

The identification of a gene regulatory network related to seed longevity in both Medicago truncatula and Arabidopsis reveals a role for biotic defense-related genes in acquisition of longevity.
Abstract
Seed longevity, the maintenance of viability during storage, is a crucial factor for preservation of genetic resources and ensuring proper seedling establishment and high crop yield. We used a systems biology approach to identify key genes regulating the acquisition of longevity during seed maturation of Medicago truncatula. Using 104 transcriptomes from seed developmental time courses obtained in five growth environments, we generated a robust, stable coexpression network (MatNet), thereby capturing the conserved backbone of maturation. Using a trait-based gene significance measure, a coexpression module related to the acquisition of longevity was inferred from MatNet. Comparative analysis of the maturation processes in M. truncatula and Arabidopsis thaliana seeds and mining Arabidopsis interaction databases revealed conserved connectivity for 87% of longevity module nodes between both species. Arabidopsis mutant screening for longevity and maturation phenotypes demonstrated high predictive power of the longevity cross-species network. Overrepresentation analysis of the network nodes indicated biological functions related to defense, light, and auxin. Characterization of defense-related wrky3 and nf-x1-like1 (nfxl1) transcription factor mutants demonstrated that these genes regulate some of the network nodes and exhibit impaired acquisition of longevity during maturation. These data suggest that seed longevity evolved by co-opting existing genetic pathways regulating the activation of defense against pathogens.
INTRODUCTION
Seed maturation occurs as a set of intricate, overlapping developmental processes that occur from the end of embryogenesis until seeds become physiologically independent of the parent plant. It comprises a phase of seed storage reserve deposition and the less characterized phase of maturation drying. Furthermore, during maturation, seeds acquire a range of physiological traits that are crucial for successful seedling establishment in the field, such as vigorous and homogenous germination. These traits rely on the remarkable capacity of seeds to undergo complete desiccation without loss of viability (desiccation tolerance [DT]) and to remain alive for extended periods of time when stored in the dry state (longevity) (Buitink and Leprince, 2010). While DT is acquired during seed filling, longevity progressively increases during the final phase of seed maturation (Probert et al., 2009; Chatelain et al., 2012). Seed longevity is the ultimate survival mechanism for the plant in order to bridge unfavorable conditions. The search for the mechanisms regulating seed longevity is a longstanding goal in agriculture because of its impact on crop yields, especially in stressful conditions, and its importance as a means to preserve plant genetic resources.
So far, it has been established that the synthesis of protective molecules such as nonreducing sugars, late embryogenesis abundant proteins, and heat shock proteins is associated with longevity (Prieto-Dapena et al., 2006; Rosnoblet et al., 2007; Hundertmark et al., 2011). Furthermore, antioxidants such as glutathione (Kranner et al., 2006), tocopherols (Sattler et al., 2004), and flavonoids present in the testa (Debeaujon et al., 2000) also play a role in longevity by alleviating oxidation occurring during storage. Seed longevity is a quantitative trait for which genetic variation is observed among naturally occurring accessions and quantitative trait loci have been obtained in several crops (Bentsink et al., 2000; Nguyen et al., 2012; Nagel et al., 2015). These quantitative trait loci explain slight genetic variations, indicating that numerous genes influence longevity.
Several transcriptional activators have been identified that act during seed development, including the LAFL network (LEAFY COTYLEDON1 [LEC1], ABA INSENSITIVE3 [ABI3], FUSCA3 [FUS3], LEC2) and more specific regulators (e.g., VP1/ABI3-LIKE1 and HEAT SHOCK FACTOR A9 [HSFA9]) that interact with signaling hormonal responses brought by abscisic acid (ABA), gibberellins, and auxins. The LAFL transcription factor network regulates diverse seed-specific processes, including deposition of storage reserves (starch, storage proteins, and lipids), acquisition of desiccation tolerance, developmental arrest of the embryo, and dormancy (Santos-Mendoza et al., 2008). The contribution of these transcriptional activators in regulating downstream targets associated with longevity is poorly understood. To better understand how longevity is regulated during seed development, a systems biology approach provides a promising means to boost the identification of key longevity genes and their regulators.
Systems biology approaches are being increasingly used to monitor the global dynamics of entire biological circuits rather than analyzing the role of a few genes (Long et al., 2008). Genes encoding proteins that participate in the same pathway or that are part of the same protein complex are often coregulated. Therefore, clusters of genes with related functions often exhibit expression patterns that are correlated under a large number of diverse conditions. Based on the so-called “guilt by association” principle, coexpression networks have been proven to be very effective to identify relevant gene interactions (Ficklin et al., 2010; Amrine et al., 2015; Li et al., 2015) and to identify sets of candidate genes underlying specific phenotypes (Lee et al., 2009; Mutwil et al., 2010). Horvath and colleagues defined a “gene significance measure” as a means to provide a simple geometric interpretation of coexpression network analysis (Horvath and Dong, 2008). A trait-based gene significance measure assesses the relationship between a quantitative trait (such as weight) and the gene expression data. They demonstrated that the gene significance measure has great practical importance, since it allows one to incorporate external gene information into the network analysis.
Networks constructed from mixed sample sets represent a “global” or meta-analysis view of gene coexpression and have been obtained for many species (Faccioli et al., 2005; Lee et al., 2009; Ficklin et al., 2010; Schaefer et al., 2014). However, targeted networks, focusing on a specific condition, organ, or developmental phase, allow the dissection of fine regulatory switches that will otherwise be obscured through global comparisons (Usadel et al., 2009). One example is the SeedNet, a genome-wide network model describing transcriptional interactions, representing a binary phase transition study, in nongerminating (or dormant) and germinating Arabidopsis thaliana seeds (Bassel et al., 2011). Integration of the expression data at the resolution of developmental stages and in different environmental contexts is expected to generate a high confidence dynamic gene regulatory model (Long et al., 2008). Developing seeds represent an excellent model to test this hypothesis. In this study, we took advantage of the long maturation phase of Medicago truncatula seeds to create a robust regulatory coexpression network of seed maturation (MatNet). This was achieved by capturing the dynamics of 104 transcriptomes of developing M. truncatula seeds from plants grown under different environmental conditions. The integration of physiological data describing longevity acquisition under the different environmental conditions into MatNet, using the trait-based significance measure, led to the identification of a highly interconnected gene module related to longevity. Translation to Arabidopsis revealed a highly conserved regulatory cross-species network that was experimentally validated. Besides the identification of a number of expected functions, numerous genes in the longevity module were related to defense, suggesting that seed longevity evolved by co-opting existing genetic pathways regulating the activation of defense against pathogens.
RESULTS
Physiological and Transcriptional Profiling of Seed Traits during Development under Contrasting Environmental Conditions
To capture the effect of different environments on seed development, plants were transferred from flowering onwards to the following growth conditions: standard conditions (20/18°C), low temperature (14/11°C), high temperature (26/24°C), osmotic stress (20/18°C; soil water potential of −0.1 MPa) and greenhouse conditions (variable temperature and light). Seed development was characterized from the end of embryogenesis until final maturation drying (Figure 1). Environmental conditions strongly influenced the total developmental time, ranging from 25 to 72 days after pollination (DAP) under high and low temperature conditions, respectively (Figures 1A and 1E). Environmental conditions also affected the length of each developmental phase (embryogenesis, seed filling, and maturation drying, indicated by different colors in Figure 1F). The duration of the maturation drying phase was the most influenced (Figure 1F). Whereas the acquisition of DT appeared to be coupled with seed filling, being acquired when seed weight reached between 1 and 2 mg per seed (Figures 1A to 1E), the acquisition of seed longevity, measured as P50 (i.e., the time required for 50% of the seed lot to die during controlled storage at 35°C and 75% RH), appeared to be uncoupled from the other seeds traits. Both the duration of longevity acquisition and the final P50 were strongly influenced by environmental conditions.
Figure 1.

Seed Maturation of M. truncatula in Different Environmental Conditions.
(A) to (E) Physiological analysis describing the acquisition of quality traits (water content [white circles], dry weight [green squares], desiccation tolerance [red triangles], and longevity [P50, black circles]) in developing seeds under the following conditions: (A) high temperature (26/24°C), (B) osmotic stress (OS), (C) greenhouse (GH), (D) standard (20/18°C), and (E) low temperature (14/11°C) conditions. Data are means from three independent biological repetitions of 50 seeds; error bars show sd.
(F) Representation of the developmental phases in different environmental conditions: embryogenesis (yellow), seed filling (green), and maturation drying (blue).
(G) and (H) Principal component analysis of the transcriptome of each developmental phase (embryogenesis [yellow], seed filling [green], and maturation drying [blue]) in different environmental conditions (osmotic stress [diamonds], standard [inverted triangle] low temperature [square], greenhouse [triangle], and high temperature condition [circles]).
In parallel to the phenotypic characterization, 104 Medicago NimbleGen arrays were analyzed for gene expression in 49 conditions resulting from different stages of development and different environmental conditions. To allow data mining of this complete data set and to aid the visualization of the gene expression data by the scientific community, a “seed” section was created in the Medicago eFP Browser that complements the already existing eFP browser framework at the Bio-Analytic Resource (http://bar.utoronto.ca/efp_medicago/cgi-bin/efpWeb.cgi?dataSource=medicago_seed). A global overview of the changes in the transcriptomes during development is presented using a principal component analysis (Figures 1G and 1H). The kinetics of seed maturation are visible in the first and second component, representing 41 and 13% of the transcriptome variation, respectively. This variation was attributed to developmental mechanisms regulating phase transitions between seed filling and maturation drying independently of the environmental conditions (Figure 1G). The third component (representing 9% of the variation) explained most of the variation observed between the different growth conditions, with the transcriptome from seeds grown under osmotic stress forming a discrete group (Figure 1H).
