

Abstract
High glucose-induced oxidative stress is a major contributing mechanism to the development of diabetic cardiomyopathy. Nrf2 is an emerging critical regulator of cellular defense against oxidative damage. The role of Nrf2 in diabetic cardiomyopathy was investigated in vivo. Streptozotocin (STZ) induced diabetes in Nrf2 knockout (KO) mice that rapidly progressed to severe conditions with high mortality within two weeks of injection; whereas, in wild type (WT) mice, diabetes was less severe with no death. Severe myocardial lesions were observed in diabetic KO mice that had high, sublethal levels of blood glucose including: (a) irregular myocardial arrangements, myofibrillar discontinuation, and cell death; (b) reduced electron density, discontinuation of myocardial fibers, and mitochondrial damage; and (c) markedly reduced contractility of the cardiomyocytes to β-agonist stimulation. Parallel to severe cardiomyopathy, the diabetic KO hearts showed: (a) increased apoptosis as revealed by TUNEL and PARP1 cleavage assays; (b) infiltration of granulocytes and macrophages as well as fibrosis indicating robust inflammatory response; and (c) heightened oxidative stress as evidenced by increased levels of 8-hydroxydeoxyquanine, free malondialdehyde, and 3-nitrotyrosine. Increased oxidative stress in the KO hearts was attributed to decrease or loss of the basal and induced expression of Nrf2-dependent cytoprotective genes. Our findings demonstrate that loss of Nrf2 function synergizes with high glucose to cause heightened oxidative stress in the heart leading to severe diabetic cardiomyopathy.
Introduction
Diabetes mellitus is the world’s fastest growing disease with high morbidity and mortality rates [1]. Epidemiological and experimental studies have consistently reported diabetes as a strong risk factor for the development of heart failure even after adjusting for concomitant risks such as coronary artery disease and hypertension. This has resulted in the recognition of a distinct disease process termed as “diabetic cardiomyopathy” [2-4]. Factors influencing the pathogenesis of diabetic cardiomyopathy remain unclear. It is generally believed that high blood glucose promotes reactive oxygen species (ROS) production resulting in oxidative stress in cells that in turn causes damage to multiple tissues including the heart in diabetes [5-9]. On the other hand, the repetitive contraction and high energy expenditure of the heart expose it to excessive ROS production and make cardiomyocytes vulnerable to oxidative damage [10]. Thus, a direct toxic effect of hyperglycemia on myocardial tissues via ROS has been suggested as a critical contributor to the development of diabetic cardiomyopathy.
The nuclear factor erythroid 2-related factor 2 (Nrf2) is a cap “n” collar basic leucine zipper transcription factor that has recently emerged as a critical regulator of the mammalian response to ROS, antioxidants, and electrophilic signals [11-13]. Nrf2 controls the basal expression and induction of a range of detoxification enzymes and antioxidant proteins that coordinately metabolize toxic chemicals, scavenge ROS and repair oxidative damage [11,14-17]. Genetic, animal, and epidemiological studies have implicated Nrf2 in the development of a range of disease and toxicity including cancer, chronic inflammation, and chronic degenerative pathology. Nrf2-deficient mice have a tendency to develop autoimmune lesions and a sponge-form leukoencephanopathy [18,19], as well as increased susceptibility to many toxicant-induced diseases, such as benzo [α] pyrene-induced cancer [20], metal toxicity [21,22], premature ovarian failure by 4-vinylcyclohexine diepoxide [23], and bleomycin-induced airway inflammation and lung fibrosis [24]. Conversely, activation of Nrf2 by natural and synthetic antioxidants/therapeutics has been shown effective for the prevention and treatment of diseases and toxicities associated with oxidative stress [15].
Nrf2 is broadly expressed in animal tissues, but is rapidly degraded through the ubiquitin-26S proteasome-mediated protein turnover. Nrf2 and its binding protein Keap1 are redox sensors. Inducers, such as phenolic antioxidants, Michael reaction acceptors, and transition metals, modify critical cysteine residues in Keap1 and Nrf2 leading to suppression of Nrf2 degradation and consequently, activation of Nrf2 [25-28]. Activated Nrf2 translocates into the nucleus, dimerizes with a small Maf protein, and binds to a common DNA sequence called “antioxidant responsive element” (ARE) to up-regulate the transcription of a battery of cytoprotective enzymes/proteins [17]. The mechanism by which Nrf2 is activated endogenously under disease conditions remains elusive. Presumably, endogenously produced inducers, such as ROS and prostaglandins, mediate the activation of Nrf2 [14].
We have previously shown that Nrf2 is critical in defense against high glucose-induced oxidative damage in cultured cardiomyocytes [10]. In this study, we analyzed the role of Nrf2 in the development of diabetic cardiomyopathy in vivo by using the Nrf2 knockout (KO) mice and streptozotocin (STZ)-induced diabetic model. We found that Nrf2 KO mice were highly sensitive to STZ-induced diabetic lesions to develop severe cardiomyopathy as compared with wild type (WT) control. Our findings revealed that loss of Nrf2 synergizes with high glucose to induce a heightened oxidative condition in the heart of Nrf2 KO mice resulting in severe myocardial lesions.
Materials and Methods
Mice and treatments
Nrf2 knockout (KO) mice were derived by targeted gene disruption as described elsewhere [29]. The mice were re-derived at Jackson Laboratory to ascertain a specific pathogen-free status and were backcrossed with C57BL/6J mice to obtain a C57BL/6 genetic background (>97%) [30]. C57BL/6J mice from Jackson Laboratory were used as controls. Mice were maintained at an environmentally controlled National Institute for Occupational Safety and Health animal facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. The mice were barrier maintained with a light/dark cycle of 12 hours at a constant temperature (22°C) in HEPA-filtered, individually ventilated microisolator cages (Thorn Caging Systems, Hazleton, PA) with irradiated food (6% fat; 7913; Harlan-Teklad, Madison, WI). Water was provided ad libitum. Sterile Beta Chips from Northeastern Products Company (Warrensburg, NY) were used as bedding.
