

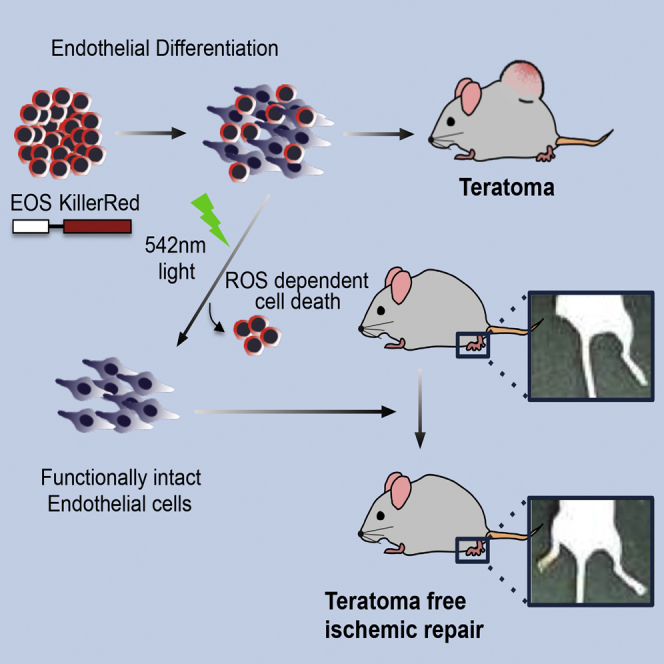
Summary
Stem-toxic small molecules have been developed to induce selective cell death of pluripotent stem cells (PSCs) to lower the risk of teratoma formation. However, despite their high efficacies, chemical-based approaches may carry unexpected toxicities on specific differentiated cell types. Herein, we took advantage of KillerRed (KR) as a suicide gene, to selectively induce phototoxicity using visible light via the production of reactive oxygen species. PSCs in an undifferentiated state that exclusively expressed KR (KR-PSCs) were eliminated by a single exposure to visible light. This highly selective cell death in KR-PSCs was exploited to successfully inhibit teratoma formation. In particular, endothelial cells from KR-mPSCs remained fully functional in vitro and sufficient to repair ischemic injury in vivo regardless of light exposure, suggesting that a genetic approach in which KR is expressed in a tightly controlled manner would be a viable strategy to inhibit teratoma formation for future safe PSC-based therapies.
Keywords: pluripotent stem cells, KillerRed, phototoxicity, teratoma, ischemic injury, vasculogenesis
Graphical Abstract
Highlights
-
•
Pluripotent specific KR induces ESC specific cell death by light exposure
-
•
Specific cell death of KR-ESCs by light exposure inhibits teratoma formation
-
•
ECs from KR-ESCs remain fully functional in vitro and in vivo by light exposure
In this article, Cha, Moon, and colleagues show that KR, of which expression is tightly controlled only in an undifferentiated state of PSCs, induces ROS-dependent cell death by visual light exposure to inhibit teratoma formation. Endothelial cells derived from PSCs remain functionally intact in vitro and in vivo after light exposure, suggesting that selective phototoxicity by KR expression would be a valid approach for teratoma-free cell therapy.
Introduction
For more than a decade, multipotent stem cells have been extensively studied for various cardiovascular diseases such as heart failure and ischemic cardiomyopathy, which was defined as the “first-generation” cell therapies (Behfar et al., 2014). The “next-generation” cell therapies include pluripotent stem cells (PSCs) such as human embryonic stem cells (hESCs) and induced PSCs (iPSCs) in an effort to maximize therapeutic value of stem cells for cardiovascular diseases (Ban et al., 2014).
However, PSCs have major safety concerns, such as teratoma formation due to possible contamination of the residual undifferentiated PSCs (Lee et al., 2013a). Teratoma is a unique characteristic of PSCs owing to their pluripotency and ability to undergo unlimited proliferation (Gruen and Grabel, 2006). These properties (e.g., pluripotency and limitless proliferation), which theoretically allow the sufficient supply of cardiomyocytes, cardiac progenitors, and endothelial cells, make PSCs the most promising cell source for regenerative medicine (Robinton and Daley, 2012). The unlimited proliferation capacity of PSCs is similar to the proliferation of cancer cells, which results from high telomerase activity (Thomson et al., 1998) and constant inhibition of retinoblastoma protein (Burdon et al., 2002). Therefore, the presence of a few undifferentiated PSCs or incompletely differentiated cells that escape the strict sorting process would be a high risk factor for tumor development in safe stem cell therapies (Masuda et al., 2014).
However, teratoma or tumor formation following xenotransplantation of human (h)PSCs into animal models (mostly rodents) has been barely reported (Kriks et al., 2011, Laflamme et al., 2007), with a few exceptions (Brederlau et al., 2006, Seminatore et al., 2010). A recent study by Doi et al. (2012) demonstrated that extended differentiation of hPSCs reduces the risk of teratoma formation in a primate model. Therefore, stringent selection of differentiated cells by flow cytometry for selective sorting (Cho et al., 2007) or prolonged differentiation (Brederlau et al., 2006, Zhang et al., 2001) would be sufficient to avoid tumorigenicity. Nevertheless, considering the “host-dependent tumorigenicity” of ESCs (e.g., xenotransplantation of hPSCs into a rodent model) (Erdö et al., 2003), the risk of teratoma formation by hPSCs in humans remains to be resolved, especially given that several clinical trials of hPSC-based therapies are ongoing (Cyranoski, 2013). Not only mouse embryonic bodies (mEBs), but also differentiated cells from mouse embryonic stem cells (mESCs) can reportedly form teratomas when transplanted into a rodent model, even after stringent cell sorting (Arnhold et al., 2004, Fujikawa et al., 2005, Moon et al., 2013).
A number of approaches have been developed to reduce the risk of teratoma formation, including antibody-based selective elimination (Tang et al., 2011), small molecules (Ben-David et al., 2013, Lee et al., 2013b), and integration of a suicide gene (Li and Xiang, 2013). Among them, the use of various chemicals to achieve selective cell death of PSCs (referred as “stemotoxic” in a review, see Knoepfler, 2009) may have unexpected caveats, despite being highly efficient (Knoepfler, 2009). Additionally, there is no evidence indicating that such chemical treatment ensures functionality of differentiated cells in vivo (Masuda et al., 2014). Therefore, we attempted to eliminate undifferentiated PSCs without using small molecules and reveal that the differentiated cells after selective ablation of undifferentiated PSCs remain fully functional in vivo. To this end, KillerRed (KR), an artificial photosensitizer protein derived from a hydrozoan chromoprotein (Bulina et al., 2006) which produces reactive oxygen species (ROS) when exposed to visible light of 540–580 nm (Wang et al., 2012), was introduced into PSCs with a pluripotent-specific promoter such that it was only expressed in undifferentiated PSCs. A single treatment with visible light successfully eliminated KR-expressing mouse PSCs (KR-mPSCs) in an ROS-dependent manner, while endothelial cells (ECs) differentiated from KR-mPSCs survived and remained functional in vitro and in vivo. The similar results were repeated in a human ESC (hESC) model. Importantly, KR-mPSCs did not form teratomas after being exposed to the light, whereas ECs differentiated from KR-mPSCs effectively regenerated blood vessels under ischemic conditions in a rodent model. Our results strongly support and provide the “proof of concept” that a KR-based suicide gene approach in which visible light selectively induces phototoxicity in PSCs would be an efficient strategy to reduce the risk of tumorigenicity in PSC-based cardiovascular repair.