Generation and Characterization of MatNet: A Robust Coexpression Network of Seed Maturation
To generate a coexpression network of seed maturation, a weighted gene coexpression network analysis was performed on differentially expressed probes (Zhang and Horvath, 2005; Glaab et al., 2009). A range of edge adjacency thresholds, controlling the trade-off between coverage of coexpression associations and strength of the associations (0 = maximum coverage; 1 = maximum association strength) were tested. An adjacency threshold value of 0.88 was found to provide both sufficient coverage and association strength for further analyses and was retained for the network generation (Supplemental Figure 1A). The resulting network (MatNet) contained 2912 nodes and 736290 weighted edges (Figure 2A; Supplemental Data Set 1). The degree distribution and power law plot indicated a resulting R2 of 0.213, which is rather low for indicating a power law typical for biological networks, but larger than for random networks (Supplemental Figure 1B) (Barabasi and Albert, 1999). However, the goal of MatNet is to provide a comprehensive collection of possible direct and indirect associations, thus focusing more on high coverage of potential interactions than on approximating the degree distribution of a typical biological network. In other words, the goal is to show all high correlations for both direct and indirect relations, not to estimate or represent direct physical interactions. The Cytoscape organic layout algorithm was used to visualize MatNet and revealed a spiral shaped network with a central cluster of interacting probes corresponding to genes with highest transcript level at the end of embryogenesis and during the seed filling phase (yellow-light green) and a tail composed of probes corresponding to genes with highest transcript level during the transition between seed filling and maturation drying phase (dark green) or increasing at final maturation (blue) (Figure 2A). The tail was connected to the central part of the network by a small number of negative connections (Figure 2A, red lines). To further characterize the network topology, transcriptional regulators were isolated from MatNet and clustered using K-means analysis on the gene expression data, revealing five main clusters (Figure 2B; Supplemental Data Set 2). These transcriptional regulators were projected onto MatNet (Figure 2C, red symbols). Using data from the Medicago Gene Expression Atlas MtGEA (He et al., 2009) and our own transcriptomic data sets (Verdier et al., 2013), we also identified probes preferentially expressed in seeds in comparison to other tissues. Out of the 2912 nodes present in MatNet, 198 had transcription levels >100-fold higher in seed versus non-seed tissues (blue symbols). Topological analysis showed that seed-specific genes were found in distinct modules (Figure 2C). The network is available for data mining at www.matnet.ml using an ad hoc interface, and the .cys file can be downloaded from https://www6.angers-nantes.inra.fr/irhs_eng/Research/Conserto.
Figure 2.

MatNet: A Stable Regulatory Coexpression Network of M. truncatula Seed Development.
(A) Weighted gene coexpression gene network of seed development of M. truncatula, visualized using an organic layout in Cytoscape. Temporal analysis of nodes in the network was obtained by coloring each gene by its specific expression profile along seed development. Numbers indicate the average time of seed development (in DAP) where the expression is maximum, derived from transcriptome data of seeds grown under standard conditions. Yellow-light green, high transcript levels during early seed development; dark green, transitory transcript levels; blue, transcript levels increasing during late seed development. Edges in red are negative connections.
(B) Expression profiles of TFs present in MatNet (cluster I to V).
(C) Projection of transcriptional regulators (red) and seed-specific genes (100× higher transcript levels in seed versus non-seed tissues [blue]). Expression profiles according to clusters I to V (B) are indicated.
A total of 250 transcription factors (TFs) and other transcriptional regulators (Supplemental Data Set 2) were distributed throughout the network, representing genes likely to exhibit sequential roles during seed development (Figure 2C). Relying on the high proportion of Arabidopsis homologous genes in MatNet, we further investigated the identity of the transcriptional regulators and their coregulated genes present in the different clusters of MatNet (Supplemental Data Set 2). Cluster I contained genes for which Arabidopsis homologous genes are implicated in proanthocyanidin and anthocyanin biosynthesis (TRANSPARENT TESTA2 [TT2], TT1, TT8, and ANTHOCYANINLESS2 [ANL2]) as well as mucilage production (MYB86), both processes being associated with testa differentiation during embryogenesis (Supplemental Data Set 2). Cluster II contained the master regulator FUS3 and several TFs implicated in the regulation of seed storage compound biosynthesis (WRINKLED1, LEC1-LIKE, and GLABRA2) and seed size (APETALA2), suggesting that this cluster represents seed filling. Cluster II also contained homologs of genes encoding two Arabidopsis ABA-responsive element binding proteins (ABSCISIC ACID RESPONSIVE ELEMENT BINDING FACTOR2 and 3) that regulate ABRE-dependent gene expression for ABA signaling. In cluster IV, TFs were mainly related to abiotic stress such as two DEHYDRATION RESPONSIVE ELEMENT BINDING PROTEIN genes (DREB2F and DREB2D) and a highly seed-specific HSF gene (HSF30). Finally, in cluster V, we found an Arabidopsis homolog of DREB2C, as well as TFs involved in flowering (putative NAC domain-containing protein 94 and FLOWERING bHLH4).
Identification of Gene Modules Related to Seed Longevity and Desiccation Tolerance
Coexpression networks have been proven to be effective to identify sets of candidate genes underlying specific phenotypes (Lee et al., 2009; Mutwil et al., 2010). Here, we investigated if MatNet could be used as a tool to identify gene modules linked to longevity. According to the trait-based gene significance measure (Horvath and Dong, 2008), P50 values obtained for the five developmental time series (Figure 3A) were used to identify transcripts with highly correlated expression profiles (Figure 3B). Depending on the environmental growth conditions, 2560 (in the standard condition) to 20,896 probes (high temperature condition) correlated with P50 (Pearson correlation coefficient [PCC] >0.85). A total of 498 probes were common to all environmental conditions (Figure 3B; Supplemental Data Set 3). When these probes were projected on MatNet, 167 of them formed a highly co-expressed module, representing 35% of cluster V (Figure 3C). We refer to this coexpression module as the “longevity” module (Figure 3D; Supplemental Data Set 3).
Figure 3.

Identification and Characterization of the Modules Related to Desiccation Tolerance and Longevity in MatNet.
(A) The evolution of longevity acquisition, expressed as P50 (days to obtain 50% germination during storage at 35°C, 75% RH), of developing seeds under the indicated environmental conditions. Data are the average of three replicates of 50 seeds (±se).
(B) Venn diagram of transcripts with expression profiles correlating with the acquisition of longevity (PCC > 0.85) for each environmental growth condition.
(C) Projection of the 498 probes common to all conditions in (B) on MatNet.
(D) Longevity module represented by the organic layout of Cytoscape. Larger nodes correspond to transcription factors.
(E) The evolution of DT acquisition, expressed as germination percentage after drying, of developing seeds under the indicated environmental conditions. Data are the average of three replicates of 50 seeds (±se).
(F) Venn diagram of transcripts that increase I(log2) > 3 upon the acquisition of DT for each environmental growth condition.
(G) Projection of the 200 probes common to all conditions in (F) on MatNet.
(H) DT module represented by the organic layout of Cytoscape. Larger nodes correspond to transcription factors.
Using a similar approach of trait-based gene significance measure, we identified a “desiccation tolerance” module. For the five developmental time series (Figure 3E), transcripts were selected whose levels increased 3-fold between desiccation sensitive and tolerant stages. A total of 200 probes were common to all environmental conditions (Figure 3F; Supplemental Data Set 4). Projection on MatNet showed that 143 probes formed the desiccation tolerance module in cluster V, localized before the longevity module (Figure 3G; Supplemental Data Set 4). The DT module contained six TFs, including three APETALA2/EREBP genes (DREB2F), a heat shock factor HSF30, TGA9, and an unknown putative TF (Figure 3H, large symbols).
Implication of Medicago ABI3 in the Regulation of the Desiccation Tolerance and Longevity Module Nodes
In Arabidopsis, both DT and longevity are under the control of the master regulator ABI3 (Ooms et al., 1993; Sugliani et al., 2009), suggesting that mechanisms conferring these two traits are controlled by the same regulatory pathways. However, our data demonstrate that the timing of the acquisition of longevity is much more influenced by the environmental conditions than DT (Figures 3A and 3E). This uncoupling suggests that the acquisition of these traits is controlled in part by independent regulatory pathways. To test this hypothesis and further elucidate the extent to which genes in the DT and longevity modules are under regulation of ABI3, we analyzed the transcriptomes of the desiccation-sensitive seeds of M. truncatula abi3 mutants at 32 and 40 DAP (Delahaie et al., 2013; Verdier et al., 2013) (Supplemental Data Set 5). These time points coincide with an increase in the transcript levels of the longevity-correlated genes in wild-type seeds. In the DT module, 85% of the nodes were significantly upregulated (Figure 4A, red nodes) or downregulated (Figure 4A, green nodes) in developing seeds of abi3 mutants, confirming the important role of ABI3 in the downstream regulation of these nodes. In the longevity module, 71 out of 167 probes were significantly downregulated in the abi3 mutants either at 32 or at 40 DAP (Figure 4A, green nodes). Also, 14 probes showed transcript levels that significantly increased in the Mtabi3 mutant at either 32 or 40 DAP (Figure 4A, red nodes). Whether the deregulation of the longevity module genes was due to a direct regulation by ABI3 or further downstream of the ABI3 pathway was investigated through the analysis of the transcriptome of hairy roots ectopically overexpressing M. truncatula ABI3 (GSE44291; Verdier et al., 2013). In the DT module, transcript levels of 43/143 (30%) nodes were significantly affected in the hairy roots, suggesting that their transcript levels are directly regulated by ABI3 (Figure 4A, blue nodes) (Supplemental Data Set 5). In contrast, in the longevity module, only five nodes had transcript levels that were significantly upregulated in the hairy roots. Overall, only 51% (85/167) of the probes of the longevity module were found to be affected in their expression in the M. truncatula abi3 seeds. This suggests that at least half of the longevity module nodes appear to be regulated via alternative pathways that are not part of the ABI3 pathway (Figure 4B).
