

Abstract
A collection of genomic DNA sequences of herpes simplex virus (HSV) strains has been defined and analyzed, and some information is available about genomic stability upon limited passage of viruses in culture. The nature of genomic change upon extensive laboratory passage remains to be determined. In this report we review the history of the HSV-1 KOS laboratory strain and the related KOS1.1 laboratory sub-strain, also called KOS (M), and determine the complete genomic sequence of an early passage stock of the KOS laboratory sub-strain and a laboratory stock of the KOS1.1 sub-strain. The genomes of the two sub-strains are highly similar with only five coding changes, 20 non-coding changes, and about twenty non-ORF sequence changes. The coding changes could potentially explain the KOS1.1 phenotypic properties of increased replication at high temperature and reduced neuroinvasiveness. The study also provides sequence markers to define the provenance of specific laboratory KOS virus stocks.
Introduction
Viruses acquire mutations as they replicate, and the viruses that emerge can be enhanced for replication, infection of the host and transmission, persistence in the host, or evasion of the host response. When viruses are taken from the normal host and placed in culture, selection for rapidly growing viruses fixes mutations that pre-exist in the population (Luria and Delbrück, 1943) or arise spontaneously in the population and that favor rapid replication in the host cells. Thus, during passage in culture viruses acquire new genetic alleles.
The herpesviruses comprise a large family of enveloped, double-stranded DNA viruses, several of which are important human pathogens (Pellett and Roizman, 2013). In addition to commonly causing labial and ocular disease, herpes simplex virus 1 (HSV-1) is the most common cause of sporadic viral encephalitis as well as a cause of severe mucocutaneous disease in immunocompromized hosts. Infection results in lifelong presence of latent virus in neural ganglia, and, because 60–90% of the world’s population is seropositive (Smith and Robinson, 2002), HSV-1 could be considered an episome of the human genome in certain neurons. The 150-kilobase pair genome of HSV-1 encodes at least eighty-four protein-coding open reading frames, as well as the long non-coding RNA latency-associated transcript and numerous miRNAs (Roizman, Knipe, and Whitley, 2013). The HSV linear double-stranded DNA genomes consist of two covalent linked components, the long (L) and short (S) components, which invert relative to each other by intramolecular recombination (Roizman, Knipe, and Whitley, 2013). The L component consists of unique sequences (UL) bounded by inverted repeats (RL and RL’), and the S component consists of the unique sequences (US) bounded by inverted repeats (RS and RS’) (Figure 1) (Roizman et al., 1979). The termini contain direct repeats of a sequence called the “a” sequence, and copies of this sequence are present in an inverted form, designated the a’ sequence, at the L-S junction (Hayward et al., 1975). The genomic structure can therefore be diagrammed aLan-RL-UL-RL’-a’m-RS’-US-RS-aS (Roizman, Knipe, and Whitley, 2013).
Figure 1. Diagram of the structure of the herpes simplex virus genome.

The top row shows the long (L) component and the short (S) component of the HSV genome. The bottom row shows the unique sequences as a line and the boxes denote the repeated sequences. UL = unique long component sequences; US = unique short component sequences; RL and RL′ = inverted repeats bounding the long component; RS and RS′ denote inverted repeats bounding the S component. a = terminal repeat also located at the L/S junction.
HSV-1 has been studied extensively in vivo and in vitro, including studies of genetic variation at the level of individual genes, and in patterns of restriction-length polymorphisms (Norberg, Bergstrom, and Liljeqvist, 2006; Norberg et al., 2004; Norberg et al., 2007). For a number of years, the only published full genome sequence, however, had been that of HSV-1 strain 17 (McGeoch et al., 1988), a laboratory strain that has undergone many passages in vitro. Recently, additional HSV-1 genomes have been reported (Szpara et al., 2014; Szpara, Parsons, and Enquist, 2010), which are beginning to reveal more about the full range of genome sequence variation among these viruses. Several different clades are apparent, largely based on geographical origin of the isolates (Szpara et al., 2014).
Recent studies have shown that limited passage of HSV strains in culture can lead to a limited number of sequence changes in the virus (Colgrove et al., 2014), but we know less about the effects of long-term passage of viruses in culture. In particular, the HSV-1 KOS strain was disseminated to different laboratories over a number of years, and a number of separate lineages or substrains have arisen as a result of passage and plaque purification in these laboratories. These sub-strains show some differences in biological properties. Because of the differences in the sub-strains, it is important to know and keep in mind the genetic background of the KOS strains in use and the source of any DNA sequences used for mutagenesis or rescue. The purpose of this report is to review the history of the HSV-1 KOS sub-strains and to provide a genome comparison of the two major laboratory sub-strains, KOS and KOS1.1.
History of the HSV-1 KOS strain
HSV-1 KOS virus was originally isolated from a lip lesion of Kendall O. Smith and first described as the SOK strain (Smith, 1964). This strain was chosen for laboratory studies because it had a low particle: PFU ratio (Smith, 1964). The original isolate of KOS was also called KOS-63 and was passaged in the laboratory. The prototype laboratory strain KOS-63 and the low-passage clinical isolate KOS-79 were isolated 16 years apart from recurrent lip lesions in the same individual and thus were thought to be related. However, biochemical analyses of these viruses revealed significant differences in the patterns of virus-induced polypeptides as well as differences in DNA restriction endonuclease cleavage patterns (Dix et al., 1983). By one of the definitions used to define HSV-1 strains, differences in DNA restriction endonuclease cleavage sites in non-repetitive sequences (Buchman et al., 1980), Dix et al. (Dix et al., 1983) concluded that “KOS-63 and KOS-79 could be considered to be unique and unrelated strains of HSV-1.”
KOS virus, presumably a derivative of the KOS-63 virus, was plaque-purified by Priscilla Schaffer three times at 35°C and then passaged three times at 40°C to “eliminate preexisting ts virions” prior to its use for genetic studies (Schaffer et al., 1970). A panel of temperature-sensitive mutant viruses was isolated and characterized by Priscilla Schaffer and colleagues in the KOS virus genetic background (Schaffer et al., 1973), so it became a common laboratory wild-type strain. This virus was also used to construct other types of mutants including drug-resistant mutants (Coen and Schaffer, 1980) and deletion mutants (DeLuca, McCarthy, and Schaffer, 1985). This laboratory KOS strain has sometimes been called KOS (H) because it was the prototype at Baylor University in Houston, or KOS (Schaffer) because it was popularized by Priscilla Schaffer with her ts mutants.