Results
Establishment of KR-mPSCs
We aimed to induce selective cell death of undifferentiated PSCs using visible light, which is supposedly harmless to normal cells. We sought to specifically express KR, a photosensitizer protein, in a pluripotency-dependent manner. To this end, we took advantage of previously developed pluripotency-specific EOS (early transposon promoter and OCT-4 and SOX2 enhancers) vector systems such as EOS-C(3+) (Hotta et al., 2009), which contains multimerized OCT-4 core enhancer element conserved region 4 (CR4) (Okumura-Nakanishi et al., 2005). KR expression was designed to localize to mitochondria by adding a mitochondrial-targeting sequence (MTS) (Figures 1A and 1B) to maximize the induction of cell death via the production of ROS (Rizzuto et al., 1995).
Figure 1.

Establishment of KR-mPSCs
(A) Schematic presentation of the vector used for stable transfection of mouse PSCs expressing KR under the control of the EOS-C(3+) promoter (mitochondrial target sequence: MTS).
(B) KR-mESCs treated with mitotracker (200 μM) for 45 min. The KR expression (red) and mitochondria (by mitotracker, green) co-localization was shown with DAPI counterstaining. The scale bar represents 10 μm.
(C) Phase contrast image and red fluorescence of KR-mESCs (KR).
(D) Reprogramming progress of reprogrammable MEFs transfected with EOS-C(3+)-KR plasmid observed by phase contrast and fluorescence microscopy (KR).
(E) Phase contrast image and red fluorescence of KR-miPSCs (KR). The scale bar in (C)–(E) represents 100 μm.
(F and G) Comparison of pluripotency marker expression between established mPSCs (Mock and KR). The results represent the mean ± SD of two independent experiments performed in triplicate.
The constructed plasmid (EOS-C(3+)-KR) was delivered to J1 mESC line (J1), and KR expressing mESCs were sorted based on their red fluorescence (Figure 1C). KR-mESCs formed teratomas consisting of ectodermal, mesodermal, and endodermal tissues, suggesting that the introduction of KR did not interfere with the pluripotency of mESCs (Figure S1A). Similarly, EOS-C(3+)-KR was introduced into reprogrammable mouse embryonic fibroblasts (MEFs) (Carey et al., 2010) and the cells were subsequently reprogrammed by doxycycline treatment (Figure 1D). Consistent to previous reports (Hotta et al., 2009), the fully reprogrammed cells were readily distinguished by their red fluorescence, owing to activation of the EOS promoter as pluripotency was acquired, in comparison with non-fluorescent control mouse iPSCs (miPSCs) (Figures 1D, 1E, and S1B). The established miPSCs expressing EOS-C(3+)-KR (KR-miPSCs) formed teratomas, similar to KR-mESCs (Figure S1C). While several typical pluripotency markers were equivalently expressed in KR-mESCs and KR-miPSCs compared with their parental cells, KR was exclusively expressed in KR-mPSCs (Figures 1F and 1G). These results indicate that the introduction of KR did not impair the pluripotency properties of PSCs.
Specific Expression of KR in a Pluripotent-Dependent Manner
Red fluorescence from KR expression driven by activation of the EOS-C(3+) promoter was distinct to KR-mPSCs (Figures 1C–1E); therefore, we next investigated whether expression of KR was suppressed as KR-mESCs underwent differentiation. Spontaneous differentiation of KR-mESCs was induced through embryonic body (EB) formation followed by monolayer culture (Figure 2A, inserted). Red fluorescence from KR was observed in the EB and gradually diminished over time (Figure 2A). Consistently, KR expression was suppressed concurrent with Nanog and Oct-4 suppression during spontaneous differentiation (Figure 2B). KR protein level was also markedly suppressed during differentiation (Figure S2A). Expression of marker genes of all three germ layers (Fgf5: ectoderm, Brachyury: mesoderm, and Sox17: endoderm) clearly increased in differentiated cells (after 6 days of differentiation) (Figure 2B). Similarly, red fluorescence from KR expression was only observed in OCT-4-positive, not in OCT-4-negative populations (cultured in a monolayer for 4 days after EB formation), indicating that KR was specifically expressed in undifferentiated PSCs (Figure 2C, white dotted line).
Figure 2.

Specific Expression of KR in a Pluripotent-Dependent Manner
KR-mESCs differentiated with monolayer-culture for 12 days after 3 days of EB formation for spontaneous differentiation (∗ indicates the day of sampling, inserted panel).
(A) Red fluorescence from KR indicative after differentiation monitored by phase contrast and fluorescence microscopy. The scale bar represents 100 μm.
(B) mRNA expression level of Oct-4, Nanog, and KR at indicative day (∗ in Figure 2A, inserted) during spontaneous differentiation of KR-mESCs by qPCR (top). The typical differentiation marker gene of three germ layers; Fgf5 for ectoderm, Brachyury for mesoderm, and Sox17 for endoderm measured at 6 days of monolayer-culture (bottom). The results represent the mean ± SD of two independent experiments performed in triplicate.
(C) KR-mESCs after 4 days of differentiation protocol (∗, inserted) were subject to immunostaining for OCT-4 with red fluorescence from KR (white dot line), DAPI for nuclear counterstaining. The scale bar represents 50 μm.
Induction of PSC-Specific Cell Death by Visible Light
KR expression was tightly controlled in a pluripotency-specific manner (Figure 2); therefore, irradiation with visible light of 540–580 nm was expected to induce cell death specifically in KR-mPSCs, not in differentiated cells. KR-mESCs displaying red fluorescence were completely eliminated within 6 hr after a single exposure to green light (1.3 W/cm2, 540 nm), whereas control mESCs that did not express KR had no apparent alteration (Figure 3A). The phototoxic effects in KR-mESCs were quantified by Annexin V/7-aminoactinomycin D (7-AAD) double staining. Around 90% of cells were positive for both Annexin V and 7-AAD at 24 hr after light exposure (Figure 3B). Apoptosis of KR-mESCs following light exposure was further validated by the dramatic increase in caspase-3 activity (Figure 3C). For clarification of the cell death rate of KR-mESCs after light exposure, dead cells were counted followed by trypan blue staining. The viability of light-exposed KR-mESCs was close to 0%, whereas that of control mESCs was slightly decreased after light exposure (Figure 3D). Morphological changes in KR-mESCs were examined at various time points after light exposure. Signs of cell death were evident within 9–10 hr (560–660 min) after light exposure in KR-mESCs, but not in control mESCs (Figure 3E; Movies S1 and S2). Similarly, KR-miPSCs were also killed by light exposure, as determined by Annexin V/7-AAD double staining (Figure S2B) and immunoblot analysis (Figure S2C).
