

Abstract
HDA6 is a RPD3-like histone deacetylase. In Arabidopsis, it mediates transgene and some endogenous target transcriptional gene silencing (TGS) via histone deacetylation and DNA methylation. Here, we characterized two hda6 mutant alleles that were recovered as second-site suppressors of the DNA demethylation mutant ros1–1. Although both alleles derepressed 35S::NPTII and RD29A::LUC in the ros1–1 background, they had distinct effects on the expression of these two transgenes. In accordance to expression profiles of two transgenes, the alleles have distinct opposite methylation profiles on two reporter gene promoters. Furthermore, both alleles could interact in vitro and in vivo with the DNA methyltransferase1 with differential interactive strength and patterns. Although these alleles accumulated different levels of repressive/active histone marks, DNA methylation but not histone modifications in the two transgene promoters was found to correlate with the level of derepression of the reporter genes between the two had6 alleles. Our study reveals that mutations in different domains of HDA6 convey different epigenetic status that in turn controls the expression of the transgenes as well as some endogenous loci.
DNA methylation and histone modifications dictate the chromatin state and greatly affect gene expression in animals as well as in plants. Unlike in mammals whose DNA methylation is largely limited to symmetrical CG dinucleotides mediated by the methyltransferases DNMT1 and DNMT31, methylation in Arabidopsis can occur on cytosine in any nucleotide context including symmetrical cytosine (CG and CHG), as well as asymmetrical cytosine (CHH) (H = A, T, and C) sites2. Symmetrical cytosine methylation in Arabidopsis is maintained by two DNA methylatransferases, MET1 (a DNMT1 homolog for CG methylation) and CMT3 (for CHG methylation)3. These methyltransferases can restore the methylation status to a newly synthesized DNA strand based on the parental strand methylation status with the help of VIM proteins4 and the KYP/SUVH4 protein5, respectively. On the other hand, asymmetrical cytosine methylation, which is mainly catalyzed by DRM2, would need de novo methylation guided by Pol IV and Pol V via the RNA-directed DNA methylation (RdDM) pathway and/or the Pol II-mediated pathway14. The plant-specific RdDM pathway uses small interfering RNA (siRNA) to guide the methylation and silencing of transposons, repetitive sequences and certain gene promoters2. RdDM involves siRNA biogenesis mediated by DCL3, RDR2, Pol IV, and/or Pol V7,8,9, scaffold RNA production via Pol II, Pol V, DRD1, DMS3 and RDM110,12,13, and formation of the guiding complex as well as DRM2 recruitment14. Besides these components that are directly involved in the RdDM pathway, forward genetic screening also identified other components with profound effects on RdDM, such as RDM4, RDM12, KTF1, and HDA615,16,17,18.
DNA methylation and histone modifications are closely linked processes19. The cross-talk between DNA methylation and histone modification was initially observed in suvh39 mutated mice in which the histone methyltransferase mutation caused failure of localization of DNMT3B to the pericentromeric heterochromatin region. As a result, DNA methylation in that region was decreased20. In Arabidopsis, mutations in the histone H3K9 methyltransferase KYP decrease not only the accumulation of histone H3 lysine 9 dimethylation (H3K9me2) but also the levels of CHG DNA methylation6,21. Recently, the structural basis for CMT3-mediated CHG methylation at the H3K9me2-containing nucleosome has been described5. Both the BAH domain and the chromodomain of CMT3 were found to bind to H3K9me2-containing nucleosomes where the unmethylated cytosine was converted to methylated cytosine in the CHG context5. More recently, the histone acetylase IDM1/ROS4 was found to play a role in active DNA demethylation, a process that eukaryotic cells employ to fine-tune the methylation status to limit the silencing of certain genes22,23.
The Arabidopsis Repressor of Silence 1 (ROS1) DNA glycosylase/lysase can remove the 5-methyl group from methylcytosine at target sites using a base excision repair mechanism24. In the ros1–1 mutant, the two-linked reporter genes, 35S promoter driven NPTII (35S::NPTII) and RD29A promoter driven firefly luciferase gene (LUC) (RD29A::LUC), were both silenced as a result of reduced demethylation in their promoters25. Nonetheless, the silencing mechanisms for these two promoters are different. The silencing of the RD29A::LUC is entirely dependent on the RdDM pathway, whereas the silencing of 35S::NPTII is not26.
To better understand the mechanisms underlying the silencing of these two reporter genes and the endogenous loci that they represent, we conducted genetic screens for ros1–1 suppressors16. Here we identified two different hda6 alleles that can release the two reporter genes albeit to different extents. One allele (hda6–9) has a stronger effect on the release of the 35S::NPTII transgene while the other allele (hda6–10) has a stronger release of the RD29A::LUC transgene. We explored the mechanisms that underlie the differential derepression on the two reporter genes between the two hda6 alleles. Our results showed that the distinct DNA methylation patterns of the reporter gene promoters between the two hda6 alleles are crucial in determining the reporter gene expression, although the elevated acetylation level caused by loss of deacetylation on the reporter gene promoters also contributes to the expression patterns of the reporter genes.
Results
Two hda6 mutations suppress ros1-mediated transcriptional gene silencing and differentially regulate reporter gene expression
The Arabidopsis repressor of silencing (ros1–1) mutation led to transcriptional silencing of two expressed reporter genes (35S::NPTII and RD29A::LUC) that were introduced into its wild-type background as a single transfer-DNA (T-DNA)25,27. In a forward genetic screen for ros1–1 suppressors, we identified two suppressors of ros1–1 that releases both the 35S::NPTII and the RD29A::LUC reporter genes (Fig. 1A). Although the mutants were generated by T-DNA mutagenesis, PCR-based methods were unsuccessful in identifying the flanking sequence of the insertion in the first mutant. We thus conducted genetic mapping and the mutation was eventually narrowed down to an interval delimited by two molecular markers MDC12–10 and MDC12–50 (Fig. 1B). By sequencing the genes within the interval, we found that there was a T-DNA inserted at 12 bp before the stop codon of the HDA6 gene, which encodes the histone deacetylase 6 (HDA6) protein. This insertion would replace the last 4 amino acids of the predicated protein with 37 new amino acids before encountering a new stop codon. Since hda6 mutants in different background have been numbered up to hda6–6 (axe1–5), we named this T-DNA insertion allele of hda6 mutants in ros1–1 background as hda6–9 (Fig. 1C). The second hda6 mutant we identified in the same genetic screen has a 13-bp deletion at the end of the first exon, which caused an alteration of the last 5 amino acids in the truncated protein and a deletion of 340 amino acids as a result of a stop codon introduced by the frame shift16. Here, we refer to this mutant as hda6–10 (Fig. 1C). This mutant, like hda6–9, also de-repressed the expression of the RD29A-LUC gene and restored kanamycin resistance (Fig. 1A).