Eight-week-old male and female WT and Nrf2 KO mice on a C57BL/6 background were given a single dose of streptozotocin (STZ, 150 mg/kg body weight, i.p.; Sigma, St. Louise, MO) dissolved in a sodium citrate buffer (0.1 M, pH 4.5); the control mice received a single injection of the sodium citrate buffer only (8 mice/group/gender). Whole blood glucose (WBG) from the mouse tail vain was measured using the One Touch Ultra Blood Glucose Monitoring System (Lifescan, Milpitas, CA) on the third day after STZ injection. Mice with WBG levels of 250 mg/dL or higher were considered diabetic. To exclude a potential direct effect of STZ on cardiotoxicity, additional groups of WT and KO mice were included; the mice were given STZ (4 mice/gender/genotype), and then insulin (Humulin U, Eli Lilly, Indianapolis, IN) at a concentration of 10 units/mouse/day immediately after hyperglycemia was diagnosed. Blood glucose levels of the mice were monitored to maintain the levels between 100 and 200 mg/dL until sacrifice. Diabetic mice were observed for health daily.
Histopathology
Heart tissues were fixed and embedded in paraffin as described previously [18]. Sections of 5 μm thickness were stained with hematoxilin and eosin (H&E) and were examined under a light microscope. Data were taken from representative cardiac sections from three mice of each group (Control, STZ, and STZ+Insulin for each gender and genotype); Magnification = 40 X.
Electron microscopy
Fresh heart samples were preserved in Karnovsky’s fixative, post fixed in osmium tetroxide, mordanted in tannic acid, and stained en bloc in uranyl acetate. The tissues were then dehydrated in alcohol and embedded in Epon. Ultrathin sections were cut at 70 nm, collected onto copper grids, and stained with uranyl acetate and lead citrate. Ultrastructural sections were viewed and photographed using a JEOL JEM-1220 analytical transmission electron microscope (Tokyo, Japan).
TUNEL assay
Cardiac sections in paraffin were processed for TUNEL assay using the DeadEnd Fluorometric TUNEL system (Promega, Madison, WI) according to the manufacturer’s instructions. Briefly, the slides were de-paraffinized, re-hydrated, fixed with 4% formaldehyde, and permeated with 20 μg/ml proteinase K and 0.2% Triton X-100 in PBS. The slides were labeled with a TdT reaction mixture (Promega) for 90 min and were mounted with a mounting solution containing 4’,6-diamidino-2-phenylindole (DAPI) (Vectorshild, Vector Laboratories, Burigame, CA). Fluorescence images of apoptotic cells (green) and cell nuclei (blue) were obtained from a Zeiss LSM510 confocal miscroscope with the fluorescein isothiocyanate (FITC)-DAPI setting. Fluorescent pictures were taken with equal exposure times.
Detection of fibrosis
Cardiac sections of 5 μm thickness embedded in paraffin were de-paraffinezed and re-hydrated in distilled water. The slides were stained with Picrosius Red (Direct Red 80, Fluka, Sigma, St Louise, MO) for 110 min, followed by dehydration as described elsewhere (www.cvm.missouri.edu/vmdl/vmdl_his_SOP). Collagen fibers were stained red. Images were taken using a light microscope with the SimplePCI 6 software (Compix Inc., Cranberry, PA).
Detection of granulocyte infiltration
The enzymatic activity of Naphthol AS-D chloroacetate esterase was detected using the Naphthol AS-D chloroacetate esterase and α-Naphthyl acetate esterase detection kit (Sigma) according to a procedure from Sigma. Cardiac sections of 5 μm thickness in paraffin were de-paraffinized and re-hydrated in distilled water. The slides were stained with a freshly prepared Naphthol AS-D chloroacetate solution at 37°C for 40 min. Esterase-positive granulation was shown on red and brown under light microscope indicating infiltration of granulocytic lineage cells in the tissue.
Immunoblotting
Hearts were taken from sacrificed mice, snap frozen in liquid nitrogen, and stored at −80°C until use. Heart tissues were homogenated with Ziroconia beads (BioSpec Products,Bartlesville, OK) and proteins were extracted with the T-PER tissue protein extraction reagent containing Halt protease inhibitors (Pierce, Rockford, IL). Extracts of 60 μg protein each sample were separated on a 4-20% SDS-PAGE gradient gel (Bio-Rad, Hercules, CA). Proteins separated were transferred to PVDF membranes. The membranes were blocked with 5% non-fat milk for 1 h at a room temperature and blotted with specific antibodies for overnight at 4°C with gentle shaking. After incubating with appropriate horseradish peroxidase-conjugated secondary antibodies, protein bands were visualized using ECL (Pierce). Anti-PARP1 and anti-3-NT antibodies were obtained from Cell Signaling (Beverly, MA).
Immunofluorescent staining
Cardiac sections in paraffin were deparaffinized, rehydrated, fixed with 4% formaldehyde, and permeated with 20 μg/ml proteinase K as described for TUNEL assay. The slides were then blocked with 5% FBS in DMEM medium for 1 h and were incubated with anti-Mac-2 (macrophage antigen-2) (Cedarlane Laboratories Ltd., Burlington, Ontariao, Canada) at a dilution of 1:500 or anti-8-OHdG(8-hydroxydeoxyguanosine) (Japan Institute for the Control of Age, Fukuro, Japan) at a dilution of 1:100 in 2% FBS DMEM for 1 h at room temperature. After washing with PBS for three times, the slides were incubated with Alexa Fluor 488-conjugated secondary antibodies (Invitrogen, Carlsbad, CA) at a dilution of 1:1000 in 2% FBS DMEM for another hour. After washing for three times, the slides were mounted with the DAPI mounting solution. Fluorescence images were obtained under Zeiss LSM 510 confocal microscope with FITC-DAPI setting.
Lipid peroxidation
Free malondialdehyde (MDA) was measured using the Bioxytech MDA-586 kit from OxisResearch (Potland, OR). Briefly, heart tissues were homogenized in the presence of 5 mmol/L butylated hydroxytoluene. Extracts were prepared by a centrifugation at 10,000 × g for 10 min. MDA contents were determined spectrophotometrically according to a procedure provided by the manufacturer.