Figure 4.

ABI3-Dependent and -Independent Regulation of Nodes in the Longevity and Desiccation Tolerance Module.
(A) Nodes of the longevity and DT module (Figure 3) that are significantly downregulated (green) or upregulated (red) in seeds of M. truncatula abi3 mutants compared with wild-type seeds at 32 and 40 DAP (P < 0.001) or upregulated by ectopic expression in hairy roots (log2 I > 1, P < 0.05) (blue).
(B) Nodes of the longevity and DT module (Figure 3) that are not deregulated in the abi3 mutants during seed development. Nodes indicated by big squares represent transcriptional factors.
Generation of a Cross-Species Longevity Network Module of M. truncatula and Arabidopsis
Whereas the long maturation phase of M. truncatula enabled us to finely decipher those genes related to longevity, functional validation of candidate genes in longevity is tedious. Since the conserved part of the longevity regulation will enable translational biology to other species, we investigated if it was possible to identify and validate an Arabidopsis-M. truncatula cross-species longevity module, taking advantage of the vast amount of coexpression data of Arabidopsis. First, we characterized when DT and longevity were acquired in Arabidopsis (Figure 5A; Supplemental Figure 2). DT was acquired between 10 and 12 DAP (Supplemental Figure 2A), while longevity (P50) was gradually acquired between 12 and 17 DAP (Supplemental Figure 2B). Further maturation until 20 DAP resulted in a slight decrease in the P50. Acquisition of both traits was thus very similar for both M. truncatula and Arabidopsis (Figures 5A and 5B). Also depicted in Figure 5 are the principal events occurring in both species during seed maturation: accumulation of storage reserves and nonreducing sugars (data from Arabidopsis taken from Baud et al., 2002). Besides the different storage compounds, one of the major differences between Arabidopsis and M. truncatula seeds is that in Arabidopsis, seed filling continues until final maturation and overlaps with the acquisition of longevity, whereas in M. truncatula, both events are separated in time.
Figure 5.

Comparative Analysis of Seed Maturation Events in M. truncatula and Arabidopsis.
(A) and (B) Characterization of the acquisition of different seed traits in Arabidopsis (A) and M. truncatula (B). Desiccation tolerance and longevity were derived from Supplemental Figure 2 and Figure 1D. Accumulation of storage reserves and nonreducing sugars is derived from Baud et al. (2002) for Arabidopsis and from Gallardo et al. (2007) for M. truncatula.
(C) Heat map of SCC values between seed developmental stages of M. truncatula (columns) and Arabidopsis (rows).
(D) Transcript levels of marker genes of seed development in M. truncatula and Arabidopsis. TT8 (At4g09820; Medtr1g072320), FUS3 (At3g26790; Medtr7g083700), ANAC100 (At5g61430; Mtr.41161.1.S1_at), EM6 (At2g40170; Medtr4g016960), and EM1 (At3g51810; Medtr7g069250). DD, dry mature seeds.
In order to identify comparable stages of seed development between Arabidopsis and M. truncatula, transcriptome data of different seed developmental stages were compared between both species by determination of the Spearman rank correlation coefficient (SCC) according to Patel et al. (2012). The SCC was calculated between nine seed developmental stages of Arabidopsis AtGenExpress Development Atlas (Schmid et al., 2005) and nine developmental stages of M. truncatula grown under standard conditions (Figure 5C). The Arabidopsis transcriptomes of the later developmental stages (stage 9 until dry seeds) overlap with a large window of M. truncatula development, between 23 and 44 DAP (Figure 5C). Comparison of transcript levels of marker genes of seed development also demonstrated that whereas expression of FUS3 is transitory in M. truncatula, levels remain high until final maturation in Arabidopsis (Figure 5D). These observations might be explained by the continuation of seed filling processes in Arabidopsis until final maturation, whereas in M. truncatula, seed filling has terminated at 28 DAP (Figures 5A and 5B).
Considering the overlap of the developmental programs, it was not possible to directly obtain the genes coregulating with longevity in Arabidopsis. For this reason, we used the nodes of the longevity module of M. truncatula and investigated whether the corresponding Arabidopsis homologous genes were connected using the extensive Arabidopsis interaction data present in Genemania (www.genemania.org). From the 167 nodes present in the M. truncatula longevity module, 130 Arabidopsis homologous genes were identified and submitted to Genemania. The final network contained 113 nodes and 497 edges that were mainly derived from coexpression data (431), together with 42 edges based on prediction and 23 edges based on colocalization. Thus, this analysis demonstrated that 113 genes were also connected with each other in Arabidopsis, demonstrating a high conservation of connectivity of the longevity module genes between both species (87%). Using these 113 genes, we generated a cross-species longevity network module with coregulated genes both for Arabidopsis and M. truncatula based on the Genemania layout (Figure 6A; Supplemental Data Set 6). A total of six TFs were present in this network (Figure 6A, large square symbols).
Figure 6.

Identification and Characterization of Cross-Species Longevity Network Module.
(A) Regulatory gene network with nodes representing both Arabidopsis and M. truncatula homologs that are coregulated in relation to longevity.
(B) Genes of colored nodes in (A) were functionally analyzed for longevity phenotype by comparing germination after 25 d of aging between mutant and wild-type seeds. The dashed line corresponds to the germination percentage of freshly harvested, non-aged seeds. The asterisk indicates a significant difference from the wild-type Col-0 seeds (χ2 test, P < 0.001).
(C) to (F) Genes with overrepresented cis-elements in M. truncatula (blue) and Arabidopsis (yellow) promoters; (C) response to auxin stimulus, (D) light regulated, (E) defense response to pathogen, and (F) SURE, sucrose binding element.
Validation and Characterization of the Cross-Species Longevity Network Module
To investigate the role of the network nodes in longevity, Arabidopsis T-DNA insertion mutants were phenotyped for 10 genes representing nodes of the cross-species network (Figure 6A, blue nodes; Supplemental Table 1). Genes were selected based on their elevated expression level in seeds compared with other tissues to reduce the risk of affecting plant development, thereby affecting the physiology of the seeds. No homozygous seeds could be obtained from the T-DNA insertion line of pre-mRNA polyadenylation factor FIP1 producing only 50% viable seeds, which were all wild type. Likewise, for bHLH25 mutants, only wild-type seeds were obtained and the remaining seeds were not viable, suggesting that these genes play an important role in seed development. For the other 10 genes, mutant plants were grown under controlled conditions together with the wild type (Col-0) and seeds were harvested at full maturity. No effect of the T-DNA insertion was observed on plant size or flowering time, with all plants flowering with less than a 7-d interval from Col-0. Freshly harvested seed lots from all lines showed no difference in dormancy, and after stratification, all seed lots germinated at 99 to 100% (Figure 6B, dashed line; Supplemental Figure 3). All together, these data indicate that general cellular integrity or fitness was not affected in the mutants. Next, longevity was determined on the freshly harvested seeds by controlled aging conditions (35°C and 75% RH). For most lines, germination percentage dropped faster during storage in the mutants than in the wild-type (Col-0) seeds (Figure 6B). For five out of eight genes, two mutant alleles showed significantly reduced survival during storage (Figure 6B). Complete survival curves are shown in Supplemental Figure 3.
Functional in silico characterization of the genes in the cross-species longevity module was performed exploiting the ATCOECIS online resource, which correlates Gene Ontology annotations with cis-regulatory elements, thereby allowing the identification of the transcriptional regulatory pathways controlling a specific set of genes (Vandepoele et al., 2009). The most representative cis-regulatory elements of the cross-species longevity module are involved in the response to pathogens, light, auxins, or sucrose (Table 1). Isolation and analysis of the promoters of M. truncatula identified similar cis-regulatory elements to be overrepresented in the M. truncatula longevity module (Table 2). Moreover, numerous genes of the cross-species network share the same cis-elements related to auxin, light, and defense between M. truncatula and Arabidopsis, suggesting a conserved regulation of the network nodes of these two species (Supplemental Data Set 6; Figures 6C to 6F). However, for the sucrose responsive binding elements, almost no overlap was found between species (Figure 6F), possibly reflecting their differential behavior in sugar accumulation (Figures 5A and 5B).
Table 1. Analysis of Significant cis-Regulatory Elements of the Genes Present in the Longevity Cross-Species Arabidopsis-M. truncatula Module.
| GO Categories Enriched with Motif (P Value < 0.001) | Motif | P Value | Enrichment Fold |
|---|---|---|---|
| Oxygen binding, transmembrane movement | ANTATTTC | 3.88E-05 | 27 |
| Oxygen binding, defense to pathogen | NGNACATA | 3.60E-04 | 24 |
| Defense response to pathogen | AANGTCAA | 4.19E-04 | 19 |
| Light-regulated genes | NNTGGATA | 6.33E-04 | 34 |
| Glucosyltransferase | AGAATATN | 6.88E-04 | 21 |
| Carboxylic acid biosynthesis | TANACGNA | 1.30E-03 | 19 |
| Carbohydrate biosynthesis | AANGNCTG | 2.53E-03 | 15 |
| Golgi apparatus | AGATCTNN | 3.45E-03 | 18 |
| Response to auxin stimulus | CTATATAN | 3.46E-03 | 22 |
| SURE2STPAT21 | AATACTAAT | 4.38E-03 | 10 |
| Response to auxin | ATANAGAG | 5.39E-03 | 17 |
| Defense response biotic | AAGTCNAC | 6.00E-03 | 7 |
| Response to wounding | ATATATNC | 7.14E-03 | 21 |
| Response to wounding | CGNNTATA | 9.27E-03 | 14 |
ATCOERCIS Web-based resource was used to determine cis elements enriched motifs using on the −1000 bp upstream of Arabidopsis promoters.
