

Abstract
Small redox active molecules such as reactive nitrogen and oxygen species and hydrogen sulfide have emerged as important biological mediators that are involved in various physiological and pathophysiological processes. Advancement in understanding of cellular mechanisms that tightly regulate both generation and reactivity of these molecules is central to improved management of various disease states including cancer and cardiovascular dysfunction. Imbalance in the production of redox active molecules can lead to damage of critical cellular components such as cell membranes, proteins and DNA and thus may trigger the onset of disease. These small inorganic molecules react independently as well as in a concerted manner to mediate physiological responses. This review provides a general overview of the redox biology of these key molecules, their diverse chemistry relevant to physiological processes and their interrelated nature in cellular signaling.
Graphical abstract
1. Introduction
Investigation of the biological effects of nitric oxide (NO), carbon monoxide (CO) and hydrogen sulfide (H2S) initially concentrated on their context in environmental toxicology. For example NO and other nitrogen oxide species (often grouped as NOx) are major components of air pollution [1], carcinogenic nitrosamines are found in food [2, 3] and H2S and CO are industrial poisons [4, 5]. Given this history, the discovery of endogenous production of NO and related nitrogen oxides for eradication of pathogens including bacteria, parasites and viruses by the immune system and for regulation of physiological functions in the circulatory and nervous system was quite surprising [6–10]. The significance of the discovery of NO biosynthesis and its role in regulation of blood pressure was recognized by award of the 1998 Nobel Prize in Physiology or Medicine to Murad, Ignarro and Furchgott [11]. More recently, other small inorganic molecules, including CO, H2S and even H2, have also been identified as essential mediators of physiological processes. Their disequilibrium has been associated with disease onset and progression in cancer, immune response and cardiovascular function among others [12–14]. In addition, the diverse pharmacology of nitroxyl (HNO), for example in treatment of heart failure, alcoholism and cancer [15–19], strongly suggests that HNO is likely biosynthesized.
These signaling molecules are increasingly been referred to as endogenous gasotransmitters since they are all gases in pure form at STP. As gases, these molecules share important physical characteristics such as low molecular weight and neutral charge, which when coupled with lipophilicity, are important to their function as diffusible signaling agents. In the diverse environments of biological systems, the term gasotransmitter is a misnomer for these solute molecules. In addition, this term overlooks the fact that signaling is often indirect, for example after conversion of NO or H2S into redox-related derivatives. Furthermore, the importance of O2 is neglected because its production is not regulated, unlike NO, CO and H2S.
Although redox activity is a major component of the signaling processes mediated by NO and H2S, other reaction types, for example association of NO with heme systems, are also essential. Moreover, CO is not redox active. Here, we collectively consider the impact of small inorganic molecules on signaling from the perspective of the direct and indirect effects of the neutral species: O2, NO, HNO, CO, H2S and H2. A heavy emphasis is placed on the involvement of these and related species in redox signaling.
Redox active molecules play diverse and critical roles in all aspects of cell biology and physiology including metabolism, cellular signaling and host defense. Redox homeostasis is maintained by regulated production of redox active molecules, redox buffering and a diverse antioxidant system. Homeostatic regulation allows for initiation of signaling processes upon the interaction of reactive redox active species with specific targets. Under stress conditions, such as those often considered in chemical toxicology, overproduction of these species can induce damage to macromolecules including proteins, lipids and DNA. Antioxidants and other repair systems can promote survival under conditions of environmental stress; however, chemically induced stress can overwhelm such protective mechanisms creating an imbalance and leading to deterioration of cellular function. Since the boundary between normal and stress conditions is not precise, pathways exist to resolve the stressful incident and restore homeostasis or to induce cell death. Induction and control of stress-induced signaling by reactive redox active species is therefore important in both normal physiology and disease processes.
While the deleterious roles of small inorganic species, particularly reactive oxygen species (ROS), have been studied extensively, including impacts on pathological conditions such as aging and neurodegenerative diseases [20], analysis of their functions as signaling molecules is relatively new [21, 22]. Understanding of the fundamental chemistry of these species as well as the kinetic factors that control their generation and consumption is critical in order to build a realistic model for their participation in biological signaling mechanisms and physiological outcome.
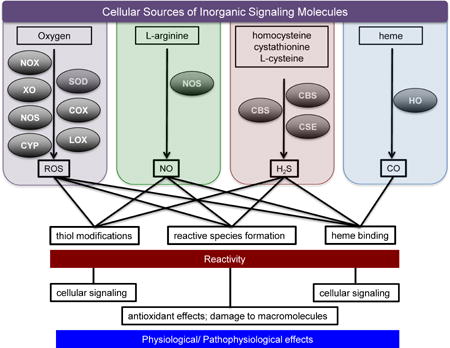
Redox biology includes reactions of these chemical species with molecular targets, which have significant biological implications, especially in the context of cellular signaling. Often, cellular signaling pathways involve receptor-ligand interactions that rely on a structure-function relationship. In contrast, signaling by small inorganic messenger molecules typically involves covalent or coordinate covalent bond formation. Specificity occurs through spatial-, temporal-, and concentration-dependent constraints, typically via regulation of their biosynthesis pathway. The sources of these endogenous species are often metalloenzymes, and their targets can be considered to be primarily, although not exclusively, metal complexes and thiols [23–26]. While the reaction sites of these species overlap, their chemical and biological signatures are distinct, due to induction of different chemical modifications. In this review we discuss and highlight their biosynthesis, chemistry, and the interplay in and between subclasses of small inorganic signaling molecules.
2. Oxygen and Reactive Oxygen Species
Molecular oxygen serves as the ultimate electron acceptor during oxidative phosphorylation. Incomplete reduction of O2 in the mitochondrial electron transport chain can lead to accumulation of ROS including superoxide (O2−), hydroxyl radical (·OH) and hydrogen peroxide (H2O2), which as highly reactive species can be damaging to cellular macromolecules including lipids, proteins and DNA when produced in an unconstrained fashion [27, 28]. A variety of antioxidants including glutathione, flavonoids and vitamins A, C and E protect against ROS toxicity. Cellular levels of ROS are also tightly regulated enzymatically (Figure 1) [29–31]. Despite the known implications of ROS imbalance in the initiation of oxidative stress and disease, ROS regulate various biological and physiological processes. Understanding of the signaling cascades mediated by these species is therefore important [32].
Figure 1.

Formation of physiologically relevant ROS by key enzymes and processes.
One-electron reduction of O2 is thermodynamically unfavorable (−0.33 V vs. NHE), which prevents indiscriminate oxidation. Direct reaction of O2 with organic substrates is also kinetically inhibited due to ground state differences (i.e., spin restrictions). These kinetic barriers can be overcome by a variety of metalloenzymes, such that activation of O2 is coupled to diverse metabolic transformations. Hydroxylation reactions, for example during purine catabolism by xanthine oxidase, are well known to lead to O2− production [33]. Rather than simply being a byproduct, O2− derived from xanthine oxidase can be utilized to oxidize other species such as cytochrome c [34]. Both xanthine oxidase and cytochrome c are commonly used experimentally to produce and detect O2−, respectively.
The toxicity of O2− is harnessed by the immune system to eradicate pathogens. NADPH oxidase is a transmembrane multienzyme complex that couples oxidation of NADPH with reduction of O2 in neutrophil phagosomes. Identification of distinct isoforms in a variety of cell types, collectively referred to as the NOX family [29, 35–37], have suggested a role for O2− production beyond host defense. Regulated production of ROS by NOX proteins has now been implicated in cell signaling processes including post-translational modifications and regulation of gene expression [38].
Oxidation or the absence of tetrahydrobiopterin, a cofactor for nitric oxide synthase (NOS), leads to uncoupling of the dimer and production of O2− [39–41]. Formation of O2− by the reductase domain of NOS is implicated in pathological conditions for example stemming from endothelial dysfunction [42–45]. More recently, a tight correlation has been demonstrated between NO biosynthesis and tetrahydrobiopterin in a number of different endothelial and vascular smooth muscle cell lines. However, the molecular mechanism of this effect is still unknown [46–48]. Uncoupling of the catalytic turnover of P450 enzymes can also lead to production of O2− by autoxidation of the ferrous dioxygen complex. Formation of ROS from P450s has been previously reviewed [49, 50].
The metabolism of arachidonic acid and polyunsaturated fatty acids by cyclooxygenases (COX) and lipoxygenases (LOX) can also generate ROS and radical species in their pathways. LOX-dependent production of O2−, initially suggested by Lynch and Thompson [51], has been demonstrated in skeletal muscle and endothelial cells [52–54]. Generation of O2− by COX has been suggested to impair K+ channels after brain injury and G protein-mediated cerebrovasodilation [55, 56]. While the mechanism of O2− formation is unknown, it is hypothesized to form by electron transfer from fatty acid hydroperoxides to O2 [57]. This reaction can be favorable if the fatty acid radical formed in the process is stabilized by resonance delocalization. The involvement of unsaturated fatty acids in production of O2− in macrophages has also been suggested [58]. In 1986 Kukreja et al. showed that NADPH or NADH is required for O2− formation by COX and LOX [59].
Three important factors that dictate whether ROS formation will lead to redox signaling or oxidative damage are the site of formation, the reaction rate with target species and the rate of detoxification. With respect to reactivity, O2− is a mild reductant, a strong oxidant and a free radical. The reduction of Fe3+ to Fe2+ ion by O2− was described by Haber and Weiss in the 1930s [60] (Eq. 1).
| 1) |
O2− can also react with a variety of both endogenous and exogenous reducing agents (e.g., thiols, ascorbate, semiquinones [61–63]) as well as other free radicals. The latter reaction is typically far more facile than the conversion of reducing agents into free radicals (e.g., near diffusion controlled reaction of O2− with NO vs. oxidation of thiols to the thiyl radical at ~103 M−1 s−1 at pH 7.4 [64]). However, the overall rate of these reactions will dependent on the concentration of the reactants. The reaction of NO for instance would only be kinetically viable in close proximity to cells that are actively producing NO while thiols are relatively stable and abundant. Either reaction type may be considered to be protective in that free radicals are consumed. However, the product of the reaction of O2− with NO is itself a reactive species (see section 3 for further details) and oxidation of redox active biomolecules can propagate free radical formation [65].
Since one-electron reduction of O2 is thermodynamically unfavorable while one-electron reduction of O2− is quite favorable (0.89 V vs. NHE), O2− is unstable to disproportionation producing O2 and H2O2 (5 × 105 M−1 s−1 at pH 7.0). Superoxide dismutases (SODs) significantly accelerate disproportionation [66], maintaining low physiological concentrations of O2−. Again, it is important to note that H2O2 is also a reactive species. H2O2 may be formed by other pathways, for instance through two-electron reduction of O2 by xanthine oxidase [67], NADPH oxidase [68] and sulfhydryl oxidase, which also converts thiols to disulfides [69], and by one-electron reduction of O2− [61, 70].
Similarly to O2−, H2O2 can react with metals and cellular reductants and can be consumed enzymatically. The interaction of H2O2 with iron complexes in particular is important. Fenton’s reagent, or hydrogen peroxide and Fe2+ (Eq. 2), was developed in the late 1800s for hydroxylation of organic compounds and oxidation of organic contaminants [71].
| 2) |
Reduction of Fe3+ to Fe2+ ion by O2− (Eq. 1) then provides a catalytic mechanism to produce powerful oxidants such as ·OH via Eq. 2 [70, 72]. Other metal ions such as Cu+, Ti3+, Cr2+, Co2+ also undergo similar reactions with H2O2 and are known as Fenton-like reagents [73].
The importance of this reaction was expanded with the discovery that oxidants derived from Fenton chemistry could alter macromolecules with pathological consequences. Fenton-like reactions are now considered to be primary causes of oxidative stress [74]. Since a rich literature was concurrently accumulating with respect to generation of oxidants and reactive molecules from ionizing radiation, it was commonly assumed that ·OH was associated with both ionizing radiation and the Fenton-like reaction. However, in the 1950s Henry Taube suggested that the oxidant was instead a hypervalent metal-oxo species, as had been earlier described by Bray and Gorin (Eq. 3) [75, 76].
| 3) |
The nature of the oxidant has since been controversial [77–79]. Formation of Fe3+ is rate-limiting, suggesting that Eq. 2 represents a concerted reaction [80]. However, in the early 1990s, stopped flow analysis led to detection of an intermediate species prior to formation of Fe3+ during oxidation of N-dimethyl nitrosamine [80–82]. This intermediate was determined to be an aqueous iron nitrosyl complex, indicating that an oxidant is produced as an intermediate prior to formation of Fe3+ [82]. These results supported prior suggestions of the metal-based oxidants during the classical Fenton reaction [75, 76]. Competition experiments also indicated that iron peroxo complexes led to similar chemical modifications as to ·OH, as proposed by Drago and colleagues with cobalt complexes [83]. The ligand environment may influence the identity of the major oxidant, with hard ligands perhaps promoting ·OH, while heme favors hypervalent species [75, 76, 80].
As with O2−, H2O2 can oxidize thiols, but the product is a sulfenic acid (Eq. 4) [84]. The reaction rate is highly dependent on the protein microenvironment (10–106 M−1 s−1), which affects thiol nucleophilicity.
| 4) |
Reductants such as ascorbate can reverse this modification through an addition/elimination mechanism [85]. Furthermore, sulfenic acids can undergo further oxidation to sulfinic acid (Eq. 5), which in turn are susceptible to oxidation to sulfonic acids (Eq. 6) [86].
| 5) |
| 6) |
Sulfenic acids can also react with excess thiols to form disulfide bonds or with proximal amines to form sulfonamides (RSNR2). The diversity of thiol modifications is extensive, and investigation of the implications of such modifications on cellular signaling is ongoing. For a review of the ROS-mediated alterations of critical cysteine residues, see Klomsiri et al. [87].
Chance demonstrated in 1949 that H2O2 was consumed in the presence of catalase [88–91] in a disproportionation reaction to water and O2. Other antioxidant enzymes, which reduce H2O2 to H2O include peroxiredoxins and glutathione peroxidases [92–96]. Although H2O2 is less reactive than O2−, control of H2O2 levels is critical given that H2O2 can readily diffuse across cellular membranes, and thus can have an extended sphere of influence. Active transport is also possible through members of the aquaporin family [97, 98].
Although production of H2O2 is well known to lead to oxidative damage of macromolecules and to be an underlying cause of numerous diseases and aging, a role for H2O2 as a messenger molecule in redox signaling is emerging. For instance post-translational modifications of cysteines lead to allosteric changes that alter protein function [31, 99]. The pathophysiological implications of H2O2 signaling in cancer, inflammation and aging have been extensively reviewed by Schieber et al. [100]. Here, we note that such pathways place metalloproteins like NADPH oxidase, which produces H2O2, and catalase, which consumes H2O2, in a highly regulated manner in a central role [101, 102].
As a highly reactive molecule, ·OH can react with a diverse variety of molecules including lipids, proteins, DNA, RNA and carbohydrates with a near diffusion controlled rate constants without specificity, thus contributing to widespread deleterious effects. However, ·OH may also be involved in cellular signaling. Reaction with lipids such as arachidonic acid leads to formation of isoprostanes [103, 104] and α,β-unsaturated aldehydes [105]. Isoprostanes have been implicated in various disorders and pathophysiological conditions [106]. Acrolein, 4-hydroxy-2-nonenal and crotonaldehyde are α,β-unsaturated aldehydes that are highly toxic and promote oxidative stress-mediated signaling associated with pathophysiological conditions by reacting with free thiol and amine groups of other macromolecules [107–109]. These reactions, along with the reactions of ·OH with proteins and DNA are well summarized by Marnett et al. [110].
3. The Chemical Biology of NO
While ROS primarily arise from reduction of O2, reactive nitrogen species (RNS) can be generated by oxidation of NO, which itself is produced by oxidation of arginine by NO synthases (NOS) (Figure 2) [111].