Cardiomyocyte contractility
Adult mouse ventricular myocytes (AMVM) were isolated as described below. Young adult mice (10 weeks old) were euthanized with sodium pentobarbital and the heart was removed rapidly. The heart was perfused with Krebs Hensleit Bicarbonate (KHB) at a constant rate of 2 ml/min using a peristaltic pump. The heart was perfused with KHB for 5 min, followed by changing to a low Ca++ KHB for an additional 10 min. The heart was then immersed in recirculating KHB with low Ca++ containing collagenase B for 30 min. The ventricle was minced and placed into a 50-ml centrifuge tube, adjusted to 25 ml with low Ca++ KHB and centrifuged at 50 × g for 2 min, and the supernatant aspirated. The concentration of Ca++ in KHB was increased in 3 increments (0.08, 0.6, 1.2 mM). Finally, the mixture was passed through a 225-μm nylon mesh and centrifuged at 50 × g for 2 min. The centrifuge procedure was repeated until the preparation was composed of 80% viable left ventricular myocytes. Only those myocytes that were rod shaped, with striations, no blebs and not spontaneously contracting were included for analysis. Myocytes typically retained their baseline fractional shortening for 4 h and were viable for biochemical and molecular analyses for 24-48 h after harvest. Only freshly isolated cells were used for physiologic experiments.
Measurements of the amplitude and velocity of unloaded single AMVM shortening and relengthening were made on the stage of an inverted phase-contrast microscope (Olympus, IX70-S1F2, Olympus Optical Co., LTD., Japan) using the Myocyte Calcium Imaging/Cell Length System in which the analog motion signal was digitized and analyzed by the EDGACQ edge detection software (Ionoptix Cor., Milton, MA). Electrical field stimulation was applied at 1 Hz and about 20 volts to achieve threshold depolarization and experiments were performed at 20% above threshold. Each cell serves as its own control by continuous superfusion of buffers and drugs [31]. Data represent the mean ± S.E.M. of 12-15 different determinations derived from 12-15 different individual myocytes from seven to nine separate myocyte preparations from seven to nine different mice.
Real-time PCR
Total RNA was reverse transcribed into single strand cDNAs, and analyzed by real-time PCR using the SYBR GREEN PCR master mix (Applied Biosystems, Foster City, CA) following standard procedures. Briefly, single strand DNA template, forward and reverse primers (10 μM each), PCR master mix, and water were added to make a final volume of 50 μl. Thermal cycling was carried out as follows: 95°C for 3 min as initial denaturing, followed by 45 cycles of 94°C for 30 sec, 60°C for 30 sec, and 72°C for 60 sec, and a final extension at 72°C for 2 min. Threshold cycles (CT values) were determined. Real-time PCR results were normalized using 1% of input as an internal control. Relative DNA amounts were calculated from CT values for each sample by interpolating into the standard curve obtained using a series of dilutions of standard DNA samples that were run under the same conditions. The sequences of the primer sets used for real-time PCR are as follows: Nrf2, forward: 5’-AGGAAGCTGGAGAACATT-3’; reverse: 5’-GTTTTTCTTTGTATCTGG-3’. Mouse NQO1: forward, 5’-CCCCACTCTATTTTGCTCCA-3’; reverse, 5’-ACTCCTTTTCCCATCCTCGT-3’. Actin: forward, 5’-GACCTCTATGCCAACACAGT-3’; reverse, 5’-ACT-CATCGTACTCCTGCTTG-3’. Representative data from three separate experiments were presented.
Statistical analysis
Data were collected from repeated experiments and were represented as means ± standard deviations. One-way ANOVA and Student’s t test were used for statistical analysis. A p value of <0.05 was considered statistically significant.
Results
Severe cardiomyopathy in diabetic Nrf2 KO mice
We have previously reported that Nrf2 is critical in defense against high glucose-induced oxidative damage in cultured cardiomyocytes, supporting the notions that diabetic cardiomyopathy results from the exposure of cardiomyocytes to high levels of blood glucose and Nrf2 has a protective role against diabetic myocardial lesions by suppressing oxidative damage [10]. In the present study, we examined if Nrf2 protects against diabetic cardiomyopathy in intact animals by comparing diabetic lesions between Nrf2 WT and KO mice. A single injection of STZ (150 mg/kg body weight, i.p.) induced diabetes in both male WT and KO mice. However, diabetes in KO mice (KO+STZ) progressed rapidly to severe conditions with blood glucose reaching or exceeding 600 mg/dL, resulting in a high mortality rate within two weeks of treatment (Figure 1). On the other hand, diabetes in WT mice (WT+STZ) was much less severe and did not cause death during the two-week treatment. The average glucose levels for control WT and KO mice were similar to each other (WT, 138.7 ± 17.5 mg/dL; KO, 139 ± 13.8 mg/dL); the average for WT+STZ was 380 ± 17.3 mg/dL (p<0.001, n=8, compared with WT); and the average for KO+STZ was 558 ± 38.3 mg/dL (p<0.001, n=8, compared with WT; p<0.01, n=8, compared with WT+STZ). Both WT and KO control mice gained body weight slightly by ~0.25 to 0.3 grams during the two-week experiment; whereas, STZ-treated WT and KO mice lost body weight by an average of 1.5 or 2.1 grams, respectively. STZ may cause tissue damage independently of pancreatic islet b cell lesions, which may have contributed to increased toxicity observed in diabetic KO mice. Therefore, additional groups of WT and KO mice were given STZ injection followed by insulin to maintain the blood glucose level within a normal range. These mice survived well and did not develop diabetes in either WT or KO groups (Figure 1B, WT+STZ+Insulin and KO+STZ+Insulin). Thus, the mortality in diabetic KO mice was caused by diabetes. These results revealed that KO mice are highly sensitive to STZ-induced diabetes and diabetic lesions compared with WT mice.
