

Abstract
Neurotropic viruses initiate infection in peripheral tissues prior to entry into the central nervous system (CNS). However, mechanisms of dissemination are not completely understood. We used genetically marked viruses to compare dissemination of poliovirus, yellow fever virus 17D (YFV-17D), and reovirus type 3 Dearing in mice from a hind limb intramuscular inoculation site to the sciatic nerve, spinal cord, and brain. While YFV-17D likely entered the CNS via blood, poliovirus and reovirus likely entered the CNS by transport through the sciatic nerve to the spinal cord. We found that dissemination was inefficient in adult immune-competent mice for all three viruses, particularly reovirus. Dissemination of all viruses was more efficient in immune-deficient mice. Although poliovirus and reovirus both accessed the CNS by transit through the sciatic nerve, stimulation of neuronal transport by muscle damage enhanced dissemination only of poliovirus. Our results suggest that these viruses access the CNS using different pathways.
Keywords: virus, neurotropic, sciatic nerve, interferon, barrier, poliovirus, reovirus, yellow fever virus 17D
Introduction
Neurotropic viruses can invade and infect the central nervous system (CNS), which includes the spinal cord and brain. These viruses come from a variety of different families and include several that infect humans (Koyuncu et al., 2013). Neurotropic viruses enter the CNS through peripheral nerves or by crossing the blood-brain barrier following hematogenous dissemination. Different viruses target different cell types within the nervous system, causing symptoms ranging from seizures to paralysis or death. Here, we investigated the dissemination patterns of three neurotropic viruses, each from a different family, to discern whether common mechanisms are employed to invade the CNS. Viral dissemination barriers can be masked by titer-based assays since post-barrier viral replication can produce high viral titers (Erickson and Pfeiffer, 2013; Kuss et al., 2008; Lancaster and Pfeiffer, 2010; Pfeiffer and Kirkegaard, 2006). Therefore, we used genetically marked virus pools to examine dissemination of poliovirus, yellow fever virus 17D (YFV-17D), and reovirus.
Poliovirus is a nonenveloped virus with a positive-sense, single-stranded RNA genome from the Picornaviridae family. It is most commonly transmitted in humans by the fecal-oral route and causes paralysis in less than 1% of infected individuals due to damage to motor neurons. Poliovirus accesses the CNS by either crossing the blood-brain barrier (Yang et al., 1997) or via retrograde axonal transport in peripheral nerves (Ohka et al., 1998; Pfeiffer, 2010; Ren and Racaniello, 1992). Young mice and immune-deficient mice engineered to express the poliovirus receptor (PVR/CD155) have enhanced susceptibly to poliovirus infection (Crotty et al., 2002; Ida-Hosonuma et al., 2005; Ohka et al., 2007). When poliovirus is inoculated into the hind limb muscle of these mice, the virus spreads to the CNS by transport through the sciatic nerve to the spinal cord and brain. Concordantly, sciatic nerve transection limits poliovirus spread to the CNS following intramuscular inoculation (Gromeier and Wimmer, 1998; Ohka et al., 1998; Ren and Racaniello, 1992). Poliovirus enters neurons and other cell types by binding PVR, followed by endocytosis and uncoating (Koike et al., 1991b; Ohka et al., 2007; Ren and Racaniello, 1992). The cytoplasmic domain of PVR interacts with Tctex-1, a light chain of cytoplasmic dynein, which facilitates transport of virus-containing endosomes through the fast retrograde transport system (Mueller et al., 2002; Ohka et al., 2004). It is unclear whether other molecules are associated with retrograde axonal transport of poliovirus in neurons.
YFV is an enveloped virus with a positive-sense, single-stranded RNA genome from the Flaviviridae family. YFV usually infects humans through the bite of a mosquito, with outcomes ranging from asymptomatic infection to severe hemorrhagic fever. YFV rarely enters the CNS, but the virus is capable of causing encephalitis (Monath and Barrett, 2003). The receptor for YFV and its mechanism of entry into the CNS are not known. YFV strain 17D (YFV-17D) is a live-attenuated vaccine strain derived from the virulent Asibi strain. It is one of the safest live-attenuated vaccines available, but in rare cases, YFV-17D can enter the CNS and induce encephalitis following vaccination (Monath and Barrett, 2003). Laboratory rodents have varying degrees of susceptibility to YFV-17D, with young and immune-deficient animals having the greatest susceptibility (Fitzgeorge and Bradish, 1980; Lee and Lobigs, 2008; Meier et al., 2009; Theiler, 1930; Thibodeaux et al., 2012). Early studies suggested that YFV disseminates to the CNS hematogenously (Mims, 1957; Sawyer and Lloyd, 1931); indeed, many flaviviruses access the CNS through blood. It is unclear whether YFV can undergo retrograde transport in neurons.
Mammalian orthoreoviruses (hereafter called reoviruses) are nonenveloped viruses with a segmented double-stranded RNA genome from the Reoviridae family. Reovirus infects humans by the fecal-oral route, but individual strains differ in their dissemination patterns and receptor specificities (Barton et al., 2003; Weiner et al., 1977; Weiner et al., 1980). Reovirus infects most humans during childhood and, in rare instances, reovirus is capable of entering the CNS in humans. The age-dependent barrier to reovirus CNS entry is recapitulated in mice, as reovirus disseminates to the brain of young but not adult mice following intramuscular inoculation (Mann et al., 2002; Tardieu et al., 1983). In mice, strain type 1 Lang (T1L) disseminates to the CNS primarily through a hematogenous route, while strain type 3 Dearing (T3D) disseminates to the CNS through neural or hematogenous pathways (Antar et al., 2009; Boehme et al., 2011; Morrison et al., 1991; Tyler et al., 1986). When reovirus T3D is inoculated into the hind limb muscle of newborn mice, the virus spreads to the CNS by trafficking through the sciatic nerve to the spinal cord. Sciatic nerve transection inhibits T3D dissemination from the hind limb muscle to the spinal cord (Boehme et al., 2011; Tyler et al., 1986). However, sciatic nerve transection delays, but does not prevent, T3D spread to the brain following intramuscular inoculation, highlighting the importance of hematogenous spread following this inoculation route (Boehme et al., 2011). T1L and T3D reoviruses attach to host cells using cell-surface glycans and junctional adhesion molecule A, followed by receptor-mediated endocytosis (Barton et al., 2001a; Barton et al., 2001b; Campbell et al., 2005; Forrest et al., 2003; Maginnis et al., 2006; Reiss et al., 2012). T3D infects neurons using the Nogo receptor NgR1, although glycan interactions are also important for viral attachment to neurons (Frierson et al., 2012; Konopka-Anstadt et al., 2014). NgR1 is a GPI-anchored protein that is unlikely to interact directly with dynein (Saha et al., 2014). However, dynein is required for reovirus entry and endocytic transport in non-neuronal cells (Mainou et al., 2013). An inhibitor of fast retrograde axonal transport inhibits T3D dissemination to the CNS in mice (Tyler et al., 1986), suggesting that dynein-mediated fast retrograde axonal transport is involved in reovirus transport in neurons.
The mouse sciatic nerve is a tractable model system to study how viruses move from the periphery to the CNS because of its length, width, and ease of dissection. Following intramuscular inoculation of the hind limb, viruses can enter the sciatic nerve and transit by retrograde fast axonal transport using dynein motors (Ohka et al., 1998; Ren and Racaniello, 1992; Tyler et al., 1986). This transport system is essential, considering that viruses travel long distances within an axon. For example, viruses inoculated into the gastrocnemius muscle of the adult mouse travel over 2 cm within a single cell prior to reaching the site of viral replication in the cell body. The sciatic nerve model has been used to examine the routes by which neurotropic viruses gain access to the CNS and to study the effects of the type I interferon (IFN) response and stimulation of retrograde axonal transport (Boehme et al., 2011; Gromeier and Wimmer, 1998; Ida-Hosonuma et al., 2005; Koyuncu et al., 2013; Lancaster and Pfeiffer, 2010; Mann et al., 2002; Tyler et al., 1986). For example, poliovirus transport in the sciatic nerve of mice is inefficient, but efficiency is enhanced by ablating the type I IFN response or increasing retrograde axonal transport via muscle damage. Importantly, virulence correlates with viral diversity in the CNS: mice with high viral diversity in the CNS have higher mortality than mice with low viral diversity in the CNS (Kuss et al., 2008; Lancaster and Pfeiffer, 2010; Pfeiffer and Kirkegaard, 2005; Vignuzzi et al., 2006).
