–8). *, p < 0.05; **, p < 0.01. C, ChIP assay. H9C2 cells were treated with glucose. ChIP analyses were performed to measure binding of Nrf2 to the putative Txnip ARE and of MondoA to ChoRE-a.
To directly examine whether Nrf2 binds to Txnip ARE in intact cells, ChIP analysis was performed. Figure 6C showed that Nrf2 binds to Txnip ARE in the absence of glucose in WT cells; high glucose further increased binding. As expected, no binding was observed in Nrf2 KO cells with or without glucose, confirming that binding is specific for Nrf2. Induction of Txnip by glucose involves binding of MondoA, a positive regulator of Txnip transcription, to ChoRE-a (−86 to −65) and ChoRE-b (−173 to −153) of the Txnip promoter (Fig. 6A, bottom) (Minn et al., 2005; Yu and Luo, 2009); therefore, binding of MondoA to ChoRE-a was compared with binding of Nrf2 to ARE. Binding of MondoA to ChoRE-a was barely detectable in the absence of glucose but was significantly increased in the presence of glucose in Nrf2 WT cells, consistent with its role for induction of Txnip by glucose (Fig. 6B, middle). In Nrf2 KO cells, binding of MondoA was readily detected and was higher than glucose-induced binding in WT cells; glucose further increased the binding. The results demonstrate that Nrf2 suppresses Txnip expression and induction by binding to Txnip ARE; moreover, Nrf2 suppression is the major mechanism responsible for the low basal expression of Txnip and for suppression of Txnip induction by glucose, both of which involve inhibition of binding of MondoA to ChoREs.
Nrf2 Stimulates Trx Activity by Inhibiting Txnip Expression
We tested whether TXNIP is responsible for high glucose-induced oxidative stress and apoptosis by knocking down TXNIP in H9C2 cells. The basal level of TXNIP protein in H9C2 was low in cells transfected with control siRNA but was induced by high glucose (Fig. 7A). Transfection of Txnip-specific siRNA into H9C2 cells markedly reduced the level of TXNIP protein in the presence of glucose. High glucose induced ROS production by 2.3-fold (Fig. 7, B and C) in control siRNA-transfected cells (Fig. 7, B and C). However, high glucose failed to significantly increase ROS production in cells transfected with Txnip siRNA. High glucose induced apoptosis of H9C2 cells in two separate assays: 1) increase of caspase 3/7 activity (Fig. 8A) and 2) increased percentage of apoptotic cells measured as PhiPhiLux G1D2-positive cells (PhiPhiLux G1D2 is a fluorescence indicator of apoptosis) (Fig. 8B). Knockdown of TXNIP blocked glucose-induced apoptosis in both assays (Fig. 8). These data indicate a critical role of TXNIP in high glucose-induced oxidative stress and apoptosis in H9C2 cells.
Fig. 7.

TXNIP mediates glucose-induced ROS production in H9C2. Cells were transfected with Txnip siRNA followed by treatment with glucose. A, immunoblotting of TXNIP and actin proteins. B, fluorescent staining of ROS. ROS production was examined with DHE fluorescent staining. DAPI was used to stain for nuclei. C, quantification of DHE fluorescence intensity. Data represent means and S.D. *, p < 0.05; **, p < 0.01.
Fig. 8.

TXNIP mediates glucose-induced apoptosis in H9C2. Cells were treated as described for Fig. 7. A, apoptosis measured for caspase 3/7 activity as luminescence intensity using Caspase-Glo. B, apoptosis measured for PhiPhiLux G1D2-positive cells (percentage) using multiparameter flow cytometry. Data represent means and S.D. from three to six samples. **, p < 0.01.