Figure 1. Luminescence and kanamycin resistance phenotypes of ros1 suppressors.

(A) WT (C24-LUC, harboring the RD29A::LUC transgene), ros1–1, and the two suppressor mutants, hda6–9 and hda6–10, were either grown on MS agar plates for 7 d and their luminescence was imaged after cold treatment (1 d, 4 °C); or they were grown on MS agar plates supplemented with 75 μg/ml kanamycin and their pictures were taken 10 days after germination. (B) Molecular mapping delimited the mutation of hda6–9 to the interval between the molecular markers MDC12-10 and MDC12-50. (C) Schematic structure of the wild type HDA6, hda6–10 and hda6–9 proteins.
We analyzed the gene expression phenotypes in these two hda6 mutants. Unexpectedly, we found that the two mutant alleles expressed the two transgenes quite differently. Quantitative RT-PCR analysis indicated that the hda6–10 mutant strongly released RD29A::LUC, with its expression level more than 10 times higher than in the hda6–9 allele (Fig. 2A, upper panel). Consistently, the endogenous RD29A expression was also enhanced to a similar extent (Fig. 2A, middle panel). In contrast, the hda6–9 allele strongly released 35S::NPTII expression, with its expression level nearly 10 times higher than in hda6–10 (Fig. 2A, lower panel). RNA blot analysis further confirmed the difference between the two alleles in NPTII expression (Fig. 2B).
Figure 2. Complementation of the two hda6 mutants.

(A) Complementation of the kanamycin resistance and luminescence expression phenotypes of the two hda6 mutants by wild-type HDA6 genomic DNA. (B) HDA6 genomic DNA-complemented hda6–9 and hda6–10 mutants restored ros1–1 molecular phenotypes.
Using random primers-prepared instead of oligo d(T)-synthesized cDNA library, we examined the expression of some endogenous targets of HDA6. We found that loci such as the 106B repeat sequence (centromere region), the 180bp satellite repeat sequence (centromere region), Amplicon 1 (a 45S rDNA, in a pericentromeric region)28 and solo LTR (A) (pericentromeric region) were more strongly released in hda6–9 than in hda6–10. However, other HDA6 target loci such as IGN6 (intergenic noncoding region 6, located in regions rich in transposon-derived elements, siRNA production and DNA hypermethylation, a Pol V specific target), IGN15 (located in gene-rich regions with relatively few transposon-related repeats, a Pol V specific target)11,29 and TA3 (a LTR retrotransposon) were expressed at far higher levels in hda6–10 than in hda6–9. On the other hand, TSI (transcriptional silencing information), G1136 (an endogenous genomic target of HDA6)30 and AtMU1 were strongly released with similar levels of expression in both hda6 mutants (Fig. 2C).
Previous studies identified a null allele of hda6, axe1–531, which was recently named hda6–632. To comparing the two hda6 alleles with the hda6 null mutant in the same background, we crossed hda6–6 (axe1–5) with ros1–1 and obtained the ros1 hda6–6 mutant (axe1–5 was introduced into the ros1 background) by genotyping. We conducted qRT-PCR analysis on the expression of the reporter genes and two endogenous targets. It was found that the two hda6 alleles had far stronger releasing effects on NPTII and SDC, but had much lower releasing effects on ERT732 relative to ros1 hda6–6 (Fig. 2D).
To confirm the suppression of transcriptional gene silencing (TGS) observed in the two hda6 alleles was caused by the respective mutations, a genomic DNA fragment corresponding to the HDA6 promoter and the coding region was introduced into these two mutants. The resulting transgenic plants showed kanamycin sensitivity and reduced LUC expression, similar to the original ros1–1 mutant (Fig. 3A), demonstrating that the hda6 mutations were responsible for the derepression of these transgenes silenced by the ros1–1 mutation. RT-PCR assays demonstrated that the full-length HDA6 transcript was detected in the transgenic plants as well as in the wild type and ros1–1 mutant, but it was absent in the two hda6 mutants (Fig. 3B). Interestingly, in both hda6 mutants, there was a simultaneous increase in the expression of its close homolog HDA7, which likely compensated for the loss of HDA6 (Fig. 3B). Interestingly, a hda7 knock-down line also had a higher expression level of HDA6 relative to the wild type33. This compensation effect also disappeared in the complemented lines, as indicated by an expression level of HDA7 similar to that in the wild type and ros1–1 (Fig. 3B). Furthermore, one of the HDA6 target genes, At5g41660, was expressed in the hda6 mutants but was silenced again in the complemented seedlings (Fig. 3B). All these lines of evidence showed that the hda6 mutations in the two mutants were responsible for reporter gene derepression in the ros1–1 background.
Figure 3. Transcriptional gene silencing is suppressed in the hda6–9 and hda6–10 mutant plants.

(A) Expression levels of LUC (upper panel), endogenous RD29A (middle panel) and NPTII (lower panel) relative to those in ros1–1 were measured with qRT-PCR using UBQ3 as an internal reference. The Y-axis represents the fold changes compared to ros1–1. Ten-day-old seedlings were treated at 4 °C for 1 day, and then total RNAs were extracted for analyzing the expression of the reporter genes and endogenous genes. (B) RNA blot assays showed the expression level of NPTII in wild type, ros1–1, hda6–9, hda6–10 and ros1 ago4. UBQ3 was used as a loading control. (C) Expression of selected endogenous targets of HDA6 determined by RT-PCR with random hexamer primers. ACT2 (ACTIN2) (its CT mean in all 4 samples with 20 cycles) was used as an internal reference. (D) Relative expression of Group E target ERT7, 35S::NPTII, and SDC among the two hda6 mutated alleles and ros1 hda6–6 (the hda6 null mutant axe1–5 in the ros1–1 background). Data are means and standard derivations based on 3 biological replicates.