Figure 2.

Biosynthesis of NO from oxidation of L-arginine by nitric oxide synthase.
Three isoforms have been identified: neuronal NOS (nNOS, NOS1), inducible NOS (iNOS, NOS2) and endothelial NOS (eNOS, NOS3). The constitutive isoforms eNOS and nNOS produce low cellular levels of NO, generally in response to transient changes in calcium levels, while induction of iNOS can lead to sustained NO fluxes in the micromolar range. Under hypoxia, reduction of nitrite (NO2−) can provide an alternative, oxygen-independent source of low levels of NO (for example, [112]).
Intense interest in the chemistry of NO and related RNS emerged following the identification of NO as an important mediator of both physiological and pathophysiological processes [7–10]. Unlike many signaling agents, which rely on receptors where structural relationships determine function, the chemistry of RNS determines biological activity. Since the lifetimes of RNS are limited, the kinetics of these reactions determine biological outcomes. Defining these reactions within the context of physiology and disease is challenging. A simplifying concept developed in the 1990s is the chemical biology of NO [113–115], which identifies two distinct reactions types based on NO concentration, reactive species and reaction kinetics (Figure 3).
Figure 3.

The Chemical Biology of NO: direct and indirect reactions of NO with kinetically important cellular targets and their biological implications.
3a. Direct effects of NO
Direct effects involve interaction of NO itself with biological targets. These reactions are facile with rate constants >105 M−1 s−1 and primarily involve heme proteins and other radical species. Perhaps the most important example of a direct effect is coordination of NO to the heme protein soluble guanylyl cyclase (sGC). This association leads to the majority of the physiological effects of NO through formation of cGMP from GTP [116]. Nitrosylation of ferrous sGC (7 × 108 M−1 s−1) [117] labilizes the axial His105 ligand [118] due to the strong trans effect of NO. The resulting structural change leads to significant enhancement of activity [119, 120].
While loss of NO from ferrous heme complexes tends to be very slow [121], release of NO from sGC is critical for reversible activation. The slow rate of NO dissociation measured with purified sGC has been puzzling [122]. Recent studies indicate a dependence on ATP and GTP [123] suggesting that sGC deactivation may be complex [122, 124, 125].
NO can interact with a number of other heme proteins including hemoglobin, cyclooxygenase, cytochrome P450 and cytochrome c oxidase [126, 127]. NO can either bind directly to the iron center or can interact with other bound ligands. For instance, the reaction of NO with oxyhemoglobin leads to formation of nitrate (NO3−) and methemoglobin (3.4 × 107 M−1s−1) [128] through interaction with ligated O2. This reaction is generally considered to serve as a major regulatory mechanism of NO flux [129]. NO can also react directly with hypervalent species formed during the Fenton reaction (Eq. 3) to reduce the metal to a normal oxidation state (Eq. 7) [130, 131].
| 7) |
The concentration and duration of NO is important with respect to interactions with metal centers. In addition to kinetic factors, the thermodynamic stability of the metal-nitrosyl bond is important in determining the extent of signaling. NO has a higher affinity for Fe2+ compared to Fe3+. Several summaries of rate constants for Fe2+ and Fe3+ heme interactions have been reported [126, 127, 132, 133]. A quick rule of thumb for the flux of NO required for activation is to take the reciprocal of the Keq, which is kon/koff. This provides a ballpark estimate for the importance of a reaction under defined conditions. For example, Keq ~109 M−1 for sGC indicates that a flux of 1–10 nM is required for pathway activation. In contrast, Fe3+ proteins such as COX2 have Keq ~102 M−1, indicating that the formation of a stable nitrosyl complex requires a 100 μM steady state NO flux, which is not readily achieved in vivo [127].
As a free radical, the reaction of NO with other free radicals is typically facile. An important exception is the dimerization of NO, which is only favored at low temperatures of high pressures. Association of NO with other free radicals in biological systems often serves an antioxidant role abating propagation cycles and associated toxicities [113, 114]. Reaction of NO with O2− and NO2 results in alteration of the ROS/RNS profile and thus is described in more detail in the following section.
3b. Indirect effects of NO
The concentration of NO produced by NOS dictates the reactive species that are produced as well as their downstream targets. Interaction of NO at nanomolar levels with sGC promotes vasorelaxation [9, 134, 135]. In contrast, higher concentrations of NO can lead to interactions with other metal centers or to formation of RNS typically by oxidation of NO. These species mediated the indirect effects of NO by modifying unique biological targets. Such reactions are critical in anti-pathogen and anti-tumor responses of the immune system [6, 136–140]. The major RNS include nitrogen dioxide (NO2), dinitrogen trioxide (N2O3), dinitrogen tetraoxide (N2O4) and ONOO−. These reactive species are potent nitrating, nitrosating and oxidizing agents [113, 141] that play important roles in redox biology and sGC-independent signaling.
RNS formation pathways include autoxidation of NO (Eq. 8)
| 8) |
The mechanism of NO autoxidation is still a matter of debate, but both NO2 and N2O3 are often invoked (Eqs. 9, 10).
| 9) |
| 10) |
Autoxidation of NO has a reasonably high rate constant (4.8 × 106 M−2 s−1 [142]) but is second order in NO. This dependence may limit the significance of the reaction to conditions of high NO such as inflammation. Furthermore, that both NO and O2 partition to nonaqueous environments suggests that NO autoxidation may be confined to hydrophobic lipid membranes.
Once formed, N2O3 can undergo rapid hydrolysis to form nitrous acid (HNO2) (Eq. 11) [143], which is a weak acid (pKa of 3.4).
| 11) |
The reversibility of Eqs. 10 and 11 leads to a mechanism for conversion of nitrite to NO, for example in acidic environments such as the stomach [144–146], and demonstrates the interrelated nature of NO and RNS.
Formation of NO2 and N2O3 can lead to oxidation/nitration/nitrosation of biomolecules such as proteins and lipids. The principal mediator of RNS-associated oxidative stress is NO2. The toxic effects of NO2 are well known for example in air pollution and cigarette smoke [1]. It is associated with irritation of the nose and eye, as well as pulmonary edema and bronchiolitis. Nonetheless, NO2 is also a key component of the immune system during pathogen eradication [147, 148]. NO2 induces nitration of tyrosine, and this post-translational modification is associated with inflammation and disease processes [149, 150]. Formation of nitrotyrosine is a central feature of NO2 chemistry and is used as a biomarker for NO2 [151, 152]. Several studies suggest that nitration occurs on specific residues rather than indiscriminately [153, 154]. While tyrosine nitration is a well-established phenomenon, the underlying routes of NO2 formation and the impacts of tyrosine nitration on protein function are less clear. Readers are referred to excellent reviews on the link between protein nitration and disease etiology [155, 156].
The primary mediator of nitrosative stress is N2O3, which promotes a variety of non-radical reactions with thiols and amines. Nitrosation of secondary amines can produce carcinogenic nitrosamines [7]. Nitrosation of proline has been observed in gastric cancer patients and may provide strong evidence of N2O3 generation in vivo under conditions of inflammation [157, 158]. Nitrosation of thiols can inhibit enzymatic function and can activate key signaling pathways under normal and pathophysiological conditions [159, 160]. For example, N2O3 inhibits DNA repair proteins by modification of key thiol residues, degradation of zinc finger proteins, and nitrosation of lysine residues for example in ligases, which results in deamination [161–166]. In contrast, nitrosation of growth factors and membrane thiols can activate important pro-growth signaling mechanisms that promote tissue restoration as well as inflammatory disease processes [167]. For review of the implications of nitrosation in physiological and pathophysiological processes see Anand et al. [168].
Formation of an S-nitrosothiol is often used as a nitrosative footprint [169], but pathways other than interaction of thiols with N2O3 can lead to S-nitrosothiol production [170]. A large body of literature suggests S-nitrosothiols as a principle mediator of sGC independent cellular signaling. A detailed discussion on RSNO is beyond the scope of this review, and readers are referred to several other reviews [171–173].
Similarly to NO autoxidation, the rapid reaction of NO and O2− (Eq. 12; 6.7 × 109 M−1 s−1 [174] is a prime example of the interrelated nature of RNS and ROS.
| 12) |
In addition to modulation of the bioavailability of both NO and O2− (thus preventing formation of H2O2), the product ONOO− is highly reactive [175–177] and can be converted to other RNS. Although ONOO− can undergo isomerization to form NO3− (1 s−1) in a deactivation pathway, homolytic cleavage to form NO2 and ·OH is significant (30% yield) [178]. Also, ONOO− reacts with CO2 leading to formation of ONOOCO2− (Eq. 13; 2.3 × 103 M−1 s−1) [179], which can further decompose to NO2 and the carbonate radical (Eq. 14), which are powerful oxidants.
| 13) |
| 14) |
In the presence of excess NO, Eq. 8 can again be invoked, leading to formation of N2O3. Such reactions impact the balance between ROS and RNS and provide a spectrum of oxidative and nitrosative chemistry. The chemistry will be modulated by the relative concentrations of NO and O2− and consumption pathways. For example, high SOD activity competes with reaction of NO.
Several studies have shown an antagonistic relationship either chemically or through regulation of enzymatic sources of ROS or NO. In the case of NADPH oxidase, a major source of ROS, NO inhibits assembly in an isoform-specific manner, leading to reduced ROS levels [180–182]. Furthermore, the interactions of NO and ROS can modulate signaling pathways. For example, both NO and ROS stabilize the stress protein p53, but NO moderates ROS–mediated p53 stabilization and similarly ROS abates NO p53 stabilization [183, 184]. Taken together this redox balance provides an important component to overall cellular and physiological signaling and toxicity. As such, the antioxidant effects of NO have been exploited in preventing and treating stroke and myocardial infarctions [185–187].
3c. Role of NO in breast cancer
NO has a concentration-dependent role in cancer biology [188]. Intermediate NO concentrations play a critical role in regulation of tumor growth, migration and metastasis [189, 190]. NO levels between 200–700 nM flux for several hours lead to cellular proliferation and properties associated with metastasis of estrogen receptor negative (ER(−)) breast cancer cells [189]. In contrast, such NO fluxes regulate wound-healing responses critical for tissue restoration in normal cells [191]. Moreover, in cancer an NO flux of ~300 nM promotes activation of the extracellular signal-regulated protein kinase (ERK) and phosphoinositide 3-kinase (PI3k)/protein kinase B (Akt) pathways, as well as stabilization of hypoxia-inducible factor-1 (HIF-1α) [192–194]. Two intriguing mechanisms lead to these cellular responses. One involves nitrosation of specific thiol residues of membrane bound proteins including SRC, EGFR, and ERK by autoxidation products of NO [193], while the other involves nitration of tyrosine residues. Interestingly, TIMP1 an endogenous inhibitor of matrix metalloproteinases (MMPs) can be nitrated at key tyrosine residues that prevent its inhibitory function of active MMPs [195] while promoting activation of PI3k/Akt pro-survival signaling [196].
3d. Delivery of exogenous NO
There are several methods for delivery of NO [197, 198]. NO-based anti-cancer therapeutic applications require >800 nM steady state NO levels for prolonged periods of time [199]. Such flux levels systematically lead to unwanted cardiovascular side effects including a hypotensive response and impaired platelet function since proper cardiovascular function requires a significantly lower flux of NO. Therefore, therapeutic levels of NO must be delivered site-specifically for oncologic applications. Several strategies exist, including development of prodrugs. One of the most reliable and versatile classes of NO donor is diazeniumdiolates, which are often referred to as NONOates [200]. These compounds spontaneously decompose in a controlled, time-dependent fashion to yield specific fluxes of NO. Prodrug technology can significantly increase the half-life of NONOates, as well as facilitate site-specific delivery. For example, liver specific enzymes activate V-PYRRO/NO while JS-K is activated by glutathione transferase and shows significant anti-cancer properties in various cell lines [201]. Recently, NO-releasing nanoparticles have shown promise due to their ability to release high payloads of NO [202–204].
Another attractive delivery strategy involves photochemical release of NO from metal nitrosyl complexes [205, 206]. A major challenge of this strategy is the design of complexes with high intensity absorption bands at low energy, since longer wavelengths of light penetrate skin to greater depths than those of higher energies. Toward this end, Ford and coworkers have shown that Fe-S complexes such as Roussin salts can radiosensitize hypoxic cells upon photochemical release of NO [207]. More recently, quantum dots have been adopted to release NO [208].
3e. NO and nitrite
Nitrite can be reduced to NO under hypoxic conditions, leading to dilation of blood vessels and increased blood flow [209, 210]. Study of the reaction of nitrite with heme proteins dates back several decades [211]. Nitrite stored in tissue is reduced by Fe2+ complexes to produce NO [212–214]. This provides an important protective mechanism against tissue ischemia because the O2-dependence of NOS activity dictates reduced levels of NO under hypoxic conditions [112, 215, 216].
One of the most important aspects for cardiovascular health is the role of oral and gastrointestinal microbiota, which provides an important nitrite/NO reduction cycle [217, 218]. Nitrate from food such as spinach or beet juice is converted to nitrite in the mouth. Upon swallowing, in the acidic environment of the stomach, disproportion of nitrous acid leads to NO, which has beneficial effects such as reducing gut ulceration. Nitrate in the circulating blood is secreted again through the saliva glands and can be reduced to nitrite. Maintenance of circulating and tissue nitrite provides a buffer against hypoxia, which can promote vasodilation and decrease thrombosis [218]. Thus, metal-mediated reduction of nitrite is important in maintaining cardiovascular health and in part explains why a vegetable rich diet is a positive health factor for prevention of cardiovascular disease and cancer.
Ford and colleagues examined the reaction of nitrite with a variety of metal and porphyrin-like complexes and have identified a novel mechanism involving the transfer of oxygen to form NO [219]. Moreover, this intriguing mechanism may produce nitroxyl (HNO) as well [220]. This may provide a unique mechanism to regulate the NO/HNO balance, which mediates different physiological outcomes.
4. The Chemical Biology of HNO
HNO is a nitrogen oxide that has been studied since the late 1800s [221–223]. In addition to extensive studies on structural and spectroscopic parameters, the number of reports on the pharmacological effects HNO has been expanding since the turn of this century. Discovery of unique biological properties have led to a resurgence of interest, particularly in understanding the chemical biology of HNO and comparison of physiological effects with the redox sibling NO [18, 224].
Although several studies postulate mammalian biosynthesis of HNO, this remains highly debated, in part due to inefficient/nonspecific in vivo detection systems. Possible routes for endogenous production of HNO include but are not limited to oxidative degradation of the NO biosynthesis intermediate Nω-hydroxy-L-arginine (NOHA), release from NOS in the absence of the cofactor tetrahydrobiopterin, reaction of S-nitrosothiols with thiols, reduction of NO by metalloproteins such as MnSOD, xanthine oxidase or cytochrome c, from the reaction of H2S with HSNO (thionitrous acid, the smallest S-nitrosothiol), and most recently from nitrite via heme-iron-catalyzed metabolism of H2S [225–234]. Regardless of the open question of biosynthesis, numerous studies have shown the potential of HNO to be used in pharmacological settings such as cardiovascular disorders, inflammation, alcoholism, cancer and pain [15–17, 235–238].
The chemistry of HNO has several interesting aspects. First, HNO is metastable and undergoes rapid dehydrative dimerization (Eq. 15; 8 × 106 M−1 s−1) [239–241].
| 15) |
This self-consumption pathway both precludes storage and limits the reactivity and lifetime of HNO. Furthermore, the production of N2O is also often considered as an indirect marker of HNO. Although HNO is a weak acid (pKa of HNO > 11 [242–244]), the rate of proton transfer between the acid-base pair is severely constrained by a spin difference (1HNO and 3NO−). The spin forbidden relationship coupled with a self-consumption pathway not only inhibits establishment of an acid-base equilibrium but also dictates that resulting chemistry is likely from the initial species (HNO or NO−) rather than from a combination of the two. For a detailed discussion, see [245].