TXNIP promotes oxidative stress by inhibiting Trx, whereas Nrf2 inhibits oxidative stress and TXNIP expression. Therefore, we examined the interplay among Nrf2, TXNIP, and Trx. High glucose induced TXNIP (Fig. 4, A and B) but inhibited Trx activity (Fig. 9A) in H9C2 cells. In the absence of high glucose, knockdown of Txnip did not significantly change Trx activity, which is due to low-level expression of Txnip in H9C2 cells under a basal condition (Fig. 9B) and is consistent with the result from other investigators (Schulze et al., 2004). Knockdown of Nrf2 significantly reduced Trx activity by 25% (Fig. 9B), which is consistent with elevated expression of TXNIP in Nrf2 knockdown cells (Fig. 4). In the presence of high glucose, Trx activity was significantly reduced because of induction of TXNIP by glucose. However, this suppression of Trx by glucose was abolished in Txnip knockdown cells, confirming that TXNIP mediates inhibition of Trx by glucose. Knockdown of Nrf2 reduced Trx activity to a level lower than that for high glucose alone, suggesting that knockdown of Nrf2 increased induction of TXNIP by glucose, which repressed Trx activity. The results indicate that Nrf2 boosts Trx activity in the absence and presence of high glucose by inhibiting Txnip expression.
Fig. 9.

Nrf2 regulates Trx activity through TXNIP. H9C2 cells were transfected with Txnip siRNA or Nrf2 siRNA followed by treatment with glucose. Cell lysate was prepared, and Trx activity was measured to show suppression of Trx by high glucose (A) or regulation of Trx activity by TXNIP and Nrf2 (B). Data represent means and STD from three samples. *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
Discussion
The GSH and the Trx systems are two major intracellular thiol-reducing and ROS-scavenging branches (Ma, 2010). In the GSH system, peroxides are reduced by glutathione peroxidase with GSH as cofactor, and oxidized glutathione is reduced by glutathione reductase. In the Trx system, Trx reduces hydrogen peroxide via Prx, and oxidized Trx is reduced by Trx reductase and with NADPH as cofactor. Trx regulates redox states in a compartment-dependent manner with Trx1 residing in the cytoplasm and nucleus and Trx2 in the mitochondria. The cyclic Trx redox reaction is inhibited by TXNIP, wherein TXNIP binds to reduced Trx and blocks Trx redox cycles. TXNIP is highly induced by glucose in diabetes, and consistently elevated expression of Txnip has been associated with the development of type 2 diabetes and several other pathological and stress conditions (Wang et al., 2002, 2006; Minn et al., 2005; Parikh et al., 2007).
Nrf2 regulates the cellular defense against oxidants and electrophiles by controlling the basal expression and induction of a battery of genes that encode drug-metabolizing enzymes, such as NAD(P)H:quinone oxidoreductase 1 and glutathione transferases, and cytoprotective proteins, such as heme oxygenase 1, through the ARE. A growing body of evidence has indicated that Nrf2 protects animals from cancer, chronic inflammation, neurodegeneration, and chemical toxicity, which are marked with prominent oxidative stress (Kensler et al., 2007; Ma, 2010; Taguchi et al., 2011; Ma and He, 2012). Given the protective role of Nrf2 in diseases associated with oxidative damage, we postulate that Nrf2 directly controls the ROS metabolism and redox state of cells in addition to drug metabolism and cytoprotective functions. The mechanism of such regulation is currently not well understood. In this study, we examined the role and mechanism of action of Nrf2 in the development of diabetic myocardial lesions responsible for the development of diabetic cardiomyopathy, a complication caused by direct toxic effects of high glucose in the heart and associated with heart failure in diabetic patients. The study demonstrated that Nrf2 is required for inhibition of oxidative stress and apoptosis in diabetic heart, and, importantly, revealed a novel molecular mechanism of redox regulation by Nrf2 via transcriptional suppression of the basal and diabetes-induced expression of Txnip in the heart.
Examination of diabetes in Nrf2 KO mice revealed severe structural and functional lesions in the heart marked with heightened oxidative stress and apoptosis in comparison with the WT control. These results are consistent with our previous report that high glucose induced severe oxidative damage and cell death in Nrf2-deficient myocardial cells (He et al., 2009), revealing a critical and intrinsic function of Nrf2 for protection against the development of diabetic lesions in the heart. Loss of Nrf2 also increased inflammation in diabetic heart that may promote oxidative stress and contribute to observed pathological changes in the heart (He and Ma, 2012).