Table 2. Analysis of Significant cis-Regulatory Elements of the Genes Present in the Longevity Module of MatNet.
| Description | cis-Element | Sequence | P Value | O | E | N |
|---|---|---|---|---|---|---|
| Auxin-responsive factors | ARFAT | TGTCTC | 1.28E-112 | 59.62 | 6.33 | 15 |
| Light response promoter regions | IBOX | GATAAG | 5.98E-103 | 57.69 | 6.42 | 28 |
| Phosphate starvation-responsive gene | P1BS | GNATATNC | 2.11E-071 | 36.54 | 3.72 | 9 |
| ABA induced | DRE2COREZMRAB17 | ACCGAC | 3.20E-049 | 25.00 | 2.53 | 7 |
| Pathogen-responsive genes | GCCCORE | GCCGCC | 2.52E-042 | 17.31 | 1.46 | 1 |
| Gibberellin-responsive element | TATCCACHVAL21 | TATCCAC | 1.48E-035 | 19.23 | 2.07 | 1 |
| Sucrose-responsive element | SURE1STPAT21 | AATAGAAAA | 5.10E-031 | 17.31 | 1.92 | 2 |
O and E, observed and expected frequency in the longevity module; N, number of promoters whose Z score was >1.5. cis-elements were determined using the PLACE Web-based resource on the −1000 bp upstream of M. truncatula promoters.
Implication of WRKY3 and NFXL1 in the Acquisition of Longevity during Maturation
Considering that defense is one of the overrepresented biological functions of the longevity module nodes, two TFs present in the cross-species network were further characterized because of their known role in biotic stress, NFXL1 and WRKY3, the expressolog of M. truncatula WRKY33, which is present in the longevity module of this species. In Arabidopsis, transcripts of both genes increased during maturation and were more abundant in seeds compared with other tissues (Figures 7A and 7B; Supplemental Figure 4). Both homologous genes of M. truncatula exhibit similar expression profiles.
Figure 7.

Characterization of nfxl1 and wrky3 Mutants in Seed Maturation and Longevity Acquisition.
(A) Absolute expression of NFXL1 (At1g10170) in Arabidopsis seed development.
(B) Absolute expression of WRKY3 (At2g03340) in Arabidopsis development. Data are taken from the eFP browser 2.0 (www.bar.utoronto.ca).
(C) Representative picture of siliques and seeds of mutants and the wild type harvested at 15 DAP.
(D) and (E) Loss of germination during storage (75% RH, 35°C) of seeds at 15 DAP (dashed lines) or at maturity (solid lines) for the wild type, nfxl1 (D), and wrky3 (E).
(F) and (G) Germination of nfxl1 (F), wrky3 (G), and wild-type seeds after 14 d of incubation in ABA. For (D) to (G), 100 seeds of three biological replicates were tested. Germination between lines is significant when >8% (χ2 test, P < 0.05)
(H) and (I) Transcript levels of four network nodes: WRKY33 (I), HSPRO2 (II), NFXL1 (III), and LZF1 (IV). The asterisk indicates a significant difference from the wild-type Col-0 seeds (t test, P < 0.01). Levels in nfxl1 mutants (H); transcript levels in wrky3 mutants (I).
(J) Wild-type, nfxl1, and wrky3 seeds after 12 h of tetrazolium staining.
(K) and (L) Number of stained (red) seeds in wild-type and nfxl1 (K) and wrky3 (L) seeds, determined on three biological replicated of 50 seeds. The asterisk indicates a significant difference from the wild type Col-0 seeds (χ2 test, P < 0.001).
Further characterization of T-DNA insertion lines in both genes showed that dry weight and soluble sugar contents of mature seeds were not significantly different between the mutant seeds and wild-type seeds (Supplemental Figure 5 and Supplemental Table 2). Likewise, onset of chlorophyll loss occurred around 15 DAP, without any apparent difference for the nfxl1 and wrky3 mutants compared with wild-type seeds (Figure 7C). At 15 DAP, longevity (P50) of wild-type seeds has been acquired for ∼40% compared with the mature wild-type seeds (Figures 7D and 7E). The acquisition of longevity was already affected in both wrky3 and nfxl1 mutants seeds that were harvested at 15 DAP (Figures 7D and 7E, dashed lines). This longevity difference between mutant and wild-type seeds increased slightly upon further maturation. Furthermore, ABA sensitivity was not significantly affected in mature seeds of both mutants (Figure 7F), albeit that there was a slight significant increase in sensitivity for seeds of the wrky3-1 line (Figure 7G).
To explore the predictive role in of the edges in the cross-species network in mediating gene-gene interactions, we investigated whether the nodes in the cross-species network were under the regulation of WRKY3 or NFXL1 by qPCR. Analysis of the presence of W-box and/or NF-x putative cis-regulatory elements in the promoter genes of the longevity module revealed that 12 of them contained both cis-elements, including HSPRO2 and WRKY33 and are first neighbors of both WRKY3 and NFXL1 in the cross-species network (Supplemental Data Set 6). In nfxl1 mutant seeds at 15 DAP, transcript levels of HSPRO2 were significantly lower than in wild-type seeds (Figures 7H, II). In 15 DAP seeds of wrky3 mutants, transcript levels of both HSPRO2 and WRKY33 were affected (Figure 7I, I and II). Promoter analysis of NFXL1 and LZF1 genes revealed the presence of the W-box element. NFXL1 transcripts were lower in the wrky3 mutants (Figure 7I, III) whereas no difference was found in the transcript levels of LZF1 in either of the mutants (Figures 7H and 7I, IV).
In Arabidopsis, seeds of mutants with defects in seed coat properties such as permeability and composition such as flavonoid pigmentation exhibit reduced storage stability (Debeaujon et al., 2000). Flavonoid pigmentation is controlled by several tt genes, some of which were found in our cross-species longevity (Supplemental Data Set 6). This prompted us to investigate whether the testa was affected in mature nfxl1 and wrky3 seeds. Seed coat permeability was evaluated in mature seeds by incubation in tetrazolium salts for 12 h. In a repeatable manner, the number of stained seeds for both the nfxl1 lines (Figures 7J and 7K) and wrky3 lines (Figures 7J and 7L) was doubled compared with the corresponding wild-type seeds.
DISCUSSION
Translational Biology between M. truncatula and Arabidopsis Infers a Cross-Species Gene Module Related to Longevity
The construction of MatNet, a robust coexpression network of seed maturation, and the subsequent integration of the acquisition of longevity using trait-based gene significance measure (Horvath and Dong, 2008) led to the discovery of a gene coexpression module from which confirmed and new genes regulating seed longevity were successfully inferred. MatNet is based on the reasoning that although environmental growth conditions will affect the timing and extent of different seed traits that are acquired during seed development, the genes that are part of the core genetic pathways governing the establishment of these traits will always be expressed. The integration of transcriptomes of developing seeds obtained from five growth conditions into the network led to the separation of functionally associated genes from those coexpressed due to overlapping developmental processes or sampling differences, thus removing biological noise and capturing the conserved backbone of seed maturation. As such, MatNet displays a higher level of internal network structure compared with the structure of a network based on a single environmental condition (Verdier et al., 2013). In addition, MatNet was successful in inferring regulatory genes controlling seed longevity due to the long maturation time frame of M. truncatula seeds, which allowed detailed mapping of physiological events from seed filling onwards (Verdier et al., 2013). In Arabidopsis, the short maturation phase results in substantial overlap between seed filling and late maturation, complicating the discrimination between processes regulating longevity and other traits (Figure 5). Indeed, in this species, a total of 8329 genes correlated with longevity (PCC > 0.85), including FUSCA3 and ANAC100, for which the M. truncatula homologs are expressed transitorily during maturation and do not correlate with longevity.
The subsequent translation of MatNet to Arabidopsis shows a high level of connectivity (87%) of the longevity module genes both for M. truncatula and Arabidopsis, suggesting conserved genetic pathways regulating longevity. This suggestion is also evident from the analysis of the promoter regions of the cross-species longevity genes showing common cis-regulatory elements related to auxin, light, and to a lesser extent biotic defense responses in both species (Figure 6). The main difference was found for sugar responsive elements. This might be related to the different sugar metabolism that takes place during final maturation. In developing seeds of M. truncatula, sucrose levels decrease to 10% of the total soluble sugar content in mature seeds, at the expense of raffinose family oligosaccharide accumulation, whereas in developing seeds of Arabidopsis, sucrose levels increase until final maturation and represent 80% of the total soluble sugars (Figure 5).
Functional validation of the genes inferred from the cross-species longevity module identified with a high success rate (5/8) new and confirmed genes controlling seed longevity. The longevity module contained a number of genes implicated in previously identified mechanisms conferring longevity, such as DNA repair (DNA LIGASE-1) and protection by the testa pigments (TT7 and mate efflux family protein with high sequence similarity to TT12). Also, the presence of several HSPs (HSP17.6, HSP21, and DnaJ) is in accordance with previous studies demonstrating the importance of these proteins for seed longevity (Almoguera et al., 2009). The identification and further characterization of an α-galactosidase, AGAL2, for which mutant seeds showed decreased longevity and increased sucrose and stachyose contents (Supplemental Figure 6) points to a role of raffinose family oligosaccharide metabolism in longevity. Nonreducing sugars are associated with survival in the dry state, although a direct role in the acquisition of longevity remains to be proven. In our study, the increased amounts of nonreducing sugar molecules in the mutant seeds cannot explain the reduced longevity via a protective function and confirmed previous genetic and biochemical studies (Bentsink et al., 2000; Buitink et al., 2000).
