

Abstract
When dealing with large scale gene expression studies, observations are commonly contaminated by sources of unwanted variation such as platforms or batches. Not taking this unwanted variation into account when analyzing the data can lead to spurious associations and to missing important signals. When the analysis is unsupervised, e.g. when the goal is to cluster the samples or to build a corrected version of the dataset—as opposed to the study of an observed factor of interest—taking unwanted variation into account can become a difficult task. The factors driving unwanted variation may be correlated with the unobserved factor of interest, so that correcting for the former can remove the latter if not done carefully. We show how negative control genes and replicate samples can be used to estimate unwanted variation in gene expression, and discuss how this information can be used to correct the expression data. The proposed methods are then evaluated on synthetic data and three gene expression datasets. They generally manage to remove unwanted variation without losing the signal of interest and compare favorably to state-of-the-art corrections. All proposed methods are implemented in the bioconductor package RUVnormalize.
Keywords: Batch effect, Control genes, Gene expression, Normalization, Replicate samples
1. Introduction
Over the last few years, microarray-based gene expression studies involving a large number of samples have been conducted (Cancer Genome Atlas Research Network, 2008), with the goal of helping understand or predict some particular factors of interest like the prognosis or the subtypes of a cancer. Such large gene expression studies are often carried out over several years, may involve several hospitals or research centers and typically contain some unwanted variation. Sources of unwanted variation can be technical elements such as batches, different platforms or laboratories, or any biological signal which is not the factor of interest of the study such as heterogeneity in ages or different ethnic groups.
Unwanted variation can easily lead to spurious associations. For example when one is looking for genes which are differentially expressed between two subtypes of cancer, the observed differential expression of some genes could actually be caused by differences between laboratories if laboratories are partially confounded with subtypes. When doing clustering to identify new subgroups of the disease, one may actually identify some of the unwanted factors if their effects on gene expression are stronger than the subgroup effect. If one is interested in predicting prognosis, one may actually end up predicting whether the sample was collected at the beginning or at the end of the study because better prognosis patients were accepted at the end of the study. In this case, the classifier obtained would have little value for predicting the prognosis of new patients.
Similar problems arise when trying to combine several smaller studies rather than working on one large heterogeneous study: in a dataset resulting from the merging of several studies the strongest effect one can observe is generally related to the membership of samples to different studies. A very important objective is therefore to remove this unwanted variation without losing the variation of interest.
A large number of methods have been proposed to tackle this problem, mostly using linear models. When both the factor of interest and the unwanted factors are observed, the problem essentially boils down to a linear regression. ComBat (Johnson and others, 2007) is an empirical Bayes version of linear regression that has been shown to be quite effective. When the factor of interest is observed but the unwanted factors are not, the latter need to be estimated before a regression is possible. This can be done using some form of factor analysis (Leek and Storey, 2007, 2008; Gagnon-Bartsch and Speed, 2012), or using the entire covariance structure of the gene expression matrix (Kang and others, 2008; Listgarten and others, 2010). Finally if the factor of interest itself is not defined, some methods (Alter and others, 2000) use singular value decomposition (SVD) on gene expression to identify and remove the unwanted variation and others (Benito and others, 2004) remove observed batches by linear regression. A more detailed overview of the literature on this subject is provided in Section 1 of the Supplementary Material.
In this paper, we focus on this latter case where there is no predefined factor of interest. This situation arises when performing unsupervised estimation tasks such as clustering or principal component analysis (PCA), in the presence of unwanted variation. It can also be the case that one needs to normalize a dataset without knowing which factors of interest will be studied. Our objective is to correct the gene expression by estimating and removing the unwanted variation, without removing the—unobserved—variation of interest.
The recent work of Gagnon-Bartsch and Speed (2012) suggests that negative control genes can be used to estimate unwanted factors. Here we propose ways to improve estimation of the effect of these sources when the factor of interest is not observed. Our contributions here are 3-fold. We propose estimators which, given the unwanted factors estimated by Gagnon-Bartsch and Speed (2012), are well suited to estimating their effect in the presence of unobserved factors of interest. We introduce a different estimator which relies on replicate samples. Finally, we systematically compare existing and proposed correction methods on an extensive set of experiments.
Section 2 recalls the model of Gagnon-Bartsch and Speed (2012) and introduces estimators of the effect of a given unwanted factors, which are suited to the case where the factor of interest is unobserved. Section 3 describes an alternative estimator of the unwanted variation using replicate samples rather than the unwanted factors previously estimated using control genes. Section 4 compares existing and proposed correction methods on synthetic data, Section 5 does the same thing on a gene expression dataset.
2. Correction using negative control genes
The removal of unwanted variation (RUV) model used by Gagnon-Bartsch and Speed (2012) is a linear model first introduced in this context by Leek and Storey (2007), with a term representing the variation of interest and another term representing the unwanted variation:
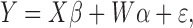 |
(2.1) |
with 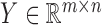 ,
,
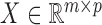 ,
, 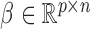 ,
,
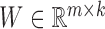 ,
, 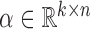 , and
, and
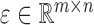 .
. 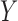 is the observed matrix of expression of
is the observed matrix of expression of
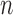 genes for
genes for 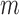 samples,
samples, 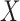 represents the
represents the
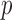 factors of interest,
factors of interest, 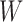 the
the
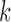 unwanted factors and
unwanted factors and 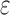 some
noise, typically
some
noise, typically 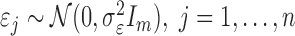 . While Leek and
Storey (2007) and Gagnon-Bartsch and Speed
(2012) use a gene-specific variance
. While Leek and
Storey (2007) and Gagnon-Bartsch and Speed
(2012) use a gene-specific variance 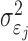 , we restrict
ourselves to a common variance in this work—Sections
, we restrict
ourselves to a common variance in this work—Sections 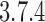 and
and
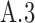 in Gagnon-Bartsch
and others (2013) provide a detailed and illustrated discussion
of why this approximation is reasonable. Both
in Gagnon-Bartsch
and others (2013) provide a detailed and illustrated discussion
of why this approximation is reasonable. Both 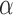 and
and 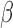 are modeled as
fixed, i.e., non-random.
are modeled as
fixed, i.e., non-random.
