

Background: The RING E3 AO7/RNF25 binds its E2 with unusually high affinity.
Results: AO7 has a secondary E2 binding site adjacent to the RING.
Conclusion: This site prevents the stimulatory effect of non-covalent backside binding of ubiquitin, has unique agonist properties, and allows for structural analysis of RING mutants.
Significance: Knowledge of how RING E3s mediate ubiquitination is critical to understanding cellular protein regulation.
Keywords: allosteric regulation, crystallography, E3 ubiquitin ligase, proteolysis, ubiquitin, ubiquitin-conjugating enzyme (E2 enzyme), RING finger
Abstract
RING proteins constitute the largest class of E3 ubiquitin ligases. Unlike most RINGs, AO7 (RNF25) binds the E2 ubiquitin-conjugating enzyme, UbcH5B (UBE2D2), with strikingly high affinity. We have defined, by co-crystallization, the distinctive means by which AO7 binds UbcH5B. AO7 contains a structurally unique UbcH5B binding region (U5BR) that is connected by an 11-amino acid linker to its RING domain, forming a clamp surrounding the E2. The U5BR interacts extensively with a region of UbcH5B that is distinct from both the active site and the RING-interacting region, referred to as the backside of the E2. An apparent paradox is that the high-affinity binding of the AO7 clamp to UbcH5B, which is dependent on the U5BR, decreases the rate of ubiquitination. We establish that this is a consequence of blocking the stimulatory, non-covalent, binding of ubiquitin to the backside of UbcH5B. Interestingly, when non-covalent backside ubiquitin binding cannot occur, the AO7 clamp now enhances the rate of ubiquitination. The high-affinity binding of the AO7 clamp to UbcH5B has also allowed for the co-crystallization of previously described and functionally important RING mutants at the RING-E2 interface. We show that mutations having marked effects on function only minimally affect the intermolecular interactions between the AO7 RING and UbcH5B, establishing a high degree of complexity in activation through the RING-E2 interface.
Introduction
Specificity in ubiquitination is conferred in large part by E3 ubiquitin ligases. Each E3 functions with a subset of the ∼40 different (in mammals) E2 ubiquitin-conjugating enzymes. E3s mediate the direct or indirect transfer of ubiquitin from the active site Cys of an activated E2 (E2∼Ub)6 to, most frequently, primary amines (Lys or the N termini of proteins). RING-type E3s (consisting primarily of RING finger and U-box proteins), which constitute the large majority of E3s, mediate the direct transfer of ubiquitin from E2 (for review, see Refs. 1 and 2). The isopeptide or peptide bonds formed with ubiquitin can be on heterologous substrates or on the E3 itself; the latter is usually referred to as autoubiquitination (Ref. 3; for review, see Ref. 4). Proteins can be modified with a single ubiquitin on one or more residues (monoubiquitination), or substrate-conjugated ubiquitin can itself be ubiquitinated on any of its seven Lys residues or its N terminus (5), forming multi- or polyubiquitin chains. Some polyubiquitin linkages represent particularly potent proteasome-targeting signals, whereas others specify different functions.
RING-type E3s bind both E2∼Ub and substrates and allosterically activate E2∼Ub (for review, see Ref. 1). Structural studies have revealed that RING-type domains bind E2s in a canonical manner using the RING's two Zn2+-coordinating loops and the intervening central α-helix. This RING-E2 binding is generally of low affinity (for review, see Ref. 1). For this reason, differentiating the activating function of specific residues at the RING-E2 interface from their effects on overall RING-E2 affinity can be problematic.
Key residues required for the allosteric activation of E2s have been identified (6). More recently, it has become clear that activation of E2∼Ub is dependent, in part, on the RING domain stabilizing a “closed” conformation of E2∼Ub that is primed for ubiquitin transfer. This conformation, where the orientation of the E2∼Ub thioester bond is optimal for nucleophilic attack, is a consequence of critical interactions of ubiquitin with both E2 and RING-type domains (Refs. 7–16; for review, see Ref. 1). Understanding precisely how this orientation and these stabilizing interactions enhance catalysis remains an area of intense investigation.
Other than at the E2 active site, ubiquitin also binds to a specific region on the “backside” of a subset of E2s (UbB binding), which enhances RING:E2 affinity and stimulates ubiquitination (17–20). Several E3s also bind their cognate E2s on secondary sites away from the RING-E2 interface. For the most part these overlap the backside UbB binding site. E2 backside binding by E3s has also been shown to have substantial effects on ubiquitination (16, 18, 19, 21–25). Backside binding to ubiquitin E2s is shared with plant proteins that target E2s to the plasma membrane (26, 27), and remarkable parallels exist in the SUMO conjugating system (Refs. 28–34; for review, see Ref. 1). Determining the varied structures, functions, and dynamics of secondary E2 binding sites, including ones that await discovery, is critical to understanding the in vivo regulation of modification by ubiquitin and ubiquitin-like proteins.