The HSV-1 KOS genomic sequence has been reported in two studies (Macdonald et al., 2012); Kinchington Genbank sequence). Interestingly, Szpara et al (2014) reported that KOS clustered with an Asian HSV-1 strain by phylogenetic analysis. Grose (2014) hypothesized that Smith acquired this virus when he served in Korea during the Korean War, which could explain the origin of this virus.
History of the KOS1.1 virus
Robert Hughes and William Munyon obtained HSV-1 KOS virus from Edmundo Kraiselburd and plaque-purified it at 39°C to generate what they named the KOS1.1 virus (Hughes and Munyon, 1975). They isolated 12 temperature-sensitive mutant viruses that represented 7 complementation groups (Hughes and Munyon, 1975). Myron (Mike) Levine’s laboratory used the KOS1.1 virus obtained from Hughes and Munyon (Adler, Glorioso, and Levine, 1978) to isolate a number of additional temperature-sensitive mutant viruses that included the ts13, ts18, ts656, and tsLG4 mutant viruses (Sandri-Goldin, Levine, and Glorioso, 1981). The KOS1.1 virus also served as the source of HSV-1 DNA for construction of the pSG plasmids, which included nearly the entire HSV-1 genome (Goldin et al., 1981). Levine provided KOS1.1 wt and ts mutants to the Knipe laboratory in 1979–1980. The Knipe laboratory constructed a series of ICP8 (Gao and Knipe, 1989) and ICP27 gene mutant viruses (Rice and Knipe, 1990) in this genetic background.
Other derivatives of the KOS1.1 virus
Joe Glorioso’s laboratory plaque-purified a virus from KOS1.1 that they called KOS 321 (Holland et al., 1983). In addition, Jack Stevens obtained the HSV-1 KOS1.1 virus from Mike Levine’s laboratory, and his laboratory plaque-purified it and named it KOS (M) to denote its origin as the University of Michigan (Thompson et al., 1986). Certain later papers referred to this virus as KOS (Yuhasz and Stevens, 1993). KOS (M) virus showed reduced neuroinvasion and reduced replication in spinal ganglia as compared with HSV-1 strain 17 virus (Javier, Sedarati, and Stevens, 1986; Thompson et al., 1986). The neuroinvasiveness defect in KOS (M) mapped to a region of the glycoprotein B gene, and two amino acid residue differences were reported in the KOS (M) gB gene relative to the strain ANG gB gene, R485H and A523 V (Yuhasz and Stevens, 1993). The KOS (M) virus was used to make various mutants in the Stevens, Wagner, Feldman, Thompson and Bloom laboratories, including latency-associated transcript coding region mutants and recombinants expressing reporter proteins from the LAT promoter (Devi-Rao et al., 1994; Dobson et al., 1989; Izumi et al., 1989; Sedarati et al., 1989). We had observed that the KOS1.1 virus established latent infection very inefficiently (L. Morrison and D. Knipe, unpublished results), so in this study we directly compared the biological properties and genome sequences of an early passage KOS virus and a KOS1.1 virus and defined their genomic sequences.
Results
Biological Properties of the HSV-1 KOS and KOS1.1 Viral Stocks
We have used the HSV-1 KOS strain extensively for latency studies in a murine model (Cliffe, Garber, and Knipe, 2009; Coen et al., 1989; Leib et al., 1989a; Leib et al., 1989b; Wang et al., 2005), but we had also used the HSV-1 KOS1.1 virus for genetic analysis (Gao and Knipe, 1989; Rice and Knipe, 1990). We therefore wanted to compare the biological properties and genome sequence of the two sub-strains. The HSV-1 KOS virus stock used for preparation of viral DNA was obtained by amplifying a passage 12 stock provided by Dr. Priscilla Schaffer, and the KOS1.1 virus stock used for preparation of viral DNA for sequencing was approximately three passages from the stock originally provided by M. Levine. To test the biological properties of these virus stocks, they were tested in the mouse corneal model of infection (Leib et al., 1990). We observed viral eye titers that were less than 2-fold higher for KOS as compared with KOS1.1 (Figure 2A), but latent viral DNA loads were 20-fold higher for KOS as compared with KOS1.1 (Figure 2B). These results showed the KOS and KOS1.1 stocks used for our studies had biological properties similar to those previously observed for KOS (M) relative to HSV-1 strain 17 virus (Javier, Sedarati, and Stevens, 1986; Thompson et al., 1986).
Figure 2. In vivo Phenotype of HSV-1 KOS and KOS1.1.
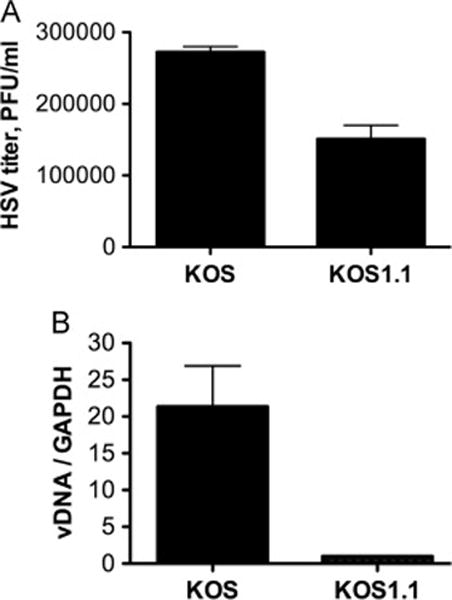
Groups of mice (n = 30/group) were infected with HSV-1 KOS or HSV-1 KOS1.1, as described in Materials and Methods. (A) Virus from eyeswabs of five HSV KOS- and five KOS1.1-infected mice during acute infection was titrated on Vero cells. Shown are mean values and standard error of the means. (B) Latent viral DNA from HSV KOS and KOS1.1-infected mice at 30 days postinfection was quantified by real-time PCR relative to a GAPDH cellular control gene during latent infection. Shown are the mean values and standard error of the means.