Although the mechanism is unclear, damage to muscle increases transport of both poliovirus and non-viral cargo protein in the sciatic nerve (Gromeier and Wimmer, 1998; Lancaster and Pfeiffer, 2010). Muscle damage also decreases the time to disease onset in poliovirus-infected mice (Gromeier and Wimmer, 1998; Lancaster and Pfeiffer, 2010). This damage response and subsequent enhanced poliovirus transport in neurons also may be operative in humans. In children, injury to muscle from trauma or intramuscular injections increases the incidence of poliovirus-induced paralysis, particularly in the damaged limb (Anderson and Skaar, 1951; Guyer et al., 1980; Hill and Knowelden, 1950; McCloskey and Melb, 1950; Strebel et al., 1995; Wyatt, 1985). The effect of muscle damage on transport of other viruses is unclear.
To investigate differences in dissemination patterns of distinct neurotropic viruses, we intramuscularly inoculated adult or newborn immune-competent or immune-deficient mice lacking the type I IFN-α/β receptor (IFNAR−/−) with genetically marked pools of poliovirus, YFV-17D, or reovirus T3D. We quantified viral dissemination at 72 hours post-inoculation (hpi) and found that dissemination of reovirus T3D was more restricted than poliovirus and YFV-17D in adult immune-competent mice. All three viruses had enhanced dissemination in IFNAR−/− mice. Stimulating retrograde axonal transport with muscle damage enhanced poliovirus dissemination but did not enhance dissemination of either YFV-17D or reovirus T3D. Dissemination of poliovirus and reovirus was substantially enhanced in newborn mice, with reovirus showing the largest differences as a function of host age. Overall, our results suggest that poliovirus, YFV-17D, and reovirus T3D disseminate to the CNS using different pathways with different efficiencies.
Materials and methods
Plasmid construction
The plasmids used to prepare the nine genetically marked reovirus strains were engineered using site-directed mutagenesis of the M1 gene segment of the reovirus T3D M1 cDNA plasmid beginning with nucleotide 426 and ending at nucleotide 444 (see Fig. 4A). PCR products were subcloned using Bgl II and Mfe I restriction sites. Fidelity of mutagenesis for each PCR-generated region was confirmed by sequencing (Sequencing Core, UT Southwestern Medical Center, Dallas, TX). The 10 poliovirus plasmids and six YFV-17D plasmids have been described (Erickson and Pfeiffer, 2013; Kuss et al., 2008; Pfeiffer and Kirkegaard, 2003).
Fig. 4.
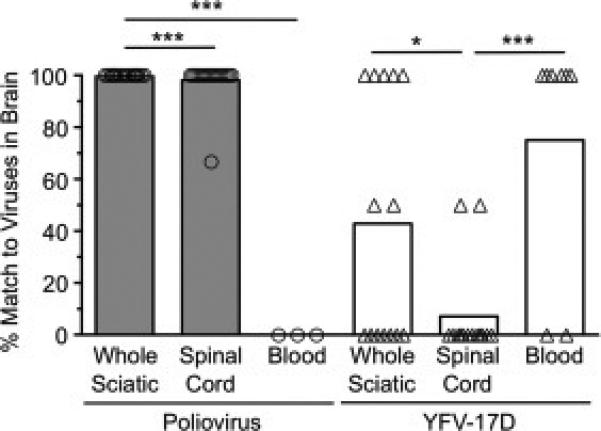
Poliovirus and YFV-17D likely reach the brain through different routes. Viral pool members detected in the brain were scored relative to the pool members detected in the sciatic nerve, spinal cord, or blood of each IFNAR+/+ or IFNAR−/− mouse to determine the degree of overlap. We used only mice in which less than or equal to half of the total pool members were detected in the brain, since animals with most or all of the pool members present in the brain would be uninformative for determining trafficking routes. Each symbol represents a single mouse. Bars represent means of 3-18 mice. Data are shown as percent match of viral pool members present in the brain to viral pool members present in each tissue. As an example, if the brain contained poliovirus pool members 2 and 4 (of 10 total) and the spinal cord contained poliovirus pool members 4, 6, 8, and 10, then the percent match of brain viruses to viral pool members present in the spinal cord is 50% ([1 matching virus/2 total viruses present in brain] × 100). Values that are significantly different, as determined by the Mann-Whitney test, are indicated by asterisks as follows: *, P < 0.05, **, P < 0.005, ***, P < 0.0005.
Viruses and cells
L929 cells (reovirus) and BHK cells (YFV-17D) were propagated in Dulbecco's modified Eagle's medium (DMEM) with 5% fetal bovine serum, and HeLa cells (poliovirus) were propagated in DMEM with 10% calf serum. Reovirus plaque assays were performed as described using 6 × 105 L929 cells seeded into wells of 6-well plates. Monolayers were stained with neutral red at 6 d post-infection, and plaques were counted at 7 d post-infection (Virgin et al., 1988). Poliovirus plaque assays were performed as described using 106 HeLa cells seeded into wells of 6-well plates. Monolayers were stained with crystal violet at 2 d post-infection (Pfeiffer and Kirkegaard, 2003). YFV-17D plaque assays were performed as described using 106 BHK cells seeded into wells of 6-well plates. Monolayers were stained with crystal violet at 5 d post-infection (Erickson and Pfeiffer, 2013). For co-infection experiments with poliovirus and reovirus (Fig. 6), we could discriminate reovirus plaques from poliovirus plaques because L929 cells do not support poliovirus replication due to the absence of PVR.
Fig. 6.
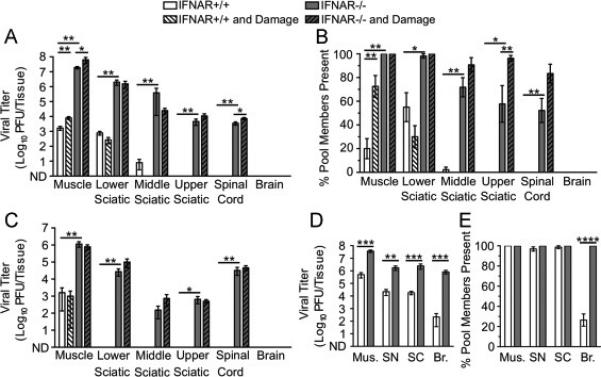
Dissemination of reovirus T3D to the CNS is limited by type I IFN responses and older age but not inefficient retrograde axonal transport. IFNAR+/+ and IFNAR−/− mice were inoculated intramuscularly with 107 PFU total of nine genetically marked reoviruses, with or without additional muscle damage. Viral titers were determined by plaque assay, and viral population diversity was determined using a hybridization-based assay. Reovirus T3D titer (A) and viral population diversity (B) in tissues harvested from adult IFNAR+/+ or IFNAR−/− mice with or without muscle damage. Tissues were collected at 72 hpi. (C) Reovirus T3D titer in tissues from adult mice collected at 7 d post-inoculation. Reovirus T3D titer (D) and viral population diversity (E) in tissues from 3-day-old IFNAR+/+ or IFNAR−/− mice. Tissues were collected at 72 hpi, prior to disease onset. Results are presented as mean +/− standard error of the mean from 5-9 mice per condition. Values that are significantly different, as determined by the Mann-Whitney test, are indicated by asterisks as follows: *, P < 0.05, **, P < 0.005, ***, P < 0.0005. Mus., Muscle, SN, Sciatic Nerve, SC, Spinal Cord, Br., Brain. ND, none detected.