The pKa of HNO is actually derived from the reduction potential for the NO/NO− couple of −0.8 V [242, 243]. At physiological pH, the reduction potential will be increased to −0.5 V. Such a potential is nearly inaccessible biologically, indicating that NO reduction is unlikely. In contrast, oxidation of HNO to NO is thermodynamically favorable [246, 247]. However, the proton-coupled process is kinetically constrained, such that other reactions are more likely, particularly with thiols and higher valent metals [245, 248]. A number of studies have now firmly established unique and distinct chemical biology of HNO compared to NO [18, 127, 245, 249]. Nonetheless, the potential for conversion of HNO to NO under biological conditions [250–252] should be considered when assessing the effects of exposure to HNO.
4a. HNO and thiols
As an electrophile, HNO can potentially react with a variety of biological nucleophiles. Unlike NO, thiols represent a major site of direct reactivity for HNO under physiological conditions [18, 229]. Association of HNO with thiols generates N-hydroxysulfenamide (Eq. 16). In the presence of excess thiol, this unstable intermediate can further react with a second thiol (Eq. 17) to produce disulfide and NH2OH. On the other hand, the N-hydroxysulfenamide can rearrange to produce a sulfinamide (Eq. 18) [229, 253]. The disulfide product represents a biologically reversible process as biological reductants regenerate the thiol while generation of sulfinamide is unique in terms of HNO chemical biology and represents an irreversible modification of thiols [249, 254].
| 16) |
| 17) |
| 18) |
Many of the physiological actions of HNO can be explained on the basis of modification of critical thiols. For instance, inhibition of enzymes such as aldehyde dehydrogenase (ALDH), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and the zinc finger protein poly(ADP-ribose) polymerase (PARP) can explain HNO effects respectively on alcohol metabolism, glycolysis and apoptosis [161, 255, 256].
4b. HNO and metal proteins
The relative stability of ferrous compared to ferric nitrosyl complexes is well recognized [121, 126]. Unlike NO, HNO does not readily form stable complexes, although Fe(II)HNO heme protein complexes have been isolated [257, 258]. Compared to NO, the association of HNO is generally more favorable with Fe3+ rather than Fe2+ [18, 259, 260]. The reaction of HNO with Fe3+ hemoproteins leads to formation of the corresponding ferrous nitrosyl species via reductive nitrosylation (Eq. 19) [261, 262].
| 19) |
The concomitant spectral shifts upon reductive nitrosylation of metmyoglobin are often used to confirm HNO production from donor compounds [260]. Other than globins, cytochromes and peroxidases also undergo reductive nitrosylation by HNO [18, 263] as do other metal complexes [264]. Reductive nitrosylation can also lead to formation of free NO depending on protein identity. For instance axial occupation in ferricytochrome c or d10 metal centers such as in Cu,Zn SOD reduces nitrosyl complex stability [250, 262].
Contrary to their different reactivity toward Fe3+ species, both NO (4 × 107 M−1 s−1) and HNO (1 × 107 M−1 s−1) react facilely with Fe(II)O2 complexes such as oxymyoglobin to give the ferric protein [18, 265, 266]. The mechanism of this reaction for HNO has not been fully established, due in part to the lack of complete end product characterization. Regardless of the mechanism, NO and HNO can be differentiated by using a scavenger such as the thiol glutathione (GSH), which quenches HNO chemistry [18].
A primary and well established biological target for NO is the Fe2+ heme protein sGC [267]. Based on the observation that HNO donors induce vasorelaxation, speculation has been offered as to whether HNO can interact with sGC in a manner similar to NO [225, 268]. HNO has recently been implicated as an activator of sGC by interacting with the ferrous heme and with thiol residues [269]. Unlike NO, the trans effect of HNO is not strong enough to cleave the distal Fe-His bond, thus implying that sGC mediated cellular signaling is tightly controlled [270]. The sGC-dependent vasodilatation and sGC-independent positive inotropy of HNO has long been studied as a viable option for treatment of cardiovascular disorders [271–273]. Recently, HNO mediated activation of sGC showed suppression of cardiomyocyte hypertrophy and O2− generation, thus further emphasizing its role in cardiovascular pharmacotherapy [274].
4c. HNO and O2
Overwhelming data suggests that HNO and O2 react to form an oxidant that differs from synthetic ONOO− [275–278]. Unlike the reaction of NO and O2− or bolus peroxynitrite, free radicals are not produced in the reaction of HNO with O2 as verified by the absence of protein nitration or dimerization of phenolic compounds [275]. However, oxidation of ferrocytochrome c by HNO indicates that the product of HNO autooxidation may induce one-electron oxidation under certain circumstances [279]. Observation of hydroxylation suggests the presence of a strong two-electron oxidant. In addition, HNO autoxidation can induce DNA double strand breaks while ONOO− does not induce similar damage [276, 280–282]. Despite these differences, use of a boronate probe has led to the proposal that the oxidation product of HNO and O2 is in fact ONOO− [283, 284]. One possible explanation is that different protonated isomers may be formed. A recent calculation suggests HN(O)(O2) as an intermediate of HNO autoxidation [285]. This species would be expected to be an oxygen atom transfer agent culminating in two electron-oxidation rather than radical chemistry. The challenge to this potential hypothesis is how fast the nitrogen deprotonates to give ONOO−. This reaction warrants further examination.
4d. HNO and NO
HNO and NO react with each other to form reactive intermediates (Eq. 20–22). However, these reactions are biologically feasible only in the presence of low concentration of scavengers such as GSH since the rate constants for reaction of HNO with GSH (2 × 106 M−1 s−1) [18] or for dimerization (Eq. 15; 8 × 106 M−1 s−1) are similar to that of reactions 20–22.
| 20) |
| 21) |
| 22) |
The discussions above indicate that the biological chemistry of HNO is rich and diverse. Effects associated with HNO must be analyzed in light of direct interaction with primary targets such as thiols and metalloproteins or indirect effects that arises from its interaction with NO and O2.
4e. Delivery of exogenous HNO
As with NO, donor compounds are extensively used to produce HNO [286, 287]. Commonly used donors include Angeli’s salt (Na2N2O3) [241, 288], Piloty’s acid (C6H5SO2NHOH) and derivatives [289, 290], acyl nitroso compounds [291–293], acyloxy nitroso compounds [294–296] and primary amine based diazeniumdiolates [RNH-N(O)=NO−] [297, 298]. Among these donor classes, Angeli’s salt is the most prevalent donor used for chemical and biological studies. Angeli’s salt has been well characterized and is a spontaneous donor of HNO [241]. Piloty’s acid and derivatives are useful base-sensitive HNO donors whose HNO release profile can be tuned by varying the organic substituent. A tendency to generate NO instead of HNO under aerobic conditions has limited usage of Piloty’s acid and derivatives [289]. More recently, HNO donors based on acyloxy nitroso compounds and diazeniumdiolates have been developed [299–301]. Recent examination of the acetoxymethyl and aspirin derivatives of isopropylamine diazeniumdiolate (IPA/NO) demonstrated that HNO could be site-specifically delivered to cancer cells [237, 301]. The clinical potential of HNO-based therapeutics warrants development of new HNO donors.
5. The Chemical Biology of H2S
H2S is mainly formed by anaerobic digestion of organic matter. The toxic and environmental effects of H2S have been extensively studied over the last century. Endogenous production of H2S in combination with its physiological and pathophysiological roles in mammalian systems has led to renewed interest in understanding its biological function [25]. H2S is endogenously produced in mammalian tissue mainly by cystathionine β-synthase (CBS) [302, 303] and cystathionine γ-lyase (CSE) [304, 305] from L-cysteine, homocysteine, and cystathione (Figure 4). H2S is also produced from 3-mercaptopyruvate by the action of 3-mercaptopyruvate-S-transferase (MST) [306].
Figure 4.

Pathways for biosynthesis of H2S from L-cysteine, homocysteine, cystathione and 3-mercaptopyruvate catalyzed by CBS, CSE and MST.
As a physiological mediator [307], H2S is involved in calcium homeostasis, neuromodulation, inflammatory response, cardiovascular and gastrointestinal function [308–314]. The physiological concentration of H2S is estimated to be on the order of 15 nM [315], and its levels are tightly controlled. With a pKa of 6.8, both H2S and HS− exist under physiological conditions. As it is not currently known whether H2S, HS− or both are biologically active, it is common to simply refer to H2S as a collective term. Like NO and HNO, H2S also has various biological targets, and thus has a diverse chemical biology (Figure 5).
Figure 5.

Overview of the chemical biology of H2S: biological targets of H2S and their biological implications.
5a. H2S and metal proteins
The physiological concentration and activity of H2S can be mediated by binding to heme proteins. Such reactions can induce cytotoxic or cytoprotective responses in a concentration-dependent manner as seen with mitochondrial cytochrome c oxidase [316, 317]. However, the interaction of H2S with cytochrome c oxidase is complex as it can act as both inhibitor and electron donor [317, 318]. At low concentrations, H2S can directly reduce the metal center. In the process, H2S is oxidized to the thiyl radical or elemental sulfur, and O2 is consumed (Eq. 23) [317, 319]. Elevated levels of H2S can inhibit cytochrome c oxidase in a noncompetitive manner (kon = 1.5 × 104 M−1 s−1, koff = 6 × 10−4 s−1) unlike NO, which is competitive with O2 binding (Eq. 24) [319, 320].
| 23) |
| 24) |
The reaction of H2S has also been studied extensively in hemoglobin and myoglobin. H2S can reduce ferric heme, thus restoring O2 binding function [319]. However, at higher concentrations, H2S can react with oxyglobins to form sulfheme derivatives, which have weak affinities for O2 [319, 321]. Formation of the sulfheme complex requires the presence of a histidine residue at the distal site [322, 323]. At physiological concentrations, the interactions of H2S with hemeproteins such as cytochrome c oxidase, myoglobin and hemoglobin are suggested to be associated with activation of ATP-sensitive potassium channels and regulation of muscle relaxation [320, 324, 325].
5b. H2S and O2
O2 is an antagonist of H2S and plays a major role in H2S bioavailability. Autoxidation, although spontaneous, is slow and leads to formation of sulfur (Eq. 25), which can then further react with excess sulfide to form polysulfides (Eq. 26).
| 25) |
| 26) |
The presence of free metals, phenols and aldehydes can accelerate this reaction [326, 327].
Similarly to thiols, H2S also reacts with various ROS and RNS [328]. The reactions of H2S with H2O2, ONOO− or alkyl peroxides to form hydrogen thioperoxide (HSOH; e.g., Eq. 27) [329] may be biologically relevant. HSOH is a reactive intermediate that can modify thiols or react with H2S to form persulfides and polysulfides (Eq. 28), which are key players in H2S-mediated signaling.
| 27) |
| 28) |
HSOH can also be further oxidized, for example to sulfite and sulfate. Such antioxidant properties of H2S can downregulate several pro-inflammatory cytokines including NF-κB, TNF-α, IL-1β, IL-6 and IL-8 [330–332].
H2S can undergo nitrosation (Eq. 29) or react with S-nitrosothiols (Eq. 30) to form the smallest nitrosothiol, HSNO [233, 333]. This reaction is not only associated with RNS scavenging but also is invoked as a possible mechanism for HNO biosynthesis as mentioned in Section 4.
| 29) |
| 30) |
Interactions of H2S with heme systems can indirectly control O2 bioavailability, leading to reduced metabolic activity [334, 335]. The interplay of H2S and O2 can have widespread implications in the pathology of ischemia-reperfusion [336]. H2S signaling may be pronounced in hypoxic environments such as tumors. Increased H2S production from CBS in colorectal and ovarian cancers promotes proliferation, angiogenesis and migration, which can be reversed by silencing of CBS both in vitro and in vivo [337]. Cancer cells that survive hypoxic or oxidative damage show rapid cell proliferation and a nicotinamide phosphoribosyltransferase-dependent increase in tolerance to higher H2S levels [338]. Thus treatment of cancer cells with an H2S donor protects cells from drug-induced damage [338]. Inhibition of CBS may therefore be a potential area for development of anticancer therapeutics [337].
5c. H2S and thiols
Fukuto and colleagues have hypothesized the formation of persulfides by the interaction of H2S with oxidized thiols such as cystine (Eq. 31) [339].
| 31) |
Ida et al. developed a detection method that demonstrated the presence of persulfide and polysulfide species at substantially higher levels in mammalian cells, tissues, and plasma [340]. Miranda and Wink recently commented on the relationship between H2S and persulfides, which are nucleophilic reductants that can modify protein structure and activity [341]. Polysulfides have emerged as a key class of reactive species in H2S biology as they exhibit antibacterial, antifungal, antiviral and anticancer properties [342, 343].
H2S can also react with key cysteine residues to undergo S-sulfhydration (-SSH) under physiological conditions. Many proteins including 20–25% of liver proteins, tubulin, actin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are sulfhydrated under physiological conditions, which may augment their activity. The role of protein sulfhydration in signaling has been reviewed by Paul et al. [344]. These findings open up a new aspect of sulfur redox biology that may impact thiol-based cellular signaling.
6. Carbon Monoxide and Hydrogen
CO is mainly formed by partial decomposition of organic matter, and similarly to H2S, the toxic and environmental effects of CO have been extensively studied. On the other hand H2 has no cellular cytotoxicity even at high concentrations. Biological H2 is mainly produced by bacteria from hydrocarbons. Gut microbiome is rich in such bacteria. While these inorganic effector molecules are known to serve in various physiological and pathophysiological settings, [26, 345–347], their mechanisms of action are not fully understood at present.
6a. Carbon Monoxide
In the early 1950s, Sjöstrand showed for the first time that CO is produced under physiological conditions by degradation of hemoglobin [348, 349]. In the 1960s, Tenhunen and colleagues identified heme oxygenase as the endogenous source of CO [350, 351] (Figure 6).
Figure 6.

Biosynthesis of CO from degradation of heme by heme oxygenase.
Ryter et al. recently reviewed the biological effects of CO and its role in various biomedical applications, including protective effects in organ transplant, inflammatory pathways, cardiovascular function and cancer biology [14]. In addition, CO has also been implicated as a modulator of intraocular pressure and can play an important role in glaucoma therapy [352] and cardiac mitochondrial biogenesis [353].
Heme proteins are among the most important cellular targets of CO. For example, CO can bind to hemoglobin with an affinity ~250 times higher than O2 [354]. Binding of CO to subunits of hemoglobin also increases the affinity for O2 binding, thus inhibiting O2 release. Even though this process is reversible, it can lead to CO poisoning [355, 356]. Another potential target of CO is the cytochrome P450 family of enzymes. P450s are involved in oxidative metabolism of drugs and xenobiotics. Physiological levels of CO are usually too low to inhibit P450s, however exogenous exposure to CO can lead to inhibition of P450s affecting drug metabolism and vascular tone [357]. Ferrous cytochrome c oxidase, which is a key respiratory chain enzyme, can also bind CO, and this may play a biological role under hypoxic conditions [358]. CO can also bind to and activate sGC similarly if less effectively than NO and induces vasorelaxation in rat-tail artery [359]; it also inhibits platelet aggregation [360].
6b. Hydrogen
Molecular hydrogen has long been considered as a viable option for renewable energy as water is the sole product of combustion of H2. While certain microorganisms are able to metabolize H2, the production of H2 in human flatus was first reported in 1862 [361]. Levitt and colleagues showed in 1969 that more than 99% of H2 production is colonic while the presence of excess small bowel bacteria increased H2 production as well [362]. In 1982, McKay and colleagues observed H2 production by human intestinal anaerobic bacteria [363].