Gagnon-Bartsch and Speed (2012) were mainly
interested in the case where 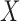 is an observed factor of interest, and the objective is
to test which genes are affected by this factor of interest—whether
is an observed factor of interest, and the objective is
to test which genes are affected by this factor of interest—whether
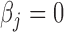 for each gene
for each gene 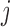 . If
. If 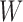 is also observed, the
maximum likelihood estimator of
is also observed, the
maximum likelihood estimator of 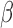 is a well studied linear regression estimator. The
major contribution of Gagnon-Bartsch and Speed
(2012) is to provide an estimator of
is a well studied linear regression estimator. The
major contribution of Gagnon-Bartsch and Speed
(2012) is to provide an estimator of 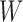 exploiting the fact that some genes are
known to be negative controls, i.e., unrelated to the factor of interest
exploiting the fact that some genes are
known to be negative controls, i.e., unrelated to the factor of interest
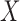 . We refer to this estimator as
. We refer to this estimator as 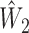 in the
remaining of this article. By contrast in this work, we are interested in the case where
in the
remaining of this article. By contrast in this work, we are interested in the case where
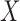 is not observed. Our objective in general will be to
estimate
is not observed. Our objective in general will be to
estimate 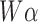 and remove it from
and remove it from 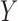 .
.
2.1. A random effect version of RUV for unobserved 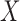
If 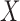 is not observed,
is not observed, 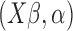 become non-identifiable even given
become non-identifiable even given 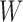 . A naive solution is to estimate
. A naive solution is to estimate
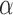 by regression of
by regression of 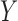 on
on
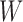 , i.e., to project
, i.e., to project 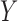 onto the orthogonal
complement of
onto the orthogonal
complement of 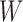 . This approach is referred to as naive
RUV-2 in Gagnon-Bartsch and Speed
(2012) and is expected to be helpful as long as
. This approach is referred to as naive
RUV-2 in Gagnon-Bartsch and Speed
(2012) and is expected to be helpful as long as 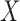 and
and
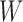 are not too correlated. If however there is some degree
of confounding between the factor of interest and the unwanted variation sources, such a
radical removal of the latter will remove too much of the former. In an extreme example,
if one studies the effect of a treatment on gene expression and all treated samples are
done on Day 1 and all untreated samples on Day 2, removing all variation along the Day
1–Day 2 axis also removes all variation between treated and untreated samples.
are not too correlated. If however there is some degree
of confounding between the factor of interest and the unwanted variation sources, such a
radical removal of the latter will remove too much of the former. In an extreme example,
if one studies the effect of a treatment on gene expression and all treated samples are
done on Day 1 and all untreated samples on Day 2, removing all variation along the Day
1–Day 2 axis also removes all variation between treated and untreated samples.
We now discuss how a random 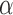 version of (2.1) could improve estimation when
version of (2.1) could improve estimation when 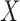 and
and
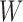 are not expected to be orthogonal.
are not expected to be orthogonal.
The naive RUV-2 estimator of 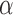 is formally given by
is formally given by
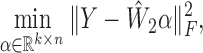 |
(2.2) |
which is the maximum likelihood
estimator of 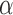 for model (2.1) if
for model (2.1) if 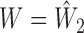 and
and 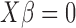 . If we keep the same model
and endow
. If we keep the same model
and endow 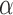 with a distribution
with a distribution 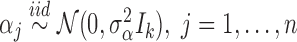 , the maximum
a posteriori estimator of
, the maximum
a posteriori estimator of 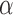 becomes:
becomes:
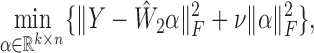 |
(2.3) |
where
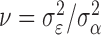 . Here again like with
. Here again like with
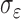 , we limit ourselves to a model where
, we limit ourselves to a model where
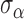 is common to all genes. Sections 14 and 15 of the Supplementary Material discuss a related model
where
is common to all genes. Sections 14 and 15 of the Supplementary Material discuss a related model
where 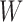 rather than
rather than 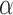 is modeled as a random
quantity.
is modeled as a random
quantity.
The only difference with the naive RUV-2 estimator (2.2) is the 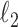 penalty term: (2.3) is a ridge regression against
penalty term: (2.3) is a ridge regression against
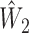 whereas (2.2) is an ordinary regression. In this context where
whereas (2.2) is an ordinary regression. In this context where
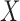 is unobserved and
is unobserved and 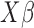 is set to 0
to estimate
is set to 0
to estimate 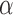 , this difference can be important if
, this difference can be important if
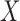 and
and 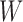 are correlated—for more detail, see
Section 3 of the Supplementary
Material.
are correlated—for more detail, see
Section 3 of the Supplementary
Material.
2.2. Generalization: joint estimation of 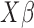 and
and 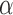
Assuming some structure is known on the unobserved 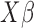 term, it is
possible to write a joint estimator of
term, it is
possible to write a joint estimator of 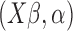 given
given
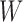 rather than fixing
rather than fixing 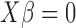 :
:
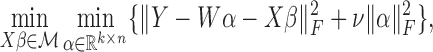 |
(2.4) |
where
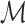 is a subset of
is a subset of 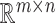 . A typical example of
. A typical example of 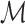 would be
a clustering structure
would be
a clustering structure 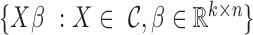 , where
, where 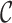 denotes the set of cluster
membership matrices
denotes the set of cluster
membership matrices 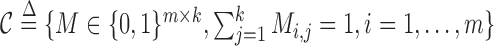 . In this case, the minimization over
. In this case, the minimization over
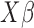 in (2.4) for a given
in (2.4) for a given 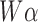 can be addressed by a k-means
algorithm over
can be addressed by a k-means
algorithm over 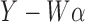 . Other typical examples include PCA where
. Other typical examples include PCA where
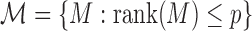 and sparse dictionary learning
(Mairal and others, 2010)
where
and sparse dictionary learning
(Mairal and others, 2010)
where 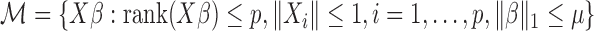 . Section 12 of the Supplementary Material provides an alternative
formulation of (2.4).
. Section 12 of the Supplementary Material provides an alternative
formulation of (2.4).