Several lines of evidence suggest that there is an ABI3-independent pathway regulating the acquisition of longevity. Half of the longevity nodes were not altered in transcript levels in the M. truncatula abi3 seeds. ABI3, together with other members of the LAFL network, is implicated in the cellular phase transition from the embryonic to the vegetative stage, regulating major developmental changes related to seed storage accumulation and DT (Santos-Mendoza et al., 2008). In Arabidopsis, weak alleles of ABI3 are fully desiccation tolerant but have reduced storability, making it one of the major regulators of longevity (Ooms et al., 1993; Sugliani et al., 2009; Verdier et al., 2013). In wkry3 and nfxl1 mutant seeds, dry weight accumulation and other developmental processes regulated by ABI3, such as chlorophyll degradation and the accumulation of nonreducing soluble sugar contents, were not affected in mature seeds, and NFXL1, WRKY3, or WRKY33 have not been identified as targets of ABI3, LEC1, or LEC2 (Braybrook et al., 2006; Junker et al., 2012; Mönke et al., 2012). In M. truncatula, our transcriptome data on abi3 mutant seeds and ectopic expression of ABI3 using a hairy root system demonstrate that NFXL and WRKY33 transcript levels were not deregulated by ABI3, making these TF putative regulators of an ABI3-independent pathway involved in longevity.
A Role for Auxin in the Regulation of Longevity
Surprisingly, genes that are present in the cross-species longevity module are highly enriched in binding sites for auxin response factors (ARFs) in their promoters (Supplemental Data Set 3). Instead of an expected 6% of genes, 60% of the M. truncatula genes of the longevity module contain the ARFAT cis-element (Table 2). Previously, a link between auxin and longevity was established by Carranco et al. (2010), who showed an interaction between the auxin/indole-3-acetic acid protein IAA27 and the heat shock factor HSFA9 of Helianthus annuus. HSFA9 activates transcription of specific small heat shock protein (sHSP) genes and is involved in the control of a genetic program that regulates seed longevity (Prieto-Dapena et al., 2006; Almoguera et al., 2009). In our study, ARF binding sites were found in the promoters of the M. truncatula and Arabidopsis HSP21 of the longevity module. An increase in auxin in maturing seeds might result in a derepression of the sHSP21 genes, possibly in interaction with HSFA9, although further work is needed to corroborate this hypothesis. Further analysis of the genes with cis-regulatory ARF elements points to a role of the degradation of Trp upstream of the indole-3-acetic acid (IAA) and indole glucosinolate biosynthetic pathways (Supplemental Data Set 6). Four nodes in the longevity module are linked to this degradation pathway: CYP79B2, which converts Trp to indo-3-acetaldoxime, a precursor of both IAA and indole glucosinolates (Kim et al., 2015); CYP83B1 (SUPERROOT2 [SUR2]), an enzyme in the metabolic branch point between IAA and indole glucosinolate biosynthesis, regulating IAA homeostasis (Bak et al., 2001); CYP76C7, involved both in the Trp catabolic process and regulating CYP79B2 and SUR2; and CYP82C2, shown to modulate indole glucosinolate biosynthesis as well as jasmonate-induced root growth inhibition and defense gene expression (Liu et al., 2010).
Link between Seed Longevity and Genetic Pathways Regulating the Activation of Defense against Necrotrophic Pathogens
Another surprising finding inferred from the cross-species network is the high number of genes related to defense against pathogens that are present in the longevity module. Several lines of evidence support this previously unidentified link between defense-associated genes and longevity. First, mutation of WRKY3 and NFXL1, two TFs previously implicated in defense against pathogens, led to reduced longevity. WRKY3 is a homolog of the M. truncatula WRKY33, present in the longevity module of this species. Although At-WRKY33 has a slightly higher sequence similarity to Mt-WRKY33, we chose to investigate WRKY3 since its expression is much more similar to that of Mt-WRKY33 than At-WRKY33, as both genes are highly expressed at the end of seed development. Interestingly, we found that transcript levels of WRKY33 decreased in the wrky3 mutants, indicating that these genes are part of an intricate regulatory network. After infection with the necrotrophic fungal pathogen Botrytis cinerea, leaves of wrky3 mutants exhibit more severe disease symptoms and support higher fungal growth than wild-type plants, whereas no effect is detected with the biotrophic pathogen Pseudomonas syringae (Lai et al., 2008). Likewise, the WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens concomitant with reduced expression of the jasmonate-regulated plant defensin gene PDF1.2 (Zheng et al., 2006). It should be noted that the M. truncatula homolog of PDF1.2 is part of the M. truncatula longevity module (Supplemental Data Set 3), although it was not retained in the cross-species longevity module. As inferred by the network, seeds of wrky3 mutants are affected in the acquisition of longevity and also exhibit a higher permeability of tetrazolium salt (Figure 7), suggesting defects in the testa (Debeaujon et al., 2000).
A second transcription factor that is part of the cross-species longevity network is NFXL1, a NF-X1-type zinc finger protein. We demonstrated that this transcription factor is also involved in seed longevity and seed coat permeability. As for WRKY3, its expression is strongest in seeds, with an increase during final seed maturation (Figure 7). In Arabidopsis, NFXL1 has been implicated in the trichothecene phytotoxin-induced response as well as in the general defense response (Asano et al., 2008). In addition, plants overexpressing NFXL1 display a higher survival when subjected to abiotic stresses such as salt, drought, or high light intensity (Lisso et al., 2006).
A search for W-box and NF-x putative cis-elements in the promoter region of the cross-species longevity module nodes revealed several putative targets for WRKY3 and NFXL1, such as CYP79B2 and CYP83B1, the two genes implicated in auxin biosynthesis and homeostasis (Supplemental Data Set 6). Here, we experimentally confirmed that NFXL1 transcript levels are deregulated in seeds of wrky3 mutants harvested at 15 DAP, demonstrating the predictive power of the cross-species longevity module. In addition, HSPRO2 transcripts were lower in both nfxl1 and wrky3 mutant seeds. HSPRO2 is a positive regulator of basal defense against pathogens, as Arabidopsis knockout mutants are more susceptible to P. syringae infection (Murray et al., 2007). HSPRO2 interacts with AKINβγ, a subunit of the Sucrose nonfermenting-Related Kinase complex (SnRK1) (Gissot et al., 2006). SnRK1 functions as a central integrator of transcription networks in plant stress and energy signaling, in particular in response to biotic and abiotic stress (Schwachtje et al., 2006; Baena-González et al., 2007). It has been suggested that HSPRO2 participates in the activation of SnRK1-mediated response to regulate changes in metabolism required for defense against pathogens. For example, HSPRO2 and GAL83, another subunit of the SnRK1 complex, are part of the same regulatory pathway, negatively regulating seedling growth during interaction with Piriformospora indica (Schuck et al. (2013). In a previous study (Rosnoblet et al., 2007), we demonstrated that silencing SNF4b, a seed-specific regulatory subunit of the SnRK1 complex in M. truncatula, results in reduced longevity. Interestingly, SNF4b regulates defense gene expression in dormant M. truncatula seeds during imbibition such as ABR18 and WRKY33, which are also present in the M. truncatula longevity module (Bolingue et al., 2010).
The presence of the defense genes in the cross-species longevity module, as well as genes implicated in the auxin and indole glucosinate biosynthesis, points to a role of passive, developmentally regulated biotic defense in the acquisition of longevity during late seed maturation. This developmentally regulated biotic defense appears to employ similar pathways as those used upon infection with necrotrophic pathogens. Mechanisms involved in passive defense include physical barriers related to thickened cell walls, or substances such as cutin, suberin, or pectin and chemical barriers, including the accumulation of compounds with microbial activity such as phytoanticipins (constitutively regulated phytoalexins), defensins, saponins, or certain defense proteins (Walters, 2010). That a preemptive strategy to protect seed embryos from pathogen attack is also serving a function in seed physiology has received little attention and is so far limited to the observation that the thick layer of β-glucans, which can defend plants against infection, also play key role in regulating seed coat-imposed dormancy (reviewed in Leubner-Metzger, 2003). In our study, the increased permeability of the wrky3 and nfxl1 seeds indicates a difference in the physical barriers that might explain the longevity phenotype. Other mutants with increased seed coat permeability that are also known to be affected in seed longevity are the tt mutants, functioning in the synthesis of the flavonoids anthocyanins and proanthocyanidins (Debeaujon et al., 2000; Clerkx et al., 2004). One of the major functions of these molecules is to provide protection against microbial pathogens, insect pests, and herbivores (Dixon, 2005). Recently, a study conducted on M. truncatula revealed the activation of tt gene expression during bacterial infection of seeds (Terrasson et al., 2015), suggesting the existence of a complex regulatory interplay involved in seed coat and stress response. The longevity module contains two tt genes (TT7 and a MATE efflux family protein), although transcript levels of TT7 were not deregulated in a consistent manner in the wrky3 and nfxl1 seeds (data not shown). Alternatively, there is compelling evidence that auxins play a role in the resistance to necrotrophic pathogens via modulation of development such as cell wall properties (Raiola et al., 2011). The presence of W-box and NF-x regulatory cis-elements in the promoter regions of IAA regulating genes CYP79B2 and CYP83B1 and the high number of ARFs in the longevity module provide another link between pathogen-related and developmental roles of auxins and offer a new research area for seed biology. Across 195 species, seed longevity is correlated to seed structure and climate origin but not with seed mass and storage composition, with early angiosperms having short-lived seeds in dry storage (Probert et al., 2009). We hypothesize that seed longevity has evolved by co-opting existing genetic pathways regulating the activation of defense against necrotrophic pathogens to ensure prolonged survival in the dry state to be ready for next generations.