Figure 1.
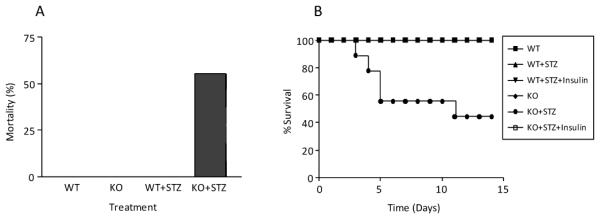
Increased sensitivity of Nrf2 KO mice to STZ-induced diabetic lesions. Eight-week-old male WT and Nrf2 KO mice were treated with 0.1 M citrate buffer (n = 8), STZ (150 mg/kg body weight, n = 8), or STZ plus insulin (n = 4). Tail blood glucose level was monitored for diagnosis of diabetic conditions. Mortality was monitored daily for 14 days. (A) Mortality at day 14. (B) Time curve of percent survival. In (B), mortality was observed only in the group of KO+STZ.
In order to better assess the role of Nrf2 in diabetic myocardial lesions in KO mice, diabetes was induced by STZ and insulin was supplemented to maintain blood glucose levels between 400 and 600 mg/dL. No lethality was observed in the mice, but diabetic KO mice were generally weaker than diabetic WT mice causing, for instance, a greater loss of body weight in diabetic KO than WT mice (data not shown). Histological examination of the mice revealed severe pathological changes in the hearts of diabetic KO mice in comparison with those of control KO and diabetic WT mice. As shown in Figure 2A, control hearts had regular and intact myocardial arrangements and clearly visible nuclei (a, d, g, and j). Diabetic WT hearts (WT+STZ, b and h) showed focal cell death and certain irregularity of the myocardial fibers. On the other hand, diabetic KO hearts from both male and female mice (KO+STZ; e and k) exhibited large areas of irregular myocardial arrangements, myofibrillar discontinuation, and cell death (karyorrhexis, pyknosis, and karyolysis). WT and KO mice injected with STZ followed by insulin (STZ+Insulin) exhibited a normal morphology of myocardial structures (c, f, i, and l), indicating that the above observed pathology was not due to a direct toxicity of STZ on the heart but reflected a diabetic complication in the heart. Ultrastructurally, reduced density, discontinuation of myocardial fibers and altered morphology of mitochondria were observed in the hearts of both WT and KO diabetic mice. However, the lesions were more diffuse and severe in KO than WT mice (Figure 2B). Additionally, more non-myocardial cells were seen in the cardiac tissues of diabetic KO mice than in control KO and diabetic WT mice.
Figure 2.

Myocardial structural damage. Hearts from Nrf2 WT and Nrf2 KO mice were collected 14 days after STZ treatment. Diabetic condition was confirmed by blood glucose detection (≥250 mg/dL). (A) H&E staining: Paraffin-embedded heart tissue (transverse section) was stained with H&E and examined under light microscope (40X). Apparently damaged areas were located inside of circles and arrows. Bar = 20 μm. (B) Ultrastructural damage: Micrographs of transmission electron microscopy of control and diabetic heart samples. Bar = 2 μm.
The prominent structural lesions in diabetic KO mouse hearts implicate impairment of myocardial functions. Therefore, we examined the contractility of cardiomyocytes of both WT and KO mice with or without diabetes. Fourteen days after STZ injection, AMVM were isolated from mouse heart and were stimulated for contraction with the β-adrenergic agonist isoproterenol. As shown in Figure 3, AMVM from control KO mice showed significantly lower contractile force (expressed as isoproterenol-evoked shortening) than AMVM from control WT mice (i.e., ~25% of WT), revealing that loss of Nrf2 reduced the contractility of cardiomyocytes in normal mouse heart. Diabetes decreased isopreterenol-evoked contraction of WT AMVM by ~30% as compared with WT control. On the other hand, AMVM from diabetic KO mice showed nearly a total loss of responsiveness to isoproterenol for contraction, indicating diabetes severely impaired the contractile function of KO hearts compared with WT, which was in agreement with the structural changes described above. Taken together, the structural and functional analyses revealed that loss of Nrf2 caused severe cardiomyopathy under diabetic conditions.
Figure 3.
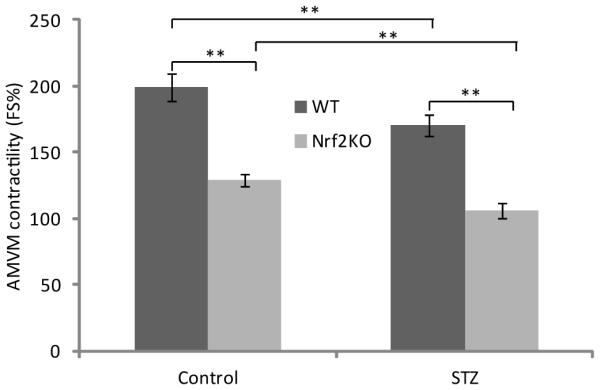
Ventricular myocyte contractility. AMVM were isolated from diabetic mice two weeks after STZ injection. Contractility was measured as percent shortening of ventricular myocytes in the presence of b-adrenergic agonist isoproterenol at 100 nM. Data represent means ± standard deviations from 12 - 15 individual myocytes isolated from eight separate myocyte preparatioins. **, p<0.01.