AO7 (RNF25) was first identified because of its unusually high-affinity binding to the E2 UbcH5B (UBE2D2) (3, 35). This binding, which is dependent on both an intact AO7 RING and a C-terminal extension, helped lead to the determination that RING proteins are generally ubiquitin ligases (3). Although its cellular role remains obscure, the high-affinity AO7-UbcH5B binding led us to revisit AO7 as a model for RING:E2 interactions.
We establish herein that the structurally atypical RING domain of AO7 is part of a unique “clamp”-like structure that is comprised of the RING and the C-terminal extension. This clamp surrounds UbcH5B and includes a secondary UbcH5B binding site that overlaps the UbB binding site on UbcH5B. The effects of disrupting this clamp provide insights into the functional consequences of both the AO7 clamp and UbB binding (17). In addition, the unique nature of the AO7 clamp has facilitated the co-crystallization of UbcH5B together with AO7 RING mutants, which are analogous to mutations in other RINGs that have been shown to result in either compromised function or E2 binding (36–39). This provides a means to begin to systematically dissociate overall RING:E2 affinity from functions of specific amino acids at the RING-E2 interface.
Experimental Procedures
Antibodies and Plasmids
Rat monoclonal HA antibody was from Roche Applied Science (catalog #11867423001). Rabbit polyclonal UbcH5 antiserum was from US Biological (catalog #U1000-86-M). Rabbit polyclonal GFP antiserum was from Santa Cruz (SC-8334). Rabbit polyclonal ubiquitin antibody has been described (40). pET15b-UbcH5B has been described (35) and was used as the template for point mutations. Plasmids encoding UbcM2/UbcH9/Ube2e3 were gifts from Scott Plafker and from Wade Harper (Addgene plasmid #15789) and have been described (41, 42). All AO7-encoding constructs were generated by PCR from human AO7 EST (Thermo Scientific Clone ID: 4811222). AO7R (Fig. 1A) and AO7 217–258 were cloned into pGEX-6P-1 (GE Healthcare) from BamHI to XhoI. Sequence-encoding AO7RE (Fig. 1A) was cloned into the first multiple cloning site of a modified pETDUET-1 (Novagen) vector containing an N-terminal GST fusion protein in place of His6 (pETDUET-GST), from BamHI to HindIII. A PreScission Protease (GE Healthcare) sequence (LEVLFQGP) was included in all N-terminal primers for AO7 constructs, enabling removal of GST. All GST fusions cleaved by PreScission Protease have residual Gly-Pro remaining at their N termini. To generate plasmids encoding both AO7RE and UbcH5B, cDNA encoding the E2 was amplified from pET15b-UbcH5B and cloned into the second multiple cloning site of pETDUET-GST from NdeI to XhoI. To generate UbcH5B for FlAsH labeling (UbcH5B-Green), an HHHHHHGSGCCPGCCGGSGGS tag was inserted directly upstream of UbcH5B, and the entire sequence cloned into pGEX-6P-1 from BamHI to XhoI. N-terminal HA-tagged human AO7 (RNF25) encoding amino acids 2–439 was generated by PCR and subcloned into pcDNA3.1 (+) (Invitrogen) from EcoRI to XhoI. Human ubiquitin was amplified by PCR from pGL-ubiquitin (a gift from R. Andrew Byrd) as template and cloned into pETDUET-GST from BamHI to HindIII with a PreScission Protease recognition sequence at the N terminus. I44A ubiquitin (UbI44A) was generated by site-specific mutagenesis of this plasmid. Point mutations and deletions were generated using QuikChange (Stratagene) and verified by sequencing. pEGFP-C1 plasmid encoding GFP was purchased from Clontech.
FIGURE 1.

A region beyond the AO7 RING finger is required for high-affinity UbcH5B binding. A, schematic of human AO7 (above). Components of the AO7 clamp are indicated in white (L, Linker). Sequence of AO7RE (below) with Zn2+-coordinating residues and structural features noted in “Results”; disordered regions are indicated with dashed lines. B, binding of in vitro transcribed and translated 35S-labeled UbcH5B to N-terminal GST fusions of AO7 prebound to GS. GST fusions were provided in excess relative to UbcH5B, and each was present in equimolar amounts as determined by Coomassie Blue. C, UbcH5B-Green modified with FlAsH was added to increasing amounts of AO7 from which GST had been cleaved. Binding was assessed by MST.