METHODS
Plant Material and Physiological Analyses
Plants of Medicago truncatula Gaertn (A17) were grown in standard conditions (20°C/18°C, 16-h photoperiod, according to Verdier et al. [2013]). At the start of flowering, plants were maintained in these conditions or transferred to different environmental conditions: high (26°C/24°C, 16 h) or low (14°C/11°C, 16 h) temperature, osmotic stress (20°C/18°C, 16 h), and maintaining a soil water potential at −0.1 MPa, greenhouse (16 to 30°C, variable photoperiod). Flowers were tagged and developing seeds were harvested at different time intervals until pod abscission and after final desiccation. Water contents were assessed gravimetrically for triplicate samples of five seeds by determination of the fresh weight and subsequent dry weight after 2 d in an oven at 96°C. Water contents are expressed on a dry weight basis. DT and longevity were determined according to Chatelain et al. (2012) on three biological replicates of 50 seeds that were rapidly dried at 43% RH under airflow and harvested from a pool of three plants each. Longevity was determined for each of the 49 developmental stages of each environmental growth condition as the storage time that was needed to decrease the germination of the seed population to 50% (P50), derived from survival curves of percentage of final germination over storage time (75% RH, 35°C). The M. truncatula abi3 mutant (NF6003) was isolated and backcrossed twice with wild-type plants as described by Delahaie et al. (2013). Seeds of abi3 mutants and the wild type (R108) were collected at 40 DAP and frozen for transcriptome analysis.
Plants of Arabidopsis thaliana (Col-0) were grown at 20°C/18°C with a 16-h photoperiod. Flowers were tagged and developing seeds were harvested at different time intervals until pod abscission and after final desiccation. After rapid drying under airflow at 43% RH, DT and longevity were determined as described above. Arabidopsis T-DNA insertion mutants (Supplemental Table 1) were obtained from the Nottingham Arabidopsis Stock Centre, and homozygous seeds were identified by PCR (for primer details, see Supplemental Table 3). Plants were grown together in the same conditions for the production of seeds. The onset of flowering and plant height was recorded for each genotype. Flowers were tagged every day, and seeds were harvested at 15 DAF and fully mature and rapidly dried for 3 d at 43% RH under constant airflow. Dry weight was assessed gravimetrically for triplicate samples of 25 seeds by determination of the weight after 2 d in an oven at 96°C. Physiological experiments were performed on three biological replicates of 100 seeds, harvested from a pool of three to six plants. Viability of the freshly harvested seeds was determined by germination after imbibition of seeds for 2 d at 4°C to remove dormancy, followed by incubation on Whatman filter paper No. 1 in 9-cm Petri dishes with 1 mL water at 20°C, 16 h light. Longevity was determined as described above. To determine ABA sensitivity, mature seeds were imbibed on filter paper on a range of ABA concentrations (mixed isomers; Sigma-Aldrich) at 20°C, 16-h light. ABA was dissolved in methanol prior to dilution in water. Control seeds were imbibed in the methanol concentration corresponding to the highest ABA concentration (0.05% methanol). Germination was scored after 14 d.
Permeability of the seed layers was assessed by incubation of three biological triplicates of 50 seeds consisting of a pool of six to eight plants each in 1% 2,3,5 triphenyltetrazolium chloride in 50 mM phosphate buffer (pH 7.0) for 12 h at 30°C, after which image analysis and percentage of red colored seeds were counted.
Transcriptome Acquisition, Data Analysis, and Bioinformatics
RNA extraction, amplification, labeling, hybridization on NimbleGen slides, and statistical data analysis were conducted as described by Verdier et al. (2013). For seeds of the 49 different developmental stages and growth conditions, two biological replicates of 50 seeds harvested from a bulk of five different plants and immediately frozen in liquid nitrogen after pod dissection were analyzed per comparison using dye switch. For the 40 DAP M. truncatula abi3 seeds, three biological replicates were used. Transcriptome data have been submitted to the Gene Expression Omnibus as SuperSeries GSE53526, containing the following subseries: 21/19°C (GSE49350), 26/24°C (GSE52832), 14/11°C (GSE53002), under greenhouse conditions (GSE52833), and osmotic stress (GSE53003); data on the ectopic expression of Mt-ABI3 in hairy roots are submitted under number GSE44291, and the abi3 mutant versus wild type transcriptome as GSE57457.
For cis-element enrichment analysis of M. truncatula promoters, DNA motifs were counted (using PLACE database resource, http://www.dna.affrc.go.jp/PLACE/signalup.html) within the region 1.5 kb upstream of the translational start site based on M. truncatula IMGAG 3.5.1 version using the Legoo Gateway (https://www.legoo.org/). The frequency of a motif in the promoters of genes of the longevity module (105 genes for which the promoter sequence was retrieved) was compared against a background frequency generated using the total genes present in MatNet (1624 genes for which the promoter sequence was retrieved). P value significance scores were calculated using a χ2 test to provide a relative ranking. An enrichment analysis was performed based on the number of elements present in the same promoter. The Z-score was calculated for each gene in the longevity module based on (X-mean)/sd, where X is the number of repetitions of a particular cis-element, and mean and sd are the average and the sd, respectively, of the element repetition in MatNet promoters. Analysis of promoter elements/Gene Ontology in Arabidopsis was conducted using the ATCOECIS online resource (http://bioinformatics.psb.ugent.be/ATCOECIS/), which determines both Gene Ontology and motif enrichment. The sequence of the NF-x regulatory element was derived from Song et al. (1994).
eFP Browser Visualization
For the integration of the Nimblegen transcriptomic data with the already existing data used for the eFP browser visualization, data were RNA-normalized and then linearized, being scaled such that expression levels of guide genes known to be involved in different stages of seed development (PHB, ATB2, AP3, SEP2, MYB3R1, ABI5, ABI3, AREB3, TT8, and HAP3) were consistent with the previous eFP browser view.
Identification of Seed-Related Genes
To identify seed related or seed specific probe sets, a Z-score was calculated using Z = (X-mean)/sd, where X is the relative expression value, mean is the average of relative expression of tissues available in MtGEA (http://mtgea.noble.org/), and sd is the sd of relative expression in all tissues available in the MtGEA. Probe sets with a Z-score above 2.0 in seeds were considered as seed-specific or preferentially expressed in seeds.
Network Generation and Inference of Desiccation Tolerance and Longevity Modules
The gene coexpression network MatNet was constructed from 104 microarray data sets corresponding to 49 developmental stages of M. truncatula seeds grown in the five different environmental conditions. Only those probes with variance >0.1 across all samples were retained to filter out genes with largely unchanged expression levels. Weighted coexpression network analysis (Zhang and Horvath, 2005) was performed using the ArrayMining Web resource (http://www.arraymining.net) (Glaab et al., 2009). The number of nodes and edges were determined for each adjacency threshold ranging from 0.8 to 0.94 with 0.2 intervals. Gene interactions were visualized using the open source software Cytoscape (version 2.8.1) using an organic layout.
Isolation of the longevity module was performed using the trait based gene significance measure, obtained by
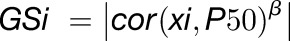 |
where the gene significance (GS) of gene i equals the absolute correlation between the gene expression profile xi and longevity expressed as P50 (days to obtain 50% germination during storage), with β = 1 (Horvath and Dong, 2008). Modulating the PCC to 0.9 or 0.8 changed the number of probes that correlated with P50, but after projection of these data lists on MatNet, the same 167 probes were identified. Equally, raising the correlation between the gene expression profiles and P50 to a power β and modulating β >1 did not affect the final gene list. For the identification of the DT module, transcripts were selected whose levels increased 3-fold between desiccation-sensitive and -tolerant stages, being at 11 and 23 DAP for high temperature, 16 and 32 DAP for greenhouse, 12 and 25 DAP for osmotic stress, 22 and 58 DAP for low temperature, and 14 and 32 DAP for standard conditions, respectively.
RT-qPCR Analysis
Reverse transcription reactions were performed on 1 µg of total RNA of 15 DAP seeds of Col-0, wrky3, and nfxl1 lines using the QuantiTect Reverse Transcription Kit (Qiagen). Quantification of transcript levels of WRKY33 (At2g38470), HSPRO2 (At2g40000), NFXL1 (At1g10170), and LZF1 (At1g78600) were performed by RT-qPCR using an ABI PRISM 7100 sequence detection system and SYBR Green as a double-stranded DNA-specific fluorescent dye, using primers presented in Supplemental Table 4. Values are based on three repetitions. TIP41-like (At4g34270), UBC21 (At5g25760), and AP2M (At5g46630) were used as reference genes.
Sugar Determination
Soluble sugar analysis on mature Arabidopsis seeds from the different lines was performed by Dionex using a Carbopac PA-1 column as previously described (Rosnoblet et al., 2007). Three biological replicates of 100 seeds that were harvested from three to six different plants were analyzed.
Accession Numbers
Sequence data from this article can be found in the Arabidopsis Genome Initiative or GenBank/EMBL databases under the following accession numbers: WRKY33 (At2g38470), HSPRO2 (At2g40000), NFXL1 (At1g10170), LZF1 (At1g78600), TIP41-like (At4g34270), UBC21 (At5g25760), AP2M (At5g46630), CAF-1 (At2g32070), NOT4 (At3g45630), AGAL2 (At5g08370), SIP2 (At3g57520), CYP81D3 (At4g37340), PPC6-1 (At3g02750), WRKY3 (At2g03340), bHLH25 (At4g37850), and FIP1 (At3g66652). Please refer to additional accession numbers in Supplemental Table 3.