Spontaneous and diabetes-induced apoptosis in KO cardiomyocytes
Because cardiomyocytes in adult hearts are non-dividable, apoptosis of the cells would result in a loss of myocardial tissues and contractility. To examine if apoptosis of cardiomyocytes contributes to the apparent pathological lesions and functional impairment in diabetic KO hearts, TUNEL assay was performed. Very few apoptotic cells were present in non-diabetic, WT hearts (Figure 4A). Diabetes induced a slight increase in the number of apoptotic cells in WT hearts. In control KO hearts, apoptosis was slightly higher than that of control WT hearts. On the other hand, apoptosis in diabetic KO hearts was markedly increased indicating that diabetes substantially increased apoptosis in the hearts of KO mice. Cells undergoing apoptosis have increased cleavage of PARP1. Immunoblotting of PARP1 cleavage product confirmed that diabetes induced apoptosis in WT hearts (Figure 4B). Moreover, control KO hearts exhibited elevated PARP1 cleavage to the level comparable to that of diabetic WT hearts indicating markedly elevated spontaneous apoptosis in mouse heart in the absence of Nrf2. Unlike the findings from TUNEL assay described above, PARP1 cleavage in diabetic KO hearts was not higher than but similar to that in control KO hearts; this difference between PARP1 cleavage and Tunnel assay could be due to differential sensitivities of the two assays to severe tissue damage in diabetic hearts. Nonetheless, the findings demonstrated a critical role of Nrf2 in protection against apoptosis in mouse heart under both basal and diabetic conditions.
Figure 4.
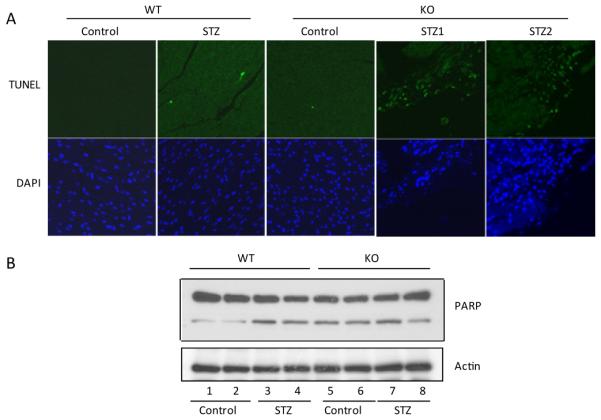
Myocardial apoptosis. (A) Paraffin sections were stained for apoptosis using the DeadEnd Fluorometric TUNEL assay (green, upper panel). DAPI (blue, lower panel) was used to stain for nuclei. Magnification = 40 X. (B) PARP cleavage was detected by immunoblotting using anti-PARP. Upper panel, PARP protein and cleavage product; lower panel, actin as loading control.
Inflammatory infiltration and fibrosis
Myocardial lesions in diabetes stimulate inflammatory responses that are important for the clearance and repair of damaged tissues. However, in many cases, inflammation could worsen the pathological lesions resulting in, for instance, fibrosis in the heart. Indeed, we found that, in agreement with the severity of pathological lesions in the heart, granulocyte infiltration was observed in diabetic hearts, but the inflammatory response was significantly more pronounced and spread-out in the hearts of Nrf2 KO than WT mice (Figure 5A and B). Similarly, diabetes stimulated macrophage infiltration in the hearts, which was significantly more evident in KO than WT (Figure 5C and D). The findings implicate Nrf2 as a critical regulator of the inflammatory response in diabetic hearts.
Figure 5.

Inflammatory infiltration and fibrosis. (A) Granulocyte infiltration. Paraffin sections of heart tissues were stained for Naphthol AS-D chloroacetate esterase activity. Increased esterase-positive granulations (red and brown) shown by arrows indicate granulocyte infiltration. Magnification = 40 X. (B) Quantitative data of experiment (A). (C) Macrophage infiltration. Mac-2-positive macrophages were shown by fluorescent staining using an anti-Mac-2 antibody of paraffin sections of mouse hearts. DAPI staining was used to show the nuclei. (D) Quantification of (C). (E) Fibrosis. Paraffin heart sections were stained with picrosirius red. Increased collagen formation (red) indicates fibrosis. Magnification = 40X. (F) Quantification of (E).*, p<0.05; **, p<0.01.
To examine fibrotic lesions, heart sections were stained for collagen fiber formation (Figure 5E and F). Fibrosis was barely detectable in non-diabetic hearts of male and female WT mice. Diabetes significantly increased collagen fiber staining in WT hearts. On the other hand, fibrosis was clearly observed in control hearts of both male and female KO mice. Fibrosis in KO hearts was drastically increased by diabetes as compared with control KO and diabetic WT hearts of both male and female mice. Increased fibrosis in diabetic KO hearts indicated that the diabetic damage in KO hearts occurred earlier and more severely than that in WT hearts in both genders.
Heightened oxidative stress
High levels of blood glucose could damage the heart by causing oxidative stress in cardiomyocytes. On the other hand, Nrf2 protects against oxidative stress by suppressing ROS production and reducing oxidative damage in a range of cell types and tissues. Therefore, high blood glucose and loss of Nrf2 function in diabetic KO mice could synergistically enhance oxidative stress in the hearts, which in turn causes severe cardiomyopathy. To directly test this possibility, we measured oxidative damage in the hearts. ROS are known to damage macromolecules including DNA, lipids, and proteins in cells. 8-OHdG, an oxidized nucleoside of nuclear and mitochondrial DNA, is the most commonly detected DNA lesion by ROS. We found that control WT mice had a low background of 8-OHdG in the heart, whereas control KO mice showed a significantly higher level of 8-OHdG in the heart indicating lack of Nrf2 promoted oxidative stress even in the absence of diabetes (Figure 6A). Diabetes significantly increased the amount of 8-OHdG in WT hearts supporting the notion that high glucose induces myocardial oxidative stress. On the other hand, 8-OHdG in diabetic KO hearts was substantially higher than that of either diabetic WT or control KO hearts, demonstrating a heightened oxidative stress state in the heart with combined high glucose and Nrf2 null function.
Figure 6.

Oxidative stress and damage. (A) Oxidative DNA damage. The 8-OHdG levels in Nrf2 WT and KO heart tissues were examined by immunofluorescent staining with an anti-8-OHdG antibody. (B) Lipid peroxidation. Nrf2 WT and KO heart tissues were homogenized and MDA production was measured. (C) Protein tyrosine nitrosation. Nrf2 WT and KO heart homogenates were immunoblotted with an anti-3NT antibody. Anti-sarcomeric acting was used as loading control. (D) Quantification of experiment (C).*, p<0.05; **, p<0.01.