Genomic Sequencing
The sequence of the KOS genome was determined by a combination of Sanger sequencing, 454 sequencing, and Illumina sequencing. The first round of sequencing employed traditional shotgun cloning and Sanger sequencing but achieved adequate coverage of only one third of the genome with good read depth and high-quality sequence. Under-represented regions showed a highly non-random distribution in areas of particularly low sequence complexity and high-GC content. Next-gen 454 sequencing (454 Life Sciences, Branford, CT) was then employed to attempt to close gaps to give near-complete genome coverage when merged with Sanger sequence data. The 454 data, however, showed a number of length variations in homopolymeric stretches within coding regions relative to the reference strain, which were not present in the Sanger sequence data. A combination of automated assembly onto partial genome scaffolds corresponding to UL, US, RL, and RS, and manual assembly of bridging reads from the raw 454 data were required to generate full genome sequences. The complete KOS sequence was derived for all 13 kilobases and 12 open reading frames of US. The remaining small gaps at the termini, the junctions between unique and repeat regions, and in ICP4 and ICP0 were closed using conventional sequencing of PCR products or from the pK1–2 plasmid encoding the ICP4 gene (DeLuca and Schaffer, 1987), and confirmed by a subsequent run of Ilumina sequencing. In the case of the “a” sequence (Varmuza and Smiley, 1985) and the region around OriS (position 131870–132250; (Weller et al., 1985), the gaps were filled in with the indicated published KOS sequence. The numbers of repeated sequences not defined by the sequence data were assigned based on the numbers in the HSV-1 strain 17 genomic sequence.
Our KOS genomic sequence (GenBank accession number KT899744) was identical to two others reported previously, GenBank accession numbers JQ673480 (Macdonald et al., 2012) and JQ780693.1 (Payne, K.M., Russell, D.A., and Kinchington, P.R., unpublished data). The sequence identity was not surprising because MacDonald et al. (2012) used a similar passage 12 virus stock from Priscilla Schaffer, and the Kinchington sequence also used an early passage stock from the Schaffer lab (personal communication). The KOS genome sequence contained a previously reported stop codon in the US9 ORF (Negatsch, Mettenleiter, and Fuchs, 2011).
Comparison of the KOS and KOS1.1 genomes
The complete KOS1.1 genome (GenBank accession # KT887224) was determined by Illumina sequencing and the numbers of repeats were assigned based on the HSV-1 strain 17 reference genome. The ORFs of the KOS and KOS1.1 genomes were highly similar. There were a small number of non-coding changes, summarized in Table 1. Only five coding changes in ORFs were observed. These included the R515H codon change in the UL27 gene encoding glycoprotein B, the T566A codon change in the UL30 gene encoding the viral DNA polymerase catalytic subunit, the UL36 sequence repeat number variation, the UL39 L383P codon change, and the S81P codon change in the ICP4 or RS1 ORF encoding the immediate-early ICP4 transcriptional regulator (Table 2). These sequence changes could contribute to the decreased neuroinvasion and increased thermostability of the KOS1.1 virus, as described in the Discussion.
Table 1.
List of non-coding changes between HSV KOS and KOS1.1
| US: | Two 1–2 nucleotide indels in homopolymeric intergenic regions |
| UL Left1: | 62,387 out of 62,399 nucleotides identical |
| Four indels in homopolymeric non-coding regions | |
| Two single-nucleotide and two 2-nucleotide silent changes | |
| UL Right: | 45,298 out of 45,303 nucleotides identical |
| No indels | |
| Five silent transitions and 1 silent transversion | |
| RS: | One silent transition in RS1 |
| RL: | One silent transition |
| Four single nucleotide indels in homopolymeric regions | |
| One 3 nucleotide deletion (Just right of LAT intron in homopolymeric run of A’s) |
UL left and UL right are divided at the repeat sequence in UL36.
Table 2.
Differences between HSV KOS and KOS1.1 coding sequences.*
| UL36 tegument | Repeat length variation | |
|---|---|---|
| KOS: | 2881 | PQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQ PQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQPQ |
| KOS1.1: | 2881 | —PQPQPQPQPQPQPQPQPQPQPQPQPQPQPXXXXXXXXPQPQPQPQPQPQPQPQPQPQ |
| UL27 gB | One codon change | |
| KOS: | 481 | PPPPGASANASVERIKTTSSIEFARLQFTYNHIQRHVNDMLGRVAIAWCELQNHELTLWN PPPPGASANASVERIKTTSSIEFARLQFTYNHIQ HVNDMLGRVAIAWCELQNHELTLWN |
| KOS1.1: | 481 | PPPPGASANASVERIKTTSSIEFARLQFTYNHIQHHVNDMLGRVAIAWCELQNHELTLWN |
| UL30 Polymerase | One codon change | |
| KOS: | 541 | NAVAEAVLKDKKKDLSYRDIPAYYATGPAQRGVIGEYCIQDSLLVGQLFFKFLPHLELSA NAVAEAVLKDKKKDLSYRDIPAYYA GPAQRGVIGEYCIQDSLLVGQLFFKFLPHLELSA |
| KOS1.1: | 541 | NAVAEAVLKDKKKDLSYRDIPAYYAAGPAQRGVIGEYCIQDSLLVGQLFFKFLPHLELSA |
| UL39 Ribonucleotide Reductase | One codon change | |
| KOS: | 361 | FCSPPRLTEDDFGLLNYALVEMQRLCLDVPPVLPNAYMPYYLREYVTRLVNGFKPLVSRS FCSPPRLTEDDFGLLNYALVEMQRLCLDVPPV PNAYMPYYLREYVTRLVNGFKPLVSRS |
| KOS1.1: | 361 | FCSPPRLTEDDFGLLNYALVEMQRLCLDVPPVPPNAYMPYYLREYVTRLVNGFKPLVSRS |
| RS1 ICP4 | One codon change | |
| KOS: | 61 | DGRAPAAGTDAGEDAGDAVSSRQLALLASMVEEAVRTIPTPDPAASPPRTPAFLADDDDG DGRAPAAGTDAGEDAGDAVS RQLALLASMVEEAVRTIPTPDPAASPPRTPAFLADDDDG |
| KOS1.1: | 61 | DGRAPAAGTDAGEDAGDAVSPRQLALLASMVEEAVRTIPTPDPAASPPRTPAFLADDDDG |
Numbers show codon position relative to the AUG. Amino acid changes are shown in boldface and indels with (—).