Figure 3.

Induction of PSC-Specific Cell Death by Visible Light
KR-mESCs exposed to visual light, and the rate of cell death 24 hr after light exposure.
(A) Number of KR expressing cells after light exposure monitored by phase contrast and fluorescence microscopy (KR). The scale bar represents 100 μm.
(B and C) Apoptotic cell death of KR-mESCs by light exposure determined by FACS analysis for Annexin V and 7-AAD staining (B) and caspase-3 enzyme activity assay (C). The results represent one of the experiments performed twice (for annexin V staining) and thrice in duplicate plates (for caspase-3 activity). The quantitative data are mean ± SD (not significant: n/s and ∗∗∗p < 0.001).
(D) Cell viability of control and light exposed KR-mESCs determined by trypan blue assay (more than 400 cells were counted at each condition.) The results represent the mean ± SD of two independent experiments performed in duplicate.
(E) Morphological change of mESCs (Mock and KR) at indicative time after light exposure observed using a JuLI Smart fluorescent cell analyzer microscope.
ROS Generation in KR-mPSCs after Light Exposure Is Responsible for Phototoxicity
The phototoxicity of KR is owing to the generation of ROS (Bulina et al., 2006). Considering the high susceptibility of PSCs to oxidative damage (Han et al., 2008), ROS generation following KR activation was expected to be strongly associated with the induction of KR-mPSCs’ cell death. The level of ROS in KR-mESCs was monitored by fluorescence microscopy (Figure 4A) and flow cytometry (Figure 4B) using dichloro-dihydro-fluorescein diacetate (DCF-DA) as a fluorescent probe. Following light exposure, ROS generation was evident in KR-mESCs, but not in control mESCs. KR-mPSCs were pretreated with N-acetyl cysteine (NAC), a well-known antioxidant, prior to light exposure in an attempt to block ROS-dependent cell death. Apoptotic KR-mPSCs after light exposure, with or without NAC pretreatment, were quantified by fluorescence-activated cell sorting (FACS). This confirmed that ROS generation following light-induced KR activation induced KR-mPSCs’ cell death (Figure 4C). Consistently, KR-mPSCs underwent cell death after light exposure as determined by the detection of cleaved caspase-3, which was prevented by pretreatment with NAC (Figure 4D).
Figure 4.

ROS Generation in KR-mPSCs after Light Exposure Is Responsible for Phototoxicity
KR-mESCs exposed to visual light.
(A and B) The cells were incubated for 1 hr then stained with DCF-DA for 30 min. ROS levels were measured by fluorescence microscopy (A) and flow cytometry (B). The scale bar in (A) represents 50 μm. The quantitative data represent mean ± SD of experiments performed in three independent studies (∗∗p < 0.01).
(C) mPSCs (Mock and KR) pretreated with 5 mM of NAC for 1 hr prior to the light exposure. Apoptotic cell death of mPSCs after light exposure was determined by FACS analysis for Annexin V and 7-AAD dual staining. The results represent one of the experiments performed twice in duplicate plates.
(D) Apoptotic cells monitored by immunostaining analysis for cleaved caspase-3 (green), DAPI for nuclear counterstaining. The scale bar represents 100 μm.
Specific Induction of Cell Death in KR-mPSC
We demonstrated that KR is specifically expressed in pluripotent cells (Figure 2) and that ROS generation by KR (Figures 4A and 4B) is responsible for phototoxicity in KR-mPSCs (Figures 4C and 4D). Accordingly, it was hypothesized that cell death effects would not be elicited in cells differentiated from KR-mPSCs, in which KR expression was strongly suppressed. Such a differential response to visible light between KR-mPSCs and differentiated cells is crucial if this approach is to be used to reduce the risk of teratoma formation in clinical application. To test this, we compared the cell death of KR-mPSCs and cells differentiated from KR-mPSCs. When KR-mPSCs were partially differentiated (Figure 5A, insert), a portion of cells remained undifferentiated, according to their red fluorescence owing to KR expression (Figure 5A, top) and positive staining for stage-specific embryonic antigen-1 (SSEA-1; Figure 5A, bottom).
Figure 5.

Specific Induction of Cell Death in KR-mPSC
(A) Immunostaining for KR or SSEA-1 (red) and cleaved caspase-3 (green) with DAPI (blue) for partially differentiated KR-mESCs (4 days after monolayer culture, ∗ inserted panel) with or without light exposure. The scale bar represents 100 μm.
(B) Apoptotic cell death analyzed by FACS analysis for cleaved caspase-3 and KR (or SSEA-1) in KR-mPSCs with or without light exposure. The quantification of cleaved caspase-3-positive and -negative populations from KR (or SSEA-1) positive or negative populations is shown.
(C) Mixed cell population in 4 days monolayer culture of KR-mESCs (with SSEA-1+ and SSEA-1− population) exposed to visual light. Shown are live cells of the SSEA-1+ and SSEA-1− population 24 hr after light exposure was determined by FACS analysis with SSEA-1 antibody (black arrow indicates SSEA-1+ population, left). The number of SSEA-1+ population was quantified as a bar graph (right). The results represent one of the experiments performed twice in duplicate plates.
(D) Introduction of KR-miPSCs with or without light exposure through subcutaneous injection. Representative images of teratomas from KR-miPSCs with light exposure (0/6: zero out of six mice) and without (5/6: five out of six mice) are shown.
(E) Images of typical three germ layers from the teratoma stained with H&E staining, Masson’s trichrome, and Alcian Blue. The scale bar represents 50 μm.
(F) Teratomas were counted and these tumor sizes were measured 30 days after injection of cells (14 and 12 mice for control and light exposed group, respectively) (∗∗∗p < 0.001).
After light exposure, only KR-expressing or SSEA-1-positive cells (undifferentiated PSCs) underwent cell death as determined by the detection of cleaved caspase-3 (Figure 5A), indicating that only KR-mPSCs underwent cell death by light exposure. Next, to quantify the light-dependent cell death of differentiated cells and KR-mPSCs, FACS analysis was performed of cells differentiated for 4 days (Figure 5A, insert). Either SSEA-1 expression or red fluorescence was used to recognize undifferentiated KR-mPSCs. Light exposure selectively induced cell death in those that exhibited red fluorescence or were SSEA-1-positive (Figure 5B). For tumor-free cell therapy to be achieved, residual undifferentiated PSCs should be depleted following differentiation, as described in a previous study (Lee et al., 2013b). At 24 hr after light exposure to cell mixture from 4 days of differentiation (Figure 5A, insert), SSEA-1-positive cells were markedly depleted by a single exposure to light (Figure 5C).