The role of H2 in human physiology remains largely unexplored. In 2007 Ohsawa et al. reported that H2 selectively reduced ·OH and ONOO− [364] although it does not scavenge radicals like NO and O2− that are involved in cellular signaling. Although facile, the reaction of ·OH with H2 (3.4 × 107 M− 1 s− 1) [365] may not be generally competitive with other pathways that consume ·OH. However, this reaction prevents an immediate-type allergic reaction in mice, suggesting the possibility of selective modulation of signaling [366].
In 2010 Hong et al. reviewed the beneficial effects of exogenous H2 in clinical and animal studies [367]. The observed antiapoptotic, antiinflammatory and antiallergy outcomes of consumption of H2 saturated water or inhalation of H2 were suggested to largely be a function of selective antioxidant effects. That H2 diffuses rapidly and can cross the blood brain barrier may be beneficial in prevention of ROS-mediated diseases [368].
The combination of NO and H2 significantly decreased cardiac infarct size and 3-nitrotyrosine formation compared to NO alone, and this effect was attributed to removal of RNS by H2 [369]. Inhalation of H2 by cardiomyopathic hamsters decreased oxidative stress and attenuated embryonic gene expression under hypoxic conditions [370]. This effect was also demonstrated in a rat model of myocardial ischemia-reperfusion injury [371]. H2 also suppressed hepatic injury caused by ischemia/reperfusion [372].
In a mouse model lacking leptin receptors, exposure to H2 induced fibroblast growth factor 21 (FGF21), which is involved in regulation of metabolism [373], This result indicates a role for H2 in treatment of obesity and diabetes. H2 may also serve as an effective and safe radioprotective agent [374]. Despite a growing literature on the protective role of H2 in various conditions, the chemical biology of H2 requires further investigation to understand the underlying mechanisms at a molecular level.
7. Crosstalk between Redox Signaling Molecules
The diverse physiological roles of O2, NO, CO and H2S pose key questions regarding their interaction with one another and the effect of such reactions on cell signaling. The reactions of O2 with NO and H2S have been described in earlier sections. While CO is not known to directly react with O2, it can indirectly effect O2-mediated cellular signaling. For example, HO-1 can modulate production of ROS from NADPH oxidase/NOX, suggesting that CO is involved in O2 sensing [375] (Figure 7). NO can upregulate HO-1 expression thus increasing endogenous levels of CO, while CO can regulate iNOS expression and activity [376, 377]. As mentioned above, CO can also activate sGC leading to smooth muscle relaxation [378]. Based on their interrelationship and the similar roles of CO and NO, CO is speculated to substitute for NO under NO-deficient conditions and to regulate the bioavailability of NO.
Figure 7.

Crosstalk and interdependence of inorganic signaling molecules.
Whiteman et al. showed that in vitro incubation of sodium hydrosulfide, an H2S donor, with NO led to formation of RSNO [379]. On the other hand, H2S can reduce GSNO to release NO [380]. Eberhardt et al. proposed that NO and H2S can generate HNO resulting in sustained calcium flux, release of calcitonin gene-related peptide (CGRP) and in turn vasodilation [381]. Recently, H2S has been shown to inhibit iNOS expression via HO-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide (LPS) [331]. On the other hand, NO can bind tightly to the heme site of human CBS (Kd ≤ 0.23 μM, kon = 8 × 103 M−1 s−1 and koff = 0.003 s−1) and regulate its activity in vivo [382]. Similarly, CO can react with the heme protein CBS, which is involved in H2S production. Therefore, while CO may not play a direct role in biological processes, it has the ability to affect O2, NO and H2S dependent processes. Thus, crosstalk between these signaling agents can significantly impact physiological and pathophysiological processes.
8. Conclusions
The goal of this review is to provide an overview of biologically relevant reactions of the gaseous signaling molecules O2, CO, NO, H2S and H2 and their interaction with each other to impact cellular signaling. These molecules have overlapping molecular targets including metalloproteins and thiols, which may lead to direct as well as indirect physiological and patho-physiological effects by modulating bioavailability. The biological roles of these signaling molecules are also both concentration and O2-dependent. The implications of formation and regulation of these species in inflammatory pathways, cardiac pathophysiology and cancer biology is not fully understood. Further research to understand availability and interrelationships under physiological conditions may allow for development of novel therapeutic agents.
Highlight.
Small inorganic molecules constitute an important class of signaling agent.
Regulated biosynthesis and kinetic constraints control signaling by these species.
Redox interchange is a key factor in signaling by inorganic effector molecules.
Different classes of inorganic molecules can interact to influence signaling.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute and Center for Cancer Research.
Abbreviations
- ALDH
aldehyde dehydrogenase
- CGRP
calcitonin gene-related peptide
- CO
carbon monoxide
- COX
cyclooxygenase
- CBS
cystathionine β-synthase
- CSE
cystathionine γ-lyase
- P450
cytochrome P450
- N2O3
dinitrogen trioxide
- N2O4
dinitrogen tetraoxide
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- H2O2
hydrogen peroxide
- H2S
hydrogen sulfide
- ·OH
hydroxyl radical
- IPA/NO
isopropylamine diazeniumdiolate
- LPS
lipopolysaccharide
- LOX
lipoxygenase
- MST
3-mercaptopyruvate-S-transferase
- NOHA
Nω-hydroxy-L-arginine
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NO
nitric oxide
- NOS
nitric oxide synthase
- NO2
nitrogen dioxide
- HNO
nitroxyl
- PARP
poly(ADP-ribose) polymerase
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- STP
standard temperature and pressure
- O2−
superoxide
- SOD
superoxide dismutase
- HSNO
thionitrous acid
Footnotes
Dedication
This review is dedicated to Prof. Peter C. Ford whose contributions to inorganic chemistry include the areas of photochemistry, catalytic reactions and transition metal complexes. Prof. Ford’s research interests span catalytic conversion of biomass feedstocks to chemicals and fuels, analysis of reactions of coordinated nitrogen oxides that are generated in mammalian biology and photochemical delivery of small bioregulatory molecules to physiological targets [121, 219, 383, 384]. His work on transition metal complexes with NO, NO2− and CO has provided model compounds for biological systems and has extended our understanding of the involvement of critical inorganic molecules in physiology and disease.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schwartz SE. Wiley; New York: 1983. [Google Scholar]
- 2.Williams DLH. Nitrosation. Cambridge University Press; Cambridge ; New York: 1988. [Google Scholar]
- 3.Williams DLH. Nitrosation reactions and the chemistry of nitric oxide. 1. Elsevier; Amsterdam ; Boston: 2004. [Google Scholar]
- 4.United States. National institutes of health. Division of industrial hygiene [from old catalog] Hydrogen sulfide: its toxicity and potential dangers. U. S. Govt. print. off; Washington: 1941. [Google Scholar]
- 5.Dwyer B. Carbon Monoxide: A clear and present danger. 3. Esco Press; Mount Prospect, Ill: 2003. [Google Scholar]
- 6.Hibbs JB, Jr, Taintor RR, Vavrin Z, Rachlin EM. Biochem Biophys Res Commun. 1988;157:87–94. doi: 10.1016/s0006-291x(88)80015-9. [DOI] [PubMed] [Google Scholar]
- 7.Miwa M, Stuehr DJ, Marletta MA, Wishnok JS, Tannenbaum SR. Carcinogenesis. 1987;8:955–958. doi: 10.1093/carcin/8.7.955. [DOI] [PubMed] [Google Scholar]
- 8.Ignarro LJ. Biochem Pharmacol. 1991;41:485–490. doi: 10.1016/0006-2952(91)90618-f. [DOI] [PubMed] [Google Scholar]
- 9.Ignarro LJ. Thromb Haemost. 1993;70:148–151. [PubMed] [Google Scholar]
- 10.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Proc Natl Acad Sci U S A. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradbury J. Lancet. 1998;352:1287. doi: 10.1016/S0140-6736(05)70495-5. [DOI] [PubMed] [Google Scholar]
- 12.Wink DA, Hines HB, Cheng RY, Switzer CH, Flores-Santana W, Vitek MP, Ridnour LA, Colton CA. J Leukocyte Biol. 2011;89:873–891. doi: 10.1189/jlb.1010550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kajimura M, Fukuda R, Bateman RM, Yamamoto T, Suematsu M. Antioxid Redox Signal. 2010;13:157–192. doi: 10.1089/ars.2009.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryter SW, Otterbein LE. Bioessays. 2004;26:270–280. doi: 10.1002/bies.20005. [DOI] [PubMed] [Google Scholar]
- 15.Paolocci N, Katori T, Champion HC, St John ME, Miranda KM, Fukuto JM, Wink DA, Kass DA. Proc Natl Acad Sci U S A. 2003;100:5537–5542. doi: 10.1073/pnas.0937302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagasawa HT, DeMaster EG, Redfern B, Shirota FN, Goon DJ. J Med Chem. 1990;33:3120–3122. doi: 10.1021/jm00174a001. [DOI] [PubMed] [Google Scholar]
- 17.Norris AJ, Sartippour MR, Lu M, Park T, Rao JY, Jackson MI, Fukuto JM, Brooks MN. Int J Cancer. 2008;122:1905–1910. doi: 10.1002/ijc.23305. [DOI] [PubMed] [Google Scholar]
- 18.Miranda KM, Paolocci N, Katori T, Thomas DD, Ford E, Bartberger MD, Espey MG, Kass DA, Feelisch M, Fukuto JM, Wink DA. Proc Natl Acad Sci U S A. 2003;100:9196–9201. doi: 10.1073/pnas.1430507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basudhar D, Cheng RC, Bharadwaj G, Ridnour LA, Wink DA, Miranda KM. Free Radic Biol Med. 2015;83:101–114. doi: 10.1016/j.freeradbiomed.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harman D. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 21.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 22.Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 23.Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, Switzer CH, Donzelli S, Hussain P, Vecoli C, Paolocci N, Ambs S, Colton CA, Harris CC, Roberts DD, Wink DA. Free Radic Biol Med. 2008;45:18–31. doi: 10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forman HJ, Torres M, Fukuto J. Mol Cell Biochem. 2002;234–235:49–62. [PubMed] [Google Scholar]
- 25.Kabil O, Motl N, Banerjee R. Biochim Biophys Acta. 2014;1844:1355–1366. doi: 10.1016/j.bbapap.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi AM, Otterbein LE. Antioxid Redox Signal. 2002;4:227–228. doi: 10.1089/152308602753666271. [DOI] [PubMed] [Google Scholar]
- 27.Cadenas E, Sies H. Adv Enzyme Regul. 1985;23:217–237. doi: 10.1016/0065-2571(85)90049-4. [DOI] [PubMed] [Google Scholar]
- 28.Costa VM, Carvalho F, Bastos ML, Carvalho RA, Carvalho M, Remiao F. Curr Med Chem. 2011;18:2272–2314. doi: 10.2174/092986711795656081. [DOI] [PubMed] [Google Scholar]
- 29.Lambeth JD. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 30.Brand MD. Exp Gerontol. 2010;45:466–472. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bindoli A, Rigobello MP. Antioxid Redox Signal. 2013;18:1557–1593. doi: 10.1089/ars.2012.4655. [DOI] [PubMed] [Google Scholar]
- 32.Finkel T. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marro PJ, Baumgart S, Delivoria-Papadopoulos M, Zirin S, Corcoran L, McGaurn SP, Davis LE, Clancy RR. Pediatr Res. 1997;41:513–520. doi: 10.1203/00006450-199704000-00010. [DOI] [PubMed] [Google Scholar]
- 34.McCord JM, Fridovich I. J Biol Chem. 1968;243:5753–5760. [PubMed] [Google Scholar]
- 35.Al Ghouleh I, Khoo NK, Knaus UG, Griendling KK, Touyz RM, Thannickal VJ, Barchowsky A, Nauseef WM, Kelley EE, Bauer PM, Darley-Usmar V, Shiva S, Cifuentes-Pagano E, Freeman BA, Gladwin MT, Pagano PJ. Free Radic Biol Med. 2011;51:1271–1288. doi: 10.1016/j.freeradbiomed.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geiszt M, Leto TL. J Biol Chem. 2004;279:51715–51718. doi: 10.1074/jbc.R400024200. [DOI] [PubMed] [Google Scholar]
- 37.Bedard K, Krause KH. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 38.Brown DI, Griendling KK. Free Radic Biol Med. 2009;47:1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pou S, Pou WS, Bredt DS, Snyder SH, Rosen GM. J Biol Chem. 1992;267:24173–24176. [PubMed] [Google Scholar]
- 41.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA., Jr Proc Natl Acad Sci U S A. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cosentino F, Hishikawa K, Katusic ZS, Luscher TF. Circulation. 1997;96:25–28. doi: 10.1161/01.cir.96.1.25. [DOI] [PubMed] [Google Scholar]
- 43.Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Circulation. 2002;105:1656–1662. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- 44.Wang Q, Yang M, Xu H, Yu J. Evid Based Complement Alternat Med. 2014;850312:4. doi: 10.1155/2014/850312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Z, Ming XF. Clin Med Res. 2006;4:53–65. doi: 10.3121/cmr.4.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Werner ER, Werner-Felmayer G, Mayer B. Proc Soc Exp Biol Med. 1998;219:171–182. doi: 10.3181/00379727-219-44331. [DOI] [PubMed] [Google Scholar]