The objective of this joint modeling can be 2-fold: one may still just want to estimate
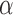 in order to return a corrected
in order to return a corrected 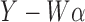 matrix,
but hope that the joint estimation will yield a better estimate of
matrix,
but hope that the joint estimation will yield a better estimate of
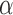 (in the sense of
(in the sense of 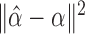 ). One may also be actually interested in estimating
the unobserved
). One may also be actually interested in estimating
the unobserved 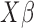 , e.g., to solve a clustering problem in the presence of
unwanted variation.
, e.g., to solve a clustering problem in the presence of
unwanted variation.
A joint solution for 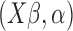 is generally not available for (2.4). A possible way of maximizing the
likelihood of
is generally not available for (2.4). A possible way of maximizing the
likelihood of 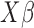 however is to alternate between a step of optimization
over
however is to alternate between a step of optimization
over 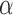 for a given
for a given 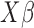 , which corresponds to a ridge
regression problem, and a step of optimization over
, which corresponds to a ridge
regression problem, and a step of optimization over 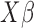 for a given
for a given
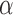 using the relevant unsupervised estimation procedure over
using the relevant unsupervised estimation procedure over
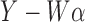 . Each step decreases the objective
. Each step decreases the objective
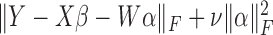 , and even if this procedure
does not converge in general to the global maximum likelihood of
, and even if this procedure
does not converge in general to the global maximum likelihood of 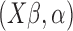 ,
it may yield better estimates than (2.3) where
,
it may yield better estimates than (2.3) where 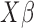 is simply assumed to be 0.
is simply assumed to be 0.
Finally, such a joint scheme can be used to build a different estimator of
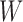 , akin to the feasible generalized least squares (Freedman, 2005) used in regression: once an
estimate of
, akin to the feasible generalized least squares (Freedman, 2005) used in regression: once an
estimate of 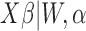 becomes available,
becomes available, 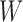 can be re-estimated
using and SVD on the residuals
can be re-estimated
using and SVD on the residuals 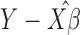 rather than the
control genes. This approach is discussed in Section 2 of the Supplementary Material.
rather than the
control genes. This approach is discussed in Section 2 of the Supplementary Material.
3. Correction using replicate samples
We now introduce an alternative estimator of the unwanted variation
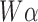 , which, unlike the ones discussed in Section 2 does not rely on a previous estimator of
, which, unlike the ones discussed in Section 2 does not rely on a previous estimator of
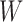 . Symmetrically to the negative control genes used to
estimate
. Symmetrically to the negative control genes used to
estimate 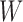 in Section 2, we now
consider negative “control samples” for which the factor of interest
in Section 2, we now
consider negative “control samples” for which the factor of interest
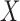 is 0.
is 0.
In practice, one way of obtaining such control samples is to use replicate samples, i.e.,
samples that come from the same tissue but which were hybridized in two different settings,
say across time or platform. The profile formed by the difference of two such replicates
should only be influenced by unwanted variation—those whose levels differ between the two
replicates. In particular, the 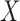 of this difference should be 0. By construction, this
approach is only able to deal with unwanted variation with respect to which replicates are
available, which is often the case for technical unwanted variation but rarely the case for
biological unwanted variation.
of this difference should be 0. By construction, this
approach is only able to deal with unwanted variation with respect to which replicates are
available, which is often the case for technical unwanted variation but rarely the case for
biological unwanted variation.
More generally when there are more than two replicates, one may take all pairwise
differences or the differences between each replicate and the average of the other
replicates. We will denote by 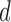 the indices of these artificial control samples formed
by differences of replicates, and we therefore have
the indices of these artificial control samples formed
by differences of replicates, and we therefore have 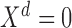 where
where
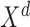 are the rows of
are the rows of 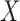 indexed by
indexed by
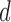 .
.
Such samples can then be used to estimate 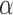 in a way that is dual to the way
(Gagnon-Bartsch and Speed, 2012) used control
genes to estimate
in a way that is dual to the way
(Gagnon-Bartsch and Speed, 2012) used control
genes to estimate 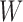 , recalled in Section 3 of the Supplementary Material. More precisely, we
consider the following algorithm:
, recalled in Section 3 of the Supplementary Material. More precisely, we
consider the following algorithm:
Use the rows of
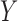 corresponding to control samples
corresponding to control samples
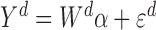 to estimate
to estimate 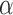 . Assuming
i.i.d. noise
. Assuming
i.i.d. noise 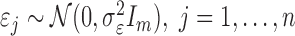 , the
, the 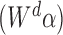 matrix maximizing the
likelihood of this model is
matrix maximizing the
likelihood of this model is 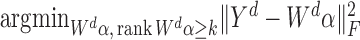 . By the same argument used to compute
. By the same argument used to compute
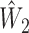 , this argmin is reached for
, this argmin is reached for
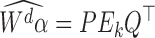 , where
, where 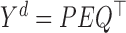 is
the SVD of
is
the SVD of 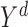 and
and 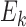 is the diagonal matrix with the
is the diagonal matrix with the
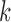 largest singular values as its
largest singular values as its
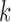 first entries and 0 on the rest of the diagonal. We
can use
first entries and 0 on the rest of the diagonal. We
can use 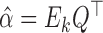 .
.Using
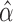 in the restriction
in the restriction 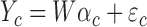 of
of 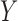 to the control gene columns, the
maximum likelihood estimate of
to the control gene columns, the
maximum likelihood estimate of 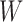 is now solved by a linear
regression,
is now solved by a linear
regression, 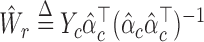 .
.Once
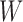 and
and 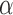 are estimated,
are estimated,
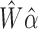 can be removed from
can be removed from 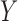 .
.
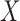 is not required in this procedure which constitutes a fully unsupervised
correction for
is not required in this procedure which constitutes a fully unsupervised
correction for 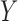 .
.
The extreme case where all genes are used as control genes is of interest, and is discussed in Section 10 of the Supplementary Material.
Finally, this replicate-based correction yields an estimator 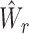 of
of
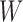 , obtained by regression of
, obtained by regression of 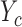 against
against
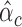 . This estimator could be used in any of the methods
described in Section 2. Section 11 of the Supplementary Material discusses the difference
between
. This estimator could be used in any of the methods
described in Section 2. Section 11 of the Supplementary Material discusses the difference
between 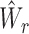 and
and 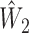 .
.