Supplemental Data
Supplemental Figure 1. Characterization of network properties related to MatNet.
Supplemental Figure 2. Longevity of developing Arabidopsis seeds.
Supplemental Figure 3. Survival curves of wild-type and T-DNA mutant seeds of Arabidopsis during storage at 35°C and 75% RH.
Supplemental Figure 4. Absolute transcript levels of NFLX1 and WRKY3 in Arabidopsis development
Supplemental Figure 5. Dry weight and sugar content in mature seeds of wkry3 and nfxl1 mutants.
Supplemental Figure 6. Soluble sugar content in mature seeds of Arabidopsis wild type Col-0, agal2, and sip2 mutants.
Supplemental Table 1. Insertion mutants (SALK, GK lines) of Arabidopsis used for the cross-species network validation.
Supplemental Table 2. Soluble sugar content in mature seeds of Col-0, nfxl1, and wrky3 mutants.
Supplemental Table 3. Primers used for T-DNA insertion screening of Arabidopsis mutants.
Supplemental Table 4. Primers used for qPCR.
Supplemental Data Set 1. Genes present in MatNet
Supplemental Data Set 2. List of transcriptional regulators present in MatNet
Supplemental Data Set 3. Genes correlating with longevity acquisition in all environmental conditions and present in MatNet.
Supplemental Data Set 4. Genes correlating with desiccation tolerance acquisition in all environmental conditions and those present in MatNet.
Supplemental Data Set 5. Regulation of genes present in the desiccation tolerance and longevity module by ABI3.
Supplemental Data Set 6. Cross-species network of Arabidopsis and Medicago truncatula genes linked to longevity.
Supplementary Material
Acknowledgments
We thank Pascal Gamas (INRA, Toulouse, France) for sharing the M. truncatula genome data provided following Illumina genome sequencing carried out in the ANR-funded “Symbimics” program ANR-08-GENO-106 that was used for the design of part of the probes. We thank the ANAN platform of the SFR Quasav, Angers, France for the access to hybridization facilities and G. Hunault (University of Angers) for help with the promoter analysis of M. truncatula. K.R. was funded by an INRA postdoctoral fellowship. This work was partially funded by a grant from the Region des Pays de la Loire (QUALISEM 2009-2013). We thank the Salk Institute Genomic Analysis Laboratory for providing the sequence-indexed Arabidopsis T-DNA insertion mutants and the Nottingham Arabidopsis Stock Centre for seed distribution.
AUTHOR CONTRIBUTIONS
K.R. performed research, analyzed the data, and wrote the article. J.L.V., B.L.V., J.V., and D.L. performed research. S.P., A.P., R.V.P., and N.J.P. analyzed data. J.B. and O.L. designed research, analyzed the data, and wrote the article.
Glossary
- DT
desiccation tolerance
- ABA
abscisic acid
- DAP
days after pollination
- TF
transcription factor
- PCC
Pearson correlation coefficient
- SCC
Spearman rank correlation coefficient
- IAA
indole-3-acetic acid
References
- Almoguera C., Prieto-Dapena P., Díaz-Martín J., Espinosa J.M., Carranco R., Jordano J. (2009). The HaDREB2 transcription factor enhances basal thermotolerance and longevity of seeds through functional interaction with HaHSFA9. BMC Plant Biol. 9: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrine K.C., Blanco-Ulate B., Cantu D. (2015). Discovery of core biotic stress responsive genes in Arabidopsis by weighted gene co-expression network analysis. PLoS One 10: e0118731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano T., Masuda D., Yasuda M., Nakashita H., Kudo T., Kimura M., Yamaguchi K., Nishiuchi T. (2008). AtNFXL1, an Arabidopsis homologue of the human transcription factor NF-X1, functions as a negative regulator of the trichothecene phytotoxin-induced defense response. Plant J. 53: 450–464. [DOI] [PubMed] [Google Scholar]
- Baena-González E., Rolland F., Thevelein J.M., Sheen J. (2007). A central integrator of transcription networks in plant stress and energy signalling. Nature 448: 938–942. [DOI] [PubMed] [Google Scholar]
- Bak S., Tax F.E., Feldmann K.A., Galbraith D.W., Feyereisen R. (2001). CYP83B1, a cytochrome P450 at the metabolic branch point in auxin and indole glucosinolate biosynthesis in Arabidopsis. Plant Cell 13: 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabasi A.L., Albert R. (1999). Emergence of scaling in random networks. Science 286: 509–512. [DOI] [PubMed] [Google Scholar]
- Bassel G.W., Lan H., Glaab E., Gibbs D.J., Gerjets T., Krasnogor N., Bonner A.J., Holdsworth M.J., Provart N.J. (2011). Genome-wide network model capturing seed germination reveals coordinated regulation of plant cellular phase transitions. Proc. Natl. Acad. Sci. USA 108: 9709–9714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud S., Boutin J.P., Miquel M., Lepiniec L., Rochat C. (2002). An integrated overview of seed development in Arabidopsis thaliana ecotype WS. Plant Physiol. Biochem. 40: 151–160. [Google Scholar]
- Bentsink L., Alonso-Blanco C., Vreugdenhil D., Tesnier K., Groot S.P., Koornneef M. (2000). Genetic analysis of seed-soluble oligosaccharides in relation to seed storability of Arabidopsis. Plant Physiol. 124: 1595–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolingue W., Rosnoblet C., Leprince O., Vu B.L., Aubry C., Buitink J. (2010). The MtSNF4b subunit of the sucrose non-fermenting-related kinase complex connects after-ripening and constitutive defense responses in seeds of Medicago truncatula. Plant J. 61: 792–803. [DOI] [PubMed] [Google Scholar]
- Braybrook S.A., Stone S.L., Park S., Bui A.Q., Le B.H., Fischer R.L., Goldberg R.B., Harada J.J. (2006). Genes directly regulated by LEAFY COTYLEDON2 provide insight into the control of embryo maturation and somatic embryogenesis. Proc. Natl. Acad. Sci. USA 103: 3468–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buitink J., Hemminga M.A., Hoekstra F.A. (2000). Is there a role for oligosaccharides in seed longevity? An assessment of intracellular glass stability. Plant Physiol. 122: 1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buitink J., Leprince O. (2010). Desiccation tolerance: From genomics to the field. Plant Sci. 179: 554–564. [Google Scholar]
- Carranco R., Espinosa J.M., Prieto-Dapena P., Almoguera C., Jordano J.R. (2010). Repression by an auxin/indole acetic acid protein connects auxin signaling with heat shock factor-mediated seed longevity. Proc. Natl. Acad. Sci. USA 107: 21908–21913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatelain E., Hundertmark M., Leprince O., Le Gall S., Satour P., Deligny-Penninck S., Rogniaux H., Buitink J. (2012). Temporal profiling of the heat-stable proteome during late maturation of Medicago truncatula seeds identifies a restricted subset of late embryogenesis abundant proteins associated with longevity. Plant Cell Environ. 35: 1440–1455. [DOI] [PubMed] [Google Scholar]
- Clerkx E.J.M., Blankestijn-De Vries H., Ruys G.J., Groot S.P.C., Koornneef M. (2004). Genetic differences in seed longevity of various Arabidopsis mutants. Physiol. Plant. 121: 448–461. [Google Scholar]
- Debeaujon I., Léon-Kloosterziel K.M., Koornneef M. (2000). Influence of the testa on seed dormancy, germination, and longevity in Arabidopsis. Plant Physiol. 122: 403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delahaie J., Hundertmark M., Bove J., Leprince O., Rogniaux H., Buitink J. (2013). LEA polypeptide profiling of recalcitrant and orthodox legume seeds reveals ABI3-regulated LEA protein abundance linked to desiccation tolerance. J. Exp. Bot. 64: 4559–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon R.A. (2005). Engineering of plant natural product pathways. Curr. Opin. Plant Biol. 8: 329–336. [DOI] [PubMed] [Google Scholar]
- Faccioli P., Provero P., Herrmann C., Stanca A.M., Morcia C., Terzi V. (2005). From single genes to co-expression networks: extracting knowledge from barley functional genomics. Plant Mol. Biol. 58: 739–750. [DOI] [PubMed] [Google Scholar]
- Ficklin S.P., Luo F., Feltus F.A. (2010). The association of multiple interacting genes with specific phenotypes in rice using gene coexpression networks. Plant Physiol. 154: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo K., Firnhaber C., Zuber H., Hericher D., Belghazi M., Henry C., Kuster H., Thompson R.D. (2007). A combined proteome and transcriptome analysis of developing. Medicago truncatula seeds. Mol. Cell. Proteomics 6: 2165–2179. [DOI] [PubMed] [Google Scholar]
- Gissot L., Polge C., Jossier M., Girin T., Bouly J.P., Kreis M., Thomas M. (2006). AKINbetagamma contributes to SnRK1 heterotrimeric complexes and interacts with two proteins implicated in plant pathogen resistance through its KIS/GBD sequence. Plant Physiol. 142: 931–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaab E., Garibaldi J.M., Krasnogor N. (2009). ArrayMining: a modular web-application for microarray analysis combining ensemble and consensus methods with cross-study normalization. BMC Bioinformatics 10: 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Benedito V.A., Wang M., Murray J.D., Zhao P.X., Tang Y., Udvardi M.K. (2009). The Medicago truncatula gene expression atlas web server. BMC Bioinformatics 10: 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S., Dong J. (2008). Geometric interpretation of gene coexpression network analysis. PLOS Comput. Biol. 4: e1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundertmark M., Dimova R., Lengefeld J., Seckler R., Hincha D.K. (2011). The intrinsically disordered late embryogenesis abundant protein LEA18 from Arabidopsis thaliana modulates membrane stability through binding and folding. Biochim. Biophys. Acta 1808: 446–453. [DOI] [PubMed] [Google Scholar]