- 47.Gross SS, Levi R. J Biol Chem. 1992;267:25722–25729. [PubMed] [Google Scholar]
- 48.Bendall JK, Douglas G, McNeill E, Channon KM, Crabtree MJ. Antioxid Redox Signal. 2014;20:3040–3077. doi: 10.1089/ars.2013.5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ortiz de Montellano PR, De Voss JJ. Nat Prod Rep. 2002;19:477–493. doi: 10.1039/b101297p. [DOI] [PubMed] [Google Scholar]
- 50.Zangar RC, Davydov DR, Verma S. Toxicol Appl Pharmacol. 2004;199:316–331. doi: 10.1016/j.taap.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 51.Lynch DV, Thompson JE. FEBS Lett. 1984;173:251–254. [Google Scholar]
- 52.Zuo L, Christofi FL, Wright VP, Bao S, Clanton TL. J Appl Physiol. 1985;97:661–668. doi: 10.1152/japplphysiol.00096.2004. [DOI] [PubMed] [Google Scholar]
- 53.Xu X, Gao X, Potter BJ, Cao JM, Zhang C. Atertio Thromb Vasc Biol. 2007;27:871–877. doi: 10.1161/01.ATV.0000259358.31234.37. [DOI] [PubMed] [Google Scholar]
- 54.Lubrano V, Balzan S. Free Radical Res. 2014;48:841–848. doi: 10.3109/10715762.2014.929122. [DOI] [PubMed] [Google Scholar]
- 55.Armstead WM. Anesthesiology. 2003;98:1378–1383. doi: 10.1097/00000542-200306000-00012. [DOI] [PubMed] [Google Scholar]
- 56.Armstead WM. J Neurotrauma. 2001;18:1039–1048. doi: 10.1089/08977150152693737. [DOI] [PubMed] [Google Scholar]
- 57.Hamilton GA. Molecular mechanisms of oxygen activation. In: Hayaishi O, editor. Mol Biol. Academic Press; New York: 1974. p. xvi.p. 678. [Google Scholar]
- 58.Bromberg Y, Pick E. Cell Immunol. 1983;79:240–252. doi: 10.1016/0008-8749(83)90067-9. [DOI] [PubMed] [Google Scholar]
- 59.Kukreja RC, Kontos HA, Hess ML, Ellis EF. Circul Res. 1986;59:612–619. doi: 10.1161/01.res.59.6.612. [DOI] [PubMed] [Google Scholar]
- 60.Haber F, Weiss J. Proceedings of the Royal Society of London A: Mathematical, Physical and Engineering Sciences. 1934;147:332–351. [Google Scholar]
- 61.Song Y, Buettner GR. Free Radic Biol Med. 2010;49:919–962. doi: 10.1016/j.freeradbiomed.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Som S, Raha C, Chatterjee IB. Acta Vitaminol Enzymol. 1983;5:243–250. [PubMed] [Google Scholar]
- 63.Winterbourn CC, Metodiewa D. Free Radic Biol Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 64.Winterbourn CC, Metodiewa D. Methods Enzymol. 1995;251:81–86. doi: 10.1016/0076-6879(95)51112-1. [DOI] [PubMed] [Google Scholar]
- 65.Thomas EL, Learn DB, Jefferson MM, Weatherred W. J Biol Chem. 1988;263:2178–2186. [PubMed] [Google Scholar]
- 66.Thannickal VJ, Fanburg BL. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 67.Kelley EE, Khoo NK, Hundley NJ, Malik UZ, Freeman BA, Tarpey MM. Free Radic Biol Med. 2010;48:493–498. doi: 10.1016/j.freeradbiomed.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dupuy C, Virion A, Ohayon R, Kaniewski J, Deme D, Pommier J. J Biol Chem. 1991;266:3739–3743. [PubMed] [Google Scholar]
- 69.Thorpe C, Hoober KL, Raje S, Glynn NM, Burnside J, Turi GK, Coppock DL. Arch Biochem Biophys. 2002;405:1–12. doi: 10.1016/s0003-9861(02)00337-5. [DOI] [PubMed] [Google Scholar]
- 70.Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. 4. Oxford University Press; Oxford ; New York: 2007. [Google Scholar]
- 71.Fenton HJH, Jones HO. J Chem Soc. 1900;77:69–76. [Google Scholar]
- 72.Haber F, Weiss J. Naturwissenschaften. 1932;20:948–950. [Google Scholar]
- 73.Goldstein S, Meyerstein D, Czapski G. Free Radic Biol Med. 1993;15:435–445. doi: 10.1016/0891-5849(93)90043-t. [DOI] [PubMed] [Google Scholar]
- 74.Halliwell B. Plant Physiol. 2006;141:312–322. doi: 10.1104/pp.106.077073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bray WC, Gorin MH. J Am Chem Soc. 1932;54:2124–2125. [Google Scholar]
- 76.Cahill AE, Taube H. J Am Chem Soc. 1952;74:2312–2318. [Google Scholar]
- 77.Koppenol WH. Redox Rep. 2001;6:229–234. doi: 10.1179/135100001101536373. [DOI] [PubMed] [Google Scholar]
- 78.Koppenol WH. Free Radic Biol Med. 1993;15:645–651. doi: 10.1016/0891-5849(93)90168-t. [DOI] [PubMed] [Google Scholar]
- 79.Yamamoto N, Koga N, Nagaoka M. J Phys Chem B. 2012;116:14178–14182. doi: 10.1021/jp310008z. [DOI] [PubMed] [Google Scholar]
- 80.Wink DA, Nims RW, Saavedra JE, Utermahlen WE, Jr, Ford PC. Proc Natl Acad Sci U S A. 1994;91:6604–6608. doi: 10.1073/pnas.91.14.6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wink DA, Nims RW, Desrosiers MF, Ford PC, Keefer LK. Chem Res Toxicol. 1991;4:510–512. doi: 10.1021/tx00023a002. [DOI] [PubMed] [Google Scholar]
- 82.Wink DA, Wink CB, Nims RW, Ford PC. Environ Health Perspect. 1994;102(Suppl 3):11–15. doi: 10.1289/ehp.94102s311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Drago RS, Corden BB, Barnes CW. J Am Chem Soc. 1986;108:2453–2454. doi: 10.1021/ja00269a057. [DOI] [PubMed] [Google Scholar]
- 84.Carballal S, Radi R, Kirk MC, Barnes S, Freeman BA, Alvarez B. Biochemistry (Mosc) 2003;42:9906–9914. doi: 10.1021/bi027434m. [DOI] [PubMed] [Google Scholar]
- 85.You KS, Benitez LV, McConachie WA, Allison WS. Biochim Biophys Acta. 1975;384:317–330. doi: 10.1016/0005-2744(75)90033-9. [DOI] [PubMed] [Google Scholar]
- 86.Poole LB, Karplus PA, Claiborne A. Annu Rev Pharmacool Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 87.Klomsiri C, Karplus PA, Poole LB. Antioxid Redox Signal. 2011;14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chance B. J Biol Chem. 1949;179:1311–1330. [PubMed] [Google Scholar]
- 89.Chance B. Arch Biochem. 1949;22:224–252. [PubMed] [Google Scholar]
- 90.Chance B. Arch Biochem. 1949;21:416–430. [PubMed] [Google Scholar]
- 91.Chance B, Herbert D. Biochem J. 1950;46:402–414. doi: 10.1042/bj0460402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rhee SG, Kang SW, Netto LE, Seo MS, Stadtman ER. BioFactors. 1999;10:207–209. doi: 10.1002/biof.5520100218. [DOI] [PubMed] [Google Scholar]
- 93.Hofmann B, Hecht HJ, Flohe L. Biol Chem. 2002;383:347–364. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- 94.Wood ZA, Schroder E, Robin Harris J, Poole LB. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 95.Brigelius-Flohe R. Free Radic Biol Med. 1999;27:951–965. doi: 10.1016/s0891-5849(99)00173-2. [DOI] [PubMed] [Google Scholar]
- 96.Ursini F, Maiorino M, Brigelius-Flohe R, Aumann KD, Roveri A, Schomburg D, Flohe L. Methods Enzymol. 1995;252:38–53. doi: 10.1016/0076-6879(95)52007-4. [DOI] [PubMed] [Google Scholar]
- 97.Bienert GP, Moller AL, Kristiansen KA, Schulz A, Moller IM, Schjoerring JK, Jahn TP. J Biol Chem. 2007;282:1183–1192. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- 98.Lee WK, Thevenod F. Am J Physiol, Cell Physiol. 2006;291:C195–202. doi: 10.1152/ajpcell.00641.2005. [DOI] [PubMed] [Google Scholar]
- 99.Paulsen CE, Carroll KS. Chem Rev. 2013;113:4633–4679. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schieber M, Chandel NS. Curr Biol. 2014;24:034. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Forman HJ, Maiorino M, Ursini F. Biochemistry (Mosc) 2010;49:835–842. doi: 10.1021/bi9020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Veal EA, Day AM, Morgan BA. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 103.Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ., 2nd Proc Natl Acad Sci U S A. 1990;87:9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Milne GL, Yin H, Hardy KD, Davies SS, Roberts LJ. Chem Rev. 2011;111:5973–5996. doi: 10.1021/cr200160h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Benedetti A, Comporti M, Esterbauer H. Biochim Biophys Acta. 1980;620:281–296. doi: 10.1016/0005-2760(80)90209-x. [DOI] [PubMed] [Google Scholar]
- 106.Montuschi P, Barnes PJ, Roberts LJ., 2nd FASEB J. 2004;18:1791–1800. doi: 10.1096/fj.04-2330rev. [DOI] [PubMed] [Google Scholar]
- 107.Grimsrud PA, Xie H, Griffin TJ, Bernlohr DA. J Biol Chem. 2008;283:21837–21841. doi: 10.1074/jbc.R700019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mohler ER, Franklin MT, Adam LP. Biochem Biophys Res Commun. 1996;225:915–923. doi: 10.1006/bbrc.1996.1272. [DOI] [PubMed] [Google Scholar]
- 109.Parola M, Robino G, Marra F, Pinzani M, Bellomo G, Leonarduzzi G, Chiarugi P, Camandola S, Poli G, Waeg G, Gentilini P, Dianzani MU. J Clin Invest. 1998;102:1942–1950. doi: 10.1172/JCI1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marnett LJ, Riggins JN, West JD. J Clin Invest. 2003;111:583–593. doi: 10.1172/JCI18022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Marletta MA. J Biol Chem. 1993;268:12231–12234. [PubMed] [Google Scholar]
- 112.Modin A, Bjorne H, Herulf M, Alving K, Weitzberg E, Lundberg JO. Acta Physiol Scand. 2001;171:9–16. doi: 10.1046/j.1365-201X.2001.00771.x. [DOI] [PubMed] [Google Scholar]
- 113.Wink DA, Grisham MB, Mitchell JB, Ford PC. Methods Enzymol. 1996;268:12–31. doi: 10.1016/s0076-6879(96)68006-9. [DOI] [PubMed] [Google Scholar]
- 114.Wink DA, Hanbauer I, Grisham MB, Laval F, Nims RW, Laval J, Cook J, Pacelli R, Liebmann J, Krishna M, Ford PC, Mitchell JB. Curr Top Cell Regul. 1996;34:159–187. doi: 10.1016/s0070-2137(96)80006-9. [DOI] [PubMed] [Google Scholar]
- 115.Wink DA, Mitchell JB. Free Radic Biol Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 116.Moncada S, Higgs EA. Eur J Clin Invest. 1991;21:361–374. doi: 10.1111/j.1365-2362.1991.tb01383.x. [DOI] [PubMed] [Google Scholar]
- 117.Stone JR, Marletta MA. Biochemistry (Mosc) 1996;35:1093–1099. doi: 10.1021/bi9519718. [DOI] [PubMed] [Google Scholar]
- 118.Zhao Y, Schelvis JP, Babcock GT, Marletta MA. Biochemistry (Mosc) 1998;37:4502–4509. doi: 10.1021/bi972686m. [DOI] [PubMed] [Google Scholar]
- 119.Stone JR, Marletta MA. Biochemistry (Mosc) 1994;33:5636–5640. doi: 10.1021/bi00184a036. [DOI] [PubMed] [Google Scholar]
- 120.Stone JR, Sands RH, Dunham WR, Marletta MA. Biochem Biophys Res Commun. 1995;207:572–577. doi: 10.1006/bbrc.1995.1226. [DOI] [PubMed] [Google Scholar]
- 121.Ford PC, Lorkovic IM. Chem Rev. 2002;102:993–1018. doi: 10.1021/cr0000271. [DOI] [PubMed] [Google Scholar]
- 122.Derbyshire ER, Marletta MA. Annu Rev Biochem. 2012;81:533–559. doi: 10.1146/annurev-biochem-050410-100030. [DOI] [PubMed] [Google Scholar]
- 123.Cary SP, Winger JA, Marletta MA. Proc Natl Acad Sci U S A. 2005;102:13064–13069. doi: 10.1073/pnas.0506289102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cary SP, Winger JA, Derbyshire ER, Marletta MA. Trends Biochem Sci. 2006;31:231–239. doi: 10.1016/j.tibs.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 125.Tsai AL, Berka V, Sharina I, Martin E. J Biol Chem. 2011;286:43182–43192. doi: 10.1074/jbc.M111.290304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ford PC. Inorg Chem. 2010;49:6226–6239. doi: 10.1021/ic902073z. [DOI] [PubMed] [Google Scholar]
- 127.Flores-Santana W, Switzer C, Ridnour LA, Basudhar D, Mancardi D, Donzelli S, Thomas DD, Miranda KM, Fukuto JM, Wink DA. Arch Pharmacal Res. 2009;32:1139–1153. doi: 10.1007/s12272-009-1805-x. [DOI] [PubMed] [Google Scholar]
- 128.Eich RF, Li T, Lemon DD, Doherty DH, Curry SR, Aitken JF, Mathews AJ, Johnson KA, Smith RD, Phillips GN, Jr, Olson JS. Biochemistry (Mosc) 1996;35:6976–6983. doi: 10.1021/bi960442g. [DOI] [PubMed] [Google Scholar]
- 129.Lancaster JR., Jr Nitric Oxide. 1997;1:18–30. doi: 10.1006/niox.1996.0112. [DOI] [PubMed] [Google Scholar]
- 130.Gorbunov NV, Osipov AN, Day BW, Zayas-Rivera B, Kagan VE, Elsayed NM. Biochemistry (Mosc) 1995;34:6689–6699. doi: 10.1021/bi00020a014. [DOI] [PubMed] [Google Scholar]
- 131.Kanner J, Harel S, Granit R. Arch Biochem Biophys. 1991;289:130–136. doi: 10.1016/0003-9861(91)90452-o. [DOI] [PubMed] [Google Scholar]
- 132.Ouellet H, Lang J, Couture M, Ortiz de Montellano PR. Biochemistry (Mosc) 2009;48:863–872. doi: 10.1021/bi801595t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Laverman LE, Wanat A, Oszajca J, Stochel G, Ford PC, van Eldik R. J Am Chem Soc. 2001;123:285–293. doi: 10.1021/ja001696z. [DOI] [PubMed] [Google Scholar]
- 134.Gruetter CA, Barry BK, McNamara DB, Gruetter DY, Kadowitz PJ, Ignarro LJ. J Cyclic Nucleotide Res. 1979;5:211–224. [PubMed] [Google Scholar]
- 135.Katsuki S, Arnold W, Mittal C, Murad F. J Cyclic Nucleotide Res. 1977;3:23–35. [PubMed] [Google Scholar]
- 136.Stuehr DJ, Nathan CF. J Exp Med. 1989;169:1543–1555. doi: 10.1084/jem.169.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hibbs JB, Jr, Westenfelder C, Taintor R, Vavrin Z, Kablitz C, Baranowski RL, Ward JH, Menlove RL, McMurry MP, Kushner JP, et al. J Clin Invest. 1992;89:867–877. doi: 10.1172/JCI115666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hibbs JB, Jr, Taintor RR, Vavrin Z. Science. 1987;235:473–476. doi: 10.1126/science.2432665. [DOI] [PubMed] [Google Scholar]
- 139.Leaf CD, Wishnok JS, Tannenbaum SR. Carcinogenesis. 1991;12:537–539. doi: 10.1093/carcin/12.3.537. [DOI] [PubMed] [Google Scholar]