ORF Analysis relative to Strain 17
We also compared the HSV-1 KOS sequence to the original HSV-1 strain 17 reference (GenBank accession # NC_001806). Of the 56 ORFs in UL, 12 ORFs in US and three in the long and short repeats in the strain 17 sequence, all are present in KOS, and there are no new large ORF’s. Overall sequence conservation is high, ranging from 95 to 100%. Nine genes show 100% identity between KOS and Strain 17 (Table 3). Of note, a majority of these (UL16, 18, 20, 21, and 28) lie within a relatively small stretch of the genome. Most of the sequence variation was accounted for by single nucleotide polymorphisms, but 17 ORFs showed small insertions or deletions, most often in areas of tandem repeats. Several genes showed closely paired insertions and deletions relative to the HSV-1 strain 17 reference sequence, which would predict internal frameshifts and subsequent restoration of the initial reading frame, resulting in short changes in predicted amino acid sequence (Table 4). An additional pair of frameshifts in UL44 are translationally silent (not shown). These sequence differences are similar to those reported by others (Szpara et al., 2010; Macdonald et al., 2012).
Table 3.
Genes with 100% identity between HSV-1 Strains KOS and 17
| Gene | Function | Length (bp) |
|---|---|---|
| UL16 | Tegument protein | 1122 |
| UL18 | Capsid protein | 957 |
| UL20 | Membrane protein | 669 |
| UL21 | Tegument protein | 1608 |
| UL28 | Terminase subunit | 2358 |
| UL35 | Capsid protein | 339 |
| UL45 | Membrane protein | 519 |
| UL55 | Nonstructural protein | 561 |
| US8 | Glycoprotein E | 1653 |
Table 4.
Alignments of genes where compensating frameshifts occur between HSV-1 KOS and 17.*
| UL2 | 17: | 61 | RSSGPAALLAALEAGPAGVTFSSSAPPDPPMDLTNGGVSPAATSAPLDWTTFRRVFLIDD RSSGPA PAGVTFSSSAPPDPPMDLTNGGVSPAATSAPLDWTTFRRVFLIDD |
| KOS: | 61 | RSSGPAGAPRRPRGCPAGVTFSSSAPPDPPMDLTNGGVSPAATSAPLDWTTFRRVFLIDD | |
| US4 | 17: | 120 | DKPNRPVVPSPDPNNSPARPETSRPKTPPTIIGPLATRPTTRLTSKGRPLVPTPQHTPLF DKPNRPVVP P PNNSPARPETSRPKTPPT IGPLATRPTT+L SKGRPLVPTPQHTPLF |
| KOS: | 120 | DKPNRPVVPPPGPNNSPARPETSRPKTPPTSIGPLATRPTTQLPSKGRPLVPTPQHTPLF | |
| US2 | 17: | 181 | PAAPNHPLETLLSRYEYQYGVVLPGTNGRERDCMRWLRSLIALHKPHPATPGPLTTSHPV PAAPNHPLETLLSRYEYQYGVVLPGTNGRERDCMRWLRSLIALHKPHPAT PLTTSH V |
| KOS: | 181 | PAAPNHPLETLLSRYEYQYGVVLPGTNGRERDCMRWLRSLIALHKPHPATSHPLTTSHSV | |
| UL17 | 17: | 1 | MNAHLANEVQTISATARVGPRSLVHVIISSECLAAAGIPLAALMRGRPGLGTAANFQVEI MNAHLANEVQ P SLVHVIISSECLAAAGIPLAALMRGRPGLGTAANFQVEI |
| KOS: | 1 | MNAHLANEVQYDLGHVPGRPSSLVHVIISSECLAAAGIPLAALMRGRPGLGTAANFQVEI | |
| UL27 | 17: | 1 | MRQGAPARGRRWFVVWALLGLTLGVLVASAAPSSPGTPGVAAATQAANGGPATPAPPAPG M QGAP GRRWFVVWALLGLTLGVLVASAAPSSPGTPGVAAATQAANGGPATPAPPA G |
| KOS: | 1 | MHQGAPSWGRRWFVVWALLGLTLGVLVASAAPSSPGTPGVAAATQAANGGPATPAPPALG | |
| UL8 | 17: | 541 | ANAVRLRHPLCLALEGVYTHAVAWSQAGVWFWNSRDNTDHLGGFPLRGPAYTTAAGVVRD AN +RLRHPLCLALEGVYTHAVAW+QAGVWFWNSRDNTDHLGGFPLRGPAYTTAAGVVRD |
| KOS: | 541 | ANGLRLRHPLCLALEGVYTHAVAWNQAGVWFWNSRDNTDHLGGFPLRGPAYTTAAGVVRD | |
| UL41 | 17: | 361 | TPPELVQVPNAQLLEEHRSYVANPRRHVIHDAPESLDWLPDPMTITELVEHRYIKYVIS LTPPELVQVPNAQLLEEHRSYVA RRHVIHDAPESLDWLPDPMTITELVEHRYIKYVIS |
| KOS: | 361 | LTPPELVQVPNAQLLEEHRSYVASRRRHVIHDAPESLDWLPDPMTITELVEHRYIKYVIS | |
| US3 | 17: | 61 | PSEAERLCHLQEILAQMYGNQDYPIEDDPSADAADDVDEDAPDDVAYPEEYAEELFLPGD P AERLCHLQEILAQMYGNQDYPIEDDPSADAADDVDEDAPDDVAYPEEYAEELFLPGD |
| KOS: | 61 | PGDAERLCHLQEILAQMYGNQDYPIEDDPSADAADDVDEDAPDDVAYPEEYAEELFLPGD | |
| UL52 | 17: | 481 | PASPGEDTAGGTPPPQTCGIVKRLLRLAATEQQGPTPPAIAALIRNAAVQTPLPVYRISM PASPGEDTAGGTPPPQTCGIVKRLLRLAATEQQ TPPAIAALIRNAAVQTPLPVYRISM |
| KOS: | 481 | PASPGEDTAGGTPPPQTCGIVKRLLRLAATEQQDTTPPAIAALIRNAAVQTPLPVYRISM |
Codon numbers marked relative to the ATG initiator. Predicted amino acid changes are in boldface.