Reoviruses harboring the nine different mutated M1 genome segments were recovered by plasmid-based rescue (Kobayashi et al., 2010). Monolayers of BHK-T7 cells at 90% confluency (~3 × 106 cells) seeded in 60-mm dishes (Costar; Corning Inc., Corning, NY) were co-transfected with 10 plasmids representing the cloned reovirus T3D genome using 3 μL of TransIT-LT1 transfection reagent (Mirus Bio LLC; Madison, WI) per μg of plasmid DNA. Following 72 h of incubation, recombinant viruses were isolated from transfected cells by plaque purification using monolayers of L929 cells. (Virgin et al., 1988). High-titer reovirus stocks were prepared by large-scale infections and purification by cesium chloride gradient centrifugation (Furlong et al., 1988).
The relative fitness of the nine genetically marked reoviruses was evaluated by a serial passage competition experiment (Erickson and Pfeiffer, 2013; Kuss et al., 2008). Each of the nine viruses at a dose of 106 PFU was mixed together and adsorbed to 106 L929 cells. After 24 h of incubation, infected cells were collected, and 25% of the harvested cells were plated on a fresh monolayer of 106 L929 cells to initiate further replication cycles. This process was repeated for a total of 7 passages, and the relative ratios of each virus in each passage were quantified by hybridization assay as described below. Pool member fitness was assessed in vivo by calculating the number of times each pool member was detected relative to the total possible number of times that the pool member could have been detected. Pool member totals were from all reovirus mouse experiments, with the exception of any experiments where all nine pool members were detected in all possible tissues.
Mouse experiments
All animals were handled in strict accordance with good animal practice as defined by the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All mouse studies were performed at the University of Texas Southwestern Medical Center (Animal Welfare Assurance no. A3472-01) using protocols approved by the UT Southwestern Institutional Animal Care and Use Committee (IACUC). All studies were performed in a manner designed to minimize pain and suffering, and any animals that exhibited severe disease signs were euthanized immediately in accordance with IACUC-approved endpoints. C57BL/6 hPVR-Tg21 mice (called IFNAR+/+ throughout) and C57BL/6 hPVR-Tg21 mice lacking the IFN-α/β receptor (called IFNAR−/− throughout) were provided by S. Koike (Tokyo, Japan) (Ida-Hosonuma et al., 2005; Koike et al., 1991a). Although PVR expression is required only for poliovirus infection, we used the PVR-transgenic mice for all viruses in this study to allow direct comparisons to be made using isogenic strains. We mixed equal PFU of each pool member for a total of 107 PFU/mouse in 30 μL for 6-10 week-old adult mice or 10 μL for 3-day-old mice and inoculated the mixture into the lower left gastrocnemius muscle (Kuss et al., 2008). For YFV-17D infection of 3-day-old mice, the inoculum was 104 PFU due to low virus concentration and the small 10 μL inoculum. Muscle damage was induced in adult mice by inserting a 29-gauge needle 5 times once daily into the muscle around the inoculation site (Fig. 1). Muscle damage was not induced in 3-day old mice due to their small size. Mice were euthanized at a predetermined endpoint or at the onset of disease signs, whichever occurred earliest, according to IACUC-approved methods.
Fig. 1.
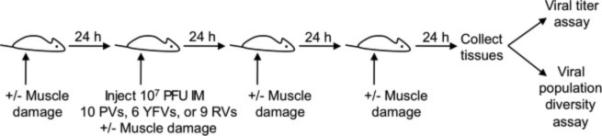
Experimental design. Mice were inoculated intramuscularly (IM) with 107 PFU of 10 poliovirus (PV), six YFV-17D, or nine reovirus (RV) pool members, and tissues were collected at 72 hpi unless otherwise indicated. Muscle damage was induced in a subset of mice before, during, and after viral inoculation to stimulate retrograde axonal transport in the sciatic nerve.
Tissue harvest, processing and viral diversity assays
Several tissues (muscle, sciatic nerve, spinal cord, brain, blood) were collected as described (Erickson and Pfeiffer, 2013; Lancaster and Pfeiffer, 2010). Adult mice infected with YFV-17D were perfused with PBS prior to tissue collection due to moderate viral titers in blood. Perfusion was not performed in poliovirus- or reovirus-infected mice due to negligible viral titers in blood. In experiments using adult mice, the sciatic nerve was sectioned into lower, middle, and upper segments (Lancaster and Pfeiffer, 2010). The upper sciatic nerve segment included both the dorsal root ganglia and the motor nerves. The spinal cord was collected as the whole spine from the lumbar vertebrae (L5-L6) to the middle of the cervical vertebrae (C4-C5). Tissues were weighed and suspended in 1-3 volumes of PBS and homogenized with a Bullet Blender (Next Advance, Inc, Averill Park, NY) according to the manufacturer's instructions. Tissues from YFV-17D or reovirus-infected mice were frozen and thawed once prior to homogenization, whereas poliovirus-infected tissues were frozen and thawed three times after homogenization. All samples were centrifuged at 13,000 rpm for 1 min to isolate virus-containing supernatant. Virus titers in supernatants were quantified by plaque assay and amplified prior to the viral population diversity assay to aid in detection of pool members. Poliovirus was amplified in HeLa cells until cytopathic effects were apparent (6-48 h, depending on the tissue). YFV-17D was amplified for 48 h in BHK cells (Erickson and Pfeiffer, 2013). Reovirus was amplified following a 1:10 dilution of supernatant and plated on L929 cells. Cells were incubated at 37°C for 12 h to 5 d, depending upon tissue titer, and monitored regularly for cytopathic effects. In all cases, cells were collected in 1 mL TRI Reagent, and RNA was extracted as described (Kuss et al., 2008; Lancaster and Pfeiffer, 2010). RT-PCR of poliovirus and YFV-17D samples was performed as described (Erickson and Pfeiffer, 2013; Kuss et al., 2008). RT-PCR of reovirus samples was performed as described (Kuss et al., 2008; Pfeiffer and Kirkegaard, 2005) with a few modifications to facilitate amplification of reovirus dsRNA. Total cellular RNA, the REO M1 614 antisense primer 5’ TAGAGTGAGGAACACGACC 3’, and all reaction components except for SSII reverse transcriptase were incubated at 100°C for 20 min to separate dsRNA, allowed to cool slowly to 42°C, and then incubated with SSII reverse transcriptase at 42°C for 1 h. PCR reactions were performed as described (Kuss et al., 2008), but with the M1 297 sense primer 5’ CGGACAACGTTGATCGTCG 3’ and the M1 542 antisense primer 5’ CGCATAGTACTTTCTAGGAGC 3’. Blotting and hybridization were performed as described (Kuss et al., 2008) but with reovirus-specific probes. The reovirus probe sequences are the reverse complement of sequences shown in Fig. 4A, but with an extra T on the 5’ end of each. Reovirus probes were radiolabeled using T4 polynucleotide kinase and [γ-32P] ATP, and excess nucleotides were removed with the Qiagen Nucleotide Removal Kit (Qiagen, Valencia, CA). Hybridization was performed at 56°C overnight, followed by washing. Membranes were exposed to a Phosphor Screen for 2 d and scanned with a STORM Scanner. Signals were normalized to a mismatched control sample on each membrane as described (Kuss et al., 2008).
Results
Immune deficiency, muscle damage, and young age enhance poliovirus transport to the CNS
Analysis of viral titer is not always sufficient to define viral dissemination barriers; in fact, robust viral replication following traverse of a barrier can mask barriers completely (Kuss et al., 2008; Lancaster and Pfeiffer, 2010; Pfeiffer and Kirkegaard, 2006). To overcome this obstacle, we inoculated mice with genetically marked viruses and used a hybridization-based assay to quantify the viral population diversity in a given tissue (Kuss et al., 2008; Lancaster and Pfeiffer, 2010). A decrease in viral population diversity from one tissue to another indicates a viral transport barrier. Using this approach, we previously found that poliovirus transport to the CNS is inefficient, but ablating the type I IFN response enhanced viral transport to the CNS. Additionally, we found that muscle damage increased poliovirus transport to the CNS by enhancing retrograde axonal transport in the sciatic nerve (Lancaster and Pfeiffer, 2010). However, our previous study analyzed viral titer and population diversity in tissues at disease onset, days 3-8 post-inoculation. Here, we examined viral transport to the CNS at an early time point, 72 hpi, to determine whether the type I IFN response, muscle damage, or host age influences viral dissemination early in infection.