Differences in DNA methylation on the transgene promoters cause the opposite expression patterns of the reporter genes in the two hda6 mutants
Transcriptional gene silencing is often caused by hypermethylation of the promoter DNA34. With the varied derepression of the two reporter genes in the two hda6 alleles, we wanted to determine whether there was any difference in the methylation status of their promoters. Indeed, bisulfite sequencing showed that methylation in the CG, CHG and CHH contexts on the 35S promoter and transgenic RD29A promoter had distinct differences among the genotypes (Fig. 4). Overall, the total DNA methylation in the CG, CHG and CHH contexts was lower in hda6–10 (48%, 35% and 18%, respectively) than in hda6–9 (64%, 61% and 27%, respectively) on the RD29A transgene promoter (Fig. 4A). In contrast, the CG and CHG methylation levels on the 35S promoter were much lower in hda6–9 (29% and 28%, respectively) than in hda6–10 (82% and 70%, respectively). Interestingly there was a higher level of methylation in the distal region (−300 to −100) than in the proximal region (−100 to 0) of the transgenic RD29A promoter except for CG methylation in ros1–1 where the methylation level was uniform across the entire promoter region (Figures S3A to S3C). A similar methylation pattern across the 35S promoter was also found for CG and CHG methylation in the hda6–9 mutant but not in ros1 or hda6–10 (Figure S4).
Figure 4. DNA methylation analysis on the RD29A promoter and the 35S promoter.
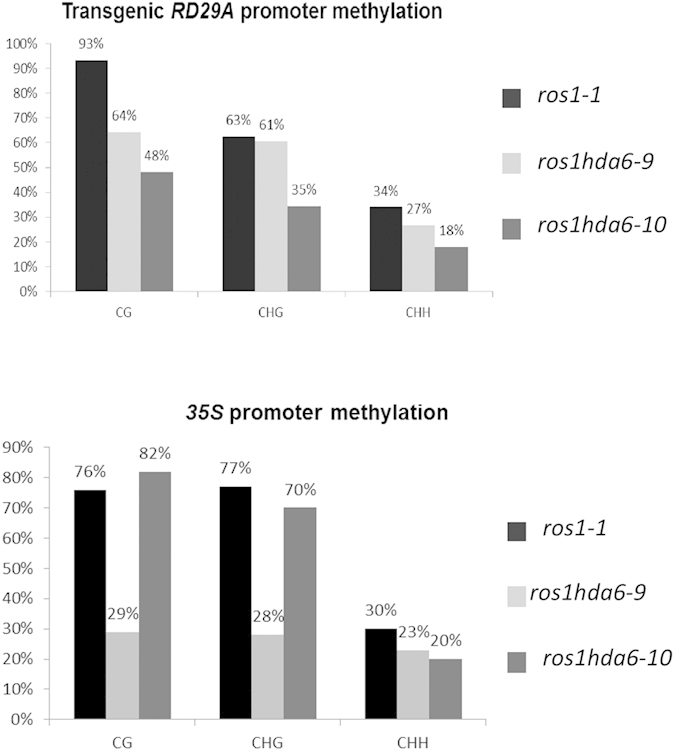
Bisulfite sequencing determination of DNA methylation levels on transgenic RD29A promoter (−270 to 0) (upper panel) and 35S promoter (−232 to 0) (lower panel) in ros1–1, hda6–9 and hda6–10. Data are based on 24 individual colonies for each genotype.
To corroborate the methylation status of the 35S promoter, we conducted Chop-PCR with methylation-dependent restriction enzyme digestion that can distinguish the CG methylation status. We found that hda6–9 had a lower DNA methylation level than the hda6–10 mutant had, which is in accordance with our bisulfite sequencing results, although both mutants had lower DNA methylation levels than ros1–1 and the wild type (Figure S3D). Moreover, our Chop-PCR results on Amplicon3, a nucleolar fragment in the 45S rDNA region28, also showed a lower CG methylation level in the hda6–9 mutant than in the hda6–10 mutant (Figure S3D). These data indicate that the DNA methylation levels of the two promoters inversely correlated with the gene expression levels in these alleles.
To determine whether these differences in DNA methylation contributed to the opposite effects of the two hda6 mutations on the expression of 35S::NPTII and RD29A::LUC genes, we used 5-aza-2′-deoxycytidine to block genomic DNA methylation, and checked the subsequent transcript levels of these two reporter genes. After a 14-day treatment with 5-aza-2′-deoxycytidine, the total RNA from the wild type, ros1–1, hda6–9 and hda6–10 were extracted, and qRT-PCR were performed with NPTII, LUC and At5g41660 (a HDA6 endogenous target gene) primers. While the 5-aza-2′-deoxycytidine treatment significantly decreased the overall methylation levels as reflected by the reporter gene expression levels, the impact on the two hda6 alleles was striking. After the treatment, the nearly 10-fold difference in the NPTII expression level between hda6–9 and hda6–10 (Fig. 2A, lower panel) was significantly reduced to less than 2-fold (Fig. 5, left, upper panel). Similarly, the over 10 times difference in LUC expression between the two hda6 mutants nearly disappeared, with the expression level in hda6–9 even slightly higher than in hda6–10 (Fig. 2A, upper panel and 5, left, lower panel). However, the relative expression level of At5g41660 among the two hda6 mutants was not significantly affected by the treatment (Fig. 5, right panels). These data showed that the differing level of DNA methylation is the main reason for the distinct effects of the two hda6 mutants on the expression pattern of 35S::NPTII and RD29A::LUC genes.
Figure 5. Target gene expression after 5-aza-deoxycytidine treatments.

Expression levels of the indicated genes relative to those in ros1–1 were determined with qRT-PCR using UBQ3 as an internal reference. Data are means and standard derivations based on 3 biological replicates.
The two hda6 alleles have different abilities to interact with MET1 in vitro and in vivo
Since the two hda6 alleles had significantly different DNA methylation patterns on the promoters of the two reporter genes as well as other target genes, we wanted to understand the molecular basis for these differences. DNA methylation is catalyzed by a group of dedicated DNA methyltransferases. Interestingly, previous research found that HDA6 and the methyltransferase MET1 shared many common targets30. Recently, it was reported that HDA6 in fact could physically interact with MET135. We thus asked whether the two hda6 alleles had different abilities to interact with MET1.
We used mutated hda6 as baits and MET1 (R2FB, aa.735–869) as the prey to perform yeast two-hybrid assays. The results showed that both hda6–9 and hda6–10 could interact with MET1 in vitro on low stringent selective plates (SD/-Trp/-Leu/X) (Fig. 6A). However, under the higher stringent selective condition (SD/-Trp/-Leu/-Ade/-His/X/A), hda6–9 lost its ability to interact with MET1, but hda6–10 still interacted with MET1 to a similar extent as the wild-type HDA6 (Fig. 6B). These results indicated that hda6–10 has a better ability than hda6–9 in interacting with MET1 in vitro.