As few as 500 mESCs are required to form a teratoma in a mouse model (Erdö et al., 2003), whereas at least 1 × 105 hESCs are required for teratoma formation in a mice xenograft model (Lee et al., 2009), possibly due to host-dependent bias (Erdö et al., 2003). For this reason, a mouse teratoma model combined with mPSC transplantation is a more suitable model to verify whether a strategy can inhibit teratoma formation. Since iPSCs develop teratomas more aggressively than ESCs, (Gutierrez-Aranda et al., 2010), we first examined whether light exposure could block teratoma formation by miPSCs. For maximization of teratoma formation, 1 × 106 of KR-miPSCs, which were exposed to light or not, were subcutaneously injected into mice and teratoma formation was monitored. If more than 500 injected cells survived following light exposure, teratomas would develop (Erdö et al., 2003). After 32 days, five of six mice injected with KR-miPSCs that had not been exposed to light developed evident teratoma-like masses, whereas none of the mice injected with KR-miPSCs that had been exposed to light developed teratoma-like masses (Figure 5D). Accordingly, the teratomas formed by KR-miPSCs contained tissues of all three germ layers (Figure 5E). In addition, 26 mice (14 and 12 mice for control and light-exposed group, respectively) were examined for determining teratoma formation. As consistently, none of the mice developed teratoma after light exposure prior to transplantation (0 of 12 mice) (Figures 5F and S5).
Light Exposure Does Not Affect the Function of ECs Differentiated from KR-mPSCs In Vitro
Although selective cell death toward mPSCs was sufficient to prevent teratoma formation (Figure 5), the function of differentiated cells must be preserved to conceptually prove that this approach has potential for future clinical applications. To prove that light exposure, which specifically induced the cell death of undifferentiated KR-mPSCs, is suitable to safely isolate differentiated cells, we differentiated KR-mESCs into ECs as described previously (Kim et al., 2008). In sorted ECs, large granules of von Willebrand factor (vWF) were dispersed throughout the cytoplasm (Figure S3A). The properties of ECs were briefly characterized by an acetylated low-density lipoprotein (Ac-LDL) uptake assay (Figure S3B). ECs derived from KR-mESCs (EC-KR-mESCs) remained functional after light exposure as determined by in vitro assays of tubule formation (Figure 6A) and Ac-LDL uptake (Figure 6B), with no obvious expression of active caspase-3 in any cells (Figure 6C). To support the selective cell death of KR-mESCs, but not EC-KR-mESCs, we mixed KR-mESCs prelabeled with CFSE fluorescent dye with EC-KR-mESCs and then subjected the cell mixture to the light exposure. Clear morphological change, indicating that cell death was manifested only in KR-mESCs after light exposure, while no sign of change was observed in EC-KR-mESCs (Movie S3). These data indicate that EC-KR-mESCs are not susceptible to light-induced cell death, unlike KR-mESCs.
Figure 6.

Functionally Intact in Endothelial Cells from KR-mESCs In Vitro and In Vivo after Light Exposure
(A) Tubule formation and (B) Ac-LDL (red) uptake assay performed 24 hr after light exposure. The scale bars represent 200 μm (A) and 100 μm (B), not significant: n/s. The results represent one of the experiments performed thrice.
(C) Cell death in ECs 24 hr after light exposure determined by immunostaining for cleaved caspase-3 and DAPI nuclear counterstaining. The scale bar represents 100 μm.
(D) Representative images of serial laser Doppler perfusion images (red to yellow) (top). Quantitative analysis of blood perfusion ratio of ischemic to non-ischemic hindlimb at indicative days is shown as a line graph (bottom). Data are mean ± SEM.
(E) Physiological status of ischemic limbs 24 days after PBS or cell transplantation. Standard scoring image of hindlimb of Limb salvage, Foot necrosis, Limb loss (top), and the recording number of cases for each condition with or without cell transplantation was summarized as a table (bottom).
(F) Histological and immunohistochemical analyses of normal limbs and ischemic limbs retrieved 24 days after treatment. Tissue regeneration and attenuated fibrosis were determined by H&E and trichrome Masson’s staining and detection of capillary formation was by anti-PECAM in serial-sectioned tissues. The scale bar represents 200 μm (Normal and EC-KR-mESCs) and 500 μm (PBS).
(G) Testis injection of KR-mESCs (1 × 106 cells containing 50% of KR-mESCs and 50% of EC-KR-mESCs) 24 hr after light exposure, 8 weeks after transplantation, representative images of teratoma mass (top) and normal testis (bottom) from mice transplanted with a cell mixture.
(H) Teratomas were counted and these tumor sizes were measured 51 days after injection of cells into total 20 mice (10 mice for each group) (∗∗∗p < 0.001).
(I) Fluorescent images of DiI (red) and DAPI (blue) for identifying ECs and nucleus, respectively. The squares with dotted lines were shown in the separate images with high-magnification field of each area (right and left). The scale bar represents 100 μm.
Intact Vasculogenesis by EC-KR-mESCs without Teratoma Formation after Light Exposure
EC-KR-mESCs were fully functional in vitro regardless of light exposure (Figures 6A and 6B), and this supports the notion that EC-KR-mESCs isolated after light exposure would be an optimal cell source for recovery from ischemic injury without the risk of teratoma formation. For investigation of the therapeutic effects of EC-KR-mESCs, the cells were injected into hindlimb ischemic models, with or without light exposure. Changes in blood flow in the ischemic hindlimb were determined by laser Doppler perfusion imaging analysis at various time points. Blood perfusion was markedly higher in ischemic hindlimbs injected with EC-KR-mESCs, both exposed to light or not, than in those injected with PBS (Figure 6D, top). At 24 days after cell transplantation, the blood flow ratio (ischemic to non-ischemic limb), both EC-KR-mESCs regardless of light exposure (n = 5), markedly increased in comparison to the PBS group (n = 5) (Figure 6D, bottom). This difference was also obvious when assessing the histological status of ischemic limbs. Mice injected with EC-KR-mESCs exposed to light showed a clear improvement in limb salvage (n = 2/5) and foot necrosis (n = 3/5) compared to mice injected with PBS, who exhibited extensive necrosis of the ischemic hindlimb and severe limb loss by autoamputation (n = 4/5) (Figure 6E). Histological analysis was performed of muscle degeneration and fibrosis in ischemic limbs at 24 days after injection of PBS (Figure 6F, PBS) or EC-KR-mESCs exposed to light or not (Figure 6F, Cont or Light exposure, respectively), as well as muscles from non-ischemic limbs (Figure 6F, Normal). This revealed that muscles of PBS-injected mice displayed massive degeneration, abnormal structures, and fibrosis in contrast to EC-KR-mESC groups (Figure 6F). Similarly, platelet EC adhesion molecule (PECAM) staining of serial-sectioned tissues was performed to assess capillary formation in ischemic areas, revealing that transplantation of EC-KR-mESCs significantly enhanced the formation of PECAM-positive capillaries (Figure 6F, white arrows). Moreover, EC-KR-mESCs, prelabeled with dialkylcarbocyanime (DiI) prior to engraftment, were observed in the ECs-transplanted tissue (Figure S3C). In particular, DiI positive cells were found in the area with smooth muscle actin (SMA; Figure S3D, open arrows) staining (Figure S3D, closed arrows), indicating that EC-KR-mESCs contribute to vasculogenesis normally even after light exposure. These results highlight the safety of light exposure to differentiated cells from KR-mESCs.