- 140.Ridnour LA, Thomas DD, Mancardi D, Espey MG, Miranda KM, Paolocci N, Feelisch M, Fukuto J, Wink DA. Biol Chem. 2004;385:1–10. doi: 10.1515/BC.2004.001. [DOI] [PubMed] [Google Scholar]
- 141.Gow A, Duran D, Thom SR, Ischiropoulos H. Arch Biochem Biophys. 1996;333:42–48. doi: 10.1006/abbi.1996.0362. [DOI] [PubMed] [Google Scholar]
- 142.Wink DA, Darbyshire JF, Nims RW, Saavedra JE, Ford PC. Chem Res Toxicol. 1993;6:23–27. doi: 10.1021/tx00031a003. [DOI] [PubMed] [Google Scholar]
- 143.Grätzel M, Taniguchi S, Henglein A. Ber Bunsenges Phys Chem. 1970;74:488–492. [Google Scholar]
- 144.Tannenbaum SR, Weisman M, Fett D. Food Cosmet Toxicol. 1976;14:549–552. doi: 10.1016/s0015-6264(76)80006-5. [DOI] [PubMed] [Google Scholar]
- 145.Tannenbaum SR, Moran D, Falchuk KR, Correa P, Cuello C. Cancer Lett. 1981;14:131–136. doi: 10.1016/0304-3835(81)90122-1. [DOI] [PubMed] [Google Scholar]
- 146.Duncan C, Dougall H, Johnston P, Green S, Brogan R, Leifert C, Smith L, Golden M, Benjamin N. Nat Med. 1995;1:546–551. doi: 10.1038/nm0695-546. [DOI] [PubMed] [Google Scholar]
- 147.Chan J, Xing Y, Magliozzo RS, Bloom BR. J Exp Med. 1992;175:1111–1122. doi: 10.1084/jem.175.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yu K, Mitchell C, Xing Y, Magliozzo RS, Bloom BR, Chan J. Tuber Lung Dis. 1999;79:191–198. doi: 10.1054/tuld.1998.0203. [DOI] [PubMed] [Google Scholar]
- 149.Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, van der Vliet A. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 150.van der Vliet A, Eiserich JP, Halliwell B, Cross CE. J Biol Chem. 1997;272:7617–7625. doi: 10.1074/jbc.272.12.7617. [DOI] [PubMed] [Google Scholar]
- 151.Gaut JP, Byun J, Tran HD, Lauber WM, Carroll JA, Hotchkiss RS, Belaaouaj A, Heinecke JW. J Clin Invest. 2002;109:1311–1319. doi: 10.1172/JCI15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sampson JB, Ye Y, Rosen H, Beckman JS. Arch Biochem Biophys. 1998;356:207–213. doi: 10.1006/abbi.1998.0772. [DOI] [PubMed] [Google Scholar]
- 153.Viner RI, Ferrington DA, Williams TD, Bigelow DJ, Schoneich C. Biochem J. 1999;340:657–669. [PMC free article] [PubMed] [Google Scholar]
- 154.Gole MD, Souza JM, Choi I, Hertkorn C, Malcolm S, Foust RF, 3rd, Finkel B, Lanken PN, Ischiropoulos H. Am J Physiol Lung Cell Mol Physiol. 2000;278:L961–967. doi: 10.1152/ajplung.2000.278.5.L961. [DOI] [PubMed] [Google Scholar]
- 155.Souza JM, Peluffo G, Radi R. Free Radic Biol Med. 2008;45:357–366. doi: 10.1016/j.freeradbiomed.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 156.Radi R. Acc Chem Res. 2013;46:550–559. doi: 10.1021/ar300234c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Stillwell WG, Xu HX, Glogowski J, Wishnok JS, Tannenbaum SR, Correa P. IARC Sci Publ. 1991;105:83–87. [PubMed] [Google Scholar]
- 158.Wishnok JS, Tannenbaum SR, Stillwell WG, Glogowski JA, Leaf CD. Environ Health Perspect. 1993;99:155–159. doi: 10.1289/ehp.9399155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Marshall HE, Merchant K, Stamler JS. FASEB J. 2000;14:1889–1900. doi: 10.1096/fj.00.011rev. [DOI] [PubMed] [Google Scholar]
- 160.Lima B, Forrester MT, Hess DT, Stamler JS. Circ Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sidorkina O, Espey MG, Miranda KM, Wink DA, Laval J. Free Radic Biol Med. 2003;35:1431–1438. doi: 10.1016/j.freeradbiomed.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 162.Wink DA, Vodovotz Y, Laval J, Laval F, Dewhirst MW, Mitchell JB. Carcinogenesis. 1998;19:711–721. doi: 10.1093/carcin/19.5.711. [DOI] [PubMed] [Google Scholar]
- 163.Laval F, Wink DA, Laval J. Rev Physiol Biochem Pharmacol. 1997;131:175–191. doi: 10.1007/3-540-61992-5_8. [DOI] [PubMed] [Google Scholar]
- 164.Graziewicz M, Wink DA, Laval F. Carcinogenesis. 1996;17:2501–2505. doi: 10.1093/carcin/17.11.2501. [DOI] [PubMed] [Google Scholar]
- 165.Kroncke KD, Fehsel K, Schmidt T, Zenke FT, Dasting I, Wesener JR, Bettermann H, Breunig KD, Kolb-Bachofen V. Biochem Biophys Res Commun. 1994;200:1105–1110. doi: 10.1006/bbrc.1994.1564. [DOI] [PubMed] [Google Scholar]
- 166.Wink DA, Laval J. Carcinogenesis. 1994;15:2125–2129. doi: 10.1093/carcin/15.10.2125. [DOI] [PubMed] [Google Scholar]
- 167.Switzer CH, Glynn SA, Cheng RYS, Ridnour LA, Green JE, Ambs S, Wink DA. Mol Cancer Res. 2012;10:1203–1215. doi: 10.1158/1541-7786.MCR-12-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Anand P, Stamler JS. J Mol Med. 2012;90:233–244. doi: 10.1007/s00109-012-0878-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Maron BA, Tang SS, Loscalzo J. Antioxid Redox Signal. 2013;18:270–287. doi: 10.1089/ars.2012.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Smith BC, Marletta MA. Curr Opin Chem Biol. 2012;16:498–506. doi: 10.1016/j.cbpa.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hogg N. Annu Rev Pharmacool Toxicol. 2002;42:585–600. doi: 10.1146/annurev.pharmtox.42.092501.104328. [DOI] [PubMed] [Google Scholar]
- 172.Williams DLH. Acc Chem Res. 1999;32:869–876. [Google Scholar]
- 173.Broniowska KA, Hogg N. Antioxid Redox Signal. 2012;17:969–980. doi: 10.1089/ars.2012.4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Huie RE, Padmaja S. Free Radic Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 175.Pryor WA, Jin X, Squadrito GL. Proc Natl Acad Sci U S A. 1994;91:11173–11177. doi: 10.1073/pnas.91.23.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kissner R, Nauser T, Bugnon P, Lye PG, Koppenol WH. Chem Res Toxicol. 1997;10:1285–1292. doi: 10.1021/tx970160x. [DOI] [PubMed] [Google Scholar]
- 177.Szabo C, Ischiropoulos H, Radi R. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 178.Koppenol WH, Moreno JJ, Pryor WA, Ischiropoulos H, Beckman JS. Chem Res Toxicol. 1992;5:834–842. doi: 10.1021/tx00030a017. [DOI] [PubMed] [Google Scholar]
- 179.Denicola A, Freeman BA, Trujillo M, Radi R. Arch Biochem Biophys. 1996;333:49–58. doi: 10.1006/abbi.1996.0363. [DOI] [PubMed] [Google Scholar]
- 180.Pleskova M, Beck KF, Behrens MH, Huwiler A, Fichtlscherer B, Wingerter O, Brandes RP, Mulsch A, Pfeilschifter J. FASEB J. 2006;20:139–141. doi: 10.1096/fj.05-3791fje. [DOI] [PubMed] [Google Scholar]
- 181.Selemidis S, Dusting GJ, Peshavariya H, Kemp-Harper BK, Drummond GR. Cardiovasc Res. 2007;75:349–358. doi: 10.1016/j.cardiores.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 182.Qian J, Chen F, Kovalenkov Y, Pandey D, Moseley MA, Foster MW, Black SM, Venema RC, Stepp DW, Fulton DJR. Free Radic Biol Med. 2012;52:1806–1819. doi: 10.1016/j.freeradbiomed.2012.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Brune B. Antioxid Redox Signal. 2005;7:497–507. doi: 10.1089/ars.2005.7.497. [DOI] [PubMed] [Google Scholar]
- 184.Thomas DD, Ridnour LA, Espey MG, Donzelli S, Ambs S, Hussain SP, Harris CC, DeGraff W, Roberts DD, Mitchell JB, Wink DA. J Biol Chem. 2006;281:25984–25993. doi: 10.1074/jbc.M602242200. [DOI] [PubMed] [Google Scholar]
- 185.Wink DA, Vodovotz Y, Grisham MB, DeGraff W, Cook JC, Pacelli R, Krishna M, Mitchell JB. Methods Enzymol. 1999;301:413–424. doi: 10.1016/s0076-6879(99)01105-2. [DOI] [PubMed] [Google Scholar]
- 186.Ma XL, Gao F, Liu GL, Lopez BL, Christopher TA, Fukuto JM, Wink DA, Feelisch M. Proc Natl Acad Sci U S A. 1999;96:14617–14622. doi: 10.1073/pnas.96.25.14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mason RB, Pluta RM, Walbridge S, Wink DA, Oldfield EH, Boock RJ. J Neurosurg. 2000;93:99–107. doi: 10.3171/jns.2000.93.1.0099. [DOI] [PubMed] [Google Scholar]
- 188.Ridnour LA, Thomas DD, Donzelli S, Espey MG, Roberts DD, Wink DA, Isenberg JS. Antioxid Redox Signal. 2006;8:1329–1337. doi: 10.1089/ars.2006.8.1329. [DOI] [PubMed] [Google Scholar]
- 189.Heinecke JL, Ridnour LA, Cheng RY, Switzer CH, Lizardo MM, Khanna C, Glynn SA, Hussain SP, Young HA, Ambs S, Wink DA. Proc Natl Acad Sci U S A. 2014;111:6323–6328. doi: 10.1073/pnas.1401799111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Glynn SA, Boersma BJ, Dorsey TH, Yi M, Yfantis HG, Ridnour LA, Martin DN, Switzer CH, Hudson RS, Wink DA, Lee DH, Stephens RM, Ambs S. J Clin Invest. 2010;120:3843–3854. doi: 10.1172/JCI42059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ridnour LA, Windhausen AN, Isenberg JS, Yeung N, Thomas DD, Vitek MP, Roberts DD, Wink DA. Proc Natl Acad Sci U S A. 2007;104:16898–16903. doi: 10.1073/pnas.0702761104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Switzer CH, Cheng RY, Ridnour LA, Glynn SA, Ambs S, Wink DA. Breast Cancer Res. 2012;14:R125. doi: 10.1186/bcr3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Switzer CH, Glynn SA, Cheng RY, Ridnour LA, Green JE, Ambs S, Wink DA. Mol Cancer Res. 2012;10:1203–1215. doi: 10.1158/1541-7786.MCR-12-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Switzer CH, Ridnour LA, Cheng R, Heinecke J, Burke A, Glynn S, Ambs S, Wink DA. For Immunopathol Dis Therap. 2012;3:117–124. doi: 10.1615/ForumImmunDisTher.2012006108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Patruno A, Pesce M, Marrone A, Speranza L, Grilli A, De Lutiis MA, Felaco M, Reale M. J Cell Physiol. 2012;227:2767–2774. doi: 10.1002/jcp.23024. [DOI] [PubMed] [Google Scholar]
- 196.Ridnour LA, Barasch KM, Windhausen AN, Dorsey TH, Lizardo MM, Yfantis HG, Lee DH, Switzer CH, Cheng RY, Heinecke JL, Brueggemann E, Hines HB, Khanna C, Glynn SA, Ambs S, Wink DA. PloS one. 2012;7:e44081. doi: 10.1371/journal.pone.0044081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Thatcher GR. Curr Top Med Chem. 2005;5:597–601. doi: 10.2174/1568026054679281. [DOI] [PubMed] [Google Scholar]
- 198.Thomas DD, Miranda KM, Espey MG, Citrin D, Jourd’Heuil D, Paolocci N, Hewett SJ, Colton CA, Grisham MB, Feelisch M, Wink DA. Nitric Oxide. 2002;359:84–105. doi: 10.1016/s0076-6879(02)59174-6. [DOI] [PubMed] [Google Scholar]
- 199.Thomas DD, Espey MG, Ridnour LA, Hofseth LJ, Mancardi D, Harris CC, Wink DA. Proc Natl Acad Sci U S A. 2004;101:8894–8899. doi: 10.1073/pnas.0400453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Davies KM, Wink DA, Saavedra JE, Keefer LK. J Am Chem Soc. 2001;123:5473–5481. doi: 10.1021/ja002899q. [DOI] [PubMed] [Google Scholar]
- 201.Maciag AE, Saavedra JE, Chakrapani H. Anticancer Agents Med Chem. 2009;9:798–803. doi: 10.2174/187152009789056949. [DOI] [PubMed] [Google Scholar]
- 202.Friedman AJ, Han G, Navati MS, Chacko M, Gunther L, Alfieri A, Friedman JM. Nitric Oxide. 2008;19:12–20. doi: 10.1016/j.niox.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 203.Cabrales P, Han G, Roche C, Nacharaju P, Friedman AJ, Friedman JM. Free Radic Biol Med. 2010;49:530–538. doi: 10.1016/j.freeradbiomed.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Riccio DA, Schoenfisch MH. Chem Soc Rev. 2012;41:3731–3741. doi: 10.1039/c2cs15272j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ford PC. Nitric Oxide. 2013;34:56–64. doi: 10.1016/j.niox.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 206.Tfouni E, Krieger M, McGarvey BR, Franco DW. Coord Chem Rev. 2003;236:57–69. [Google Scholar]
- 207.Conrado CL, Wecksler S, Egler C, Magde D, Ford PC. Inorg Chem. 2004;43:5543–5549. doi: 10.1021/ic049459a. [DOI] [PubMed] [Google Scholar]
- 208.Burks PT, Garcia JV, GonzalezIrias R, Tillman JT, Niu M, Mikhailovsky AA, Zhang J, Zhang F, Ford PC. J Am Chem Soc. 2013;135:18145–18152. doi: 10.1021/ja408516w. [DOI] [PubMed] [Google Scholar]
- 209.Gladwin MT, Raat NJ, Shiva S, Dezfulian C, Hogg N, Kim-Shapiro DB, Patel RP. Am J Physiol Heart Circ Physiol. 2006;291:H2026–2035. doi: 10.1152/ajpheart.00407.2006. [DOI] [PubMed] [Google Scholar]
- 210.Gladwin MT. J Clin Invest. 2004;113:19–21. doi: 10.1172/JCI200420664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Doyle MP, Pickering RA, DeWeert TM, Hoekstra JW, Pater D. J Biol Chem. 1981;256:12393–12398. [PubMed] [Google Scholar]
- 212.Wink DA. Nat Med. 2003;9:1460–1461. doi: 10.1038/nm1203-1460. [DOI] [PubMed] [Google Scholar]
- 213.Lundberg JO, Gladwin MT, Ahluwalia A, Benjamin N, Bryan NS, Butler A, Cabrales P, Fago A, Feelisch M, Ford PC, Freeman BA, Frenneaux M, Friedman J, Kelm M, Kevil CG, Kim-Shapiro DB, Kozlov AV, Lancaster JR, Jr, Lefer DJ, McColl K, McCurry K, Patel RP, Petersson J, Rassaf T, Reutov VP, Richter-Addo GB, Schechter A, Shiva S, Tsuchiya K, van Faassen EE, Webb AJ, Zuckerbraun BS, Zweier JL, Weitzberg E. Nat Chem Biol. 2009;5:865–869. doi: 10.1038/nchembio.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO, 3rd, Gladwin MT. Nat Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 215.Stuehr DJ. J Nutr. 2004;134:2765S–2767S. doi: 10.1093/jn/134.10.2748S. [DOI] [PubMed] [Google Scholar]
- 216.Stuehr DJ. Biochim Biophys Acta-Bioenerg. 1999;1411:217–230. doi: 10.1016/s0005-2728(99)00016-x. [DOI] [PubMed] [Google Scholar]
- 217.Wink DA, Paolocci N. Hypertension. 2008;51:617–619. doi: 10.1161/HYPERTENSIONAHA.107.106617. [DOI] [PubMed] [Google Scholar]