Diabetes significantly increased lipid peroxidation as measured by increased formation of free MDA, a marker of lipid peroxidation, in the hearts of WT mice (Figure 6B). In control KO mice, MDA was significantly higher than that of the control WT mice. Diabetes markedly increased MDA production in the KO hearts, which was higher than either control KO or diabetic WT hearts. Thus, oxidative lipid damage measured by MDA in the hearts was in agreement with oxidative DNA damage measured with 8-OHdG.
Oxidative stress is often accompanied with increased nitrosative damage to proteins from reactive nitrogen species (RNS). Nitrosative protein damage can be measured as increased formation of 3-NT in tissues, one of the most common markers of nitric oxide (•NO)-dependent oxidative damage. Immunoblotting of 3-NT in the hearts revealed that STZ injection induced formation of 3-NT in WT mice (Figure 6C and 6D), but the highest amount of 3-NT was observed in diabetic KO mice. The results indicated that increased nitrosative damage occurred in diabetic cardiac tissues and that Nrf2 protected the heart from nitrosative damage.
Given the important role of Nrf2 in protection against oxidative damage, the heightened cardiac oxidative stress and inflammatory changes indicate that lack of Nrf2 in the heart resulted in intrinsic lesions in cellular oxidative defense that synergize with hyperglycemia to cause the severe myocardial lesions. To analyze the mechanism of action of Nrf2 in protection against diabetic cardiomyopathy at a molecular level, transcriptional regulation of the cytoprotective genes in the heart was examined. Nrf2 mRNA was expressed in the hearts of the WT but not KO mice (Figure 7) because a major portion of the Nrf2 gene was not transcribed in Nrf2 KO cells. Nqo1 is a prototype of Nrf2-controlled, ARE-dependent gene encoding the cytoplasmic detoxification enzyme NQO1. Nqo1mRNA was detected in WT but not Nrf2 KO hearts confirming that Nrf2 is required for the basal expression of the gene in the heart. Diabetes significantly induced Nqo1 mRNA expression in WT hearts. However, induction of Nqo1 was lost in Nrf2 KO mice. Expression of heme oxygenase 1 (Ho1), another Nrf2 and ARE-dependent gene, mRNA followed a similar pattern to Nqo1 (data not shown). Thus, loss of Nrf2 disrupted the protective, transcriptional response to oxidative stress in diabetic heart to promote oxidative damage.
Figure 7.

Induction of cytoprotective genes. Total RNA was prepared from hearts of control and diabetic Nrf2 WT and KO mice. Expression of mRNA of Nqo1, Nrf2, and Action was expressed as fold change over control.**, p<0.01.
Discussion
Diabetic cardiomyopathy can occur clinically in the absence of coronary artery disease and hypertension and thus, is recognized as a distinct diabetic complication that contributes significantly to heart failure among diabetic patients [2-4]. Specific therapeutic and preventive measures for diabetic cardiomyopathy are currently lacking [32]. The mechanism by which high blood glucose damages myocardial tissues to cause the disease and the factors that influence this pathogenic process remain elusive. We have previously reported that high glucose induced elevated production of ROS in cultured neonatal and adult ventricular myocytes, which correlated with increased apoptosis and mitochondrial damage [10]. These findings support the notion that oxidative damage is a major contributor to high glucose-induced myocardial lesions in diabetes [5-9]. Presumably, a hyperglycemia-induced process of overproduction of superoxide by the mitochondrial electron-transport chain leads to multiple molecular and biochemical changes in cells and tissues that may involve the activation of multiple signaling pathways [5,7,33]. Such changes would also include the activation of cellular defensive mechanisms against oxidative stress and tissue damage important in the development of diabetic lesions.
In the previous study, we found that Nrf2, an oxidant/antioxidant/electrophile-activated receptor/transcription factor, is important in defense against high glucose-induced oxidative damage in cardiomyocytes [10]. Cardiomyocytes isolated from Nrf2 KO mice had significantly higher levels of ROS production than WT controls under basal conditions and high glucose (20 to 40 mM) increased ROS production, which was significantly higher in KO than WT cells. Parallel to increased oxidative stress, the KO cells had a higher percentage of cells undergoing apoptosis, increased sensitivity to mitochondrial damage by mitochondrial complex II inhibitor 3-nitropropionic acid, reduced response to β-agonist for contraction, and decrease or total loss of induction of certain cytoprotective enzymes and proteins upon stimulation with high glucose. The current study was designed to examine if Nrf2 is important in protection against diabetic myocardial lesions in intact animals. Our results demonstrated that Nrf2 KO mice were highly sensitive to STZ-induced diabetic cardiomyopathy, which resulted from heightened oxidative stress in the heart of diabetic KO mice.
Nrf2 has been implicated in a wide range of diseases, such as cancer and chronic degenerative diseases, and toxicities, such as metal toxicity, polycyclic aromatic hydrocarbon-induced cancer, and chemical/particle/fiber-induced lung fibrosis [18-24,34]. A common theme emerged from the diseases was that they all have oxidative stress as a component in disease pathogenesis and Nrf2 appears to suppress ROS production and oxidative lesions in various experimental systems. In this study, we found that loss of Nrf2 caused markedly increased oxidative DNA damage, lipid peroxidation, and protein nitrosation in the heart of diabetic KO mice, all indicating exacerbated oxidative stress in the heart.
Three mechanisms may account for increased oxidative damage in Nrf2 KO hearts. First, loss of Nrf2 in the heart resulted in the loss or reduction of the expression and induction of ARE-dependent cytoprotective genes including NQO1 and HO1. Furthermore, we found that the expression of a number of enzymes important in ROS metabolism including glutathione peroxidase 2, 6, and 8 were down-regulated, whereas expression of several enzymes that promote ROS production was increased in the Nrf2 KO heart (He and Ma, unpublished observation). These findings are consistent with our previous conclusion that loss of Nrf2 caused intrinsic defect in the defense against oxidative damage in cardiomyocytes due to the lack or reduced expression of ARE-dependent genes. Second, STZ induced higher levels of glucose in KO mice than in WT mice, raising the possibility of increased sensitivity of pancreatic islet β-cells to STZ-induced apoptosis in Nrf2 KO mice; further studies are underway to analyze the role of Nrf2 in β-cell function. Third, cardiomyocytes are prone to damage by ROS due to their continuous and repetitive contraction for pumping blood and high energy demand from mitochondrial respiration, both of which may lead to increased ROS production. Therefore, the high sensitivity of the KO heart to diabetes likely reflects a synergistic effect among hyperglycemia, Nrf2 null function, and vulnerability of cardiomyocytes to ROS, which leads to a heightened oxidative stress state and structural and functional lesions in the heart.