4. Result on synthetic data
We start with a set of experiments on synthetic data, where we generate the data ourselves.
In this case, we are able to measure how well each correction method recovers the true
matrix 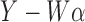 and it makes sense to talk about “right” or “best”
corrections.
and it makes sense to talk about “right” or “best”
corrections.
4.1. Protocol
We generate data according to model (2.1). 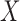 has two columns: one is a binary variable, the other
an associated continuous variable. One can think of the binary variable as some clinical
grouping of the tumor, such as the ER status for breast cancer. The continuous variable
could be survival time. More precisely the survival covariate is sampled from an
exponential law with parameter
has two columns: one is a binary variable, the other
an associated continuous variable. One can think of the binary variable as some clinical
grouping of the tumor, such as the ER status for breast cancer. The continuous variable
could be survival time. More precisely the survival covariate is sampled from an
exponential law with parameter 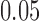 for ER
for ER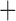 samples and
samples and
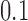 for ER
for ER samples.
samples. 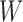 also has two
columns, a binary one which could be the technical platform, and a continuous one which
could be the RNA quality. RNA quality is sampled from a normal distribution—independently
from the technical platform.
also has two
columns, a binary one which could be the technical platform, and a continuous one which
could be the RNA quality. RNA quality is sampled from a normal distribution—independently
from the technical platform.
The columns of 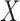 and
and 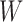 are then transformed to have norm 1.
We generate
are then transformed to have norm 1.
We generate 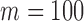 samples with
samples with 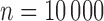 genes each
using model (2.1). The
genes each
using model (2.1). The
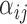 and
and 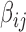 parameters are iid sampled
from a normal distribution with mean 0 and variance 1, except for the 100 control genes,
for which
parameters are iid sampled
from a normal distribution with mean 0 and variance 1, except for the 100 control genes,
for which 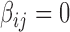 . The
. The 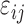 parameters are i.i.d.
sampled from a normal distribution with mean 0 and variance
parameters are i.i.d.
sampled from a normal distribution with mean 0 and variance 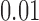 . We also
generate 10 additional samples with 2 replicates each, with the same values of
. We also
generate 10 additional samples with 2 replicates each, with the same values of
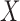 but different values of
but different values of 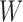 .
.
We compare the performances of ComBat (Johnson
and others, 2007), quantile normalization (QN, Bolstad and others, 2003), naive
RUV-2, the random effect model above, and the replicate-based model on simulated data.
ComBat cannot deal with continuous unwanted variation factors and is only given the binary
unwanted one. Each method using negative control genes is given either the actual negative
control genes, or everything but the negative control genes (“poor control genes”
version). Furthermore, we try two iterative methods, as described in 2.2. We perform 100 iterations: in the first version,
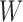 is estimated once and for all from control genes like
in the non-iterative random effect estimator, and in the second version it is re-estimated
from the
is estimated once and for all from control genes like
in the non-iterative random effect estimator, and in the second version it is re-estimated
from the 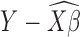 as detailed in Section 2 of the Supplementary Material after every 34
iterations.
as detailed in Section 2 of the Supplementary Material after every 34
iterations.
Table 1 shows the performances of each
correction method in three different settings: canonical correlations (Hotelling, 1936) between 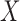 and
and
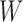 equal to
equal to 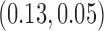 (Independent),
(Independent), 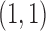 (Confounded),
and
(Confounded),
and 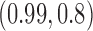 (Moderate). The normalized
reconstruction error is measured by
(Moderate). The normalized
reconstruction error is measured by 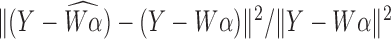 : our objective is to remove the unwanted variation
from
: our objective is to remove the unwanted variation
from 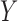 , and only the unwanted variation. These performances
are those obtained when using the hyperparameter used to generate the data for each
method. The effect of misspecification for
, and only the unwanted variation. These performances
are those obtained when using the hyperparameter used to generate the data for each
method. The effect of misspecification for 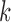 and
and 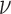 is discussed in
Section 8 of the Supplementary Material.
is discussed in
Section 8 of the Supplementary Material.
Table 1.
Simulations: reconstruction error after various corrections in the three studies settings
| Independent | Confounded | Moderate | |
|---|---|---|---|
| Uncorrected | 0.67 | 0.68 | 0.67 |
| QN | 0.74 | 0.66 | 0.63 |
| ComBat | 0.34 | 0.66 | 0.76 |
| naive RUV2 | 0.17 | 0.67 | 0.53 |
| naive RUV2 poor control | 0.74 | 0.67 | 0.67 |
| random effect | 0.23 | 0.34 | 0.31 |
| random poor control | 0.48 | 0.37 | 0.38 |
| Iterative | 0.11 | 0.46 | 0.19 |
| Iterative update | 0.06 | 0.40 | 0.16 |
| Replicates | 0.32 | 0.30 | 0.30 |
| Replicates poor control | 0.28 | 0.28 | 0.28 |
4.2. Result
In the case, where 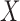 and
and 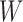 are generated independently, the
QN-corrected matrix yields a larger reconstruction error than the uncorrected one. ComBat
helps, because it removes all of the platform effect without affecting much the signal
along
are generated independently, the
QN-corrected matrix yields a larger reconstruction error than the uncorrected one. ComBat
helps, because it removes all of the platform effect without affecting much the signal
along 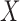 . Naive RUV-2 gets even better results because it also
accounts for the continuous unwanted factor, but its performance is severely reduced if we
use non-control genes: in this case, the estimated
. Naive RUV-2 gets even better results because it also
accounts for the continuous unwanted factor, but its performance is severely reduced if we
use non-control genes: in this case, the estimated 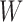 —by PCA on the
non-control genes—is associated with the true
—by PCA on the
non-control genes—is associated with the true 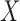 , which leads to removing too much
variance along
, which leads to removing too much
variance along 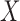 .
.
The random effect model yields similar performances as naive RUV-2, slightly worse
because it uses 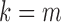 and therefore shrinks the signal in every
direction. It is also affected by the use of non-control genes, but less dramatically than
naive RUV-2 because it does not remove all the signal along the estimated
and therefore shrinks the signal in every
direction. It is also affected by the use of non-control genes, but less dramatically than
naive RUV-2 because it does not remove all the signal along the estimated
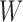 . The iterative methods greatly improves the
performances.