A pathway-focused gene expression array identified 24 genes in the oxidative stress and antioxidant defense pathways whose mRNA expressions were altered in the heart of Nrf2 KO mice, supporting the notion that Nrf2 regulates ROS and redox homeostasis by controlling gene expression. Among the genes, Txnip was up-regulated by 4-fold in Nrf2 KO mice. These and subsequent analyses of Txnip mRNA and protein expression demonstrated that 1) loss of Nrf2 markedly increased the basal expression of Txnip in the heart, 2) diabetes induced Txnip in WT heart and significantly further increased Txnip expression in Nrf2 KO heart, 3) overexpression of Nrf2 suppressed the basal expression and induction of Txnip by glucose, and 4) siRNA knockdown of Nrf2 boosted the basal and glucose-induced expression of TXNIP. These findings, for the first time, identified Nrf2 as a critical regulator of Txnip expression that negatively controls the basal and high glucose-induced expression of Txnip in the heart. This conclusion does not exclude other Nrf2-regulated, redox-active, genes, such as those shown in Tables 1 and 2, for the protective effect of Nrf2 on the heart against oxidative stress.
Induction of Txnip by glucose and certain other inducers, such as adenosine-containing compounds, involves activation of the MondoA-Mlx complex; activated MondoA-Mix dimer accumulates in the nucleus, binds to ChoRE-a (−86 to −65) and ChoRE-b (−173 to −153), and mediates Txnip induction (Minn et al., 2005; Yu and Luo, 2009; Yu et al., 2009). Full induction also requires recruitment of histone H3 acetyl-transferase to promoter-bound MondoA-Mlx as well as nuclear factor Y to two CCAAT boxes of the Txnip promoter (Yu and Luo, 2009; Peterson et al., 2010). The Txnip upstream region (from −1 to −2000) is sufficient to replicate induction of Txnip promoter transcription by glucose and suppression of the basal expression and induction by Nrf2 (Fig. 5), confirming a transcriptional mechanism of Txnip regulation by glucose and Nrf2. Inspection of the Txnip promoter sequence revealed a putative ARE from −1286 to −1276 of the mouse Txnip promoter. The Txnip ARE is identical to the known ARE core sequence and is highly similar to an ARE-like sequence consensus (Fig. 6A). The ARE sequence is required for basal and glucose-induced transcription activities of the Txnip promoter as shown by mutational analysis of the Txnip promoter (Fig. 6B).
Nrf2 binds to Txnip ARE in untreated WT cells and high glucose further increased the binding in intact cells, as demonstrated by ChIP assay (Fig. 6C). Binding was not observed in Nrf2 KO cells with or without high glucose, confirming that binding is Nrf2-specific. Binding of MondoA to Txnip ChoRE in WT cells was detected when cells were treated with high glucose; this result is in agreement with a Txnip induction model in which Txnip is expressed at a low level under basal conditions in many cell types but is induced by glucose through MondoA-driven transcription. In Nrf2 KO cells, binding of MondoA was markedly increased in the absence of high glucose, to a level even higher than that of glucose-induced binding in WT cells, which is consistent with increased basal expression of Txnip in KO cells; these observations indicate that Nrf2 is a principal regulator of Txnip expression under basal conditions by suppressing MondoA binding to ChoRE. In the presence of high glucose, binding of MondoA in KO cells was the highest, indicating that Nrf2 suppresses glucose-stimulated binding of MondoA to ChoRE. Taken together, these data revealed that Nrf2 functions as a key gatekeeper of Txnip transcription, wherein Nrf2 binds to ARE and blocks MondoA binding to ChoRE to keep Txnip expression at a low level under basal conditions; high glucose activates Nrf2 to increase its binding to ARE, which in turn suppresses the binding of high glucose-activated MondoA to ChoRE to inhibit Txnip induction by glucose. Suppression of glucose-induced MondoA binding by Nrf2 may help keep the induction of Txnip at a submaximal level under diabetic and stress conditions, which may be advantageous for cells to survive in the presence of oxidative challenge.