For this strategy to be applicable to cell therapy, transplantation of a mixed population of cells containing undifferentiated PSCs should not lead to teratoma formation following light exposure. Finally, to verify that light exposure prevents teratoma formation by KR-mESCs, a mixed cell population (1 × 106 cells, 50% EC-KR-mESCs and 50% KR-mESCs) was exposed to light or not then transplanted into the mouse testis. As predicted, light exposure prior to transplantation successfully inhibited the formation of teratoma mass including all three germ-layer tissues at 8 weeks after transplantation, whereas such masses were seen in mice injected with cells that had not been exposed to light (Figures 6G and S4). For statistical validation, the same experiments were repeated with total 20 mice (10 mice for each group) and showed that none of mice developed teratoma after light exposure (Figures 6H and S6). Most importantly, DiI prelabeled ECs were found in the testis tissues from one with teratoma and without teratoma (Figure 6I), suggesting that light exposure prior to cell injection, selectively ablates KR-mESCs, but not EC-KR-mESCs (Figure 6H).
Selective Cell Death by Light Exposure of KR-hESCs
We also established KR-expressing hESCs (KR-hESCs) based on H9 hESCs using lentiviral transduction system as described in experimental procedures (Figure 7A). For an efficient sorting, EGFP was conjugated along with KR. As shown in Figures 7B–7D, KR expression in KR-hESCs was confirmed at mRNA level (Figure 7B) and protein level (Figures 7C and 7D) after cell sorting. As similar to the case of mPSCs (Figures 1F and 1G), KR expression in hESCs gave a marginal effect on the expression level of typical pluripotency specific transcription factors such as OCT-4, SOX2, and NANOG (Figure S7A). KR expression was suppressed during differentiation as OCT-4 and NANOG expression were decreased (Figure S7B). Upon visible light exposure, KR-hESCs underwent cell death in a comparable manner to that observed with mESCs (Figure 3), which was determined by morphological changes (Figure 7E), immunoblotting analysis (Figure 7F), and Annexin V staining (Figure 7G). These data indicate that selective cell death with KR expression and visual light exposure can be also achieved in a hESC model.
Figure 7.

Selective Cell Death by Light Exposure of KR-hESCs
(A) Schematic presentation of the lentiviral vector used for stable transduction of hESCs expressing KR under the control of the EOS-S(4+) promoter (mitochondrial target sequence: MTS).
(B) Comparison of KR expression between established hESCs (GFP and KR) by qPCR. The results represent the mean ± SD of three independent experiments (∗∗∗p < 0.001).
(C) Green and red fluorescence of KR-hESCs with DAPI counterstaining. The scale bar represents 10 μm.
(D) Relative expression level of pluripotency markers (OCT-4, SOX2, and NANOG) between GFP and KR-hESCs by immunoblotting analysis.
(E) Light microscopic images of hESCs (GFP and KR) 24 hr after light exposure. The scale bar represents 500 μm.
(F) Cell death was determined by immunoblotting for PARP cleavage and α-tubulin was used as an equal loading control.
(G) Flow cytometry plots of Annexin V and 7-AAD staining 24 hr after light exposure (left) and a bar graph of relative level of cell death population (right). The results represent one of the experiments performed twice.
Discussion
Many small molecules, including two reported by our group (Lee et al., 2013b), have been suggested to be selectively toxic to PSCs. However, as described previously (Knoepfler, 2009), despite the high potency and efficacy of small molecules that target hPSC-specific genes (e.g., BIRC5) (Lee et al., 2013b), there may be unexpected side effects on certain differentiated cell types owing to unexpected expression of the particular target protein (e.g., BIRC5 expression in hematopoietic stem cells, see Leung et al., 2007). Although the small molecules used in previous studies had no significant effects on cellular functions, at least those of dopaminergic neurons and smooth muscle cells (Lee et al., 2013b), such possible side effects need to be carefully examined for other small molecules.
As selective cell death of undifferentiated hPSCs is crucial to guarantee functional safety of differentiated cells during a treatment, development of alternative strategies based on unique gene expression and properties of undifferentiated PSCs is required. In this respect, KR, which produces ROS upon exposure to visible light of 540–580 nm (Bulina et al., 2006), was synergized with the unique features of PSCs, namely, their high sensitivity to oxidative stress (Han et al., 2008) and high susceptibility to mitochondrial-dependent apoptosis (Lee et al., 2013b) to develop an alternative strategy.
To this end, KR was expressed at the mitochondria (Figure 1B), and this expression was tightly controlled by EOS-C(3+), an artificial pluripotent-specific promoter (Okumura-Nakanishi et al., 2005) (Figure 2). ROS generated upon exposure of KR to light specifically induced mPSC cell death (Figure 5), and the PSCs specific cell death by KR expression blocked teratoma formation in a mouse model (Figures 5 and 6). More importantly, EC-KR-mESCs were functional in vitro and in vivo (Figure 6) after exposure to light at an intensity that was sufficient to inhibit teratoma formation (Figures 6G and 6H). This approach does not use additional chemicals, unlike the typical suicide gene-based approach, which uses thymidine kinase and ganciclovir, which may have cell-type-dependent cytotoxicity (Rong et al., 2012, Janoly-Dumenil et al., 2009) or hepatotoxicity (Shea et al., 1987).
Despite numerous studies demonstrating that teratomas do not form following xenotransplantation of cells differentiated from hPSCs into a rodent model (Cho et al., 2007, Kriks et al., 2011), review articles continue to warn of the risk of teratoma or tumor formation in hPSC-based therapies (Cunningham et al., 2012, Lee et al., 2013a). Cells derived from mPSCs are apparently more prone to develop tumors in a mouse model (Arnhold et al., 2004, Moon et al., 2013), consistent with host-dependent bias (Erdö et al., 2003). Thereby, it is most appropriate to verify a teratoma inhibition strategy using mPSCs in a mouse model to exclude the host-dependent bias. Once the strategy is validated, it can be applied to hPSCs.