- 218.Webb AJ, Patel N, Loukogeorgakis S, Okorie M, Aboud Z, Misra S, Rashid R, Miall P, Deanfield J, Benjamin N, MacAllister R, Hobbs AJ, Ahluwalia A. Hypertension. 2008;51:784–790. doi: 10.1161/HYPERTENSIONAHA.107.103523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Lim MD, Lorkovic IM, Ford PC. J Inorg Biochem. 2005;99:151–165. doi: 10.1016/j.jinorgbio.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 220.Heinecke JL, Khin C, Pereira JC, Suarez SA, Iretskii AV, Doctorovich F, Ford PC. J Am Chem Soc. 2013;135:4007–4017. doi: 10.1021/ja312092x. [DOI] [PubMed] [Google Scholar]
- 221.Brown HW, Pimentel GC. J Chem Phys. 1958;29:883–888. [Google Scholar]
- 222.Dalby FW. Can J Phys. 1958;36:1336–1371. [Google Scholar]
- 223.Salotto AW, Burnelle L. Chem Phys Lett. 1969;3:80–83. [Google Scholar]
- 224.Paolocci N, Jackson MI, Lopez BE, Miranda K, Tocchetti CG, Wink DA, Hobbs AJ, Fukuto JM. Pharmacol Ther. 2007;113:442–458. doi: 10.1016/j.pharmthera.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Fukuto JM, Chiang K, Hszieh R, Wong P, Chaudhuri G. J Pharmacol Exp Ther. 1992;263:546–551. [PubMed] [Google Scholar]
- 226.Fukuto JM, Wallace GC, Hszieh R, Chaudhuri G. Biochem Pharmacol. 1992;43:607–613. doi: 10.1016/0006-2952(92)90584-6. [DOI] [PubMed] [Google Scholar]
- 227.Schmidt HH, Hofmann H, Schindler U, Shutenko ZS, Cunningham DD, Feelisch M. Proc Natl Acad Sci U S A. 1996;93:14492–14497. doi: 10.1073/pnas.93.25.14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Adak S, Wang Q, Stuehr DJ. J Biol Chem. 2000;275:33554–33561. doi: 10.1074/jbc.M004337200. [DOI] [PubMed] [Google Scholar]
- 229.Wong PS, Hyun J, Fukuto JM, Shirota FN, DeMaster EG, Shoeman DW, Nagasawa HT. Biochemistry (Mosc) 1998;37:5362–5371. doi: 10.1021/bi973153g. [DOI] [PubMed] [Google Scholar]
- 230.Niketic V, Stojanovic S, Nikolic A, Spasic M, Michelson AM. Free Radic Biol Med. 1999;27:992–996. doi: 10.1016/s0891-5849(98)00256-1. [DOI] [PubMed] [Google Scholar]
- 231.Saleem M, Ohshima H. Biochem Biophys Res Commun. 2004;315:455–462. doi: 10.1016/j.bbrc.2004.01.081. [DOI] [PubMed] [Google Scholar]
- 232.Sharpe MA, Cooper CE. Biochem J. 1998;332:9–19. doi: 10.1042/bj3320009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Filipovic MR, Miljkovic J, Nauser T, Royzen M, Klos K, Shubina T, Koppenol WH, Lippard SJ, Ivanovic-Burmazovic I. J Am Chem Soc. 2012;134:12016–12027. doi: 10.1021/ja3009693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Miljkovic J, Kenkel I, Ivanovic-Burmazovic I, Filipovic MR. Angew Chem. 2013;52:12061–12064. doi: 10.1002/anie.201305669. [DOI] [PubMed] [Google Scholar]
- 235.Paolocci N, Saavedra WF, Miranda KM, Martignani C, Isoda T, Hare JM, Espey MG, Fukuto JM, Feelisch M, Wink DA, Kass DA. Proc Natl Acad Sci U S A. 2001;98:10463–10468. doi: 10.1073/pnas.181191198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Pagliaro P, Mancardi D, Rastaldo R, Penna C, Gattullo D, Miranda KM, Feelisch M, Wink DA, Kass DA, Paolocci N. Free Radic Biol Med. 2003;34:33–43. doi: 10.1016/s0891-5849(02)01179-6. [DOI] [PubMed] [Google Scholar]
- 237.Basudhar D, Bharadwaj G, Cheng RY, Jain S, Shi S, Heinecke JL, Holland RJ, Ridnour LA, Caceres VM, Spadari-Bratfisch RC, Paolocci N, Velázquez-Martínez CA, Wink DA, Miranda KM. J Med Chem. 2013;56:7804–7820. doi: 10.1021/jm400196q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Zarpelon AC, Souza GR, Cunha TM, Schivo IR, Marchesi M, Casagrande R, Pinge-Filho P, Cunha FQ, Ferreira SH, Miranda KM, Verri WA., Jr Neuropharmacology. 2013;71:1–9. doi: 10.1016/j.neuropharm.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Smith PAS, Hein GE. J Am Chem Soc. 1960;82:5731–5740. [Google Scholar]
- 240.Kohout FC, Lampe FW. J Am Chem Soc. 1965;87:5795–5796. [Google Scholar]
- 241.Hughes MN, Wimbledon PE. J Chem Soc, Dalton Trans. 1976:703–707. [Google Scholar]
- 242.Shafirovich V, Lymar SV. Proc Natl Acad Sci U S A. 2002;99:7340–7345. doi: 10.1073/pnas.112202099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Bartberger MD, Liu W, Ford E, Miranda KM, Switzer C, Fukuto JM, Farmer PJ, Wink DA, Houk KN. Proc Natl Acad Sci U S A. 2002;99:10958–10963. doi: 10.1073/pnas.162095599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Shafirovich V, Lymar SV. J Am Chem Soc. 2003;125:6547–6552. doi: 10.1021/ja034378j. [DOI] [PubMed] [Google Scholar]
- 245.Miranda KM. Coord Chem Rev. 2005;249:433–455. [Google Scholar]
- 246.Dixon RN. J Chem Phys. 1996;104:6905–6906. [Google Scholar]
- 247.Gomes JRB, Ribeiro da Silva MDMC, Ribeiro da Silva MAV. J Phys Chem A. 2004;108:2119–2130. [Google Scholar]
- 248.Flores-Santana W, Salmon DJ, Donzelli S, Switzer CH, Basudhar D, Ridnour L, Cheng R, Glynn SA, Paolocci N, Fukuto JM, Miranda KM, Wink DA. Antioxid Redox Signal. 2011;14:1659–1674. doi: 10.1089/ars.2010.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Fukuto JM, Switzer CH, Miranda KM, Wink DA. Annu Rev Pharmacool Toxicol. 2005;45:335–355. doi: 10.1146/annurev.pharmtox.45.120403.095959. [DOI] [PubMed] [Google Scholar]
- 250.Murphy ME, Sies H. Proc Natl Acad Sci U S A. 1991;88:10860–10864. doi: 10.1073/pnas.88.23.10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Fukuto JM, Hobbs AJ, Ignarro LJ. Biochem Biophys Res Commun. 1993;196:707–713. doi: 10.1006/bbrc.1993.2307. [DOI] [PubMed] [Google Scholar]
- 252.Pino RZ, Feelisch M. Biochem Biophys Res Commun. 1994;201:54–62. doi: 10.1006/bbrc.1994.1668. [DOI] [PubMed] [Google Scholar]
- 253.Shoeman DW, Shirota FN, DeMaster EG, Nagasawa HT. Alcohol. 2000;20:55–59. doi: 10.1016/s0741-8329(99)00056-7. [DOI] [PubMed] [Google Scholar]
- 254.Johnson GM, Chozinski TJ, Gallagher ES, Aspinwall CA, Miranda KM. Free Radic Biol Med. 2014;76:299–307. doi: 10.1016/j.freeradbiomed.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.DeMaster EG, Redfern B, Nagasawa HT. Biochem Pharmacol. 1998;55:2007–2015. doi: 10.1016/s0006-2952(98)00080-x. [DOI] [PubMed] [Google Scholar]
- 256.Lopez BE, Rodriguez CE, Pribadi M, Cook NM, Shinyashiki M, Fukuto JM. Arch Biochem Biophys. 2005;442:140–148. doi: 10.1016/j.abb.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 257.Farmer PJ, Sulc F. J Inorg Biochem. 2005;99:166–184. doi: 10.1016/j.jinorgbio.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 258.Kumar MR, Pervitsky D, Chen L, Poulos T, Kundu S, Hargrove MS, Rivera EJ, Diaz A, Colon JL, Farmer PJ. Biochemistry (Mosc) 2009;48:5018–5025. doi: 10.1021/bi900122r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Ford PC, Fernandez BO, Lim MD. Chem Rev. 2005;105:2439–2455. doi: 10.1021/cr0307289. [DOI] [PubMed] [Google Scholar]
- 260.Miranda KM, Nims RW, Thomas DD, Espey MG, Citrin D, Bartberger MD, Paolocci N, Fukuto JM, Feelisch M, Wink DA. J Inorg Biochem. 2003;93:52–60. doi: 10.1016/s0162-0134(02)00498-1. [DOI] [PubMed] [Google Scholar]
- 261.Bazylinski DA, Hollocher TC. J Am Chem Soc. 1985;107:7982–7986. [Google Scholar]
- 262.Doyle MP, Mahapatro SN, Broene RD, Guy JK. J Am Chem Soc. 1988;110:593–599. [Google Scholar]
- 263.Buyukafsar K, Nelli S, Martin W. Br J Pharmacol. 2001;132:165–172. doi: 10.1038/sj.bjp.0703812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Bari SE, Marti MA, Amorebieta VT, Estrin DA, Doctorovich F. J Am Chem Soc. 2003;125:15272–15273. doi: 10.1021/ja036370f. [DOI] [PubMed] [Google Scholar]
- 265.Doyle MP, Pickering RA, Cook BR. J Inorg Biochem. 1983;19:329–338. [Google Scholar]
- 266.Doyle MP, Hoekstra JW. J Inorg Biochem. 1981;14:351–358. doi: 10.1016/s0162-0134(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 267.Ignarro LJ. Adv Pharmacol. 1994;26:35–65. doi: 10.1016/s1054-3589(08)60050-2. [DOI] [PubMed] [Google Scholar]
- 268.Vanin AF, Vedernikov Iu I, Galagan ME, Kubrina LN, Kuzmanis Ia A, Kalvin’sh I, Mordvintsev PI. Biokhimiia. 1990;55:1408–1413. [PubMed] [Google Scholar]
- 269.Miller TW, Cherney MM, Lee AJ, Francoleon NE, Farmer PJ, King SB, Hobbs AJ, Miranda KM, Burstyn JN, Fukuto JM. J Biol Chem. 2009;284:21788–21796. doi: 10.1074/jbc.M109.014282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Goodrich LE, Lehnert N. J Inorg Biochem. 2013;118:179–186. doi: 10.1016/j.jinorgbio.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 271.Zhu G, Groneberg D, Sikka G, Hori D, Ranek MJ, Nakamura T, Takimoto E, Paolocci N, Berkowitz DE, Friebe A, Kass DA. Hypertension. 2015;65:385–392. doi: 10.1161/HYPERTENSIONAHA.114.04285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Irvine JC, Cao N, Gossain S, Alexander AE, Love JE, Qin C, Horowitz JD, Kemp-Harper BK, Ritchie RH. Am J Physiol Heart Circ Physiol. 2013;305:H365–H377. doi: 10.1152/ajpheart.00495.2012. [DOI] [PubMed] [Google Scholar]
- 273.Andrews KL, Lumsden NG, Farry J, Jefferis AM, Kemp-Harper BK, Chin-Dusting JP. Clin Sci. 2015;129:179–187. doi: 10.1042/CS20140759. [DOI] [PubMed] [Google Scholar]
- 274.Lin EQ, Irvine JC, Cao AH, Alexander AE, Love JE, Patel R, McMullen JR, Kaye DM, Kemp-Harper BK, Ritchie RH. PLoS One. 2012;7:10. doi: 10.1371/journal.pone.0034892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Miranda KM, Espey MG, Yamada K, Krishna M, Ludwick N, Kim S, Jourd’heuil D, Grisham MB, Feelisch M, Fukuto JM, Wink DA. J Biol Chem. 2001;276:1720–1727. doi: 10.1074/jbc.M006174200. [DOI] [PubMed] [Google Scholar]
- 276.Miranda KM, Yamada K, Espey MG, Thomas DD, DeGraff W, Mitchell JB, Krishna MC, Colton CA, Wink DA. Arch Biochem Biophys. 2002;401:134–144. doi: 10.1016/S0003-9861(02)00031-0. [DOI] [PubMed] [Google Scholar]
- 277.Jorolan JH, Buttitta LA, Cheah C, Miranda KM. Nitric Oxide. 2014;10:39–46. doi: 10.1016/j.niox.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 278.Katori T, Donzelli S, Tocchetti CG, Miranda KM, Cormaci G, Thomas DD, Ketner EA, Lee MJ, Mancardi D, Wink DA, Kass DA, Paolocci N. Free Radic Biol Med. 2006;41:1606–1618. doi: 10.1016/j.freeradbiomed.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 279.Liochev SI, Fridovich I. Arch Biochem Biophys. 2002;402:166–171. doi: 10.1016/S0003-9861(02)00074-7. [DOI] [PubMed] [Google Scholar]
- 280.Wink DA, Feelisch M, Fukuto J, Chistodoulou D, Jourd’heuil D, Grisham MB, Vodovotz Y, Cook JA, Krishna M, DeGraff WG, Kim S, Gamson J, Mitchell JB. Arch Biochem Biophys. 1998;351:66–74. doi: 10.1006/abbi.1997.0565. [DOI] [PubMed] [Google Scholar]
- 281.Chazotte-Aubert L, Oikawa S, Gilibert I, Bianchini F, Kawanishi S, Ohshima H. J Biol Chem. 1999;274:20909–20915. doi: 10.1074/jbc.274.30.20909. [DOI] [PubMed] [Google Scholar]
- 282.Ohshima H, Gilibert I, Bianchini F. Free Radic Biol Med. 1999;26:1305–1313. doi: 10.1016/s0891-5849(98)00327-x. [DOI] [PubMed] [Google Scholar]
- 283.Smulik R, Debski D, Zielonka J, Michalowski B, Adamus J, Marcinek A, Kalyanaraman B, Sikora A. J Biol Chem. 2014;289:35570–35581. doi: 10.1074/jbc.M114.597740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Zielonka J, Sikora A, Joseph J, Kalyanaraman B. J Biol Chem. 2010;285:14210–14216. doi: 10.1074/jbc.M110.110080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Guardia CMA, Lebrero MCG, Bari SE, Estrin DA. Chem Phys Lett. 2008;463:112–116. [Google Scholar]
- 286.Miranda KM, Nagasawa HT, Toscano JP. Curr Top Med Chem. 2005;5:647–664. doi: 10.2174/1568026054679290. [DOI] [PubMed] [Google Scholar]
- 287.DuMond JF, King SB. Antioxid Redox Signal. 2011;14:1637–1648. doi: 10.1089/ars.2010.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Dutton AS, Fukuto JM, Houk KN. J Am Chem Soc. 2004;126:3795–3800. doi: 10.1021/ja0391614. [DOI] [PubMed] [Google Scholar]
- 289.Zamora R, Grzesiok A, Weber H, Feelisch M. Biochem J. 1995;312:333–339. doi: 10.1042/bj3120333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Bonner FT, Ko YH. Inorg Chem. 1992;31:2514–2519. [Google Scholar]
- 291.Atkinson RN, Storey BM, King SB. Tetrahedron Lett. 1996;37:9287–9290. [Google Scholar]
- 292.Cohen AD, Zeng BB, King SB, Toscano JP. J Am Chem Soc. 2003;125:1444–1445. doi: 10.1021/ja028978e. [DOI] [PubMed] [Google Scholar]
- 293.Howard JAK, Ilyashenko G, Sparkes HA, Whiting A. Dalton Trans. 2007:2108–2111. doi: 10.1039/b704728b. [DOI] [PubMed] [Google Scholar]
- 294.Lown JW. J Chem Soc B. 1966:441–446. [Google Scholar]
- 295.Rehse K, Herpel M. Arch Pharm. 1998;331:104–110. doi: 10.1002/(sici)1521-4184(199803)331:3<104::aid-ardp104>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 296.Rehse K, Herpel M. Arch Pharm. 1998;331:111–117. doi: 10.1002/(sici)1521-4184(199803)331:3<111::aid-ardp111>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 297.Drago RS, Karstett Br. J Am Chem Soc. 1961;83:1819–1822. [Google Scholar]
- 298.Bharadwaj G, Benini PG, Basudhar D, Ramos-Colon CN, Johnson GM, Larriva MM, Keefer LK, Andrei D, Miranda KM. Nitric Oxide. 2014;2:70–78. doi: 10.1016/j.niox.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Sha X, Isbell TS, Patel RP, Day CS, King SB. J Am Chem Soc. 2006;128:9687–9692. doi: 10.1021/ja062365a. [DOI] [PubMed] [Google Scholar]