To examine viral transport early in infection, IFNAR+/+ or IFNAR−/− mice were inoculated intramuscularly with 10 genetically marked polioviruses (Fig. 1). Tissues were harvested at 72 hpi, and viral titer and population diversity were quantified. Similar to previous studies at late time points (Kuss et al., 2008; Lancaster and Pfeiffer, 2010), we found increased viral titers in all tissues of IFNAR−/− mice relative to IFNAR+/+ mice (Fig. 2A, white vs. gray bars). Similarly, viral population diversity was greater in IFNAR−/− mice relative to IFNAR+/+ mice, and viral population diversity was less in brain compared with muscle (Fig. 2B, white vs. gray bars). In contrast to previous studies at later time points where viral brain titers were high (Lancaster and Pfeiffer, 2010), viral titers in the brain were very low at 72 hpi in immune-competent mice. However, mean poliovirus titers in the brain were >8,000-fold higher in IFNAR−/− mice compared to IFNAR+/+ mice. Overall, these data suggest that poliovirus transport to the CNS is inefficient, particularly in adult immune-competent mice, at 72 hpi.
Fig. 2.
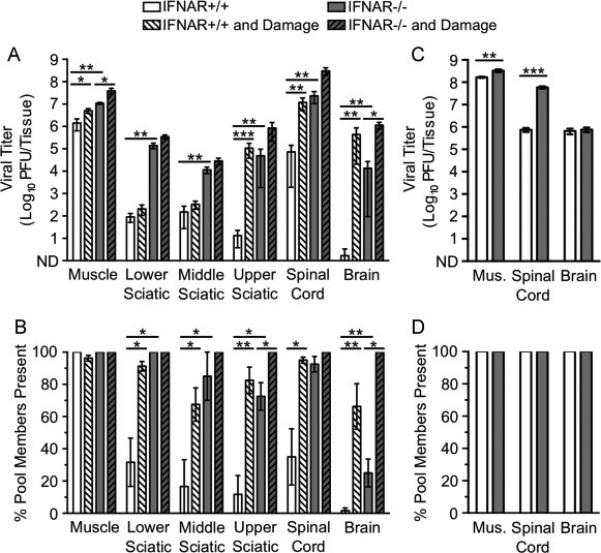
Dissemination of poliovirus to the CNS is limited by inefficient retrograde axonal transport, type I IFN responses, and older age. IFNAR+/+ and IFNAR−/− mice were inoculated intramuscularly with 107 PFU total of 10 genetically marked polioviruses, with or without additional muscle damage. Tissues were collected, viral titers were determined by plaque assay, and viral population diversity was determined using a hybridization-based assay (Kuss et al., 2008). Poliovirus titer (A) and viral population diversity (B) in tissues harvested from adult IFNAR+/+ or IFNAR−/− mice with or without muscle damage. Tissues were collected at 72 hpi, prior to disease onset. Poliovirus titer (C) and viral population diversity (D) in tissues harvested from 3-day-old IFNAR+/+ or IFNAR−/− mice. Tissues were collected at 48 hpi in C and D due to the onset of disease. Results are presented as mean +/− standard error of the mean from 4-8 mice per condition. Values that are significantly different, as determined by the Mann-Whitney test, are indicated by asterisks as follows: *, P < 0.05, **, P < 0.005, ***, P < 0.0005. Mus., muscle; ND, none detected.
We next examined the effect of muscle damage on poliovirus transport to the CNS at 72 hpi. Muscle damage was induced daily starting 1 d before viral inoculation and concluding 1 d prior to tissue collection (Fig. 1). Titers of poliovirus in tissues were generally higher in IFNAR+/+ mice with damage compared with those in IFNAR+/+ mice without damage (Fig. 2A, white vs. white hatched bars). In fact, mean poliovirus titers in brain were >270,000-fold higher in mice with muscle damage compared with mice lacking muscle damage. The muscle and brain tissues of IFNAR−/− mice with muscle damage also show increased titer compared with IFNAR−/− mice without damage (Fig. 2A, gray vs. gray hatched bars). Not surprisingly, viral population diversity was also higher for mice with muscle damage (Fig. 2B). Strikingly, the effect of muscle damage on viral titer and viral population diversity in the brain was greater than ablation of the type I IFN response. Although poliovirus replicated more efficiently in IFNAR−/− mice, trafficking to the CNS was still restricted by a barrier that was overcome by muscle damage. These results suggest that early in infection inefficient retrograde axonal transport and type I IFN restrict poliovirus trafficking to the CNS by different mechanisms. Furthermore, inefficient retrograde axonal transport is a stronger barrier for poliovirus dissemination to the CNS than the type I IFN response.
To determine whether barriers to poliovirus CNS dissemination exist in very young mice, we inoculated 3-day-old IFNAR+/+ or IFNAR−/− mice with the pool of genetically marked viruses and assessed viral titers and population diversity. Infected mice were moribund at 48 hpi; therefore, tissues were collected at this earlier time point. Viral titers were very high in muscle, spinal cord, and brain in both IFNAR+/+ and IFNAR−/− mice, and all pool members were present in all tissues (Fig. 2C-D). In agreement with previous studies, these data suggest that a major barrier limiting viral dissemination develops as mice age (Crotty et al., 2002). Overall, poliovirus dissemination is limited most by age-specific factors, followed by inefficient retrograde axonal transport, and the type I IFN response. Importantly, young mice may have altered IFN responses and retrograde axonal transport compared with adult mice, which may contribute to the enhanced dissemination of poliovirus in these animals.
Immune deficiency, but not muscle damage, enhances YFV-17D transport to the CNS
Using a pool of six genetically marked viruses, we previously found that YFV-17D inoculated intramuscularly disseminated to the brain relatively efficiently in IFNAR−/− mice at 7-8 dpi (Erickson and Pfeiffer, 2013). Here, we examined YFV-17D dissemination at 72 hpi to better understand the kinetics of viral transport by evaluating the effect of immune deficiency, muscle damage, and host age on viral dissemination. We found that immune-competent adult IFNAR+/+ mice had moderate titers of YFV-17D in all tissues tested (Fig. 3A, white bars). While immune-deficient IFNAR−/− mice had higher YFV-17D titers in several tissues, surprisingly, viral titers in brain were only 1.3-fold higher than those in IFNAR+/+ mice (Fig. 3A, white vs. gray bars). For most tissues, viral population diversity was greater in IFNAR−/− mice compared with that in IFNAR+/+ mice (Fig. 3B, white vs. gray bars). Although viral titers in the brain were similar in IFNAR−/− and IFNAR+/+ mice, viral population diversity was greater in IFNAR−/− mice. These results suggest that YFV-17D may encounter a barrier in IFNAR+/+ mice en route to the brain that was overcome in IFNAR−/− mice or that clearance of the virus differs in the two mouse strains.
Fig. 3.
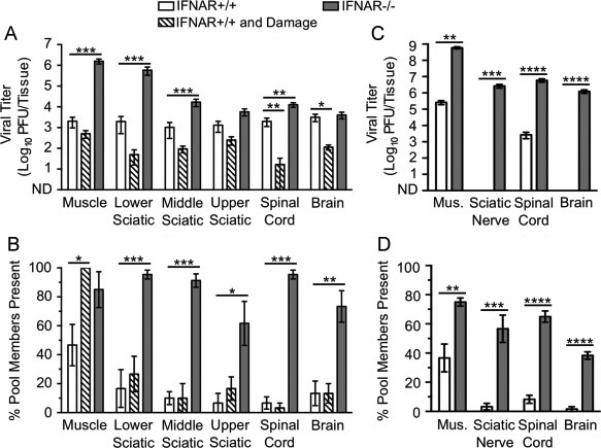
Dissemination of YFV-17D to the CNS is limited by type I IFN responses but not inefficient retrograde axonal transport. IFNAR+/+ and IFNAR−/− mice were inoculated intramuscularly with 107 PFU total of six genetically marked YFV-17D viruses, with or without additional muscle damage for IFNAR+/+ mice. Tissues were collected at 72 hpi, prior to disease onset. Viral titers were determined by plaque assay, and viral population diversity was determined using a hybridization-based assay. YFV-17D titer (A) and viral population diversity (B) in tissues harvested from adult IFNAR+/+ or IFNAR−/− mice with or without muscle damage. YFV-17D titer (C) and viral population diversity (D) in tissues harvested from 3-day-old IFNAR+/+ or IFNAR−/− mice infected with 104 PFU. Results are presented as mean +/− standard error of the mean from 6-8 mice per condition. Values that are significantly different, as determined by the Mann-Whitney test, are indicated by asterisks as follows: *, P < 0.05, **, P < 0.005, ***, P < 0.0005. Mus., Muscle; ND, none detected.