Knockdown of Txnip in H9C2 cells blocked high glucose-induced ROS production and apoptosis, demonstrating that high glucose-induced oxidative stress and apoptosis in cardiomyocytes are, at least in part, attributable to elevated expression of TXNIP (Figs. 7 and 8). Knockdown of TXNIP also blocked glucose-induced inhibition of Trx activity, indicating that TXNIP mediates Trx inhibition in the presence of high glucose (Fig. 9). On the other hand, knockdown of Nrf2 reduced Trx activity in the absence of high glucose and further reduced the activity in the presence of high glucose. These results are in agreement with the notion that high glucose induces TXNIP to inhibit Trx, whereas Nrf2 boosts Trx activity by suppressing Txnip expression under both basal and high glucose conditions.
Identification of TXNIP as a direct target gene of Nrf2 revealed a novel aspect of Nrf2 function in combating oxidative stress and apoptosis in diabetes. In this scenario, the interplay among TXNIP, Trx, and Nrf2 regulates the redox state and oxidative stress to influence the development of diabetes and diabetes complications including cardiomyopathy. Because activation of Nrf2 by natural and synthetic inducers has been shown to be efficacious for the therapy and prevention of cancer and certain chronic diseases, our demonstration of redox regulation by Nrf2 in diabetes suggests the Nrf2-TXNIP-Trx axis as a target for drug development for the treatment of diabetes and potentially other metabolic diseases.
Acknowledgments
This work was supported by the National Institute for Occupational Safety and Health.
ABBREVIATIONS
- ROS
reactive oxygen species
- Trx
thioredoxin
- Prx
peroxiredoxin
- TXNIP
thioredoxin interacting protein
- ChoRE
carbohydrate response element
- Nrf2
nuclear factor erythroid 2-related factor 2
- ARE
antioxidant response element
- KO
knockout
- WT
wild-type
- STZ
streptozotocin
- AMVM
adult mouse ventricular myocytes
- KHB
Krebs-Henseleit bicarbonate buffer
- 8-OHdG
8-hydroxydeoxyquanine
- DAPI
4′,6-diamidino-2-phenylindole
- DHE
dihydroethium
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick-end labeling
- siRNA
small interfering RNA
- ChIP
chromatin immunoprecipitation
Footnotes
Authorship Contributions
Participated in research design: He and Ma.
Conducted experiments: He and Ma.
Contributed new reagents or analytic tools: He and Ma.
Performed data analysis: He and Ma.
Wrote or contributed to the writing of the manuscript: He and Ma.
References
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci USA. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Saxena G, Mungrue IN, Lusis AJ, Shalev A. Thioredoxin-interacting protein: a critical link between glucose toxicity and β-cell apoptosis. Diabetes. 2008;57:938–944. doi: 10.2337/db07-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KS, DeLuca HF. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochim Biophys Acta. 1994;1219:26–32. doi: 10.1016/0167-4781(94)90242-9. [DOI] [PubMed] [Google Scholar]
- Chutkow WA, Birkenfeld AL, Brown JD, Lee HY, Frederick DW, Yoshioka J, Patwari P, Kursawe R, Cushman SW, Plutzky J, et al. Deletion of the α-arrestin protein Txnip in mice promotes adiposity and adipogenesis while preserving insulin sensitivity. Diabetes. 2010;59:1424–1434. doi: 10.2337/db09-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chutkow WA, Patwari P, Yoshioka J, Lee RT. Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J Biol Chem. 2008;283:2397–2406. doi: 10.1074/jbc.M708169200. [DOI] [PubMed] [Google Scholar]
- Francis GS. Diabetic cardiomyopathy: fact or fiction? Heart. 2001;85:247–248. doi: 10.1136/heart.85.3.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SH, Jeon JH, Ju HR, Jung U, Kim KY, Yoo HS, Lee YH, Song KS, Hwang HM, Na YS, et al. VDUP1 upregulated by TGF-beta1 and 1,25-dihydorxyvitamin D3 inhibits tumor cell growth by blocking cell-cycle progression. Oncogene. 2003;22:4035–4046. doi: 10.1038/sj.onc.1206610. [DOI] [PubMed] [Google Scholar]
- Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal. 2010;13:1713–1748. doi: 10.1089/ars.2010.3221. [DOI] [PubMed] [Google Scholar]
- He X, Chen MG, Lin GX, Ma Q. Arsenic induces NAD(P)H-quinone oxidoreductase I by disrupting the Nrf2 · Keap1 · Cul3 complex and recruiting Nrf2 · Maf to the antioxidant response element enhancer. J Biol Chem. 2006;281:23620–23631. doi: 10.1074/jbc.M604120200. [DOI] [PubMed] [Google Scholar]
- He X, Kan H, Cai L, Ma Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J Mol Cell Cardiol. 2009;46:47–58. doi: 10.1016/j.yjmcc.2008.10.007. [DOI] [PubMed] [Google Scholar]
- He X, Ma Q. NRF2 cysteine residues are critical for oxidant/electrophile-sensing, Kelch-like ECH-associated protein-1-dependent ubiquitination-proteasomal degradation, and transcription activation. Mol Pharmacol. 2009;76:1265–1278. doi: 10.1124/mol.109.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Ma Q. Disruption of Nrf2 synergizes with high glucose to cause heightened myocardial oxidative stress and severe cardiomyopathy in diabetic mice. J Diabet Metab. 2012;S7:002. doi: 10.4172/2155-6156.S7-002. http://dx.doi.org/10.4172/2155-6156.S7-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren A. Thioredoxin. Annu Rev Biochem. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- Hu X, Roberts JR, Apopa PL, Kan YW, Ma Q. Accelerated ovarian failure induced by 4-vinyl cyclohexene diepoxide in Nrf2 null mice. Mol Cell Biol. 2006;26:940–954. doi: 10.1128/MCB.26.3.940-954.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon JH, Lee KN, Hwang CY, Kwon KS, You KH, Choi I. Tumor suppressor VDUP1 increases p27(kip1) stability by inhibiting JAB1. Cancer Res. 2005;65:4485–4489. doi: 10.1158/0008-5472.CAN-04-2271. [DOI] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Lee KN, Kang HS, Jeon JH, Kim EM, Yoon SR, Song H, Lyu CY, Piao ZH, Kim SU, Han YH, et al. VDUP1 is required for the development of natural killer cells. Immunity. 2005;22:195–208. doi: 10.1016/j.immuni.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Ma Q. Xenobiotic-activated receptors: from transcription to drug metabolism to disease. Chem Res Toxicol. 2008;21:1651–1671. doi: 10.1021/tx800156s. [DOI] [PubMed] [Google Scholar]
- Ma Q. Transcriptional responses to oxidative stress: pathological and toxicological implications. Pharmacol Ther. 2010;125:376–393. doi: 10.1016/j.pharmthera.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Ma Q, He X. Molecular basis of electrophilic and oxidative defense: promises and perils of Nrf2. Pharmacol Rev. 2012 doi: 10.1124/pr.110.003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Kinneer K, Bi Y, Chan JY, Kan YW. Induction of murine NAD(P)H: quinone oxidoreductase by 2,3,7,8-tetrachlorodibenzo-p-dioxin requires the CNC (cap ‘n’ collar) basic leucine zipper transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2): cross-interaction between AhR (aryl hydrocarbon receptor) and Nrf2 signal transduction. Biochem J. 2004;377:205–213. doi: 10.1042/BJ20031123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AH, Hafele C, Shalev A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces β-cell apoptosis. Endocrinology. 2005;146:2397–2405. doi: 10.1210/en.2004-1378. [DOI] [PubMed] [Google Scholar]
- Muoio DM. TXNIP links redox circuitry to glucose control. Cell Metab. 2007;5:412–414. doi: 10.1016/j.cmet.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Murarka S, Movahed MR. Diabetic cardiomyopathy. J Card Fail. 2010;16:971–979. doi: 10.1016/j.cardfail.2010.07.249. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, Takagi Y, Sono H, Gon Y, Yodoi J. Identification of thioredoxin-binding protein-2/vitamin D3 up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem. 1999;274:21645–21650. doi: 10.1074/jbc.274.31.21645. [DOI] [PubMed] [Google Scholar]