Discussion
The HSV-1 KOS strain has been utilized by a number of labs and passaged and re-plaque-purified as it has been disseminated into new labs. The KOS1.1 or KOS (M) sub-strain was selected for growth at higher temperature and has been shown to be less neuroinvasive than the original KOS virus, but the genetic basis for the phenotypic differences has not been defined completely. Here we report the genome sequence for HSV KOS and its derivative KOS1.1 sub-strain, which are closely related but show a few significant genomic changes, consistent with the phenotypic differences. Five protein coding changes were observed, which will be discussed individually below. Furthermore, KOS1.1 showed only two-dozen mutations relative to KOS, principally single nucleotide insertions or deletions in homopolymeric stretches. Again, the highest density of changes was noted toward the middle of UL (Table 1).
The UL27 R515H change alters an arginine to histidine on the long alpha helix of domain III of glycoprotein B (gB) (Heldwein et al., 2006). Based on this location, the mutation could have an effect on the refolding of gB from the pre-fusion to the postfusion form (E. Heldwein, personal communication). If the prefusion structure of gB, particularly its helical core, resembles that of VSV G, then R515 would be in a region that undergoes refolding from unstructured to helical during fusion, and thus the histidine substitution might reduce fusogenicity (E. Heldwein, personal communication). Lower fusogenicity of the H515 variant could contribute to an attenuated phenotype. The R515 prefusion form may be more thermostable and in becoming more thermostable, gB may have become less able to function in some way that negatively affects its function in neuronal cells.
The RS1 S81P change alters the serine 81 residue of ICP4 to proline. ICP4 is known to be temperature-sensitive in the F strain (Knipe and Roizman, unpublished results; (Leopardi and Roizman, 1996). Therefore, this change could have been selected in the KOS1.1 genome to make the virus better able to replicate at higher temperatures.
The UL30 T566A change in KOS1.1 alters a threonine to alanine in the 3′–5′-exonuclease domain of the DNA polymerase catalytic subunit (Liu et al., 2006). Certain changes in the DNA polymerase affect the ability of the virus to replicate in sensory neurons (Pelosi et al., 1998; Terrell, Pesola, and Coen, 2014). Most importantly, the 3′–5′ exonuclease domain is known to affect the stability of hyperthermophilic archea DNA polymerases (Rodriguez et al., 2000), so this amino acid residue change could affect the thermostability of the HSV DNA polymerase.
The UL39 ribonucleotide reductase L393P change could affect the thermostability of the ribonucleotide reductase large subunit; however, this enzyme is not required for replication in cell culture, so this change was not likely to have been selected by plaque purification at high temperature. This change may make KOS1.1 less able to replicate in neurons. Finally, it is difficult to predict the effect of a change in the repeat length in the UL36 tegument protein.
In addition to the ORF coding changes, the most common were sequence length variations within the many stretches of repetitive or homopolymeric nucleotide sequence found throughout the HSV-1 genome. In part, the high frequency of these regions simply reflects the low sequence complexity that results from the high-GC content of the herpes simplex viruses. In addition, some of the sequence-length variation may result from homologous recombination, or polymerase slipping during viral replication. This feature could be significant for the host immune response during exposure to a second strain, either via natural superinfection or challenge of a vaccinated host with a new virus.
We recently observed a pattern of nearby compensating frameshift mutations in the HSV-2 HG52 reference sequence (Colgrove et al., 2014), in which a single-nucleotide insertion in one genome is balanced by single nucleotide deletion a short distance away, resulting in a run of a few completely different amino-acids flanked by highly homologous sequence on either side. Such double mutations would be expected to occur rarely by chance in a DNA virus such as HSV, and given their absence from other HSV genomes they are most likely sequencing errors resulting from the limitations of older sequencing technology with GC-rich, low-complexity regions in HSV. These changes were also recently noted in the HSV-1 strain 17 sequence (Szpara et al., 2014) and used to correct the HSV-1 strain 17 reference sequence (Genbank accession # JN555585.1). This emphasizes the importance of resequencing and correcting older reference genome sequences, particularly where high-GC content and low sequence complexity are issues. Of note, in the particular case of length variation in homopolymeric runs in 454 sequences, we observed both true biological variation (i.e. confirmed independently with Sanger sequences) and sequencing artifact due to limitations of the technology. Examination of the raw 454 reads in the latter case showed variation in number of bases called among individual reads covering the same position in the genome. The combined use of Sanger, 454, and Illumina sequencing here (and comparison with older, manually-generated sequences)—as well as automated and manual genome assembly—to generate a finished sequence for a genome of high GC content with significant repetitive sequence highlights the evolving state of sequencing technology, with particular strengths and limitations for each (Parsons et al., 2015).
Identification of parental KOS viruses
Comparison of the complete genomic sequences of KOS and KOS1.1 has provided sub-strain specific genome sequences that will allow the identification of the origin of KOS virus-derived recombinant or mutant strains. For example, the sequence changes in the UL27, UL30, UL35, or RS1 genes of the KOS1.1 genome provide a means to define whether specific viruses are derived from a parental virus similar to KOS or KOS1.1. For example, this could confirm that the KOS (M) viruses are derived from and similar to KOS1.1 rather than KOS.
In summary, this study defines the potential genetic basis for the different biological phenotypes of the HSV-1 KOS lab strain and its derivative KOS1.1 sub-strain. The results illustrate the types of genetic diversity that can be introduced into lab strains as they are passaged in culture and further plaque-purified. However, they also provide the information to determine the provenance of the parental KOS viruses used for the construction of specific KOS-derived recombinant and mutant viruses, thus allowing the use of the appropriate wild-type parent virus for comparison. These results illustrate the need for caution in the comparison of mutant viruses constructed in different KOS sub-strain backgrounds.
Materials and Methods
Viruses
HSV-1 KOS passage 12 virus was provided by Priscilla Schaffer. HSV-1 KOS1.1 was provided by Mike Levine. Both viruses were grown and titrated on Vero cells.