Figure 6. Yeast two-hybrid assays to detect the interaction between MET1 (R2FB) and hda6 mutant alleles.

(A) Negative control (pGBKT7), wild-type HDA6 (1–471aa), mutated hda6–10 (1–131aa), HDA6 (1–467) (1–467aa) and hda6–9 (1–504aa) on the less stringent medium SD/-Trp/-Leu/X. (B) The interaction assay on the more stringent medium SD/-Trp/-Leu/-Ade/-His/X/A. The pictures were taken 2 days after streaking on the shown media.
To investigate whether the two mutated hda6 alleles also interact with MET1 in vivo, we performed Bimolecular fluorescence complementation (BiFC) assays using the full-length MET1 tagged with nYFP, and the wild-type HDA6, HDA6–9, or HDA6–10 tagged with cYFP. After combining and introducing respectively the above construct pairs into the leaves of tobacco (N. benthamiana) via Agrobacterium-mediated infiltration. We found that the interaction between HDA6–9 and MET1 was far weaker than that of the wild-type HDA6 and MET1, and also weaker than that of HDA6–10 and MET1 (Fig. 7, low panel). Unlike the interaction between the wild-type HDA6 and MET1 which showed a concentrated interactive area in the nucleoplasm, or that between HDA6–10 and MET1 which showed a more limited area (strong dots in the nucleoplasm), the interaction between HDA6–9 and MET1 occurred throughout the nucleoplasm except for the nucleolus (Fig. 7 lower panel, middle). This pattern of interaction between hda6–9 and MET1 is similar to that of the localization of hda6–9 (Fig. 7, upper panel, middle).
Figure 7. Subcellular localization of the full-length HDA6 and its mutated proteins and their in vivo interaction with MET1.

Upper panels: Subcellular localization of the full-length HDA6 (wild type, left), hda6–9 (middle), and hda6–10 (right). Lower panels: BiFC interactions between MET1 and full-length HDA6 (wild type, left), hda6–9 (middle), and hda6–10 (right).
Histone modifications in the two hda6 mutants
Chromatin status determines the accessibility of transcription factors to the target gene promoter and is regulated by DNA methylation and histone modifications. We found that the different methylation levels on the two reporter gene promoters contributed greatly to the difference in the reporter gene expression in the two hda6 alleles. To test whether histone modifications were also involved in the different expressions of the two reporter genes in these alleles, we performed chromatin immunoprecipitation (ChIP) analysis with common repressive and active histone modification marks. The results showed that there is a higher methylation level and lower acetylation level on H3K9 in ros1–1 compared with the wild type on all the promoters examined (Fig. 8). However, the levels of H3K9 methylation and H3K9 and H3K27 acetylation on the RD29A promoter were similar between the two hda6 alleles although both alleles had higher acetylation and lower methylation levels compared with the ros1–1 mutant. On the other hand, the levels of H3K9 and H3K27 acetylation on the 35S promoter were quite different between the two hda6 alleles. hda6–10 had higher levels of H3K9 and H3K27 acetylation than hda6–9, ros1–1 and the wild type (Fig. 8, right, upper and middle panels).
Figure 8. Chromatin Immunoprecipitation (ChIP) with repressive marks H3K9me2 and active marks H3K9ac and H3K27ac.

H3K9me2 accumulation on the 35S promoter (right, lower panel) and the transgenic RD29A promoter (left, lower panel); H3K9ac accumulation on the 35S promoter (right, upper panel) and the RD29A transgene promoter (left, upper panel); H3K27ac accumulation on the 35S promoter (right, middle panel) and the RD29A transgene promoter (left, middle panel). Data are means and standard derivations based on 3 technical replicates.
Transcriptome profiles of the two hda6 mutants reveal their differential regulation of endogenous genes
To investigate the scope of TGS in the two hda6 alleles, we performed microarray experiments with Agilent microarrays. Compared with the ros1–1 single mutant, 1938 targets were up regulated by more than 2-fold in hda6–10, and 1707 targets were up regulated in hda6–9. Among these up-regulated targets, 1371 were common targets shared by both hda6 mutants (Figure S5). Although a significant number of targets were up regulated, 421 targets in hda6–10 and 611 targets in hda6–9 were down regulated (less than -2 fold) relative to the ros1–1 single mutant. There were 217 common targets that were down regulated in both hda6 mutants (Figure S5). With the two hda6 alleles, we detected 461 targets having more than 2-fold changes in expression in hda6–10 relative to hda6–9 (Table S1). Among these up-regulated targets, 256 corresponded to AGI genes, 32 corresponded to non-AGI regions and 29 corresponded to transposable elements. Among down-regulated targets, 127 corresponded to AGI genes and 16 corresponded to non-AGI regions (Table S1). We also detected 321 targets having a greater than 2 or less than 2-fold change in hda6–9 compared to hda6–10 (Table S1).
Since transposable elements are direct targets of epigenetic modifications, we tried to map to each chromosome the transposons with greater than 2-fold changes in expression in both hda6 alleles. Our results showed that most of these up-regulated transposons were located near pericentromeric or telomeric regions (Fig. 9). In addition, some transposons were preferentially expressed in one allele relative to the other (Fig. 9B–D).
Figure 9. Chromosomal localization of up- or down-regulated transposons in hda6 mutants relative to the ros1–1 mutant.

(A) Common up-regulated transposons in hda6–10 and hda6–9 relative to the ros1–1 single mutant. (B) hda6–10 specifically up-regulated transposons. (C) hda6–10 specifically down-regulated transposons. (D) hda6–9 specifically up-regulated transposons.