- 300.Miranda KM, Katori T, Torres de Holding CL, Thomas L, Ridnour LA, McLendon WJ, Cologna SM, Dutton AS, Champion HC, Mancardi D, Tocchetti CG, Saavedra JE, Keefer LK, Houk KN, Fukuto JM, Kass DA, Paolocci N, Wink DA. J Med Chem. 2005;48:8220–8228. doi: 10.1021/jm050151i. [DOI] [PubMed] [Google Scholar]
- 301.Andrei D, Salmon DJ, Donzelli S, Wahab A, Klose JR, Citro ML, Saavedra JE, Wink DA, Miranda KM, Keefer LK. J Am Chem Soc. 2010;132:16526–16532. doi: 10.1021/ja106552p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Chen X, Jhee KH, Kruger WD. J Biol Chem. 2004;279:52082–52086. doi: 10.1074/jbc.C400481200. [DOI] [PubMed] [Google Scholar]
- 303.Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R. J Biol Chem. 2009;284:22457–22466. doi: 10.1074/jbc.M109.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Stipanuk MH. Annu Rev Nutr. 2004;24:539–577. doi: 10.1146/annurev.nutr.24.012003.132418. [DOI] [PubMed] [Google Scholar]
- 306.Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, Kimura H. Antioxid Redox Signal. 2009;11:703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- 307.Wang R. FASEB J. 2002;16:1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- 308.Abe K, Kimura H. J Neurosci. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Hosoki R, Matsuki N, Kimura H. Biochem Biophys Res Commun. 1997;237:527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- 310.Lee SW, Hu YS, Hu LF, Lu Q, Dawe GS, Moore PK, Wong PT, Bian JS. Glia. 2006;54:116–124. doi: 10.1002/glia.20362. [DOI] [PubMed] [Google Scholar]
- 311.Tamizhselvi R, Moore PK, Bhatia M. J Cell Mol Med. 2007;11:315–326. doi: 10.1111/j.1582-4934.2007.00024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Yang G, Cao K, Wu L, Wang R. J Biol Chem. 2004;279:49199–49205. doi: 10.1074/jbc.M408997200. [DOI] [PubMed] [Google Scholar]
- 313.Zhao W, Zhang J, Lu Y, Wang R. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Predmore BL, Lefer DJ, Gojon G. Antioxid Redox Signal. 2012;17:119–140. doi: 10.1089/ars.2012.4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Furne J, Saeed A, Levitt MD. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1479–1485. doi: 10.1152/ajpregu.90566.2008. [DOI] [PubMed] [Google Scholar]
- 316.Nicholls P. Biochem Soc Trans. 1975;3:316–319. doi: 10.1042/bst0030316. [DOI] [PubMed] [Google Scholar]
- 317.Nicholls P, Kim JK. Can J Biochem. 1982;60:613–623. doi: 10.1139/o82-076. [DOI] [PubMed] [Google Scholar]
- 318.Nicholls P, Marshall DC, Cooper CE, Wilson MT. Biochem Soc Trans. 2013;41:1312–1316. doi: 10.1042/BST20130070. [DOI] [PubMed] [Google Scholar]
- 319.Pietri R, Roman-Morales E, Lopez-Garriga J. Antioxid Redox Signal. 2011;15:393–404. doi: 10.1089/ars.2010.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Cooper CE, Brown GC. J Bioenerg Biomembr. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- 321.Carrico RJ, Blumberg WE, Peisach J. J Biol Chem. 1978;253:7212–7215. [PubMed] [Google Scholar]
- 322.Rios-Gonzalez BB, Roman-Morales EM, Pietri R, Lopez-Garriga J. J Inorg Biochem. 2014;133:78–86. doi: 10.1016/j.jinorgbio.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Roman-Morales E, Pietri R, Ramos-Santana B, Vinogradov SN, Lewis-Ballester A, Lopez-Garriga J. Biochem Biophys Res Commun. 2010;400:489–492. doi: 10.1016/j.bbrc.2010.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Hughes MN, Centelles MN, Moore KP. Free Radic Biol Med. 2009;47:1346–1353. doi: 10.1016/j.freeradbiomed.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 325.Wagner F, Asfar P, Calzia E, Radermacher P, Szabo C. Crit Care. 2009;13:213. doi: 10.1186/cc7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Chen KY, Morris JC. Environ Sci Technol. 1972;6:529–537. [Google Scholar]
- 327.Luther GW, 3rd, Findlay AJ, Macdonald DJ, Owings SM, Hanson TE, Beinart RA, Girguis PR. Front Microbiol. 2011;2 doi: 10.3389/fmicb.2011.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Li Q, Lancaster JR., Jr Nitric Oxide. 2013;35:21–34. doi: 10.1016/j.niox.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Carballal S, Trujillo M, Cuevasanta E, Bartesaghi S, Moller MN, Folkes LK, Garcia-Bereguiain MA, Gutierrez-Merino C, Wardman P, Denicola A, Radi R, Alvarez B. Free Radic Biol Med. 2011;50:196–205. doi: 10.1016/j.freeradbiomed.2010.10.705. [DOI] [PubMed] [Google Scholar]
- 330.Kloesch B, Liszt M, Steiner G, Broll J. Rheumatol Int. 2012;32:729–736. doi: 10.1007/s00296-010-1682-0. [DOI] [PubMed] [Google Scholar]
- 331.Oh GS, Pae HO, Lee BS, Kim BN, Kim JM, Kim HR, Jeon SB, Jeon WK, Chae HJ, Chung HT. Free Radic Biol Med. 2006;41:106–119. doi: 10.1016/j.freeradbiomed.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 332.Pan LL, Liu XH, Gong QH, Wu D, Zhu YZ. PloS one. 2011;6:e19766. doi: 10.1371/journal.pone.0019766. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 333.Bruce KS. Free Radic Biol Med. 2013;55:1–7. doi: 10.1016/j.freeradbiomed.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Stein A, Bailey SM. Redox Biol. 2013;1:32–39. doi: 10.1016/j.redox.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Blackstone E, Morrison M, Roth MB. Science. 2005;308:518. doi: 10.1126/science.1108581. [DOI] [PubMed] [Google Scholar]
- 336.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, Kimura H, Chow CW, Lefer DJ. Proc Natl Acad Sci U S A. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Hellmich MR, Coletta C, Chao C, Szabo C. Antioxid Redox Signal. 2014;22:424–48. doi: 10.1089/ars.2014.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Sanokawa-Akakura R, Ostrakhovitch EA, Akakura S, Goodwin S, Tabibzadeh S. PloS one. 2014;9:e108537. doi: 10.1371/journal.pone.0108537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Francoleon NE, Carrington SJ, Fukuto JM. Arch Biochem Biophys. 2011;516:146–153. doi: 10.1016/j.abb.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 340.Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T, Yamamoto M, Ono K, Devarie-Baez NO, Xian M, Fukuto JM, Akaike T. Proc Natl Acad Sci U S A. 2014;111:7606–7611. doi: 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Miranda KM, Wink DA. Proc Natl Acad Sci U S A. 2014;111:7505–7506. doi: 10.1073/pnas.1405665111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Munchberg U, Anwar A, Mecklenburg S, Jacob C. Org Biomol Chem. 2007;5:1505–1518. doi: 10.1039/b703832a. [DOI] [PubMed] [Google Scholar]
- 343.Greiner R, Palinkas Z, Basell K, Becher D, Antelmann H, Nagy P, Dick TP. Antioxid Redox Signal. 2013;19:1749–1765. doi: 10.1089/ars.2012.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Paul BD, Snyder SH. Nat Rev Mol Cell Biol. 2012;13:499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 345.Domoki F, Olah O, Zimmermann A, Nemeth I, Toth-Szuki V, Hugyecz M, Temesvari P, Bari F. Pediatr Res. 2010;68:387–392. doi: 10.1203/PDR.0b013e3181f2e81c. [DOI] [PubMed] [Google Scholar]
- 346.Eckermann JM, Chen W, Jadhav V, Hsu FP, Colohan AR, Tang J, Zhang JH. Med Gas Res. 2011;1:7. doi: 10.1186/2045-9912-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Qian L, Shen J, Chuai Y, Cai J. Int J Biol Sci. 2013;9:887–894. doi: 10.7150/ijbs.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Sjostrand T. Acta Physiol Scand. 1951;22:137–141. doi: 10.1111/j.1748-1716.1951.tb00762.x. [DOI] [PubMed] [Google Scholar]
- 349.Sjostrand T. Acta Physiol Scand. 1952;26:338–344. doi: 10.1111/j.1748-1716.1952.tb00915.x. [DOI] [PubMed] [Google Scholar]
- 350.Tenhunen R, Marver HS, Schmid R. Proc Natl Acad Sci U S A. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Landaw SA, Callahan EW, Jr, Schmid R. J Clin Invest. 1970;49:914–925. doi: 10.1172/JCI106311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Bucolo C, Drago F. Pharmacol Ther. 2011;130:191–201. doi: 10.1016/j.pharmthera.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 353.Suliman HB, Carraway MS, Tatro LG, Piantadosi CA. J Cell Sci. 2007;120:299–308. doi: 10.1242/jcs.03318. [DOI] [PubMed] [Google Scholar]
- 354.Springer BA, Sligar SG, Olson JS, Phillips GN. Chem Rev. 1994;94:699–714. [Google Scholar]
- 355.Von Burg DR. J Appl Toxicol. 1999;19:379–386. doi: 10.1002/(sici)1099-1263(199909/10)19:5<379::aid-jat563>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 356.Gorman D, Drewry A, Huang YL, Sames C. Toxicology. 2003;187:25–38. doi: 10.1016/s0300-483x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 357.Leemann T, Bonnabry P, Dayer P. Life Sci. 1994;54:951–956. doi: 10.1016/0024-3205(94)00496-x. [DOI] [PubMed] [Google Scholar]
- 358.Alonso JR, Cardellach F, Lopez S, Casademont J, Miro O. Pharmacol Toxicol. 2003;93:142–146. doi: 10.1034/j.1600-0773.2003.930306.x. [DOI] [PubMed] [Google Scholar]
- 359.Wang R, Wang Z, Wu L. Br J Pharmacol. 1997;121:927–934. doi: 10.1038/sj.bjp.0701222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Brune B, Ullrich V. Mol Pharmacol. 1987;32:497–504. [PubMed] [Google Scholar]
- 361.Beitrage RE. Chem Zentrabl. 1862;7:347–351. [Google Scholar]
- 362.Levitt MD. New Engl J Med. 1969;281:122–127. doi: 10.1056/NEJM196907172810303. [DOI] [PubMed] [Google Scholar]
- 363.McKay LF, Holbrook WP, Eastwood MA. Acta Pathol Microbiol Immunol Scand [B] 1982;90:257–260. doi: 10.1111/j.1699-0463.1982.tb00114.x. [DOI] [PubMed] [Google Scholar]
- 364.Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, Katsura K, Katayama Y, Asoh S, Ohta S. Nat Med. 2007;13:688–694. doi: 10.1038/nm1577. [DOI] [PubMed] [Google Scholar]
- 365.Christensen H, Sehested K. The Journal of Physical Chemistry. 1983;87:118–120. [Google Scholar]
- 366.Itoh T, Fujita Y, Ito M, Masuda A, Ohno K, Ichihara M, Kojima T, Nozawa Y. Biochem Biophys Res Commun. 2009;389:651–656. doi: 10.1016/j.bbrc.2009.09.047. [DOI] [PubMed] [Google Scholar]
- 367.Hong Y, Chen S, Zhang JM. J Int Med Res. 2010;38:1893–1903. doi: 10.1177/147323001003800602. [DOI] [PubMed] [Google Scholar]
- 368.Ohta S. Curr Pharm Des. 2011;17:2241–2252. doi: 10.2174/138161211797052664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Shinbo T, Kokubo K, Sato Y, Hagiri S, Hataishi R, Hirose M, Kobayashi H. Am J Physiol Heart Circ Physiol. 2013;305:H542–550. doi: 10.1152/ajpheart.00844.2012. [DOI] [PubMed] [Google Scholar]
- 370.Kato R, Nomura A, Sakamoto A, Yasuda Y, Amatani K, Nagai S, Sen Y, Ijiri Y, Okada Y, Yamaguchi T, Izumi Y, Yoshiyama M, Tanaka K, Hayashi T. Am J Physiol Heart Circ Physiol. 2014;307:H1626–33. doi: 10.1152/ajpheart.00228.2014. [DOI] [PubMed] [Google Scholar]
- 371.Hayashida K, Sano M, Ohsawa I, Shinmura K, Tamaki K, Kimura K, Endo J, Katayama T, Kawamura A, Kohsaka S, Makino S, Ohta S, Ogawa S, Fukuda K. Biochem Biophys Res Commun. 2008;373:30–35. doi: 10.1016/j.bbrc.2008.05.165. [DOI] [PubMed] [Google Scholar]
- 372.Fukuda K, Asoh S, Ishikawa M, Yamamoto Y, Ohsawa I, Ohta S. Biochem Biophys Res Commun. 2007;361:670–674. doi: 10.1016/j.bbrc.2007.07.088. [DOI] [PubMed] [Google Scholar]
- 373.Kamimura N, Nishimaki K, Ohsawa I, Ohta S. Obesity. 2011;19:1396–1403. doi: 10.1038/oby.2011.6. [DOI] [PubMed] [Google Scholar]
- 374.Qian L, Cao F, Cui J, Huang Y, Zhou X, Liu S, Cai J. Free Radical Res. 2010;44:275–282. doi: 10.3109/10715760903468758. [DOI] [PubMed] [Google Scholar]
- 375.Chan EC, Dusting GJ, Liu GS, Jiang F. J Hypertens. 2014;32:1379–1386. doi: 10.1097/HJH.0000000000000179. [DOI] [PubMed] [Google Scholar]
- 376.Jiang F, Roberts SJ, Datla, Dusting GJ. Hypertension. 2006;48:950–957. doi: 10.1161/01.HYP.0000242336.58387.1f. [DOI] [PubMed] [Google Scholar]
- 377.Kim HS, Loughran PA, Billiar TR. Nitric Oxide. 2008;18:256–265. doi: 10.1016/j.niox.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Utz J, Ullrich V. Biochem Pharmacol. 1991;41:1195–1201. doi: 10.1016/0006-2952(91)90658-r. [DOI] [PubMed] [Google Scholar]
- 379.Whiteman M, Li L, Kostetski I, Chu SH, Siau JL, Bhatia M, Moore PK. Biochem Biophys Res Commun. 2006;343:303–310. doi: 10.1016/j.bbrc.2006.02.154. [DOI] [PubMed] [Google Scholar]
- 380.Ondrias K, Stasko A, Cacanyiova S, Sulova Z, Krizanova O, Kristek F, Malekova L, Knezl V, Breier A. Pflugers Arch. 2008;457:271–279. doi: 10.1007/s00424-008-0519-0. [DOI] [PubMed] [Google Scholar]
- 381.Eberhardt M, Dux M, Namer B, Miljkovic J, Cordasic N, Will C, Kichko TI, de la Roche J, Fischer M, Suarez SA, Bikiel D, Dorsch K, Leffler A, Babes A, Lampert A, Lennerz JK, Jacobi J, Marti MA, Doctorovich F, Hogestatt ED, Zygmunt PM, Ivanovic-Burmazovic I, Messlinger K, Reeh P, Filipovic MR. Nat Commun. 2014;5 doi: 10.1038/ncomms5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Vicente JB, Colaco HG, Mendes MI, Sarti P, Leandro P, Giuffre A. J Biol Chem. 2014;289:8579–8587. doi: 10.1074/jbc.M113.507533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Garcia JV, Yang J, Shen D, Yao C, Li X, Wang R, Stucky GD, Zhao D, Ford PC, Zhang F. Small. 2012;8:3800–3805. doi: 10.1002/smll.201201213. [DOI] [PubMed] [Google Scholar]
- 384.Barta K, Ford PC. Acc Chem Res. 2014;47:1503–1512. doi: 10.1021/ar4002894. [DOI] [PubMed] [Google Scholar]