To this end, we also established KR-hESCs to examine selective cell death by light exposure. Of interest, we noticed that cell death rate by light exposure in KR-hESCs was significantly lower compared to ∼100% cell death achieved in mESCs under the identical condition (Figure 3). We reasoned that, unlike in mESCs (Figure S7D), KR protein in hESCs underwent a consistent proteasome-dependent degradation despite its high mRNA expression level (Figure S7C). This notion was further supported by pretreatment of MG132, a proteasome inhibitor, which significantly increased KR protein level in KR-hESCs (Figure S7E). Recently, it has been reported that hESCs show high proteasome activity due to high expression of 19S proteasome subunit (Vilchez et al., 2012). Considering a variety of cellular and molecular differences between hESCs and mESCs have been addressed by distinct characteristics of the primed (for hESCs) and naive (for mESCs) state, respectively (Nichols and Smith, 2009), the different protein stability of KR between hESCs and mESCs might result from the increased proteasome activity in hESCs, which may be linked to distinct characteristics of the primed state of ESC.
A recently developed gene-editing technology using RNA-guided endonuclease (Gaj et al., 2013), which allows the desired gene to be inserted into a genomic safe harbor (GSH) area (Papapetrou et al., 2011), should be considered to avoid undesirable random genomic insertion of EOS-C(3+)-KR in the generation of hPSCs that do not give rise to teratomas by simple light exposure. Especially, when applied to generate KR-human iPSCs, guided insertion of EOS-C(3+)-KR into the GSH area of parental cells allowed for red fluorescence to be emitted when reprogramming was complete to properly identify iPSC colonies (Hotta et al., 2009), similar to the mouse model reported in the current study (Figure 1D).
In summary, we demonstrated that expression of EOS-KR in PSCs induces selective cell death specifically in undifferentiated PSCs upon exposure to visible light and inhibits teratoma formation without any additional chemical treatment. ECs differentiated from KR-mESCs were able to repair ischemic damage after light exposure. Therefore, this suicide gene-based approach represents a proof of concept to generate a safe source of hPSCs that do not give rise to teratomas or tumors.
Experimental Procedures
Details of the methods are available in the Supplemental Experimental Procedures.
Generation of Transgenic PSC Clones
The KR backbone vector, designed to express at the mitochondria (Bulina et al., 2006), was purchased from Evrogen (cat# FP964) for transgenic mPSC clones. Another plasmid was reconstructed for transgenic hESC clones using CSII-EF-EGFP lentiviral vector as a backbone. A vector containing the coding sequence for a EOS-C(3+) and -S(4+) cassettes (Hotta et al., 2009) were provided by Dr. Kwang-Soo Kim (Harvard Medical School). The EOS-C(3+) coding sequence was amplified by PCR and sub-cloned into the linearized KR backbone after the removal of CMV promoter in backbone vector. The EOS-S(4+) sequence was sub-cloned at the front of the KR sequence and then the EOS-S(4+)-KR cassette was sub-cloned into the CSII-EF-EGFP lentiviral backbone vector. In the resulting gene cassette, the EOS-C(3+) and -S(4+) promoters drive KR expression.
MEFs from the reprogrammable mice Col1a1tm3(tetO-Pou5f1,-Sox2,-Klf4, and -Myc)Jae/J) (Carey et al., 2010) was isolated in a standard protocol. J1 mouse ES cells and reprogrammable MEFs were transfected with a EOS-C(3+)-KR plasmid that had a neomycin gene that allowed for cell selection. Cell transfection was performed using Neon Transfection System (Life Technologies) according to the manufacturer’s instructions. Reprogrammable MEFs were transfected with EOS-C(3+)-KR plasmid to induce KR and treated with 2 μg/ml doxycycline (Sigma-Aldrich) to induce reprogramming. Culture medium was replaced every 2 days by mouse ESC medium supplemented with 2 μg/ml doxycycline (Sigma-Aldrich). Positive colonies were selected for mESC expressing KR using 200 μg/ml of neomycin, and selection of colony emitting red fluorescent colonies, and were maintained. H9 hESCs were transduced with a CSII-EF-EGFP-EOS-S(4+)-KR plasmid using lentiviral transduction system. EGFP and KR positive cells were sorted by flow cytometry and maintained as previously described (Lee et al., 2013b).
Endothelial Tube Formation and Ac-LDL Uptake Assay
Endothelial tube formation assay was performed by sorting EC-KR-mESCs (5 × 104 cells), which were subsequently seeded on 4-well matrigel-coated plates (BD Biosciences) and incubated at 37°C for 12 hr. For the Ac-LDL uptake assay, EC-KR-mESCs were incubated with 10 mg/ml DiI-labeled Ac-LDL (Biomedical Technologies) at 37°C for 4 hr. After the cells were washed three times with PBS, images were analyzed using fluorescence microscopy (Nikon).
Teratoma Formation
The pluripotency of KR-mPSCs were examined in vivo through a teratoma formation assay using two distinct methods: subcutaneous inoculation and direct testicular injection. First, 5 × 106 undifferentiated KR-miPSCs (with or without light-exposure) were inoculated subcutaneously under the right and left front leg of female BALB/c-nude mice. There are two groups of light-exposed KR-miPSCs (n = 12) and control KR-miPSCs (n = 14). For direct testicular injection, undifferentiated KR-mESCs were mixed with EC-KR-mESCs for a total count of 5 × 106 cells. Cell types were categorized as KR-mESCs + EC-KR-mESCs (n = 10), and KR-mESCs + EC-KR-mESCs (light, n = 10), and this routine was performed twice. All light-exposed groups did not form teratomas except the control KR-miPSCs group and KR-mESCs + EC-KR-mESCs group that underwent subcutaneous injection and testicular injection, respectively. All mice were euthanized 30 days or 51 days after the inoculation for KR-miPSCs or mixed population, respectively. Tumor volumes were calculated using the formula: V = a × b × c × π / 6 (a = length, b = width, and c = depth). All animal care and experimental procedures were performed under the approval of the animal care committees in Konkuk University (IACUC No. KU 13125-1).
Hindlimb Ischemia Model and Cell Transplantation
The 6-week-old female nude mice (body weight 25-30 g) (Orient bio) were anesthetized with rompun (20 mg/kg) and ketamine (100 mg/kg). The femoral artery and its branches were ligated through a skin incision with a 6-0 silk (Ethicon). The external iliac artery and all of the above arteries were then ligated. The femoral artery was excised from its proximal origin as a branch of the external iliac artery to the distal point where it bifurcates into the saphenous and popliteal arteries. Prior to transplantation, cells were labeled with CM-DiI (DiI, Molecular Probes) to detect survival and engraftment in vivo. The number of mES-ECs (1 × 105 cells/mouse) were suspended in 200 μl of PBS then injected intramuscularly into four sites of the gracilis muscle in the medical thigh with a 29-gauge insulin syringe.