Discussion
In a genetic screening for ros1–1 suppressors, we identified two hda6 alleles that suppressed ros1–1 induced transcriptional gene silencing (TGS) of both the RD29A::LUC and the 35S::NPTII reporter genes. It is known that these two reporter genes are silenced differently in the ros1–1 mutant: the RD29A::LUC is silenced via the RdDM pathway in that mutants defective in the RdDM pathway all derepress the RD29A::LUC reporter gene but they do not show obvious derepression of the 35S::NPTII reporter gene8,15,16,17,36,37. So far, only mutations of HDA6 and the SWI2/SNF2-like chromatin-remodeling protein DDM1 were found to derepress both the RD29A::LUC and the 35S::NPTII transgenes16. Although it is unknown why these two reporter genes are silenced differently, notably, both DDM1 and HDA6 directly regulate the chromatin status. In this study, while it is not unexpected that the two hda6 mutants derepressed both the RD29A::LUC and 35S::NPTII reporter genes compared to the ros1–1 mutant, surprisingly they showed distinct preferences in releasing these two reporter genes. While hda6–9 strongly derepressed 35S::NPTII expression, hda6–10 strongly derepressed RD29A::LUC expression, suggesting that the two allelic mutations may have different impacts on DNA methylation and/or histone modifications. Interestingly, although these two hda6 alleles had preferential releasing on transgene expression, none of them really behaved like the hda6 null mutant ros1 hda6–6 (axe1–5 in ros1 background) (Fig. 2D). The expression level of 35S::NPTII in the ros1 hda6–6 mutant was far lower than in hda6–9 (100 times less) or hda6–10 (10 times less). Nonetheless, the expression of an endogenous target ERT7 (a HDA6 and NRPE1 epistasis target)32 in ros1 hda6–6 was much higher than that in hda6–9 or hda6–10 (nearly 10 times higher). These results show that the two hda6 alleles are weaker, rather than null, alleles of HDA6 and that they may have the ability to discriminate and work on transgenes and endogenous genes.
Our two alleles produced two different domain disrupted protein since the two mutations occurred in different domains of the HDA6 protein. The hda6–9 mutant has a T-DNA insertion at 12 bp before the stop codon, thus causing the C-terminal disruption. On the other hand, the hda6–10 allele has a 13-bp deletion in the first exon, which creates a premature stop codon and produces a truncated peptide lacking both the HD domain and the C-terminal domain (Fig. 1C). Given that the C-terminal domain was considered the mere interaction domain with MET135, one might predict that the two hda6 alleles should have the same or similar DNA methylation patterns since both of them have a disrupted/deleted C-terminal domain. However, the two alleles exhibited significant differences in DNA methylation at the 35S promoter and the RD29A promoter, which indicated that the HD domain of HDA6 may have certain roles in DNA methylation as well. The prediction has been confirmed by a new hda6–8 allele which has a single amino acid substitution at ER motif of histone deacetylase domain38. Although yeast two-hybrid assays showed that the whole HD domain did not interact with MET135, part of the HD domain of the mammalian HDAC1 (a homolog of HDA6) was found to interact with DNMT1 (a mammal homolog of MET1)39. Furthermore, it was reported that the single point mutation (D186N) in the HD domain of HDA6 led to reduced CG, CHG and CHH methylation levels that were even lower than in the null mutant of HDA640. In addition, a point mutation (D186A) of HDA6 could not rescue the phenotypes of its axe1–5 mutant allele28. Besides the HD domain and the C-terminal domain, the N-terminal domain of HDA6 also seems to have some effects on DNA methylation, especially on rDNA repeats since the sil1 mutant with a point mutation (Gly 16 Arg) at the N-terminus of HDA6 also caused decreased DNA methylation in the rDNA region41. Thus, although the C-terminus of HDA6 can physically interact with MET1, it seems that any part of the HDA6 protein is indispensable for maintaining heterochromatin status and transgene silencing via the effects on deacetylation and DNA methylation.
It is intriguing that the two hda6 mutants would differently affect the methylation status of the two reporter genes. This may have to do, at least partly, with the difference in the interaction between the hda6 mutants and MET1, which controls much of DNA methylation. Although the two hda6 mutant proteins both interacted with MET1 on the less stringent SD/–Leu/–Trp/X media, hda6–10 but not hda6–9 interacted with MET1 on the more stringent SD/–Ade/–His/–Leu/–Trp/X/A media. Under these conditions, the interaction strength of hda6–10 and MET1 was similar to if not higher than that of the wild-type HDA6 protein and MET1 (Fig. 6B). BiFC experiments using full-length MET1 tagged with the N-terminal YFP (i.e., nYFP) and wild-type HDA6 (or HDA6–9 or HDA6–10) tagged with the C-terminal YFP (i.e., cYFP) confirmed that MET1 and different HDA6 interacted in vivo, although the intensity of the interaction signal and patterns of interaction differed among the different HDA6 proteins (Fig. 7). The reason for the interaction between hda6–10 (a truncated protein only with N-terminal and partial HD domain) and MET1 may depend on the final 5 mutated amino acids. The wild type HDA6 without last 4 amino acid (PPSS) severely impair its interaction with MET1 (e.g hda6–9), while the last 5 mutated amino acids (VLPSN) in hda6–10 are similar to wild type last 5 amino acids (NPPSS), may thus make the truncated protein has ability to strongly interact with MET1 in vitro.
A previous study on the 35S::NPTII expression in the wild type and ros1–1 mutant showed that ROS4/IDM1, a PHD zinc-finger domain-containing histone acetyltransferase, is crucial for NPTII transcription23. The knockout line of this gene cannot release the NPTII expression in the ros1–1 background even with a low DNA methylation level in the 35S promoter23, demonstrating a coordinated and interdependent regulation of the NPTII expression by both DNA methylation and histone acetylation. In hda6–10 mutant, although the 35S promoter is hypermethylated (Fig. 4, lower panel), its H3K9 and H3K27 acetylation levels were higher (Fig. 8, right, upper and middle panels) than those in the other hda6 allele, wild type as well as ros1–1. This enhanced histone acetylation may lead to a strong release of NPTII in hda6–10 relative to ros1–1 and the wild type despite of its hypermethylated promoter. Nonetheless, between the two hda6 mutants, the level of histone acetylation at the examined sites (Fig. 8) did not correlate with the level of derepression of the NPTII gene (Figs 2C and S5). Rather, the DNA methylation levels of the 35S promoter correlated well with the expression level of NPTII in the two hda6 mutants. These data raised interesting questions regarding how the nature of these promoters affects their chromatin states and how these epigenetic modifications affect gene transcription. Previous literatures showed that heterochromatic transcription is dependent on Pol IVB/V11. Interestingly, the hda6–10 mutant also showed the enhanced transcription of Pol V targets (e.g. IGN6 and IGN15, Fig 2C).