Mouse infections
Mice were housed in accordance with institutional and NIH guidelines on care and use of animals in research, and all procedures were approved by the Intitutional Animal Care and Use Committee of Harvard Medical School. Six-week old CD1 male mice (Charles River Laboratories) were anesthetized in an isoflurane chamber followed by deep anaesthesia through intraperitoneal injections of ketamine (3.7 mg/mouse) and xylazine hydrochloride (0.5 mg/mouse). Mouse corneas were scarified and infections were carried out at a dose of 2 × 106 pfu/eye of HSV-1 KOS or HSV-1 KOS 1.1 viruses. Eyeswabs were performed using sterile polyester applicators (Puritan) at two days post infection, and virus shed in tear film was titrated on Vero cells. At 30 days post infection, latently-infected trigeminal ganglia were harvested, from which DNA was isolated using the Qiagen DNeasy kit and quantified using realtime PCR with primers specific for viral DNA and a cellular control (Cliffe, Garber, and Knipe, 2009; Wang et al., 2005).
|
| |
| Primer | Sequence |
|
| |
| ICP8 F | GAGACCGGGGTTGGGGAATGAATC |
| ICP8 R | CCCCGGGGGTTGTCTGTGAAGG |
| GAPDH F | CAGGCGCCCAATACGACCAAAATC |
| GAPDH R | TTCGACAGTCAGTCAGCCGCATCTTCTT |
|
| |
Viral DNA preparation
HSV-1 KOS DNA was prepared as described elsewhere (Colgrove et al., 2014). Briefly, virus was grown on Vero cells and depending on the preparation, viral genomes were isolated either by cell-lysis, or pelleting of extracellular virus from culture supernatants, followed by SDS/proteinase-K degradation, and DNA banding in NaI gradients.
Shotgun Cloning, Sequencing and initial assembly
Early rounds of sequencing were carried out using the Qiagen TOPO Shotgun Subcloning kit, as per the manufacturers instructions [Qiagen, Germantown, MD]. Subclones were sequenced at the Harvard Medical School Biopolymers Facility. Initial assembly was performed using the base-calling, assembly, and finishing programs, Phred, Phrap, and Consed.
454 Sequencing and Assembly
Twenty micrograms of KOS DNA were prepared as described (Colgrove et al., 2014) and submitted for 454 Sequencing to the Broad Institute [www.broadinstitute.org]. The 454 reads were assembled using the Newbler suite of tools from Roche/454, using the runMapping module. The runMapping module performs a reference-based assembly with the provided reads. The repetitive nature of the HSV genome made a reference- based approach better than de novo assembly but still challenged the algorithm. We broke the reference genome into the major genome components: UL, US, RL, and RS. Each component was then used as a reference in runMapping. The resulting contigs from the repeat components were duplicated and joined with the unique component contigs to form the final assembly.
Computer assembly of the genome resulted in creation of a substantial number of disconnected contigs. Analysis of the contig ends showed them to be in areas of repetitive sequence where the software assembler was not able to correctly identify flanking sequence, either being blocked or adding an incorrect read at the end. These contigs were joined manually by choosing sequences a short way in from the end of a contig, searching the raw 454 reads for that sequence, and then adding reads as appropriate to the contig in an iterative fashion analogous to chromosomal “walking” in conventional sequencing. In addition, areas of sequence length polymorphism in an assembled contig were analyzed by looking directly at the population of 454 reads covering the corresponding areas of sequence, typically in homopolymeric runs.
Illumina sequencing and genome finishing
Ilumina sequencing reads were assembled at the Broad Institute using ALLPATHS-LG as described previously (Newman et al., 2015) against scaffolds composed of fragments of the strain 17 sequence corresponding to UL, US, RL, and RS, with 100–200 nucleotides of contiguous sequence at each end. This resulted in six contigs corresponding to these pieces with breaks in repetitive sequence in UL36 and RS. Contigs were further refined by manual insertion of 454 reads at their ends until joining of contigs were achieved. Gaps in known repeat regions were filled by adding repeat numbers by convention with the strain 17 reference sequence (GenBank accession # NC_001806).
Sequence Analysis
Whole genome alignments were performed with Mummer [http://mummer.sourceforge.net]. Genome annotation was generated using the online Virus Pathogen Resource [www.viprbc.org], using HSV-1 Strain 17 as a reference. Individual protein alignments were done with BLAST [http://blast.ncbi.nlm.nih.gov/Blast.cgi]. Further analysis was carried out using the sequence analysis package EMBOSS 3.1 [http://emboss.sourceforge.net].
Research Highlights (For Review).
“History and Genomic Sequence Analysis of the Herpes Simplex Virus 1 KOS and KOS1.1 Sub-Strains,” will be useful for the HSV field because there are several different HSV-1 KOS viruses being used in different labs, and this manuscript provides the history of them.
The manuscript compares the genomic sequence of the two major sub-strains, KOS and KOS1.1 and provides possible explanations for the differences in biological properties.
The genomes of the two sub-strains are highly similar with only five coding changes, 20 non-coding changes, and about twenty non-ORF sequence changes.
The coding changes could potentially explain the KOS1.1 phenotypic properties of increased replication at high temperature and reduced neuroinvasiveness. It also provides sequence identifiers for viruses derived from these two sub-strains so the provenance of viruses can be identified.