Because YFV and YFV-17D are thought to enter the CNS predominantly via the hematogenous route (Mims, 1957; Sawyer and Lloyd, 1931), and muscle damage stimulates transport in neurons, we hypothesized that muscle damage would not alter YFV-17D dissemination to the brain. Indeed, viral titers were not higher in IFNAR+/+ mouse tissues at 72 hpi following muscle damage (Fig. 3A, white vs. hatched bars). In fact, viral titers were lower in mice with muscle damage, suggesting that muscle injury reduced viral replication and dissemination. Muscle damage was associated with increased YFV-17D population diversity in muscle tissue but not in any of the peripheral or central nervous system tissues tested (Fig. 3B, white vs. hatched bars). It is possible that inflammation associated with muscle damage reduced YFV-17D replication.
To determine whether YFV-17D disseminates more efficiently in very young mice, we inoculated 3-day-old IFNAR+/+ or IFNAR−/− mice with the pool of genetically marked viruses and assessed viral titers and population diversity in tissues harvested at 72 hpi. Due to the small inoculation volume required for infant mice and low YFV-17D concentration, mice were inoculated with 104 PFU in these experiments rather than the 107 PFU inoculum used in experiments with adult animals. Therefore, direct comparisons between YFV-17D dissemination in adult vs. young mice could not be made. However, we were able to evaluate the relative viral dissemination efficiencies in IFNAR+/+ and IFNAR−/− infant mice. Not surprisingly, YFV-17D titers were higher and population diversity was greater in IFNAR−/− mice compared with those parameters in IFNAR+/+ mice (Fig. 3C-D). Strikingly, YFV-17D titers were undetectable in the brain of immune-competent young mice, while immune-deficient mice contained >104 PFU in the brain. Therefore, the type I IFN response is a major barrier in young mice to dissemination of YFV-17D.
Tracing individual viral pool members reveals different transport routes of poliovirus and YFV-17D to the CNS
A major strength of using genetically marked viruses for viral dissemination studies is that they can be used to determine viral transport routes from the periphery to the CNS. To determine whether intramuscularly inoculated YFV-17D disseminated to the brain through the blood rather than through peripheral nerves, we compared specific pool members found in tissues of adult mice. YFV-17D pool members detected in the brain were scored relative to the pool members detected in the sciatic nerve, spinal cord, or blood of each mouse to determine the degree of overlap. For this analysis, we used only mice in which less than or equal to half of the total pool members were detected in the brain, since animals with most or all of the pool members present in the brain would be uninformative for determining trafficking routes. We found that, on average, a YFV-17D pool member detected in the brain was detected in the sciatic nerve in 70% of mice, in the spinal cord in 45% of mice, and in the blood in 75% of mice (Fig. 4). Therefore, viruses found in the brain matched viruses found in the blood to a greater extent than viruses found in the sciatic nerve or spinal cord. In contrast, poliovirus pool members detected in the brain matched the pool members detected in the sciatic nerve and spinal cord in 98-100% of cases, and poliovirus was undetectable in the blood (Fig. 4). These data are consistent with previous studies showing that poliovirus disseminates through nerves to reach the mouse brain (Ohka et al., 2004; Ohka et al., 1998; Ren and Racaniello, 1992) and support the idea that YFV-17D disseminates primarily through the blood to infect the CNS (Mims, 1957; Sawyer and Lloyd, 1931).
Development of a reovirus T3D population diversity assay
To examine reovirus dissemination in mice, we engineered a panel of nine genetically marked reovirus strains that can be distinguished using a hybridization-based population diversity assay. The strains each contain a combination of unique silent mutations in the M1 gene segment such that RT-PCR products of each pool member are recognized by specific oligonucleotide probes (Fig. 5A). The specificity of each probe was confirmed by blotting RT-PCR products from individual strains on membranes, followed by hybridization with each 32P-labeled oligonucleotide probe (Fig. 5B). To determine whether any of the strains have altered replication efficiency compared with the others, we performed an in vitro viral serial passage experiment. This type of analysis is more sensitive than single-cycle replication assays for detection of subtle replication differences (Kuss et al., 2008). After an initial infection with a mixture containing equal aliquots of each pool member, we used the infected cells to initiate viral replication in naïve cells, and relative levels of each pool member were quantified over seven serial passages. In this experiment, all pool members were present at similar levels in the initial and final passages (Fig. 5C), suggesting that the pool members have similar fitness in vitro. To determine whether the genetically marked reovirus strains differ in fitness in vivo, we inoculated mice intramuscularly with a mixture of all nine pool members and examined viral population diversity at 72 hpi. A blot of the reovirus pool members in peripheral and CNS tissues of a representative IFNAR−/− mouse is shown in Fig. 5D. To confirm that reovirus pool members have equivalent fitness in vivo, viral population diversity data for all reovirus mouse experiments were compiled to determine the relative number of times each pool member was detected. All nine reovirus pool members were detected with similar frequency (Fig. 5E), providing strong evidence that the pool members have comparable fitness in vivo.
Fig. 5.
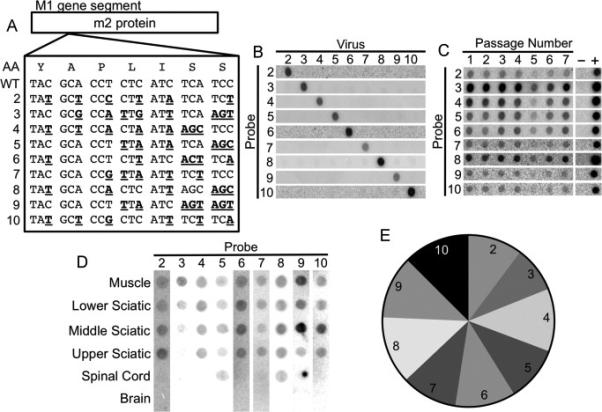
Development of a hybridization-based reovirus T3D viral population diversity assay. (A) Sequence alignment showing groups of silent point mutations introduced into the reovirus T3D M1 gene segment. The amino acid sequence is shown at the top. (B) Blots demonstrating specificity of each probe for its cognate viral RT-PCR product. (C) Serial passage competition experiment showing maintenance of all pool members. L929 cells were adsorbed with equivalent PFU of each reovirus pool member. Infected cells were collected at 24 hpi and used to initiate another cycle of replication in naïve cells. Following seven passages, the ratios of viruses were compared by hybridization assay. (D) Representative blot of RT-PCR products from a reovirus-infected IFNAR−/− mouse following intramuscular inoculation. Mice were inoculated intramuscularly with 107 PFU total of the nine genetically marked reoviruses. Tissues were collected at 72 hpi, and viral population diversity was assessed using the hybridization assay. (E) Relative prevalence of each reovirus pool member in all mouse tissues collected during this study. Data were derived from tissues of 63 mice. The relatively equal prevalence of each pool member indicates that pool members do not have significant fitness differences in vivo.
Young age and immune deficiency, but not muscle damage, enhances reovirus T3D transport to the CNS
To define factors that influence reovirus dissemination from the periphery to the CNS in mice, we examined trafficking of the nine genetically marked reoviruses following intramuscular inoculation. In accordance with previous work (Dionne et al., 2011), viral titers in muscle, sciatic nerve, and spinal cord were significantly higher in adult IFNAR−/− mice than those in adult IFNAR+/+ mice (Fig. 6A, white vs. gray bars). Remarkably, reovirus dissemination to the upper sciatic nerve and spinal cord was observed only in IFNAR−/− mice (Fig. 6A). Viral population diversity was modest in IFNAR+/+ tissues and greater in IFNAR−/− tissues (Fig. 6B, white vs. gray bars). Reovirus was not detected in the brain of adult mice of either strain at 72 hpi, which was not surprising given the well-established restriction of reovirus dissemination to the CNS in adult mice (Mann et al., 2002; Tardieu et al., 1983).