. The iterative methods greatly improves the
performances.
The replicate-based method obtains a performance similar to that of ComBat. Interestingly, its results are slightly improved by using non-control genes, see Section 10 of the Supplementary Material for a detailed discussion of this phenomenon.
When 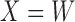 (Confounded), QN, ComBat and naive
RUV-2 lead to the same reconstruction error as the uncorrected matrix. They actually fail
for opposite reasons: the unwanted variation along
(Confounded), QN, ComBat and naive
RUV-2 lead to the same reconstruction error as the uncorrected matrix. They actually fail
for opposite reasons: the unwanted variation along 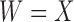 adds variance
along
adds variance
along 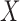 , so the uncorrected matrix has too much variance along
this direction. By contrast, ComBat/naive RUV-2 remove too much variance along
, so the uncorrected matrix has too much variance along
this direction. By contrast, ComBat/naive RUV-2 remove too much variance along
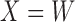 , because they treat all of it as unwanted variation.
The random effect method performs a bit less well than in the independent case, but is not
as dramatically affected by the confounding as the fixed effect naive RUV-2 method.
Importantly, the iterative methods decrease the performance with respect to the
non-iterative random effect estimator. The iterative methods only help if they manage to
estimate
, because they treat all of it as unwanted variation.
The random effect method performs a bit less well than in the independent case, but is not
as dramatically affected by the confounding as the fixed effect naive RUV-2 method.
Importantly, the iterative methods decrease the performance with respect to the
non-iterative random effect estimator. The iterative methods only help if they manage to
estimate 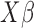 well enough to improve the estimation of
well enough to improve the estimation of
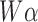 . In this setting,
. In this setting, 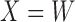 so removing
so removing
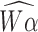 decreases the variance along
decreases the variance along 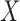 too much, and
makes it harder to identify it properly.
too much, and
makes it harder to identify it properly.
Finally, as expected from the discussion of Section 11 of Supplementary Material, the replicate-based method is not affected by the confounding at all: by construction, it only considers the variance coming from the technical unwanted variation.
The last column illustrates an intermediate case with moderate confounding. Random effect
still works much better than uncorrected, QN, ComBat and naive RUV-2, and is less affected
by the use of non-control genes. It is worth noting that even if 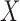 and
and
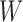 are highly correlated, the iterative methods yield a
much lower error than the non-iterative one, suggesting that they only reduce performances
in extreme cases like the total confounding setting.
are highly correlated, the iterative methods yield a
much lower error than the non-iterative one, suggesting that they only reduce performances
in extreme cases like the total confounding setting.
We now summarize our observations on these synthetic experiments. ComBat performs well to
remove an observed batch if it is largely independent from the signal of interest. Naive
RUV-2 performs well to remove a batch—observed or not—if it is given good control genes
and if the batch is not too associated with the factor of interest. The random effect
model performs like naive RUV-2 but is much less affected by confounding and poor control
genes. Iterating the estimation of 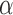 given
given 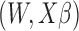 and
and
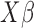 given
given 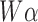 improves the estimate of
improves the estimate of
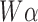 , even more so if the residuals
, even more so if the residuals 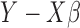 are used
to update the estimate
are used
to update the estimate 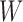 . This strategy fails however when
. This strategy fails however when
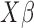 is too hard to estimate, in which case iterations can even
reduce the performances. Finally, the replicate-based method is not affected at all by
control gene quality or confounding, it only depends on the number/coverage of
replicates—see Sections 10 and 13 of the Supplementary Material for additional discussion of these points.
is too hard to estimate, in which case iterations can even
reduce the performances. Finally, the replicate-based method is not affected at all by
control gene quality or confounding, it only depends on the number/coverage of
replicates—see Sections 10 and 13 of the Supplementary Material for additional discussion of these points.
5. Result on real data
On real data, we have no way to measure how close we are to the true
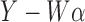 matrix. As a surrogate, we choose datasets for which we know
a factor of interest
matrix. As a surrogate, we choose datasets for which we know
a factor of interest 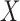 , and measure how well this factor is recovered by
clustering on each corrected gene expression matrix. The correction methods are not allowed
to use the known groups, to emulate a problem where the factor of interest is not observed.
This surrogate is admittedly imperfect, as other factors of interest may be present in these
datasets. Whether or not removing the effect of these (possibly biological) other factors is
desirable depends on whether they are considered “wanted” or “unwanted” in any particular
analysis. We consider these surrogates to be complementary to the synthetic datasets of
Section 4—which are not real data but where we can
define what the right correction is.
, and measure how well this factor is recovered by
clustering on each corrected gene expression matrix. The correction methods are not allowed
to use the known groups, to emulate a problem where the factor of interest is not observed.
This surrogate is admittedly imperfect, as other factors of interest may be present in these
datasets. Whether or not removing the effect of these (possibly biological) other factors is
desirable depends on whether they are considered “wanted” or “unwanted” in any particular
analysis. We consider these surrogates to be complementary to the synthetic datasets of
Section 4—which are not real data but where we can
define what the right correction is.
This section presents results on the microarray gene expression datasets studied in Gagnon-Bartsch and Speed (2012). We use the gender
partition as the factor of interest to be recovered, implicitly treating other factors as
unwanted variation, but discuss other options in Section 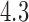 of the Supplementary Material. Two additional datasets
are studied in Sections 5 and 6 of the Supplementary
Material, and Section 7 of the Supplementary Material shows the importance of having good negative control genes
on correction quality.
of the Supplementary Material. Two additional datasets
are studied in Sections 5 and 6 of the Supplementary
Material, and Section 7 of the Supplementary Material shows the importance of having good negative control genes
on correction quality.
5.1. Protocol
For each of the correction methods that we evaluate, we apply the correction method to
the expression matrix 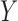 and then estimate the clustering using a
and then estimate the clustering using a
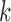 -means algorithm. To quantify how close each clustering
gets to the objective partition, we adopt the following squared distance between two given
partitions
-means algorithm. To quantify how close each clustering
gets to the objective partition, we adopt the following squared distance between two given
partitions 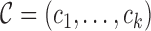 and
and 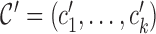 of the samples into
of the samples into 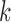 clusters:
clusters:
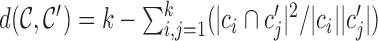 , where
, where 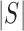 denotes the
cardinal of a set
denotes the
cardinal of a set 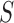 . This score ranges between 0 when the two partitions
are equivalent, and
. This score ranges between 0 when the two partitions
are equivalent, and 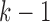 when the two partitions are completely different.