Acknowledgments
This research is dedicated to our late colleague and collaborator, Priscilla Schaffer, who established KOS as an important laboratory strain and encouraged us in the original efforts to sequence the genome of KOS. This research was supported by NIH grants AI 098681 to DMC and DMK and grant AI 057552 to DMK. This project was also funded in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. 541 HHSN272200900018C to the Broad Institute’s Genomic Sequencing Center for Infectious Diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler R, Glorioso JC, Levine M. Infection by herpes simplex virus and cells of nervous system origin: characterization of a non-permissive interaction. J Gen Virol. 1978;39:9–20. doi: 10.1099/0022-1317-39-1-9. [DOI] [PubMed] [Google Scholar]
- Buchman TG, Simpson T, Nosal C, Roizman B, Nahmias AJ. The structure of herpes simplex virus DNA and its application to molecular epidemiology. Ann NY Acad Sci. 1980;354:279–290. doi: 10.1111/j.1749-6632.1980.tb27972.x. [DOI] [PubMed] [Google Scholar]
- Cliffe AR, Garber DA, Knipe DM. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol. 2009;83:8182–90. doi: 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coen DM, Kosz-Vnenchak M, Jacobson JG, Leib DA, Bogard CL, Schaffer PA, Tyler KL, Knipe DM. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc Natl Acad Sci U S A. 1989;86:4736–4740. doi: 10.1073/pnas.86.12.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coen DM, Schaffer PA. Two distinct loci confer resistance to acycloguanosine in herpes simplex virus type 1. Proc Natl Acad Sci U S A. 1980;77:2265–2269. doi: 10.1073/pnas.77.4.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgrove R, Diaz F, Newman R, Saif S, Shea T, Young S, Henn M, Knipe DM. Genomic sequences of a low passage herpes simplex virus 2 clinical isolate and its plaque-purified derivative strain. Virology. 2014:450–451. doi: 10.1016/j.virol.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca NA, McCarthy AM, Schaffer PA. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J Virol. 1985;56:558–570. doi: 10.1128/jvi.56.2.558-570.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca ND, Schaffer PA. Activities of herpes simplex virus type 1 (HSV-1) ICP4 genes specifying nonsense polypeptides. Nucleic Acids Res. 1987;15:4491–4511. doi: 10.1093/nar/15.11.4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi-Rao GB, Bloom DC, Stevens JG, Wagner EK. Herpes simplex virus type 1 DNA replication and gene expression during explant-induced reactivation of latently infected murine sensory ganglia. J Virol. 1994;68:1271–1282. doi: 10.1128/jvi.68.3.1271-1282.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix RD, Lukes S, Pulliam L, Baringer JR. DNA restriction enzyme analysis of viruses isolated from cerebrospinal fluid and brain-biopsy tissue in a patient with herpes simplex encephalitis [letter] N Engl J Med. 1983;308:1424. doi: 10.1056/NEJM198306093082321. [DOI] [PubMed] [Google Scholar]
- Dobson AT, Sederati F, Devi-Rao G, Flanagan J, Farrell MJ, Stevens JG, Wagner EK, Feldman LT. Identification of the latency-associated transcript promotor by expression of rabbit beta-globin mRNA in mouse sensory nerve ganglia latently infected with a recombinant herpes simplex virus. J Virol. 1989;65:3844–3851. doi: 10.1128/jvi.63.9.3844-3851.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Knipe DM. Genetic evidence for multiple nuclear functions of the herpes simplex virus ICP8 DNA-binding protein. J Virol. 1989;63:5258–67. doi: 10.1128/jvi.63.12.5258-5267.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL, Sandri-Goldin RM, Levine M, Glorioso JC. Cloning of herpes simplex virus type 1 sequences representing the whole genome. J Virol. 1981;38:50–58. doi: 10.1128/jvi.38.1.50-58.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward GS, Jacob RJ, Wadsworth SC, Roizman B. Anatomy of herpes simplex virus DNA: evidence for four populations of molecules that differ in the relative orientations of their long and short components. Proc Natl Acad Sci U S A. 1975;72:4243–4247. doi: 10.1073/pnas.72.11.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. Crystal structure of glycoprotein B from herpes simplex virus 1. Science. 2006;313:217–20. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- Holland TC, Marlin SD, Levine M, Glorioso J. Antigenic variants of herpes simplex virus selected with glycoprotein-specific monoclonal antibodies. J Virol. 1983;45:672–682. doi: 10.1128/jvi.45.2.672-682.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RG, Jr, Munyon WH. Temperature-sensitive mutants of herpes simplex virus type 1 defective in lysis but not in transformation. J Virol. 1975;16:275–283. doi: 10.1128/jvi.16.2.275-283.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi KM, McKelvey AM, Devi-Rao G, Wagner EK, Stevens JG. Molecular and biological characterization of a type 1 herpes simplex virus (HSV-1) specifically deleted for expression of the latency-associated transcript (LAT) Microb Pathog. 1989;7:121–134. doi: 10.1016/0882-4010(89)90031-4. [DOI] [PubMed] [Google Scholar]
- Javier RT, Sedarati F, Stevens JG. Two avirulent herpes simplex viruses generate lethal recombinants in vivo. Science. 1986;234:746–748. doi: 10.1126/science.3022376. [DOI] [PubMed] [Google Scholar]
- Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol. 1989a;63:2893–2900. doi: 10.1128/jvi.63.7.2893-2900.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leib DA, Coen DM, Bogard CL, Hicks KA, Yager DR, Knipe DM, Tyler KL, Schaffer PA. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J Virol. 1989b;63:759–768. doi: 10.1128/jvi.63.2.759-768.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leib DA, Ruffner KL, Hildebrand C, Schaffer PA, Wright GE, Coen DM. Specific inhibitors of herpes simplex virus thymidine kinase diminish reactivation of latent virus from explanted murine ganglia. Antimicrob Agents Chemother. 1990;34:1285–1286. doi: 10.1128/aac.34.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopardi R, Roizman B. The herpes simplex virus major regulatory protein ICP4 blocks apoptosis induced by the virus or by hyperthermia. Proc Natl Acad Sci U S A. 1996;93:9583–9587. doi: 10.1073/pnas.93.18.9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Knafels JD, Chang JS, Waszak GA, Baldwin ET, Deibel MR, Jr, Thomsen DR, Homa FL, Wells PA, Tory MC, Poorman RA, Gao H, Qiu X, Seddon AP. Crystal structure of the herpes simplex virus 1 DNA polymerase. J Biol Chem. 2006;281:18193–200. doi: 10.1074/jbc.M602414200. [DOI] [PubMed] [Google Scholar]