Some of the complications of differing histone acetylation among the hda6 alleles may also have to do with their impact on the expression of other HDAs. HDA6 and HDA7 belong to the same Class I HDA1/RPD3 family42. HDA7 is normally not expressed in any part of the plant. However, light, heat and biotic stress could induce its expression42. Our data showed that HDA7 was highly expressed in the two hda6 mutants after a 24-hour cold-treatment (Fig. 3B and S2). This compensation effect may suggest that HDA7 might play some of the histone deacetylation functions of HDA6 when HDA6 is not available. Recently, the compensation effects between HDA6 and HDA7 was also found in the HDA7 knock-down line where the expression of HDA6 and HDA9 was significantly increased33.
It is known that histone modification and DNA methylation cooperatively maintain heterochromatin status to silence transgenes, transposable elements, repetitive sequences and rDNA repeats5,28,29. In mammalian systems, DNA methylation was thought to determine the chromatin structure via methyl DNA-specific binding proteins to recruit enzymatic machinery capable of locally altering histones43. On the other hand, histone modifications also determine DNA methylation via histone methylase G9a recruitment of the de novo DNA methylases DNMT3A and DNMT3B to methylate the underlying DNA44,45. In Arabidopsis, both the BAH domain and chromo domain of CMT3 form an aromatic cage to function as an interactive interface to capture H3K9me2 containing nucleosomes5. Similarly, KYP/SUVH4, a histone H3K9 methyltransferase, has a domain binding to CMT3 methylated CHG. The discoveries of different DNA methylation and histone modification of two hda6 alleles in the current study indicate that similar coordinated functions between DNA methylation and histone modification mediated by HDA6 may also exist in plants.
Material and Methods
Plant growth, mutant screening and gene cloning
The Arabidopsis wild-type C24 (WT) and the ros1–1 mutant that both harbor the homozygous RD29A-LUC transgene were described previously27. A T-DNA-mutagenized population in the ros1–1 mutant background26 was used for screening for ros1–1 suppressors. Plants were grown at 21 °C with 16 h of light and 8 h of darkness. To analyze RD29A-LUC expression, 7-d-old seedlings on MS plates were sprayed with luciferin (Promega) for luminescence imaging using a CCD camera as described27. The putative ros1–1 suppressors that emitted higher levels of luminescence than ros1–1 were transferred to soil for seed setting. These plants were further tested on Murashige and Skoog (MS) media supplement with 75 μg/ml kanamycin. The map-based cloning method was used to identify the mutated genes. The ros1 hda6–6 double mutant was obtained by crossing axe1–5 to ros1–1 mutant and F2 population was planted on the MS agar medium supplemented with 50 μg/ml kanamycin, and the resistant seedlings were genotyped to obtain homozygous ros1–1 and axe1–5 seedlings.
The genomic sequence of HDA6 from wild-type C24 was amplified and cloned into the Gateway vector PMDC99 for complementation assays. The full-length HDA6 cDNA, truncated HDA6 cDNA (for amino acids 1–467) and mutated hda6 cDNAs (hda6–9 and hda6–10) were cloned from the wild type, hda6–9, and hda6–10, respectively. These cDNAs were further cloned into the pGBKT7 vector for yeast two-hybrid assays, and also cloned into pENTRY1A before being introduced into the binary vectors pEG104 (for localization) and pEG202-cYFP (for BiFC). The plasmid for pGADT7-MET1 (R2FB), pEG201-MET1(FL)-nYFP, and pEG202-HDA6(FL)-CYFP are kindly provided by Dr. Keqiang Wu.
DNA methylation assays
For Chop-PCR analysis, genomic DNA was extracted using a plant DNeasy extraction kit (Qiagen) and 200 ng of genomic DNA was linearized with 20 U BamHI (NEB, high fidelity) for 3 h at 37 °C. The digested DNA was purified with a Qiagen spin column and further treated with 10 U of McrBC per 100 ng of BamHI-digested genomic DNA at 37 °C for 16 hours before PCR amplification. Controls also were similarly performed using H2O instead of McrBC. PCR amplification was conducted with Ex-Taq for 30 cycles, and the amplified fragments were analyzed on 1.7% agarose gel.
For bisulfite sequencing, 150 ng of genomic DNA were converted using a Qiagen-Epitect bisulfite kit, and the converted genomic DNA was amplified with bisulfite sequencing primers for the transgenic RD29A promoter, and then the amplified DNA was cloned into a pGEM-T vector (Promega). Twenty-four colonies were chosen for sequencing and DNA methylation analysis.
The 5-aza-2′-deoxycytidine treatment was performed as described25. Briefly, seeds were planted on MS plates supplemented with 50 μg/ml kanamycin and 7 μg/ml 5-aza-2′-deoxycytidine. Fourteen days after germination, the seedlings were treated at 4 °C for 24 h before being harvested for total RNA extraction.
RNA analysis
For total RNA extraction, 100 mg of seedlings were harvested after cold treatment (4 °C, 1 d) and extracted using an RNeasy plant kit (Qiagen). The extracted total RNAs were treated with DNAse I and cleaned up with an RNA mini-column or treated with a Ribo-minus kit to remove rRNA from total RNA. For cDNA synthesis, 2–3 μg total RNAs with oligo d(T) or 300 ng ribo-minus RNAs with random hexamer primers were used for reverse transcription using the Invitrogen Superscript III reverse transcriptase kit. The RT solution was diluted 10 times and used for RT-PCR/qRT-PCR. For RNA blot analyses, 12 μg total RNAs from each sample were loaded, and UBQ3 was used as an endogenous reference.
Microarray analysis was performed using the Arabidopsis Gene Expression Microarray (V4, Agilent), which contains 43,803 Arabidopsis gene probes and 1,417 Agilent control probes. There are 4 biological replicates for each sample. Total RNA (150 ng) was used to prepare Cy3-labeled probe using the low-RNA-input linear amplification/labeling kit (Agilent). The dye incorporation and copy RNA yield were measured using the Nanodrop-ND 8000 spectrophotometer (Thermo Fisher). Labeled RNA probes (1.65 mg) were fragmented using the fragmentation buffer (Agilent) and hybridized to the Arabidopsis arrays in the presence of the Gene Expression Hybridization buffer HI-RPM and the blocking agent (Agilent) for 17 h at 65 °C with a 10-rpm rotation speed in a hybridization oven (Agilent). The arrays were then washed using low stringency wash buffer 1 (Agilent) at room temperature for 1 min followed by a high-stringency wash using wash buffer 2 (Agilent) at 37 °C. The arrays were air-dried and scanned using the high-resolution array scanner (Agilent) with the appropriate settings for one-color gene expression arrays. The signal intensities were extracted from the scanned images with the aid of Feature extraction software 10.7.1.1 (Agilent) and subjected to background subtraction and spatial detrending. The outliers and the abnormal features were flagged, and the data were normalized using intraarray percentile shift normalization (threshold of 75 and above) and median-based interarray normalization. GeneSpring GX (Agilent) was used to calculate the intensity ratios and fold changes. Genes with P < 0.05 and change above 2-fold were chosen for the enrichment analysis. The microarray data were submitted to GEO(NCBI) database, the accession number is GSE73716.