To give a visual impression of the effect of the corrections on the data, we also plot the
data in the space spanned by the first two principal components.
when the two partitions are completely different.
To give a visual impression of the effect of the corrections on the data, we also plot the
data in the space spanned by the first two principal components.
We evaluate two basic correction methods: the replicate-based procedure described in
Section 3 and the random 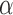 model of
Section 2.1 with
model of
Section 2.1 with 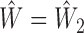 . For each of these two methods, we also evaluate
iterative versions as described in Section 2.2.
. For each of these two methods, we also evaluate
iterative versions as described in Section 2.2.
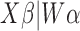 is estimated using the sparse dictionary estimator of
Mairal and others (2010),
which minimizes
is estimated using the sparse dictionary estimator of
Mairal and others (2010),
which minimizes 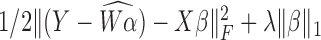 under the constraint that
under the constraint that 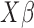 has rank
has rank
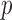 and the columns of
and the columns of 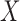 have norm 1.
have norm 1.
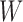 is re-estimated using the
is re-estimated using the 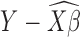 residuals every 10 iterations as discussed in Section 2 of the Supplementary Material. In addition, we
consider as baselines (i) an absence of correction, (ii) a centering of the data by level
of the known unwanted factors—similar to the correction provided by ComBat, and (iii) the
naive RUV-2 procedure.
residuals every 10 iterations as discussed in Section 2 of the Supplementary Material. In addition, we
consider as baselines (i) an absence of correction, (ii) a centering of the data by level
of the known unwanted factors—similar to the correction provided by ComBat, and (iii) the
naive RUV-2 procedure.
Some of the methods we compare require the user to choose some hyperparameters: the ranks
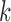 of
of 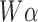 and
and 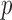 of
of
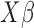 , the ridge parameter
, the ridge parameter 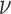 and the
strength
and the
strength 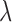 of the
of the 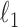 penalty on
penalty on
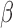 . On synthetic data, it makes sense to define which
hyperparameters yield the best correction. On real data by contrast, different choices of
these hyperparameters may lead to throwing away or keeping different signals, which can be
a good or a bad thing depending on what downstream analysis is decided afterward. This
point is illustrated in Section
. On synthetic data, it makes sense to define which
hyperparameters yield the best correction. On real data by contrast, different choices of
these hyperparameters may lead to throwing away or keeping different signals, which can be
a good or a bad thing depending on what downstream analysis is decided afterward. This
point is illustrated in Section  of the Supplementary Material: large values of
of the Supplementary Material: large values of
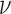 lead to a clustering by brain region while smaller values
lead to a clustering by gender.
lead to a clustering by brain region while smaller values
lead to a clustering by gender.
No single rule can be therefore given as to hyperparameter choice and judgment is
necessary each time adjustments are performed without a specified factor of interest. We
suggest using relative log expression (RLE) plots, clustering with respect to a known
factor of interest, or known differentially expressed genes with respect to a known factor
of interest as positive controls. In this experiment, we compare normalization methods
based on how well they allow clustering to recover the gender partition, so it would not
make sense to use the same criterion to choose 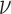 . In Section 4
of the Supplementary Material, we use RLE
plots to pick
. In Section 4
of the Supplementary Material, we use RLE
plots to pick 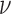 for this dataset, and also discuss the other criteria
(see Table 1 of the Supplementary
Material).
for this dataset, and also discuss the other criteria
(see Table 1 of the Supplementary
Material).
Since 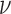 acts on the eigenvalues of
acts on the eigenvalues of 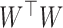 , we
recommend considering a grid of powers of 10 of the largest of them—the discussion in the
Supplementary Material regards how to
choose the power. The rank
, we
recommend considering a grid of powers of 10 of the largest of them—the discussion in the
Supplementary Material regards how to
choose the power. The rank 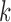 of
of 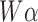 was chosen to be close to
was chosen to be close to
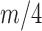 , or to the number of replicate samples when the
latter was smaller than the former. For methods using
, or to the number of replicate samples when the
latter was smaller than the former. For methods using 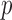 , we chose
, we chose
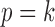 . For random
. For random 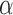 models, we
use
models, we
use 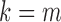 : the model is regularized by the ridge
: the model is regularized by the ridge
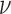 and we do not combine it with a regularization of the
rank. Finally, in order to make iterative and non-iterative methods comparable, we choose
and we do not combine it with a regularization of the
rank. Finally, in order to make iterative and non-iterative methods comparable, we choose
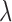 for each method such that
for each method such that 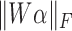 is
close to the one obtained with its non-iterative counterpart.
is
close to the one obtained with its non-iterative counterpart.
5.2. Result
Vawter and others (2004) studied differences in gene expression between male and female patients. Gagnon-Bartsch and Speed (2012) used the resulting dataset to study the performances of RUV-2.
This gender study is an interesting benchmark for methods aiming at removing unwanted variation as it expected to be affected by several technical and biological factors: two microarray platforms, three different labs, three tissue localizations in the brain. Most of the 10 patients involved in the study had samples taken from the anterior cingulate cortex (a), the dorsolateral prefontal cortex (d), and the cerebellar hemisphere (c). Most of these samples were sent to three independent labs: UC Irvine (I), UC Davis (D) and University of Michigan, Ann Arbor (M).
Gene expression was measured using either HGU-95A or HGU-95Av2 Affymetrix arrays with
12 600 genes shared between the two platforms. Six of the 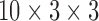 combinations were missing, leading to 84 samples. We use as control genes the same 799
housekeeping probesets, which were used in Gagnon-Bartsch and Speed (2012). The proportion of genes on the sex chromosomes
is similar in the housekeeping genes (
combinations were missing, leading to 84 samples. We use as control genes the same 799
housekeeping probesets, which were used in Gagnon-Bartsch and Speed (2012). The proportion of genes on the sex chromosomes
is similar in the housekeeping genes (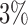 ) and other genes
(
) and other genes
(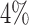 ).
).