- Luria SE, Delbrück M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald SJ, Mostafa HH, Morrison LA, Davido DJ. Genome sequence of herpes simplex virus 1 strain KOS. J Virol. 2012;86:6371–2. doi: 10.1128/JVI.00646-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch DJ, Dalrymple MA, Davison AJ, Dolan A, Frame MC, McNab D, Perry LJ, Scott JE, Taylor P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol. 1988;69(Pt 7):1531–74. doi: 10.1099/0022-1317-69-7-1531. [DOI] [PubMed] [Google Scholar]
- Negatsch A, Mettenleiter TC, Fuchs W. Herpes simplex virus type 1 strain KOS carries a defective US9 and a mutated US8A gene. J Gen Virol. 2011;92:167–72. doi: 10.1099/vir.0.026484-0. [DOI] [PubMed] [Google Scholar]
- Newman RM, Lamers SL, Weiner B, Ray SC, Colgrove RC, Diaz F, Jing L, Wang K, Saif S, Young S, Henn M, Laeyendecker O, Tobian AA, Cohen JI, Koelle DM, Quinn TC, Knipe DM. Genome Sequencing and Analysis of Geographically Diverse Clinical Isolates of Herpes Simplex Virus 2. J Virol. 2015 doi: 10.1128/JVI.01303-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norberg P, Bergstrom T, Liljeqvist JA. Genotyping of clinical herpes simplex virus type 1 isolates by use of restriction enzymes. J Clin Microbiol. 2006;44:4511–4. doi: 10.1128/JCM.00421-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norberg P, Bergstrom T, Rekabdar E, Lindh M, Liljeqvist JA. Phylogenetic analysis of clinical herpes simplex virus type 1 isolates identified three genetic groups and recombinant viruses. J Virol. 2004;78:10755–64. doi: 10.1128/JVI.78.19.10755-10764.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norberg P, Olofsson S, Tarp MA, Clausen H, Bergstrom T, Liljeqvist JA. Glycoprotein I of herpes simplex virus type 1 contains a unique polymorphic tandem-repeated mucin region. J Gen Virol. 2007;88:1683–8. doi: 10.1099/vir.0.82500-0. [DOI] [PubMed] [Google Scholar]
- Parsons LR, Tafuri YR, Shreve JT, Bowen CD, Shipley MM, Enquist LW, Szpara ML. Rapid genome assembly and comparison decode intrastrain variation in human alphaherpesviruses. MBio. 2015;6 doi: 10.1128/mBio.02213-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellett P, Roizman B. Herpesviridae. In: DM K, PM H, editors. Fields Virology. 6th. Lippincott Williams & Wilkins; Philadelphia: 2013. pp. 1802–1823. [Google Scholar]
- Pelosi E, Rozenberg F, Coen DM, Tyler KL. A herpes simplex virus DNA polymerase mutation that specifically attenuates neurovirulence in mice. Virology. 1998;252:364–72. doi: 10.1006/viro.1998.9447. [DOI] [PubMed] [Google Scholar]
- Rice SA, Knipe DM. Genetic evidence for two distinct transactivation functions of the herpes simplex virus alpha protein ICP27. J Virol. 1990;64:1704–1715. doi: 10.1128/jvi.64.4.1704-1715.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AC, Park HW, Mao C, Beese LS. Crystal structure of a pol alpha family DNA polymerase from the hyperthermophilic archaeon Thermococcus sp. 9 degrees N-7. J Mol Biol. 2000;299:447–62. doi: 10.1006/jmbi.2000.3728. [DOI] [PubMed] [Google Scholar]
- Roizman B, Jacob RJ, Knipe DM, Morse LS, Ruyechan WT. On the structure, functional equivalence, and replication of the four arrangements of herpes simplex virus DNA. Cold Spring Harb Symp Quant Biol. 1979;43 Pt 2:809–826. doi: 10.1101/sqb.1979.043.01.088. [DOI] [PubMed] [Google Scholar]
- Roizman B, Knipe DM, Whitley RJ. Herpes Simplex Viruses. In: Knipe DM, Howley PM, editors. Fields Virology. 6th. Lippincott Williams & Wilkins; Philadelphia: 2013. pp. 1823–1897. [Google Scholar]
- Sandri-Goldin RM, Levine M, Glorioso JC. Method for induction of mutations in physically defined regions of the herpes simplex virus genome. J Virol. 1981;38:41–49. doi: 10.1128/jvi.38.1.41-49.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer P, Vonka V, Lewis R, Benyesh-Melnick M. Temperature-sensitive mutants of herpes simplex virus. Virology. 1970;42:1144–1146. doi: 10.1016/0042-6822(70)90364-8. [DOI] [PubMed] [Google Scholar]
- Schaffer PA, Aron GM, Biswal N, Benyesh-Melnick M. Temperature-sensitive mutants of herpes simplex virus type 1: isolation, complementation and partial characterization. Virology. 1973;52:57–71. doi: 10.1016/0042-6822(73)90398-x. [DOI] [PubMed] [Google Scholar]
- Sedarati F, Izumi KM, Wagner EK, Stevens JG. Herpes simplex virus type 1 latency-associated transcription plays no role in establishment or maintenance of a latent infection in murine sensory neurons. J Virol. 1989;63:4455–4458. doi: 10.1128/jvi.63.10.4455-4458.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Robinson NJ. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: A global review. J Infect Dis. 2002;186 Suppl 1:S3–28. doi: 10.1086/343739. [DOI] [PubMed] [Google Scholar]
- Smith KO. Relationship between the envelope and the infectivity of herpes simplex virus. Proc Soc Exp Biol Med. 1964;115:814–6. doi: 10.3181/00379727-115-29045. [DOI] [PubMed] [Google Scholar]
- Szpara ML, Gatherer D, Ochoa A, Greenbaum B, Dolan A, Bowden RJ, Enquist LW, Legendre M, Davison AJ. Evolution and diversity in human herpes simplex virus genomes. J Virol. 2014;88:1209–27. doi: 10.1128/JVI.01987-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szpara ML, Parsons L, Enquist LW. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J Virol. 2010;84:5303–13. doi: 10.1128/JVI.00312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrell SL, Pesola JM, Coen DM. Roles of conserved residues within the pre-NH2-terminal domain of herpes simplex virus 1 DNA polymerase in replication and latency in mice. J Gen Virol. 2014;95:940–7. doi: 10.1099/vir.0.061903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Cook ML, Devi-Rao GB, Wagner EK, Stevens JG. Functional and molecular analyses of the avirulent wild-type herpes simplex virus type 1 strain KOS. J Virol. 1986;58:203–211. doi: 10.1128/jvi.58.1.203-211.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varmuza SL, Smiley JR. Signals for site-specific cleavage of HSV DNA: maturation involves two separate cleavage events at sites distal to the recognition sequences. Cell. 1985;41:793–802. doi: 10.1016/s0092-8674(85)80060-x. [DOI] [PubMed] [Google Scholar]
- Wang Q-Y, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci U S A. 2005;102:16055–16059. doi: 10.1073/pnas.0505850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller SK, Spadaro A, Schaffer JE, Murray AW, Maxam AM, Schaffer PA. Cloning, sequencing, and functional analysis of oriL, a herpes simplex virus type 1 origin of DNA synthesis. Mol Cell Biol. 1985;5:930–942. doi: 10.1128/mcb.5.5.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuhasz SA, Stevens JG. Glycoprotein B is a specific determinant of herpes simplex virus type 1 neuroinvasiveness. J Virol. 1993;67:5948–5954. doi: 10.1128/jvi.67.10.5948-5954.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]