Yeast two-hybrid assays
For yeast two-hybrid assays, the Matchmaker Gold yeast two-hybrid system was used to detect the interaction between HDA6 or its mutated versions and MET1. Wild type or mutant hda6 were cloned into pGBKT7 and transformed into Y2HGold, and pGADT7-MET1 (aa. 735–869 from Dr. Keqiang Wu) was transformed into Y187. The mating and tests were done strictly following the manual provided by the manufacturer (Clontech laboratories, Inc). X-α-gal, but not X-β-gal, was used for the reporter gene assay. The lower stringent selective plates included Minimal Media Double Dropouts (SD/-Trp/-Leu) and X-α-gal, while higher stringent selective plates included Minimal Media Quadruple Dropouts (SD/-Trp/-Leu/-Ade/-His), X-α-gal, and the antibiotic Aureobasidin A.
BiFC assays
The binary vectors harboring the wild-type HDA6 or mutant (hda6–9 or hda6–10) cDNAs tagged with C-terminal half of YFP (pEG202-HDA6/hda6–9/hda6–10-cYFP), and the binary vector harboring full-length MET1with N-terminal half of YFP (pEG201-MET1-nYFP) were introduced into GV3101 via electroporation. Single colonies of each construct were inoculated in LB medium with 50 μg/ml kanamycin, and the overnight cultures were pelleted and resuspended in a buffer (10 mM MgCl, 10 mM MES-K (pH5.6) ) supplemented with 100 μM acetosyringone, and put on bench overnight, and then the overnight cultures were Agro-infiltrated into leaves of N. benthamiana. Two days later, the fluorescence signals were documented with Zeiss confocal microscopy.
Chromatin immunoprecipitation
Chromatin immunoprecipitaion assays for reporter gene promoters and a HDA6 endogenous target (At5g41660) were performed with 2-week-old seedlings. Around 1 g of fresh seedlings was harvested and cross-linked with 1% formaldehyde under vacuum for 15 minutes, and then quenched with a 2 M glycine solution and vacuumed for additional 5 minutes. After removing the formaldehyde, the sample was washed with sterilized deionized water three times. Further grinding, immunoprecipitation as well as reverse cross-linking were performed using an EpiQuick plant ChiP kit (Epigentek Inc, Farmingdale, NY) following the manual provided by the manufacturer. Anti-H3K9me2 was from Epigentek, and antibodies against H3K9ac and H3K27ac were from Millipore. The purified genomic DNA fragments were used as templates for qPCR46,47,48,49,50,51.
Additional Information
How to cite this article: Zhang, S. et al. Two domain-disrupted hda6 alleles have opposite epigenetic effects on transgenes and some endogenous targets. Sci. Rep. 5, 17832; doi: 10.1038/srep17832 (2015).
Supplementary Material
Acknowledgments
We thank the KAUST Bioscience Core Facility for help with sequencing and microarray experiments. We also thank Dr. Keqiang Wu for providing plasmids. This study was supported by King Abdullah University of Science and Technology Baseline Grant BAS/1/1007-01-01 (to L.X), the Chinese Academy of Sciences and National Institutes of Health Grants R01GM070795 and R01GM059138 (to J.-K.Z.) and a research start-up fund from Hong Kong Baptist University to SZ.
Footnotes
Author Contributions J.-K.Z. conceived the project, S.Z., L.X. and Y.X. designed the experiments and wrote the manuscript, S.Z. performed all of the experiments, X.Z. and X.X. helped identify the hda6 mutants, and cloned the gene, P.C. helped analyse sequenced data.
References
- Lyko F. et al. Mammalian (cytosine-5) methyltransferases cause genomic DNA methylation and lethality in Drosophila. Nat Genet 23, 363–366 (1999). [DOI] [PubMed] [Google Scholar]
- Henderson I. R. & Jacobsen S. E. Tandem repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes Dev 22, 1597–1606 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X. & Jacobsen S. E. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc Natl Acad Sci USA 99 Suppl 4, 16491–16498 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo H. R., Dittmer T. A. & Richards E. J. Three SRA-domain methylcytosine-binding proteins cooperate to maintain global CpG methylation and epigenetic silencing in Arabidopsis. PLoS Genet 4, e1000156 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J. et al. Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell 151, 167–180 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H., Greenberg M. V., Feng S., Bernatavichute Y. V. & Jacobsen S. E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152, 352–364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosher R. A., Schwach F., Studholme D. & Baulcombe D. C. PolIVb influences RNA-directed DNA methylation independently of its role in siRNA biogenesis. Proc Natl Acad Sci USA 105, 3145–3150 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z. et al. An RNA polymerase II- and AGO4-associated protein acts in RNA-directed DNA methylation. Nature 465, 106–109 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorkovic Z. J., Naumann U., Matzke A. J. & Matzke M. Involvement of a GHKL ATPase in RNA-directed DNA methylation in Arabidopsis thaliana. Curr Biol 22, 933–938 (2012). [DOI] [PubMed] [Google Scholar]
- Zheng B. et al. Intergenic transcription by RNA polymerase II coordinates Pol IV and Pol V in siRNA-directed transcriptional gene silencing in Arabidopsis. Genes Dev 23, 2850–2860 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicki A. T., Haag J. R. & Pikaard C. S. Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell 135, 635–648 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S. W. et al. RNAi, DRD1, and histone methylation actively target developmentally important non-CG DNA methylation in arabidopsis. PLoS Genet 2, e83 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausin I., Mockler T. C., Chory J. & Jacobsen S. E. IDN1 and IDN2 are required for de novo DNA methylation in Arabidopsis thaliana. Nat Struct Mol Biol 16, 1325–1327 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Zhu J. K. RNA-directed DNA methylation. Curr Opin Plant Biol 14, 142–147 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z. et al. An SGS3-like protein functions in RNA-directed DNA methylation and transcriptional gene silencing in Arabidopsis. Plant J 62, 92–99 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X. J. et al. NRPD4, a protein related to the RPB4 subunit of RNA polymerase II, is a component of RNA polymerases IV and V and is required for RNA-directed DNA methylation. Genes Dev 23, 318–330 (2009a). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X. J. et al. An effector of RNA-directed DNA methylation in arabidopsis is an ARGONAUTE 4- and RNA-binding protein. Cell 137, 498–508 (2009c). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aufsatz W. et al. HDA6, a putative histone deacetylase needed to enhance DNA methylation induced by double-stranded RNA. EMBO J 21, 6832–6841 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. A. DNA methylation and cancer. Oncogene 21, 5358–5360 (2002). [DOI] [PubMed] [Google Scholar]