For the replicate-based method of Section 3, we use all possible pairs that either differ in lab, but are otherwise identical in terms of chip type, the patient, and brain region, or (ii) differ in brain region but are otherwise identical in terms of chip type, the patient, and lab, leading to 106 differences. Finally as a pre-processing, we also mean-center the samples per array type.
Since most genes function irrespective of gender, clustering by gender gives better results in general when removing genes with low variance before clustering. For each method, we therefore apply clustering after filtering different numbers of genes based on their variance after correction.
Figure 1 shows the clustering error for the
methods against the number of genes retained. The uncorrected and mean-centering cases are
not displayed to avoid cluttering the plot, but give values above
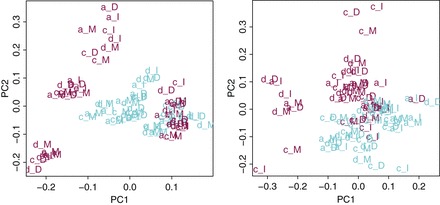
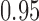 for all numbers of genes retained. Figure 2 shows the samples in the space of the first two
principal components in these two cases, keeping the 1260 genes with highest variance. On
the uncorrected data (left panel), it is clear that the samples first cluster by lab which
is the main source of variance, then by brain region which is the second main source of
variance. This explains why the clustering on uncorrected data is far away from a
clustering by gender. Mean-centering samples by region-lab (right panel) removes all
clustering per brain region or lab, but does not make the samples cluster by gender.
for all numbers of genes retained. Figure 2 shows the samples in the space of the first two
principal components in these two cases, keeping the 1260 genes with highest variance. On
the uncorrected data (left panel), it is clear that the samples first cluster by lab which
is the main source of variance, then by brain region which is the second main source of
variance. This explains why the clustering on uncorrected data is far away from a
clustering by gender. Mean-centering samples by region-lab (right panel) removes all
clustering per brain region or lab, but does not make the samples cluster by gender.
Fig. 1.
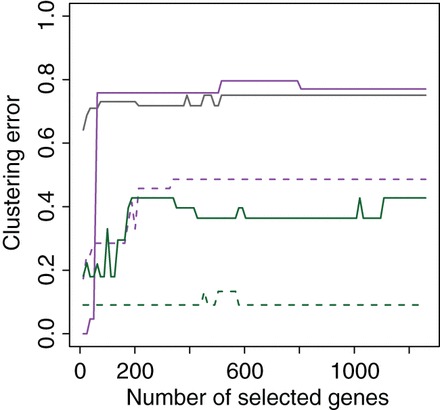
Clustering error against number of genes selected (based on variance) before
clustering. From top to bottom at 1260 genes: replicate-based correction (full
purple); naive RUV-2 (full gray); iterated replicate-based correction (dashed purple);
random 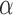 model using
model using 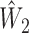 (full
green); iterated random
(full
green); iterated random 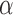 model using
model using 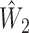 (dashed green).
(dashed green).
Fig. 2.
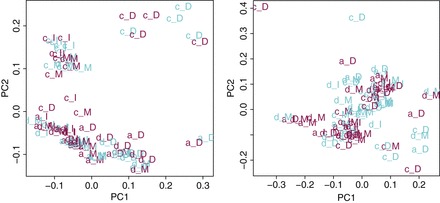
Samples of the gender study represented in the space of their first two principal components before correction (left panel) and after centering by lab plus brain region (right panel). Light blue samples are males, dark pink samples are females. The labels indicate the laboratory and brain region of each sample. The capital letter is the laboratory and the lowercase one is the brain region.
The gray line of Figure 1 shows the
performance of naive RUV-2 for  . Since naive RUV-2 is a radical
correction which removes all variance along some directions, it is expected to be more
sensitive to the choice of
. Since naive RUV-2 is a radical
correction which removes all variance along some directions, it is expected to be more
sensitive to the choice of  . The estimation is damaged by using
. The estimation is damaged by using
 (clustering error
(clustering error 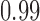 ). Using
). Using
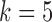 also degrades the performances, except when very few
genes are kept.
also degrades the performances, except when very few
genes are kept.
The purple lines of Figure 1 represent the replicate-based corrections. The solid line shows the performances of the non-iterative method described in Section 3. When very few genes are selected, it leads to a perfect clustering by gender, which no other method achieves regardless of the number of genes they retain. When considering more genes, however, its performance become similar to the one of naive RUV-2, suggesting that additional genes are influenced by non-gender variation which the replicate-based method does not remove. It is expected that a few genes are more strongly affected by gender than the others, so it makes sense for a correction method to recover a better clustering by gender after restriction to a small number of high variance genes. In addition, Table 1 of the Supplementary Material shows that even though the replicate-based method has a large clustering error, it actually performs as well as or better than other methods in terms of number of differentially expressed genes on the sex chromosomes.
The iterative version in dotted line leads to much better clustering except again when very few genes are selected. Figure 3 shows the samples in the space of the first two principal components after applying the non-iterative (left panel) and iterative (right panel) replicate-based method. The correction shrinks the replicates together, leading to a new variance structure, more driven by gender although not separating perfectly males and females.
Fig. 3.
Using replicates. Left: no iteration and right: with iterations.
The green lines of Figure 1 correspond to the
random 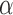 -based corrections. The solid line shows the results for
the non-iterative method. These results are good, as illustrated by the reasonably good
separation obtained in the space spanned by the first two principal components after
correction on the left panel of Figure 4. The
dotted green line corresponds to the random
-based corrections. The solid line shows the results for
the non-iterative method. These results are good, as illustrated by the reasonably good
separation obtained in the space spanned by the first two principal components after
correction on the left panel of Figure 4. The
dotted green line corresponds to the random 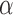 -based corrections with
iterations plus sparsity, which leads to an even lower clustering error.
-based corrections with
iterations plus sparsity, which leads to an even lower clustering error.
Fig. 4.
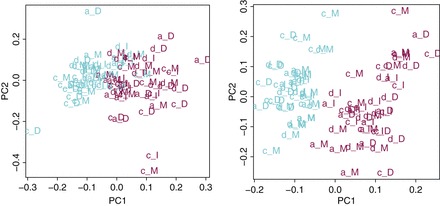
Random alpha with control genes only. Left: no iteration and right: with iterations.