- Nishizawa K, Nishiyama H, Matsui Y, Kobayashi T, Saito R, Kotani H, Masutani H, Oishi S, Toda Y, Fujii N, et al. Thioredoxin-interacting protein suppresses bladder carcinogenesis. Carcinogenesis. 2011;32:1459–1466. doi: 10.1093/carcin/bgr137. [DOI] [PubMed] [Google Scholar]
- Nordberg J, Arnér ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- Oka S, Liu W, Masutani H, Hirata H, Shinkai Y, Yamada S, Yoshida T, Nakamura H, Yodoi J. Impaired fatty acid utilization in thioredoxin binding protein-2 (TBP-2)-deficient mice: a unique animal model of Reye syndrome. FASEB J. 2006;20:121–123. doi: 10.1096/fj.05-4439fje. [DOI] [PubMed] [Google Scholar]
- Parikh H, Carlsson E, Chutkow WA, Johansson LE, Storgaard H, Poulsen P, Saxena R, Ladd C, Schulze PC, Mazzini MJ, et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007;4:e158. doi: 10.1371/journal.pmed.0040158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwari P, Chutkow WA, Cummings K, Verstraeten VL, Lammerding J, Schreiter ER, Lee RT. Thioredoxin-independent regulation of metabolism by the alpha-arrestin proteins. J Biol Chem. 2009;284:24996–25003. doi: 10.1074/jbc.M109.018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CW, Stoltzman CA, Sighinolfi MP, Han KS, Ayer DE. Glucose controls nuclear accumulation, promoter binding, and transcriptional activity of the MondoA-Mlx heterodimer. Mol Cell Biol. 2010;30:2887–2895. doi: 10.1128/MCB.01613-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rösen P, Nawroth PP, King G, Möller W, Tritschler HJ, Packer L. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a Congress Series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab Res Rev. 2001;17:189–212. doi: 10.1002/dmrr.196. [DOI] [PubMed] [Google Scholar]
- Saxena G, Chen J, Shalev A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J Biol Chem. 2010;285:3997–4005. doi: 10.1074/jbc.M109.034421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbai O, Devi TS, Melone MA, Feron F, Khrestchatisky M, Singh LP, Perrone L. RAGE-TXNIP axis is required for S100B-promoted Schwann cell migration, fibronectin expression and cytokine secretion. J Cell Sci. 2010;123:4332–4339. doi: 10.1242/jcs.074674. [DOI] [PubMed] [Google Scholar]
- Schulze PC, Yoshioka J, Takahashi T, He Z, King GL, Lee RT. Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J Biol Chem. 2004;279:30369–30374. doi: 10.1074/jbc.M400549200. [DOI] [PubMed] [Google Scholar]
- Sheth SS, Bodnar JS, Ghazalpour A, Thipphavong CK, Tsutsumi S, Tward AD, Demant P, Kodama T, Aburatani H, Lusis AJ. Hepatocellular carcinoma in Txnip-deficient mice. Oncogene. 2006;25:3528–3536. doi: 10.1038/sj.onc.1209394. [DOI] [PubMed] [Google Scholar]
- Stoltzman CA, Peterson CW, Breen KT, Muoio DM, Billin AN, Ayer DE. Glucose sensing by MondoA:Mlx complexes: a role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc Natl Acad Sci USA. 2008;105:6912–6917. doi: 10.1073/pnas.0712199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123–140. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, De Keulenaer GW, Lee RT. Vitamin D3-up-regulated protein-1 is a stress-responsive gene that regulates cardiomyocyte viability through interaction with thioredoxin. J Biol Chem. 2002;277:26496–26500. doi: 10.1074/jbc.M202133200. [DOI] [PubMed] [Google Scholar]
- Wang Z, Rong YP, Malone MH, Davis MC, Zhong F, Distelhorst CW. Thioredoxin-interacting protein (txnip) is a glucocorticoid-regulated primary response gene involved in mediating glucocorticoid-induced apoptosis. Oncogene. 2006;25:1903–1913. doi: 10.1038/sj.onc.1209218. [DOI] [PubMed] [Google Scholar]
- Yu FX, Goh SR, Dai RP, Luo Y. Adenosine-containing molecules amplify glucose signaling and enhance txnip expression. Mol Endocrinol. 2009;23:932–942. doi: 10.1210/me.2008-0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Luo Y. Tandem ChoRE and CCAAT motifs and associated factors regulate Txnip expression in response to glucose or adenosine-containing molecules. PLoS One. 2009;4:e8397. doi: 10.1371/journal.pone.0008397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]