- Lehnertz B. et al. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13, 1192–1200 (2003). [DOI] [PubMed] [Google Scholar]
- Johnson L., Cao X. & Jacobsen S. Interplay between two epigenetic marks. DNA methylation and histone H3 lysine 9 methylation. Curr Biol 12, 1360–1367 (2002). [DOI] [PubMed] [Google Scholar]
- Qian W. et al. A histone acetyltransferase regulates active DNA demethylation in Arabidopsis. Science 336, 1445–1448 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. et al. Antisilencing role of the RNA-directed DNA methylation pathway and a histone acetyltransferase in Arabidopsis. Proc Natl Acad Sci USA 109, 11425–11430 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J. Kapoor A., Sridhar V. V., Agius F. & Zhu J. K. The DNA glycosylase/lyase ROS1 functions in pruning DNA methylation patterns in Arabidopsis. Curr Biol 17, 54–59 (2007). [DOI] [PubMed] [Google Scholar]
- Gong Z. et al. ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell 111, 803–814 (2002). [DOI] [PubMed] [Google Scholar]
- Kapoor A. et al. Mutations in a conserved replication protein suppress transcriptional gene silencing in a DNA-methylation-independent manner in Arabidopsis. Curr Biol 15, 1912–1918 (2005). [DOI] [PubMed] [Google Scholar]
- Ishitani M., Xiong L., Stevenson B. & Zhu J. K. Genetic analysis of osmotic and cold stress signal transduction in Arabidopsis: interactions and convergence of abscisic acid-dependent and abscisic acid-independent pathways. Plant Cell 9, 1935–1949 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley K. et al. Mechanisms of HDA6-mediated rRNA gene silencing: suppression of intergenic Pol II transcription and differential effects on maintenance versus siRNA-directed cytosine methylation. Genes Dev 24, 1119–1132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley K. et al. Erasure of histone acetylation by Arabidopsis HDA6 mediates large-scale gene silencing in nucleolar dominance. Genes Dev 20, 1283–1293 (2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- To T. K. et al. Arabidopsis HDA6 regulates locus-directed heterochromatin silencing in cooperation with MET1. PLoS Genet 7, e1002055 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murfett J., Wang X. J., Hagen G. & Guilfoyle T. J. Identification of Arabidopsis histone deacetylase HDA6 mutants that affect transgene expression. Plant Cell 13, 1047–1061 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins T. et al. A two-step process for epigenetic inheritance in Arabidopsis. Mol Cell 54, 30–42 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cigliano R. A. et al. Histone deacetylase AtHDA7 is required for female gametophyte and embryo development in Arabidopsis. Plant Physiol 163, 431–440 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agius F., Kapoor A. & Zhu J. K. Role of the Arabidopsis DNA glycosylase/lyase ROS1 in active DNA demethylation. Proc Natl Acad Sci USA 103, 11796–11801 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. et al. HDA6 directly interacts with DNA methyltransferase MET1 and maintains transposable element silencing in Arabidopsis. Plant Physiol 158, 119–129 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. et al. An atypical component of RNA-directed DNA methylation machinery has both DNA methylation-dependent and -independent roles in locus-specific transcriptional gene silencing. Cell Res 21, 1691–1700 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X., Zhu J., Kapoor A. & Zhu J. K. Role of Arabidopsis AGO6 in siRNA accumulation, DNA methylation and transcriptional gene silencing. EMBO J 26, 1691–1701 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristova E., Fal K., Klemme L., Windels D. & Bucher E. HDA6 controls gene expression patterning and DNA methylation-independent euchromatic silencing. Plant physiol. 168(4), 1298–308 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F., Burgers W. A., Brehm A., Hughes-Davies L. & Kouzarides T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet 24, 88–91 (2000). [DOI] [PubMed] [Google Scholar]
- Aufsatz W., Stoiber T., Rakic B. & Naumann K. Arabidopsis histone deacetylase 6: a green link to RNA silencing. Oncogene 26, 5477–5488 (2007). [DOI] [PubMed] [Google Scholar]
- Probst A. V. et al. Arabidopsis histone deacetylase HDA6 is required for maintenance of transcriptional gene silencing and determines nuclear organization of rDNA repeats. Plant Cell 16, 1021–1034 (2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alinsug M. V., Yu C. W. & Wu K. Phylogenetic analysis, subcellular localization, and expression patterns of RPD3/HDA1 family histone deacetylases in plants. BMC Plant Biol 9, 37 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T., Zhang J., Keshet I., Bustin M. & Cedar H. The role of DNA methylation in setting up chromatin structure during development. Nat Genet 34, 187–192(2003). [DOI] [PubMed] [Google Scholar]
- Epsztejn-Litman S. et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol 15, 1176–1183 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman N. et al. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol 8, 188–194 (2006). [DOI] [PubMed] [Google Scholar]
- Bender J. DNA methylation and epigenetics. Annual review of plant biology 55, 41–68 (2004). [DOI] [PubMed] [Google Scholar]
- Chinnusamy V. & Zhu J. K. RNA-directed DNA methylation and demethylation in plants. Sci China C Life Sci 52, 331–343 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X. J. et al. A conserved transcriptional regulator is required for RNA-directed DNA methylation and plant development. Genes Dev 23, 2717–2722 (2009b). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nature reviews Genetics 3(9), 662–673 (2002). [DOI] [PubMed] [Google Scholar]
- Vaillant I. & Paszkowski J. Role of histone and DNA methylation in gene regulation. Curr Opin Plant Biol 10(5), 528–533 (2007). [DOI] [PubMed] [Google Scholar]
- Wierzbicki A. T., Ream T. S., Haag J. R. & Pikaard C. S. RNA polymerase V transcription guides ARGONAUTE4 to chromatin. Nat Genet 41, 630–634 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.