We observed significantly increased infiltration of neutrophils and macrophages in diabetic KO hearts. Furthermore, collagen fiber formation, an indication of fibrosis that generally occurs during chronic tissue damage, was markedly elevated as compared with WT, suggesting that myocardial lesions in KO hearts occurred earlier and more severely than in WT. Increased inflammatory response is commonly seen in animal models of disease and toxicity with Nrf2 knockout [18]. Conversely, many Nrf2 activators potently inhibit inflammation in multiple tissues [35]. Mechanistically, Nrf2 may suppress inflammation via two separate, but not mutually exclusive, mechanisms: (a) inhibition of oxidative damage to tissues, which stimulates inflammation; and (b) inhibition of the intrinsic functions of inflammatory cells. In the latter case, Nrf2 may regulate inflammatory and immune function by controlling the transcription of inflammation and immune-specific genes. Further studies are needed to distinguish the possibilities.
Both Nrf2 and its associated protein Keap1 are redox sensors. Studies on ligand-Keap1 interaction by mass spectrometry and on Keap1 structure by crystallography, NMR, and single particle electron microscopy provided significant insights into the mechanism by which Keap1 and Nrf2 interact with electrophilic inducers to activate Nrf2 [28]. In this model, Nrf2 is constantly degraded with a half life of only 20 min under basal conditions [25]. Degradation is mediated through the Keap1/Cul3 dependent E3-controlled ubiquitination and subsequent proteasomal degradation of Nrf2 [16]. Keap1 contains ~25 cysteine residues, whereas Nrf2 has 7 highly conserved cysteine residues [26,27]. Electrophilic inducers interact with the thiol groups of critical cysteine residues in both Keap1 and Nrf2 to trigger inhibition of Nrf2 ubiquitination and degradation. Stabilized Nrf2 translocates into the nucleus and mediates the basal and induced expression of target genes through ARE-dependent transcripton. The mechanism by which oxidants, such as ROS, activate Nrf2 is not clear. It is believed that ROS and other oxidants oxidize critical cysteine residues of Keap1 and Nrf2 to activate Nrf2. Alternatively, substances, such as prostaglandins, produced from oxidative and inflammatory tissue damage interact with Keap1 and Nrf2 to activate Nrf2 analogously to electrophilic inducers.
Induction of ARE-controlled cytoprotective genes by natural and synthetic antioxidants has been shown to be effective for the prevention of cancer and certain chronic diseases for several decades [15]. Recent discoveries of highly potent inducers, such as the synthetic oleanane triterpenoid 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl] imidazole (CDDO-Im), opened new opportunities for disease prevention and therapy [35]. For instance, CDDO-Im has a CD value of 0.0033 μmol/L (concentration required to double the specific enzyme activity of NQO1), and has been shown to effectively inhibit aflatoxin-induced hepatic tumorigenesis [34], vinyl carbamate-induced lung tumors [36], and cigarette smoke-induced emphysema and cardiac dysfunction [37]. Given that diabetic cardiomyopathy is a chronic complication of diabetes and no effective prevention and therapy are available, the current finding that Nrf2 plays a critical role in protection against diabetic myocardial lesions suggest that Nrf2 activators, such as CDDO-Im, can be expoited for the prevention and treatment of the disease in the future.
Acknowledgement
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the National Institute for Occupational Safety and Health.
Abbreviations
- DAPI
4’,6-diamidino-2-phenylindole
- DHE
Dihydroethium
- HO1
Heme oxygenase 1
- Mac-2
Macrophage Antigen 2
- MDA
Malondialdehyde
- NQO1
NAD(P)H:quinoneoxidoreductase 1
- Nrf2
Nuclear Factor Erythroid 2-related Factor 2
- 3-NT
3-nitrotyrosine
- 8-OHdG
8-hydroxydeoxyguanosine
- STZ
Streptozotocin
References
- 1.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 2.Murarka S, Movahed MR. Diabetic cardiomyopathy. J Card Fail. 2010;16:971–979. doi: 10.1016/j.cardfail.2010.07.249. [DOI] [PubMed] [Google Scholar]
- 3.Khavandi K, Khavandi A, Asghar O, Greenstein A, Withers S, et al. Diabetic cardiomyopathy--a distinct disease? Best Pract Res Clin Endocrinol Metab. 2009;23:347–360. doi: 10.1016/j.beem.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 4.Francis GS. Diabetic cardiomyopathy: fact or fiction? Heart. 2001;85:247–248. doi: 10.1136/heart.85.3.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes. 1999;48:1–9. doi: 10.2337/diabetes.48.1.1. [DOI] [PubMed] [Google Scholar]
- 7.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 8.Cai L, Kang YJ. Oxidative stress and diabetic cardiomyopathy: a brief review. Cardiovasc Toxicol. 2001;1:181–193. doi: 10.1385/ct:1:3:181. [DOI] [PubMed] [Google Scholar]
- 9.Cai L, Wang J, Li Y, Sun X, Wang L, et al. Inhibition of superoxide generation and associated nitrosative damage is involved in metallothionein prevention of diabetic cardiomyopathy. Diabetes. 2005;54:1829–1837. doi: 10.2337/diabetes.54.6.1829. [DOI] [PubMed] [Google Scholar]
- 10.He X, Kan H, Cai L, Ma Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J Mol Cell Cardiol. 2009;46:47–58. doi: 10.1016/j.yjmcc.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 11.Ma Q. Xenobiotic-activated receptors: from transcription to drug metabolism to disease. Chem Res Toxicol. 2008;21:1651–1671. doi: 10.1021/tx800156s. [DOI] [PubMed] [Google Scholar]