6. Discussion
We introduced methods to estimate and remove unobserved unwanted variation from gene expression data when the factor of interest is also unobserved. One method uses the negative control gene-based estimator of unwanted factors introduced in Gagnon-Bartsch and Speed (2012), and estimates the effect of these factors on gene expression using a random effect model. The second method relies on replicate samples and estimates the unwanted variation using the variation observed in differences of replicates. Both estimators can be improved by joint modeling of the variation of interest and the unwanted variation. All the methods we introduce are available in the bioconductor package RUVnormalize (Jacob, 2014).
We systematically compared the proposed correction techniques with state-of-the-art methods on both synthetic and real gene expression data. On synthetic data, we knew what the correct signal was, and could measure how well each correction method recovered this signal. When good control genes were available, the random effect estimator performed much better than existing correction methods in the presence of confounding. The replicate-based method performed less well than the control gene based one—unless a really large number of replicates was available—but was unaffected by poor quality control genes and to large confounding level. We were able to verify that both proposed methods provide a better correction even in the case where the factor of interest and the unwanted factors are totally confounded.
On real gene expression data where it did not make sense to define a single correct signal to be recovered, we assessed how well we were able to rediscover by clustering a known factor of interest which was unspecified at correction time. Here again, the proposed methods lead to better reconstruction than existing corrections.
Assessing how well each unsupervised correction method works on a new real dataset is
problematic, since the factor of interest is not observed. Clustering with respect to a
known biological factor, like we do with gender, is one option to perform this assessment.
Other options include using positive control genes and RLE plots, like we do in the Supplementary Material. None of these options is
perfect but they can be used as guidelines, to monitor whether too much variance is being
removed by any correction method. In particular, they can and should be used to choose
regularization parameters such as the rank 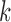 of
of 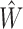 and the ridge
and the ridge
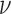 of random
of random 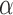 approaches. In any case, one
should keep in mind that optimizing for one known thing may not optimize for another: in our
gender data example, the parameters which were chosen by RLE and behaved well for gender
recovery are not optimal for recovering a partition by brain region.
approaches. In any case, one
should keep in mind that optimizing for one known thing may not optimize for another: in our
gender data example, the parameters which were chosen by RLE and behaved well for gender
recovery are not optimal for recovering a partition by brain region.
To conclude, our results suggest that it is possible to remove unwanted variation from gene expression without losing the signal of interest, provided enough controls are available: negative control genes which are affected by the unwanted factors only, or replicate samples. Together with other researchers in our groups we have also started applying some of the methods that we introduce here to RNA-Seq (Risso and others, 2014), metabolomics (Livera and others, 2015) and expression array data (Jacob and others, 2015) and obtained consistently good results. We hope these extensive evaluations and comparisons will be helpful to future researchers trying to remove unwanted variation from their data.
Supplementary material
Supplementary material is available at http://biostatistics.oxfordjournals.org.
Funding
This work was funded by the SU2C-AACR-DT0409 grant. Funding to pay the Open Access publication charges for this article was provided by Australian National Health and Medical Research Council Program Grant APP1054618.
Supplementary Material
Acknowledgments
The authors thank Julien Mairal, Anne Biton, Leming Shi, Jennifer Fostel, Minjun Chen, and Moshe Olshansky for helpful discussions. Conflict of Interest: None declared.
References
- Alter O., Brown P. O., Botstein D. (2000). Singular value decomposition for genome-wide expression data processing and modeling. PNAS 97(18), 10101–10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito M., Parker J., Du Q., Wu J., Xiang D., Perou C. M., Marron J. S. (2004). Adjustment of systematic microarray data biases. Bioinformatics 20(1), 105–14. [DOI] [PubMed] [Google Scholar]
- Bolstad B. M., Irizarry R. A., Astr M., Speed T. P. (2003). A comparison of normalization methods for high density. Bioinformatics 19, 185–193. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455(7216), 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Livera A. M., Sysi-Aho M., Jacob L., Gagnon-Bartsch J. A., Castillo S., Simpson J. A., Speed T. P. (2015). Statistical methods for handling unwanted variation in metabolomics data. Analytical Chemistry 87(7), 3606–3615. PMID: 25692814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman D. (2005) Statistical Models: Theory And Practice. Cambridge: Cambridge University Press. [Google Scholar]
- Gagnon-Bartsch J., Jacob L., Speed T. P. (2013). Removing unwanted variation from high dimensional data with negative controls. Technical Report, UC Berkeley. Technical report 820. Monograph in preparation.
- Gagnon-Bartsch J. A., Speed T. P. (2012). Using control genes to correct for unwanted variation in microarray data. Biostatistics 13(3), 539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotelling H. (1936). Relation between two sets of variates. Biometrika 28, 322–377. [Google Scholar]
-
Jacob L. (2014). RUV for Normalization of Expression
Array Data. Bioconductor
 3.0.
3.0. - Jacob L., Van Den Akker J., Witteveen A., Goosens I., Speed T. P., Glas A., Veer L. V. (2015). A blueprint for managing microarray technical variations and data processing in the large randomized MINDACT trial (in preparation). [Google Scholar]
- Johnson W. E., Li C., Biostatistics, Department, Biology, Computational, Rabinovic A. (2007). Adjusting batch effects in microarray expression data using empirical bayes methods. Biostatistics 1(8), 118–127. [DOI] [PubMed] [Google Scholar]
- Kang H. M., Ye C., Eskin E. (2008). Accurate discovery of expression quantitative trait loci under confounding from spurious and genuine regulatory hotspots. Genetics 180(4), 1909–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek J. T., Storey J. D. (2007). Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genetics 3(9), 1724–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek J. T., Storey J. D. (2008). A general framework for multiple testing dependence. PNAS 105(48), 18718–18723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listgarten J., Kadie C., Schadt E. E., Heckerman D. (2010). Correction for hidden confounders in the genetic analysis of gene expression. PNAS 107(38), 16465–16470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mairal J., Bach F., Ponce J., Sapiro G. (2010). Online learning for matrix factorization and sparse coding. Journal of Machine Learning Research 11, 19–60. [Google Scholar]
- Risso D., Ngai J., Speed T. P, Dudoit S. (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nature Biotechnology 32(9), 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vawter M. P. and others. (2004). Gender-specific gene expression in post-mortem human brain: localization to sex chromosomes. Neuropsychopharmacology 29(2), 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.