Statistical Analysis
The graphical data were presented as mean ± SD (error bars represent the SD). Statistical significance among groups and between groups were determined using one-way or two-way ANOVA following Bonferroni post-test and Student’s t test, respectively. Significance was assumed for p < 0.05(∗), p < 0.01 (∗∗), and p < 0.001 (∗∗∗).
Acknowledgments
We thank Jae-Ung Lee and Seoncheol Cha for providing technical support of light exposure. We also thank Dr. Min-Joon Hahn and Yong-Hwan Kim for useful comments and suggestions. This work was supported by the National Research Foundation of Korea (NRF) grant (2014R1A2A2A01005970) and the Korea Health Industry Development Institute (KHIDI) grants (HI14C3365 and HI14C2755020014), funded by the Korea government.
Published: November 12, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and three movies and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.10.004.
Contributor Information
Sung-Hwan Moon, Email: sunghwanmoon@kku.ac.kr.
Hyuk-Jin Cha, Email: hjcha@sogang.ac.kr.
Supplemental Information
{kind=link}
{kind=link}
The initial green fluorescence signal results from CFSE prelabeled KR-mESCs.
{kind=link}
References
- Arnhold S., Klein H., Semkova I., Addicks K., Schraermeyer U. Neurally selected embryonic stem cells induce tumor formation after long-term survival following engraftment into the subretinal space. Invest. Ophthalmol. Vis. Sci. 2004;45:4251–4255. doi: 10.1167/iovs.03-1108. [DOI] [PubMed] [Google Scholar]
- Ban K., Park H.J., Kim S., Andukuri A., Cho K.W., Hwang J.W., Cha H.J., Kim S.Y., Kim W.S., Jun H.W., Yoon Y.S. Cell therapy with embryonic stem cell-derived cardiomyocytes encapsulated in injectable nanomatrix gel enhances cell engraftment and promotes cardiac repair. ACS Nano. 2014;8:10815–10825. doi: 10.1021/nn504617g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behfar A., Crespo-Diaz R., Terzic A., Gersh B.J. Cell therapy for cardiac repair--lessons from clinical trials. Nat. Rev. Cardiol. 2014;11:232–246. doi: 10.1038/nrcardio.2014.9. [DOI] [PubMed] [Google Scholar]
- Ben-David U., Gan Q.F., Golan-Lev T., Arora P., Yanuka O., Oren Y.S., Leikin-Frenkel A., Graf M., Garippa R., Boehringer M. Selective elimination of human pluripotent stem cells by an oleate synthesis inhibitor discovered in a high-throughput screen. Cell Stem Cell. 2013;12:167–179. doi: 10.1016/j.stem.2012.11.015. [DOI] [PubMed] [Google Scholar]
- Brederlau A., Correia A.S., Anisimov S.V., Elmi M., Paul G., Roybon L., Morizane A., Bergquist F., Riebe I., Nannmark U. Transplantation of human embryonic stem cell-derived cells to a rat model of Parkinson’s disease: effect of in vitro differentiation on graft survival and teratoma formation. Stem Cells. 2006;24:1433–1440. doi: 10.1634/stemcells.2005-0393. [DOI] [PubMed] [Google Scholar]
- Bulina M.E., Chudakov D.M., Britanova O.V., Yanushevich Y.G., Staroverov D.B., Chepurnykh T.V., Merzlyak E.M., Shkrob M.A., Lukyanov S., Lukyanov K.A. A genetically encoded photosensitizer. Nat. Biotechnol. 2006;24:95–99. doi: 10.1038/nbt1175. [DOI] [PubMed] [Google Scholar]
- Burdon T., Smith A., Savatier P. Signalling, cell cycle and pluripotency in embryonic stem cells. Trends Cell Biol. 2002;12:432–438. doi: 10.1016/s0962-8924(02)02352-8. [DOI] [PubMed] [Google Scholar]
- Carey B.W., Markoulaki S., Beard C., Hanna J., Jaenisch R. Single-gene transgenic mouse strains for reprogramming adult somatic cells. Nat. Methods. 2010;7:56–59. doi: 10.1038/nmeth.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.W., Moon S.H., Lee S.H., Kang S.W., Kim J., Lim J.M., Kim H.S., Kim B.S., Chung H.M. Improvement of postnatal neovascularization by human embryonic stem cell derived endothelial-like cell transplantation in a mouse model of hindlimb ischemia. Circulation. 2007;116:2409–2419. doi: 10.1161/CIRCULATIONAHA.106.687038. [DOI] [PubMed] [Google Scholar]
- Cunningham J.J., Ulbright T.M., Pera M.F., Looijenga L.H. Lessons from human teratomas to guide development of safe stem cell therapies. Nat. Biotechnol. 2012;30:849–857. doi: 10.1038/nbt.2329. [DOI] [PubMed] [Google Scholar]
- Cyranoski D. Stem cells cruise to clinic. Nature. 2013;494:413. doi: 10.1038/494413a. [DOI] [PubMed] [Google Scholar]
- Doi D., Morizane A., Kikuchi T., Onoe H., Hayashi T., Kawasaki T., Motono M., Sasai Y., Saiki H., Gomi M. Prolonged maturation culture favors a reduction in the tumorigenicity and the dopaminergic function of human ESC-derived neural cells in a primate model of Parkinson’s disease. Stem Cells. 2012;30:935–945. doi: 10.1002/stem.1060. [DOI] [PubMed] [Google Scholar]
- Erdö F., Bührle C., Blunk J., Hoehn M., Xia Y., Fleischmann B., Föcking M., Küstermann E., Kolossov E., Hescheler J. Host-dependent tumorigenesis of embryonic stem cell transplantation in experimental stroke. J. Cereb. Blood Flow Metab. 2003;23:780–785. doi: 10.1097/01.WCB.0000071886.63724.FB. [DOI] [PubMed] [Google Scholar]
- Fujikawa T., Oh S.H., Pi L., Hatch H.M., Shupe T., Petersen B.E. Teratoma formation leads to failure of treatment for type I diabetes using embryonic stem cell-derived insulin-producing cells. Am. J. Pathol. 2005;166:1781–1791. doi: 10.1016/S0002-9440(10)62488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T., Gersbach C.A., Barbas C.F., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruen L., Grabel L. Concise review: scientific and ethical roadblocks to human embryonic stem cell therapy. Stem Cells. 2006;24:2162–2169. doi: 10.1634/stemcells.2006-0105. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Aranda I., Ramos-Mejia V., Bueno C., Munoz-Lopez M., Real P.J., Mácia A., Sanchez L., Ligero G., Garcia-Parez J.L., Menendez P. Human induced pluripotent stem cells develop teratoma more efficiently and faster than human embryonic stem cells regardless the site of injection. Stem Cells. 2010;28:1568–1570. doi: 10.1002/stem.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M.K., Song E.K., Guo Y., Ou X., Mantel C., Broxmeyer H.E. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008;2:241–251. doi: 10.1016/j.stem.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta A., Cheung A.Y., Farra N., Vijayaragavan K., Séguin C.A., Draper J.S., Pasceri P., Maksakova I.A., Mager D.L., Rossant J. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat. Methods. 2009;6:370–376. doi: 10.1038/nmeth.1325. [DOI] [PubMed] [Google Scholar]