- 12.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 13.Leung L, Kwong M, Hou S, Lee C, Chan JY. Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J Biol Chem. 2003;278:48021–48029. doi: 10.1074/jbc.M308439200. [DOI] [PubMed] [Google Scholar]
- 14.Ma Q. Transcriptional responses to oxidative stress: pathological and toxicological implications. Pharmacol Ther. 2010;125:376–393. doi: 10.1016/j.pharmthera.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Talalay P, Dinkova-Kostova AT, Holtzclaw WD. Importance of phase 2 gene regulation in protection against electrophile and reactive oxygen toxicity and carcinogenesis. Adv Enzyme Regul. 2003;43:121–134. doi: 10.1016/s0065-2571(02)00038-9. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi A, Ohta T, Yamamoto M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004;378:273–286. doi: 10.1016/S0076-6879(04)78021-0. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 18.Ma Q, Battelli L, Hubbs AF. Multiorgan Autoimmune Inflammation, Enhanced Lymphoproliferation, and Impaired Homeostasis of Reactive Oxygen Species in Mice Lacking the Antioxidant-Activated Transcription Factor Nrf2. Am J Pathol. 2006;168:1960–1974. doi: 10.2353/ajpath.2006.051113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubbs AF, Benkovic SA, Miller DB, O’Callaghan JP, Battelli L, et al. Vacuolar leukoencephalopathy with widespread astrogliosis in mice lacking transcription factor Nrf2. Am J Pathol. 2007;170:2068–2076. doi: 10.2353/ajpath.2007.060898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci USA. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He X, Lin GX, Chen MG, Zhang JX, Ma Q. Protection against chromium (VI)-induced oxidative stress and apoptosis by Nrf2. Recruiting Nrf2 into the nucleus and disrupting the nuclear Nrf2/Keap1 association. Toxicol Sci. 2007;98:298–309. doi: 10.1093/toxsci/kfm081. [DOI] [PubMed] [Google Scholar]
- 22.He X, Chen MG, Ma Q. Activation of Nrf2 in defense against cadmium-induced oxidative stress. Chem Res Toxicol. 2008;21:1375–1383. doi: 10.1021/tx800019a. [DOI] [PubMed] [Google Scholar]
- 23.Hu X, Roberts JR, Apopa PL, Kan YW, Ma Q. Accelerated ovarian failure induced by 4-vinyl cyclohexene diepoxide in Nrf2 null mice. Mol Cell Biol. 2006;26:940–954. doi: 10.1128/MCB.26.3.940-954.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho HY, Reddy SP, Yamamoto M, Kleeberger SR. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004;18:1258–1260. doi: 10.1096/fj.03-1127fje. [DOI] [PubMed] [Google Scholar]
- 25.He X, Chen MG, Lin GX, Ma Q. Arsenic induces NAD(P)H-quinone oxidoreductase I by disrupting the Nrf2 × Keap1 × Cul3 complex and recruiting Nrf2 × Maf to the antioxidant response element enhancer. J Biol Chem. 2006;281:23620–23631. doi: 10.1074/jbc.M604120200. [DOI] [PubMed] [Google Scholar]
- 26.He X, Ma Q. NRF2 cysteine residues are critical for oxidant/electrophile-sensing, kelch-like ECH-associated protein-1-dependent ubiquitination-proteasomal degradation, and transcription activation. Mol Pharmacol. 2009;76:1265–1278. doi: 10.1124/mol.109.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He X, Ma Q. Critical cysteine residues of Kelch-like ECH-associated protein 1 in arsenic sensing and suppression of nuclear factor erythroid 2-related factor 2. J Pharmacol Exp Ther. 2010;332:66–75. doi: 10.1124/jpet.109.160465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123–140. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- 29.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci USA. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma Q, Kinneer K, Bi Y, Chan JY, Kan YW. Induction of murine NAD(P) H:quinone oxidoreductase by 2,3,7,8-tetrachlorodibenzo-p-dioxin requires the CNC (cap ‘n’ collar) basic leucine zipper transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2): cross-interaction between AhR (aryl hydrocarbon receptor) and Nrf2 signal transduction. Biochem J. 2004;377:205–213. doi: 10.1042/BJ20031123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kan H, Xie Z, Finkel MS. iPLA2 inhibitor blocks negative inotropic effect of HIV gp120 on cardiac myocytes. J Mol Cell Cardiol. 2006;40:131–137. doi: 10.1016/j.yjmcc.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Dobrin JS, Lebeche D. Diabetic cardiomyopathy: signaling defects and therapeutic approaches. Expert Rev Cardiovasc Ther. 2010;8:373–391. doi: 10.1586/erc.10.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duncan JG. Mitochondrial dysfunction in diabetic cardiomyopathy. Biochim Biophys Acta. 2011;1813:1351–1359. doi: 10.1016/j.bbamcr.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yates MS, Kwak MK, Egner PA, Groopman JD, Bodreddigari S, et al. Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole. Cancer Res. 2006;66:2488–2494. doi: 10.1158/0008-5472.CAN-05-3823. [DOI] [PubMed] [Google Scholar]
- 35.Dinkova-Kostova AT, Liby KT, Stephenson KK, Holtzclaw WD, Gao X, et al. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci USA. 2005;102:4584–4589. doi: 10.1073/pnas.0500815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liby K, Royce DB, Williams CR, Risingsong R, Yore MM, et al. The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Res. 2007;67:2414–2419. doi: 10.1158/0008-5472.CAN-06-4534. [DOI] [PubMed] [Google Scholar]
- 37.Sussan TE, Rangasamy T, Blake DJ, Malhotra D, El-Haddad H, et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc Natl Acad Sci USA. 2009;106:250–255. doi: 10.1073/pnas.0804333106. [DOI] [PMC free article] [PubMed] [Google Scholar]