Because reovirus T3D and poliovirus both infect neurons and are transported by fast retrograde axonal transport, we hypothesized that muscle damage would enhance reovirus transport as observed in the case of poliovirus transport. Although damage induced small increases in reovirus titers in muscle of both IFNAR+/+ and IFNAR−/− mice, muscle injury was not sufficient to enhance reovirus dissemination to the sciatic nerve, spinal cord, or brain in IFNAR+/+ mice (Fig. 6A, white vs. white hatched bars). Muscle damage was associated with increased viral population diversity in muscle of IFNAR+/+ mice but generally not in other tissues (Fig. 6B). In IFNAR−/− mice, increases in viral population diversity following muscle damage were only significant in the upper sciatic nerve (Fig. 6B). Overall, these experiments show that dissemination of intramuscularly inoculated reovirus is severely restricted in immune-competent adult mice and that muscle damage does not enhance viral dissemination from the inoculation site.
We next determined whether a longer infectious time course would allow increased reovirus dissemination to the CNS. Reovirus replicates more slowly than poliovirus and may require a longer interval to reach the brain. Therefore, we compared viral titers in adult IFNAR+/+ and IFNAR−/− mice with and without muscle damage at 7 d post-inoculation. In most tissues tested, viral titers were lower at 7 days post-inoculation than at 72 hpi (Fig. 6C vs. Fig. 6A). Reovirus was not detected in the brain of adult mice under any of the conditions tested (Fig. 6C). Therefore, the lack of reovirus dissemination to the brain of adult mice at 72 hpi is not simply due to the relatively early time point of tissue collection.
To determine whether reovirus T3D disseminates more efficiently in very young mice, we inoculated 3-day-old IFNAR+/+ or IFNAR−/− mice with the pool of genetically marked viruses and assessed viral titers and population diversity. Reovirus titers were detectable in all tissues of young mice, including brain, with significantly higher titers in IFNAR−/− mice compared with those in INFAR+/+ mice (Fig. 6D). Notably, all reovirus pool members were detected in the brains of INFAR−/− mice (Fig. 6E). However, only about 25% of pool members were detected in the brains of IFNAR+/+ mice, revealing an IFN-mediated barrier limiting reovirus infection of the brain in young mice. Collectively, these experiments are consistent with previous findings on age-dependent and IFN-dependent barriers to reovirus dissemination (Dionne et al., 2011; Mann et al., 2002; Sherry et al., 1998; Tardieu et al., 1983).
Co-infection with poliovirus does not enhance reovirus dissemination to the CNS
Since poliovirus is more efficiently transported than reovirus in neurons of IFNAR+/+ and IFNAR−/− mice, we were curious about whether co-infection with poliovirus could enhance reovirus transport. We thought it possible that poliovirus infection might stimulate host pathways that mediate viral transport in neurons, which in turn would enhance transport of other types of cargoes. To test this hypothesis, adult mice were inoculated intramuscularly with 107 PFU of the 10 genetically marked polioviruses and 107 PFU of the nine genetically marked reoviruses for a total inoculum of 2 × 107 PFU. Tissues were resected at 72 hpi and processed for virus titer determination and population diversity assay using L929 cells, which do not support poliovirus replication. When titer and diversity data from co-infected mice (‘co’) are compared with data from individually infected mice (‘ind’, from Fig. 6A-B), we found no detectable change (enhancement or diminishment) in reovirus transport following co-infection with poliovirus (Fig. 7A-B). This finding suggests that poliovirus and reovirus disseminate to the CNS using distinct pathways.
Fig. 7.
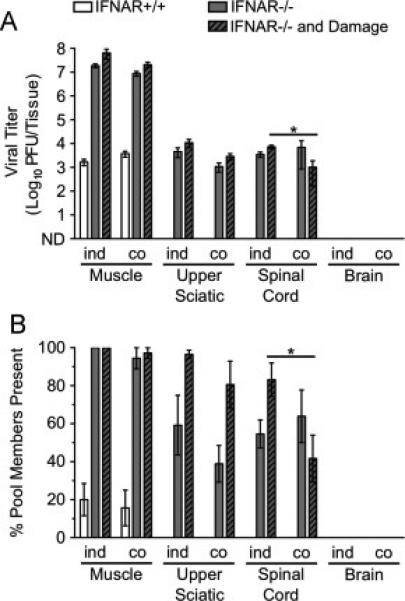
Co-infection with poliovirus does not enhance reovirus dissemination to the CNS. IFNAR+/+ and IFNAR−/− mice were inoculated intramuscularly with 107 PFU total of nine genetically marked reoviruses and 107 PFU total of 10 genetically marked polioviruses. Tissues were collected at 72 hpi. Reovirus titers and population diversity were determined by plaque assay and hybridization-based assay, respectively, using L929 cells, which do not support poliovirus replication. Reovirus T3D titer (A) and viral population diversity (B) in tissues harvested from adult IFNAR+/+ or IFNAR−/− mice with or without muscle damage. ‘ind’ indicates data from infections with reovirus only (from Fig. 6A-B data), and ‘co’ indicates data from the reovirus-poliovirus co-infection. Results are presented as mean +/− standard error of the mean from 5-7 mice per condition. Values that are significantly different, as determined by the Mann-Whitney test, are indicated by asterisks as follows: *, P < 0.05. ND, none detected.
Discussion
Knowledge about how different viruses disseminate to the CNS is essential for a comprehensive understanding of the pathogenesis of neurotropic viral infections. In this study, we compared the transport of three different neurotropic viruses following intramuscular inoculation of mice using viral titer and population diversity assays. We found that poliovirus, YFV-17D, and reovirus T3D disseminate to the CNS with varying efficiencies and use distinct mechanisms that are affected differently by host damage responses (Table 1).
TABLE 1.
Comparing CNS dissemination of three distinct neurotropic viruses
| Reovirus T3D | Poliovirus | YFV-17D | |
|---|---|---|---|
| Viral titer in brain of adult IFNAR+/+ mice | Undetectable | Very low | Moderate |
| Probable dissemination route to brain | Peripheral neurons | Peripheral neurons | Blood |
| Primary barrier | Age | Age | Interferon |
| Secondary barrier | Interferon | Inefficient retrograde axonal transport | Age |
| Tertiary barrier | --- | Interferon | --- |
| Effect of muscle damage | ~ No effect | ↓ Dissemination | ↓ Dissemination |
| Effect of IFNAR−/− | ↑ Dissemination | ↑ Dissemination | ↑ Dissemination |
Although injury to the muscle at the inoculation site enhanced neuronal transport of poliovirus in both IFNAR+/+ and IFNAR−/− mice, muscle damage did not enhance transport of either YFV-17D or reovirus T3D. In fact, muscle damage reduced YFV-17D dissemination. YFV-17D likely enters the brain by crossing the blood-brain barrier following hematogenous dissemination rather than by transport through peripheral nerves into the spinal cord. Therefore, enhancement of retrograde axonal transport in neurons induced by muscle damage would be less likely to increase YFV-17D dissemination to the CNS. Interestingly, blood-brain barrier disruption by needle puncture enhances dissemination of peripherally inoculated YFV to the brain, but peripheral tissue damage does not (Mims, 1957; Sawyer and Lloyd, 1931). Poliovirus and reovirus share many biological properties such as fecal-oral transmission, dissemination in peripheral nerves, dependence on dynein for transport, and spread in neurons by fast retrograde axonal transport. Therefore, it was somewhat surprising that muscle damage enhanced neuronal transport of poliovirus but not reovirus. Damage-dependent enhancement of retrograde axonal transport occurs for wheat germ agglutinin (Lancaster and Pfeiffer, 2010), indicating that accelerated transport as a consequence of muscle injury can occur with cargoes other than viruses. Collectively, these observations provide additional evidence that poliovirus and reovirus traffic in the nervous system using different mechanisms.