- Janoly-Dumenil A., Rouvet I., Bleyzac N., Bertrand Y., Aulagner G., Zabot M.T. Effect of duration and intensity of ganciclovir exposure on lymphoblastoid cell toxicity. Antivir. Chem. Chemother. 2009;19:257–262. doi: 10.1177/095632020901900605. [DOI] [PubMed] [Google Scholar]
- Kim G.D., Kim G.J., Seok J.H., Chung H.M., Chee K.M., Rhee G.S. Differentiation of endothelial cells derived from mouse embryoid bodies: a possible in vitro vasculogenesis model. Toxicol. Lett. 2008;180:166–173. doi: 10.1016/j.toxlet.2008.05.023. [DOI] [PubMed] [Google Scholar]
- Knoepfler P.S. Deconstructing stem cell tumorigenicity: a roadmap to safe regenerative medicine. Stem Cells. 2009;27:1050–1056. doi: 10.1002/stem.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S., Shim J.W., Piao J., Ganat Y.M., Wakeman D.R., Xie Z., Carrillo-Reid L., Auyeung G., Antonacci C., Buch A. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme M.A., Chen K.Y., Naumova A.V., Muskheli V., Fugate J.A., Dupras S.K., Reinecke H., Xu C., Hassanipour M., Police S. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat. Biotechnol. 2007;25:1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- Lee A.S., Tang C., Cao F., Xie X., van der Bogt K., Hwang A., Connolly A.J., Robbins R.C., Wu J.C. Effects of cell number on teratoma formation by human embryonic stem cells. Cell Cycle. 2009;8:2608–2612. doi: 10.4161/cc.8.16.9353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.S., Tang C., Rao M.S., Weissman I.L., Wu J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013;19:998–1004. doi: 10.1038/nm.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.O., Moon S.H., Jeong H.C., Yi J.Y., Lee T.H., Shim S.H., Rhee Y.H., Lee S.H., Oh S.J., Lee M.Y. Inhibition of pluripotent stem cell-derived teratoma formation by small molecules. Proc. Natl. Acad. Sci. USA. 2013;110:E3281–E3290. doi: 10.1073/pnas.1303669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung C.G., Xu Y., Mularski B., Liu H., Gurbuxani S., Crispino J.D. Requirements for survivin in terminal differentiation of erythroid cells and maintenance of hematopoietic stem and progenitor cells. J. Exp. Med. 2007;204:1603–1611. doi: 10.1084/jem.20062395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Xiang A.P. Safeguarding clinical translation of pluripotent stem cells with suicide genes. Organogenesis. 2013;9:34–39. doi: 10.4161/org.24317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda S., Miyagawa S., Fukushima S., Sougawa N., Ito E., Takeda M., Saito A., Sawa Y. Emerging innovation towards safety in the clinical application of ESCs and iPSCs. Nat. Rev. Cardiol. 2014;11:553–554. doi: 10.1038/nrcardio.2014.9-c1. [DOI] [PubMed] [Google Scholar]
- Moon J., Lee H.S., Kang J.M., Park J., Leung A., Hong S., Chung S., Kim K.S. Stem cell grafting improves both motor and cognitive impairments in a genetic model of Parkinson’s disease, the aphakia (ak) mouse. Cell Transplant. 2013;22:1263–1279. doi: 10.3727/096368912X657242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J., Smith A. Naive and primed pluripotent states. Cell Stem Cell. 2009;4:487–492. doi: 10.1016/j.stem.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Okumura-Nakanishi S., Saito M., Niwa H., Ishikawa F. Oct-3/4 and Sox2 regulate Oct-3/4 gene in embryonic stem cells. J. Biol. Chem. 2005;280:5307–5317. doi: 10.1074/jbc.M410015200. [DOI] [PubMed] [Google Scholar]
- Papapetrou E.P., Lee G., Malani N., Setty M., Riviere I., Tirunagari L.M., Kadota K., Roth S.L., Giardina P., Viale A. Genomic safe harbors permit high β-globin transgene expression in thalassemia induced pluripotent stem cells. Nat. Biotechnol. 2011;29:73–78. doi: 10.1038/nbt.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R., Brini M., Pizzo P., Murgia M., Pozzan T. Chimeric green fluorescent protein as a tool for visualizing subcellular organelles in living cells. Curr. Biol. 1995;5:635–642. doi: 10.1016/s0960-9822(95)00128-x. [DOI] [PubMed] [Google Scholar]
- Robinton D.A., Daley G.Q. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481:295–305. doi: 10.1038/nature10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Z., Fu X., Wang M., Xu Y. A scalable approach to prevent teratoma formation of human embryonic stem cells. J. Biol. Chem. 2012;287:32338–32345. doi: 10.1074/jbc.M112.383810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminatore C., Polentes J., Ellman D., Kozubenko N., Itier V., Tine S., Tritschler L., Brenot M., Guidou E., Blondeau J. The postischemic environment differentially impacts teratoma or tumor formation after transplantation of human embryonic stem cell-derived neural progenitors. Stroke. 2010;41:153–159. doi: 10.1161/STROKEAHA.109.563015. [DOI] [PubMed] [Google Scholar]
- Shea B.F., Hoffman S., Sesin G.P., Hammer S.M. Ganciclovir hepatotoxicity. Pharmacotherapy. 1987;7:223–226. doi: 10.1002/j.1875-9114.1987.tb03529.x. [DOI] [PubMed] [Google Scholar]
- Tang C., Lee A.S., Volkmer J.P., Sahoo D., Nag D., Mosley A.R., Inlay M.A., Ardehali R., Chavez S.L., Pera R.R. An antibody against SSEA-5 glycan on human pluripotent stem cells enables removal of teratoma-forming cells. Nat. Biotechnol. 2011;29:829–834. doi: 10.1038/nbt.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson J.A., Itskovitz-Eldor J., Shapiro S.S., Waknitz M.A., Swiergiel J.J., Marshall V.S., Jones J.M. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Vilchez D., Boyer L., Morantte I., Lutz M., Merkwirth C., Joyce D., Spencer B., Page L., Masliah E., Berggren W.T. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature. 2012;489:304–308. doi: 10.1038/nature11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Nartiss Y., Steipe B., McQuibban G.A., Kim P.K. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy. 2012;8:1462–1476. doi: 10.4161/auto.21211. [DOI] [PubMed] [Google Scholar]
- Zhang S.C., Wernig M., Duncan I.D., Brüstle O., Thomson J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001;19:1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The initial green fluorescence signal results from CFSE prelabeled KR-mESCs.