- Junker A., et al. (2012). Elongation-related functions of LEAFY COTYLEDON1 during the development of Arabidopsis thaliana. Plant J. 71: 427–442. [DOI] [PubMed] [Google Scholar]
- Kim J.I., Dolan W.L., Anderson N.A., Chapple C. (2015). Indole glucosinolate biosynthesis limits phenylpropanoid accumulation in Arabidopsis thaliana. Plant Cell 27: 1529–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranner I., Birtić S., Anderson K.M., Pritchard H.W. (2006). Glutathione half-cell reduction potential: a universal stress marker and modulator of programmed cell death? Free Radic. Biol. Med. 40: 2155–2165. [DOI] [PubMed] [Google Scholar]
- Lai Z., Vinod K., Zheng Z., Fan B., Chen Z. (2008). Roles of Arabidopsis WRKY3 and WRKY4 transcription factors in plant responses to pathogens. BMC Plant Biol. 8: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.-H., Kim Y.-K., Pham T.T.M., Song S.I., Kim J.-K., Kang K.Y., An G., Jung K.-H., Galbraith D.W., Kim M., Yoon U.-H., Nahm B.H. (2009). RiceArrayNet: a database for correlating gene expression from transcriptome profiling, and its application to the analysis of coexpressed genes in rice. Plant Physiol. 151: 16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leubner-Metzger G. (2003). Functions and regulation of b-1,3-glucanases during seed germination, dormancy release and after-ripening. Seed Sci. Res. 13: 17–34. [Google Scholar]
- Li H., Wang L., Yang Z.M. (2015). Co-expression analysis reveals a group of genes potentially involved in regulation of plant response to iron-deficiency. Gene 554: 16–24. [DOI] [PubMed] [Google Scholar]
- Lisso J., Altmann T., Müssig C. (2006). The AtNFXL1 gene encodes a NF-X1 type zinc finger protein required for growth under salt stress. FEBS Lett. 580: 4851–4856. [DOI] [PubMed] [Google Scholar]
- Liu F., Jiang H., Ye S., Chen W.P., Liang W., Xu Y., Sun B., Sun J., Wang Q., Cohen J.D., Li C. (2010). The Arabidopsis P450 protein CYP82C2 modulates jasmonate-induced root growth inhibition, defense gene expression and indole glucosinolate biosynthesis. Cell Res. 20: 539–552. [DOI] [PubMed] [Google Scholar]
- Long T.A., Brady S.M., Benfey P.N. (2008). Systems approaches to identifying gene regulatory networks in plants. Annu. Rev. Cell Dev. Biol. 24: 81–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mönke G., Seifert M., Keilwagen J., Mohr M., Grosse I., Hähnel U., Junker A., Weisshaar B., Conrad U., Bäumlein H., Altschmied L. (2012). Toward the identification and regulation of the Arabidopsis thaliana ABI3 regulon. Nucleic Acids Res. 17: 8240–8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray S.L., Ingle R.A., Petersen L.N., Denby K.J. (2007). Basal resistance against Pseudomonas syringae in Arabidopsis involves WRKY53 and a protein with homology to a nematode resistance protein. Mol. Plant Microbe Interact. 20: 1431–1438. [DOI] [PubMed] [Google Scholar]
- Mutwil M., Usadel B., Schütte M., Loraine A., Ebenhöh O., Persson S. (2010). Assembly of an interactive correlation network for the Arabidopsis genome using a novel heuristic clustering algorithm. Plant Physiol. 152: 29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel M., Kranner I., Neumann K., Rolletschek H., Seal C.E., Colville L., Fernández-Marín B., Börner A. (2015). Genome-wide association mapping and biochemical markers reveal that seed ageing and longevity are intricately affected by genetic background and developmental and environmental conditions in barley. Plant Cell Environ. 38: 1011–1022. [DOI] [PubMed] [Google Scholar]
- Nguyen T.-P., Keizer P., van Eeuwijk F., Smeekens S., Bentsink L. (2012). Natural variation for seed longevity and seed dormancy are negatively correlated in Arabidopsis. Plant Physiol. 160: 2083–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooms J., Leon-Kloosterziel K.M., Bartels D., Koornneef M., Karssen C.M. (1993). Acquisition of desiccation tolerance and longevity in seeds of Arabidopsis thaliana (A comparative study using abscisic acid-insensitive abi3 mutants). Plant Physiol. 102: 1185–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R.V., Nahal H.K., Breit R., Provart N.J. (2012). BAR expressolog identification: expression profile similarity ranking of homologous genes in plant species. Plant J. 71: 1038–1050. [DOI] [PubMed] [Google Scholar]
- Prieto-Dapena P., Castaño R., Almoguera C., Jordano J. (2006). Improved resistance to controlled deterioration in transgenic seeds. Plant Physiol. 142: 1102–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probert R.J., Daws M.I., Hay F.R. (2009). Ecological correlates of ex situ seed longevity: a comparative study on 195 species. Ann. Bot. (Lond.) 104: 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiola A., Lionetti V., Elmaghraby I., Immerzeel P., Mellerowicz E.J., Salvi G., Cervone F., Bellincampi D. (2011). Pectin methylesterase is induced in Arabidopsis upon infection and is necessary for a successful colonization by necrotrophic pathogens. Mol. Plant Microbe Interact. 24: 432–440. [DOI] [PubMed] [Google Scholar]
- Rosnoblet C., Aubry C., Leprince O., Vu B.L., Rogniaux H., Buitink J. (2007). The regulatory gamma subunit SNF4b of the sucrose non-fermenting-related kinase complex is involved in longevity and stachyose accumulation during maturation of Medicago truncatula seeds. Plant J. 51: 47–59. [DOI] [PubMed] [Google Scholar]
- Santos-Mendoza M., Dubreucq B., Baud S., Parcy F., Caboche M., Lepiniec L. (2008). Deciphering gene regulatory networks that control seed development and maturation in Arabidopsis. Plant J. 54: 608–620. [DOI] [PubMed] [Google Scholar]
- Sattler S.E., Gilliland L.U., Magallanes-Lundback M., Pollard M., DellaPenna D. (2004). Vitamin E is essential for seed longevity and for preventing lipid peroxidation during germination. Plant Cell 16: 1419–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer R.J., Briskine R., Springer N.M., Myers C.L. (2014). Discovering functional modules across diverse maize transcriptomes using COB, the Co-expression Browser. PLoS One 9: e99193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M., Davison T.S., Henz S.R., Pape U.J., Demar M., Vingron M., Schölkopf B., Weigel D., Lohmann J.U. (2005). A gene expression map of Arabidopsis thaliana development. Nat. Genet. 37: 501–506. [DOI] [PubMed] [Google Scholar]
- Schuck S., Baldwin I.T., Bonaventure G. (2013). HSPRO acts via SnRK1-mediated signaling in the regulation of Nicotiana attenuata seedling growth promoted by Piriformospora indica. Plant Signal. Behav. 8: e23537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwachtje J., Minchin P.E., Jahnke S., van Dongen J.T., Schittko U., Baldwin I.T. (2006). SNF1-related kinases allow plants to tolerate herbivory by allocating carbon to roots. Proc. Natl. Acad. Sci. USA 103: 12935–12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z., Krishna S., Thanos D., Strominger J.L., Ono S.J. (1994). A novel cysteine-rich sequence-specific DNA-binding protein interacts with the conserved X-box motif of the human major histocompatibility complex class II genes via a repeated Cys-His domain and functions as a transcriptional repressor. J. Exp. Med. 180: 1763–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugliani M., Rajjou L., Clerkx E.J.M., Koornneef M., Soppe W.J.J. (2009). Natural modifiers of seed longevity in the Arabidopsis mutants abscisic acid insensitive3-5 (abi3-5) and leafy cotyledon1-3 (lec1-3). New Phytol. 184: 898–908. [DOI] [PubMed] [Google Scholar]
- Terrasson E., Darrasse A., Righetti K., Buitink J., Lalanne D., Ly Vu B., Pelletier S., Bolingue W., Jacques M.-A., Leprince O. (2015). Identification of a molecular dialogue between developing seeds of Medicago truncatula and seedborne xanthomonads. J. Exp. Bot. 66: 3737–3752. [DOI] [PubMed] [Google Scholar]
- Usadel B., Obayashi T., Mutwil M., Giorgi F.M., Bassel G.W., Tanimoto M., Chow A., Steinhauser D., Persson S., Provart N.J. (2009). Co-expression tools for plant biology: opportunities for hypothesis generation and caveats. Plant Cell Environ. 32: 1633–1651. [DOI] [PubMed] [Google Scholar]
- Vandepoele K., Quimbaya M., Casneuf T., De Veylder L., Van de Peer Y. (2009). Unraveling transcriptional control in Arabidopsis using cis-regulatory elements and coexpression networks. Plant Physiol. 150: 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdier J., et al. (2013). A regulatory network-based approach dissects late maturation processes related to the acquisition of desiccation tolerance and longevity of Medicago truncatula seeds. Plant Physiol. 163: 757–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters D. (2010). Plant Defense: Warding Off Attack by Pathogens, Herbivores and Parasitic Plants. (Hoboken, NJ: Wiley-Blackwell).
- Zhang B., Horvath S. (2005). A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4: Article17. [DOI] [PubMed] [Google Scholar]
- Zheng Z., Qamar S.A., Chen Z., Mengiste T. (2006). Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 48: 592–605. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.