Injury to muscle at the inoculation site enhanced YFV-17D and reovirus population diversity in the muscle of IFNAR+/+ mice as well as reovirus titer in the muscle of IFNAR+/+ and IFNAR−/− animals. However, increased viral titer and population diversity in muscle as a consequence of damage did not enhance transport through peripheral nerves for either virus, suggesting that the effect may be due to muscle-specific factors that enhance viral replication or decrease viral clearance.
Co-infection with poliovirus did not accelerate neuronal transport of reovirus, likely due to differences in the host transport mechanisms used by these viruses. We think it possible that each virus associates with different neuronal compartments during transport. Differences in receptor utilization also may control transport efficiency. The PVR cytoplasmic domain associates with dynein light chain Tctex-1 to transport poliovirus using dynein motors (Mueller et al., 2002; Ohka et al., 2004). Reovirus T3D requires NgR1 to infect neurons, but NgR1 is GPI-anchored and therefore not likely to associate directly with any dynein components outside of reovirus-containing endosomes (Konopka-Anstadt et al., 2014; Saha et al., 2014). NgR1 is expressed at high levels in the adult brain but, because of myelination in the adult, the receptor may not be available for reovirus T3D binding and entry (Konopka-Anstadt et al., 2014). This aspect of NgR1 physiology could explain the age-specific barrier to reovirus infection. In contrast, PVR is widely expressed and available for poliovirus binding and entry into the brain.
Findings made in this study show that poliovirus, YFV-17D, and reovirus T3D disseminate to the CNS by distinct mechanisms. By using assays of viral load and viral population diversity, we uncovered dissemination routes to the CNS and identified barriers to dissemination for each of these viruses. Enhancement of fast retrograde axonal transport by muscle damage increased dissemination of poliovirus but not YFV-17D or reovirus T3D. These findings highlight the complexity of neurotropic virus access to the CNS and provide a framework to define virus-specific neural transportation routes.
Highlights.
We examined trafficking of neurotropic viruses using genetically marked virus pools.
We compared dissemination of poliovirus, yellow fever virus 17D and reovirus in mice.
Each of the three viruses had distinct modes of transport to the CNS.
Acknowledgments
We thank Satoshi Koike for the PVR-transgenic mice and Charlie Rice for the YFV-17D infectious clone.
This work was supported by United States Public Health Service awards AI074668 (J.K.P) and AI038296 (T.S.D.) from the National Institute of Allergy and Infectious Diseases and a Burroughs Wellcome Foundation Investigators in the Pathogenesis of Infectious Diseases award (J.K.P.). Additional support was provided by the Elizabeth B. Lamb Center for Pediatric Research.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson GW, Skaar AE. Poliomyelitis occurring after antigen injections. Pediatrics. 1951;7:741–759. [PubMed] [Google Scholar]
- Antar AA, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell host & microbe. 2009;5:59–71. doi: 10.1016/j.chom.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem. 2001a;276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. Junction adhesion molecule is a receptor for reovirus. Cell. 2001b;104:441–451. doi: 10.1016/s0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- Barton ES, Youree BE, Ebert DH, Forrest JC, Connolly JL, Valyi-Nagy T, Washington K, Wetzel JD, Dermody TS. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. The Journal of clinical investigation. 2003;111:1823–1833. doi: 10.1172/JCI16303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehme KW, Frierson JM, Konopka JL, Kobayashi T, Dermody TS. The reovirus sigma1s protein is a determinant of hematogenous but not neural virus dissemination in mice. J Virol. 2011;85:11781–11790. doi: 10.1128/JVI.02289-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JA, Schelling P, Wetzel JD, Johnson EM, Forrest JC, Wilson GA, Aurrand-Lions M, Imhof BA, Stehle T, Dermody TS. Junctional adhesion molecule a serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J Virol. 2005;79:7967–7978. doi: 10.1128/JVI.79.13.7967-7978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S, Hix L, Sigal LJ, Andino R. Poliovirus pathogenesis in a new poliovirus receptor transgenic mouse model: age-dependent paralysis and a mucosal route of infection. J Gen Virol. 2002;83:1707–1720. doi: 10.1099/0022-1317-83-7-1707. [DOI] [PubMed] [Google Scholar]
- Dionne KR, Galvin JM, Schittone SA, Clarke P, Tyler KL. Type I interferon signaling limits reoviral tropism within the brain and prevents lethal systemic infection. Journal of neurovirology. 2011;17:314–326. doi: 10.1007/s13365-011-0038-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson AK, Pfeiffer JK. Dynamic viral dissemination in mice infected with yellow fever virus strain 17D. J Virol. 2013;87:12392–12397. doi: 10.1128/JVI.02149-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgeorge R, Bradish CJ. The in vivo differentiation of strains of yellow fever virus in mice. J Gen Virol. 1980;46:1–13. doi: 10.1099/0022-1317-46-1-1. [DOI] [PubMed] [Google Scholar]
- Forrest JC, Campbell JA, Schelling P, Stehle T, Dermody TS. Structure-function analysis of reovirus binding to junctional adhesion molecule 1. Implications for the mechanism of reovirus attachment. J Biol Chem. 2003;278:48434–48444. doi: 10.1074/jbc.M305649200. [DOI] [PubMed] [Google Scholar]
- Frierson JM, Pruijssers AJ, Konopka JL, Reiter DM, Abel TW, Stehle T, Dermody TS. Utilization of sialylated glycans as coreceptors enhances the neurovirulence of serotype 3 reovirus. J Virol. 2012;86:13164–13173. doi: 10.1128/JVI.01822-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong DB, Nibert ML, Fields BN. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol. 1988;62:246–256. doi: 10.1128/jvi.62.1.246-256.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromeier M, Wimmer E. Mechanism of injury-provoked poliomyelitis. J Virol. 1998;72:5056–5060. doi: 10.1128/jvi.72.6.5056-5060.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyer B, Bisong AA, Gould J, Brigaud M, Aymard M. Injections and paralytic poliomyelitis in tropical Africa. Bulletin of the World Health Organization. 1980;58:285–291. [PMC free article] [PubMed] [Google Scholar]
- Hill AB, Knowelden J. Inoculation and poliomyelitis; a statistical investigation in England and Wales in 1949. British medical journal. 1950;2:1–6. doi: 10.1136/bmj.2.4669.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ida-Hosonuma M, Iwasaki T, Yoshikawa T, Nagata N, Sato Y, Sata T, Yoneyama M, Fujita T, Taya C, Yonekawa H, Koike S. The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J Virol. 2005;79:4460–4469. doi: 10.1128/JVI.79.7.4460-4469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Ooms LS, Ikizler M, Chappell JD, Dermody TS. An improved reverse genetics system for mammalian orthoreoviruses. Virology. 2010;398:194–200. doi: 10.1016/j.virol.2009.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike S, Ise I, Nomoto A. Functional domains of the poliovirus receptor. Proc Natl Acad Sci U S A. 1991a;88:4104–4108. doi: 10.1073/pnas.88.10.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike S, Taya C, Kurata T, Abe S, Ise I, Yonekawa H, Nomoto A. Transgenic mice susceptible to poliovirus. Proc Natl Acad Sci U S A. 1991b;88:951–955. doi: 10.1073/pnas.88.3.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka-Anstadt JL, Mainou BA, Sutherland DM, Sekine Y, Strittmatter SM, Dermody TS. The Nogo receptor NgR1 mediates infection by mammalian reovirus. Cell host & microbe. 2014;15:681–691. doi: 10.1016/j.chom.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyuncu OO, Hogue IB, Enquist LW. Virus infections in the nervous system. Cell host & microbe. 2013;13:379–393. doi: 10.1016/j.chom.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuss SK, Etheredge CA, Pfeiffer JK. Multiple host barriers restrict poliovirus trafficking in mice. PLoS Pathog. 2008;4:e1000082. doi: 10.1371/journal.ppat.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster KZ, Pfeiffer JK. Limited trafficking of a neurotropic virus through inefficient retrograde axonal transport and the type I interferon response. PLoS Pathog. 2010;6:e1000791. doi: 10.1371/journal.ppat.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Lobigs M. E protein domain III determinants of yellow fever virus 17D vaccine strain enhance binding to glycosaminoglycans, impede virus spread, and attenuate virulence. J Virol. 2008;82:6024–6033. doi: 10.1128/JVI.02509-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maginnis MS, Forrest JC, Kopecky-Bromberg SA, Dickeson SK, Santoro SA, Zutter MM, Nemerow GR, Bergelson JM, Dermody TS. Beta1 integrin mediates internalization of mammalian reovirus. J Virol. 2006;80:2760–2770. doi: 10.1128/JVI.80.6.2760-2770.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainou BA, Zamora PF, Ashbrook AW, Dorset DC, Kim KS, Dermody TS. Reovirus cell entry requires functional microtubules. mBio. 2013;4 doi: 10.1128/mBio.00405-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann MA, Knipe DM, Fischbach GD, Fields BN. Type 3 reovirus neuroinvasion after intramuscular inoculation: direct invasion of nerve terminals and age-dependent pathogenesis. Virology. 2002;303:222–231. doi: 10.1006/viro.2002.1699. [DOI] [PubMed] [Google Scholar]
- McCloskey BP, Melb MB. The relation of prophylactic inoculations to the onset of poliomyelitis. Lancet. 1950;255:659–663. doi: 10.1002/(sici)1099-1654(199910/12)9:4<219::aid-rmv249>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Meier KC, Gardner CL, Khoretonenko MV, Klimstra WB, Ryman KD. A mouse model for studying viscerotropic disease caused by yellow fever virus infection. PLoS Pathog. 2009;5:e1000614. doi: 10.1371/journal.ppat.1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mims CA. The invasion of the brain by yellow fever virus present in the blood of mice. British journal of experimental pathology. 1957;38:329–338. [PMC free article] [PubMed] [Google Scholar]
- Monath TP, Barrett AD. Pathogenesis and pathophysiology of yellow fever. Adv Virus Res. 2003;60:343–395. doi: 10.1016/s0065-3527(03)60009-6. [DOI] [PubMed] [Google Scholar]
- Morrison LA, Sidman RL, Fields BN. Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc Natl Acad Sci U S A. 1991;88:3852–3856. doi: 10.1073/pnas.88.9.3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller S, Cao X, Welker R, Wimmer E. Interaction of the poliovirus receptor CD155 with the dynein light chain Tctex-1 and its implication for poliovirus pathogenesis. J Biol Chem. 2002;277:7897–7904. doi: 10.1074/jbc.M111937200. [DOI] [PubMed] [Google Scholar]
- Ohka S, Igarashi H, Nagata N, Sakai M, Koike S, Nochi T, Kiyono H, Nomoto A. Establishment of a poliovirus oral infection system in human poliovirus receptor-expressing transgenic mice that are deficient in alpha/beta interferon receptor. J Virol. 2007;81:7902–7912. doi: 10.1128/JVI.02675-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohka S, Matsuda N, Tohyama K, Oda T, Morikawa M, Kuge S, Nomoto A. Receptor (CD155)-dependent endocytosis of poliovirus and retrograde axonal transport of the endosome. J Virol. 2004;78:7186–7198. doi: 10.1128/JVI.78.13.7186-7198.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohka S, Yang WX, Terada E, Iwasaki K, Nomoto A. Retrograde transport of intact poliovirus through the axon via the fast transport system. Virology. 1998;250:67–75. doi: 10.1006/viro.1998.9360. [DOI] [PubMed] [Google Scholar]
- Pfeiffer JK. Innate host barriers to viral trafficking and population diversity: lessons learned from poliovirus. Adv Virus Res. 2010;77:85–118. doi: 10.1016/B978-0-12-385034-8.00004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer JK, Kirkegaard K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc Natl Acad Sci U S A. 2003;100:7289–7294. doi: 10.1073/pnas.1232294100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer JK, Kirkegaard K. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog. 2005;1:e11. doi: 10.1371/journal.ppat.0010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer JK, Kirkegaard K. Bottleneck-mediated quasispecies restriction during spread of an RNA virus from inoculation site to brain. Proc Natl Acad Sci U S A. 2006;103:5520–5525. doi: 10.1073/pnas.0600834103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss K, Stencel JE, Liu Y, Blaum BS, Reiter DM, Feizi T, Dermody TS, Stehle T. The GM2 glycan serves as a functional coreceptor for serotype 1 reovirus. PLoS Pathog. 2012;8:e1003078. doi: 10.1371/journal.ppat.1003078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren R, Racaniello VR. Poliovirus spreads from muscle to the central nervous system by neural pathways. J Infect Dis. 1992;166:747–752. doi: 10.1093/infdis/166.4.747. [DOI] [PubMed] [Google Scholar]
- Saha N, Kolev M, Nikolov DB. Structural features of the Nogo receptor signaling complexes at the neuron/myelin interface. Neuroscience research. 2014;87:1–7. doi: 10.1016/j.neures.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Sawyer WA, Lloyd W. The Use of Mice in Tests of Immunity against Yellow Fever. The Journal of experimental medicine. 1931;54:533–555. doi: 10.1084/jem.54.4.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry B, Torres J, Blum MA. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J Virol. 1998;72:1314–1323. doi: 10.1128/jvi.72.2.1314-1323.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strebel PM, Ion-Nedelcu N, Baughman AL, Sutter RW, Cochi SL. Intramuscular injections within 30 days of immunization with oral poliovirus vaccine--a risk factor for vaccine-associated paralytic poliomyelitis. The New England journal of medicine. 1995;332:500–506. doi: 10.1056/NEJM199502233320804. [DOI] [PubMed] [Google Scholar]
- Tardieu M, Powers ML, Weiner HL. Age dependent susceptibility to Reovirus type 3 encephalitis: role of viral and host factors. Annals of neurology. 1983;13:602–607. doi: 10.1002/ana.410130604. [DOI] [PubMed] [Google Scholar]
- Theiler M. Susceptibility of White Mice to the Virus of Yellow Fever. Science. 1930;71:367. doi: 10.1126/science.71.1840.367. [DOI] [PubMed] [Google Scholar]
- Thibodeaux BA, Garbino NC, Liss NM, Piper J, Blair CD, Roehrig JT. A small animal peripheral challenge model of yellow fever using interferon-receptor deficient mice and the 17D-204 vaccine strain. Vaccine. 2012;30:3180–3187. doi: 10.1016/j.vaccine.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler KL, McPhee DA, Fields BN. Distinct pathways of viral spread in the host determined by reovirus S1 gene segment. Science. 1986;233:770–774. doi: 10.1126/science.3016895. [DOI] [PubMed] [Google Scholar]
- Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin H.W.t., Bassel-Duby R, Fields BN, Tyler KL. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J Virol. 1988;62:4594–4604. doi: 10.1128/jvi.62.12.4594-4604.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner HL, Drayna D, Averill DR, Jr., Fields BN. Molecular basis of reovirus virulence: role of the S1 gene. Proc Natl Acad Sci U S A. 1977;74:5744–5748. doi: 10.1073/pnas.74.12.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner HL, Powers ML, Fields BN. Absolute linkage of virulence and central nervous system cell tropism of reoviruses to viral hemagglutinin. J Infect Dis. 1980;141:609–616. doi: 10.1093/infdis/141.5.609. [DOI] [PubMed] [Google Scholar]
- Wyatt HV. Provocation of poliomyelitis by multiple injections. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1985;79:355–358. doi: 10.1016/0035-9203(85)90379-7. [DOI] [PubMed] [Google Scholar]
- Yang WX, Terasaki T, Shiroki K, Ohka S, Aoki J, Tanabe S, Nomura T, Terada E, Sugiyama Y, Nomoto A. Efficient delivery of circulating poliovirus to the central nervous system independently of poliovirus receptor. Virology. 1997;229:421–428. doi: 10.1006/viro.1997.8450. [DOI] [PubMed] [Google Scholar]