Protein Expression and Purification
Except where noted, Escherichia coli BL21 (DE3) (Invitrogen) transformed with expression plasmids were grown at 37 °C until A600 0.8–1.0. Protein expression was induced with isopropyl β-d-1-thiogalactopyranoside (0.15 mm) at 23 °C for 20 h. When the RING finger of AO7 was expressed, 50–500 μm ZnSO4 was included. For GST fusion proteins and protein complexes with UbcH5B, cell lysis was performed using an APV-2000 homogenizer in binding buffer (50 mm Tris, pH 7.5, 150 mm NaCl) with 1 mm PMSF. For UbcH5B-Green, 1 mm DTT was included in the binding buffer. After centrifugation at 27,000 × g for 30 min, supernatants were collected and incubated with glutathione-Sepharose (GS) 4B (GE Healthcare) for 2 h at 4 °C, after which beads were collected by centrifugation and thoroughly washed with binding buffer. GST fusion proteins were either eluted using elution buffer (50 mm Tris-HCl, 10 mm reduced glutathione, pH 8.0) and thoroughly dialyzed against binding buffer, or the GST tag was removed by on-column cleavage by incubating the GST fusion protein bound to GS 4B with PreScission Protease (GE Healthcare) overnight at 4 °C. After concentrating to about 5 ml using Vivaspin 20 (10-kDa cut off) (GE Healthcare) or Ultra-15 Centrifugal Filter Units (3-kDa cut off) (Millipore), target proteins were further purified by gel filtration chromatography (HiPrep 16/60 Sephacryl S-300 HR, GE Healthcare) on an Akta FPLC system using binding buffer as necessary. To generate UbcM2 for ubiquitination reactions from GST-UbcM2, bacterial lysate was passed over GS 4B followed by cleavage using 5 units of thrombin (Amersham Biosciences) for 4 h. Thrombin was removed using benzamidine-Sepharose 6B (GE Healthcare) followed by passage through a Pierce Spin Cup (Thermo Scientific) to remove residual beads.
For purification of UbcH5B, cell lysis was performed as described above in 50 mm acetic acid-sodium acetate, pH 5.4. Purification from supernatant was performed by cation exchange chromatography (HiTrap SP HP 5 ml, GE Healthcare) using a gradient of 0–1.0 M NaCl. The peak was subjected to further separation by gel filtration (HiPrep 16/60 Sephacryl S-300 HR, GE Healthcare) using Akta FPLC with binding buffer.
UbcH5B for NMR was transformed, expressed, and lysed as described (22) except that 50 mm acetic acid-sodium acetate, pH 5.4, 4 mm DTT was used for lysis. The supernatant was separated as described (22) by sequential cation exchange and gel filtration chromatography carried out on an S200 column (GE Healthcare) in 50 mm Tris-HCl, pH 7.5, 2 mm tris(2-carboxyethyl)phosphine as described (22). The extent of 15N incorporation was determined by mass spectroscopy to be >98%.
Freshly purified samples of WT AO7RE (Fig. 1A) and its mutants, in complex with UbcH5B, were used for crystallization. Samples were otherwise immediately frozen and stored at −80 °C for further functional assays.
Crystallization, Data Collection, and Structure Determination
Crystals of AO7RE·UbcH5B were obtained using the hanging drop vapor diffusion method at 19 ± 1 °C by mixing 1 μl of protein solution and 2 μl of reservoir solution (Table 2), and the premixed 3-μl droplets were equilibrated against 200 μl of reservoir solution. The diamond-shaped crystals grew to full size (∼0.1 mm × 0.1 mm × 0.1 mm) in 3 days. Crystals were flash-cooled in cold nitrogen stream after a brief soak in 100% paraffin oil as the cryo-protectant. X-ray diffraction data were collected from a single crystal on a MAR345 image plate detector mounted on a RIGAKU MicroMaxTM-007 HF microfocus x-ray generator and processed using the HKL-3000 program suite (43). Crystals of the AO7REY165A·UbcH5B and AO7REP199A·UbcH5B complexes were obtained using a similar protocol, and the data were collected on the same diffractometer system. Crystallization conditions and data collection statistics of the three samples are summarized in Table 2.
TABLE 2.
Data collection and refinement statistics
Values in parentheses are for the highest resolution shell. PS, protein solution; RS, reservoir solution; r.m.s.d., root mean square deviation.
| AO7RE·UbcH5B | AO7REY165A·UbcH5B | AO7REP199A·UbcH5B | |
|---|---|---|---|
| Crystallization | |||
| Protein solution | 12 mg/ml protein, 150 mm NaCl in 50 mm Tris-HCl, pH 7.5 | 12 mg/ml protein, 150 mm NaCl in 50 mm Tris-HCl, pH 7.5 | 12 mg/ml protein, 150 mm NaCl in 50 mm Tris-HCl, pH 7.5 |
| Reservoir solution | 20% PEG 10000, 8% Ethylene glycol in 0.1 m HEPES, pH 7.5 | 15% PEG 20000 in 0.1 m MES, pH 6.5 | 20% PEG 10000, 8% Ethylene glycol in 0.1 m HEPES, pH 7.5 |
| Drop size (μl) | 1 PS + 2 RS | 1 PS + 2 RS | 1 PS + 2 RS |
| Data collection | |||
| Cryo-protectant | 100% Paraffin oil | 100% immersion oil | 100% immersion oil |
| Space group | C2 | C2 | C2 |
| Cell dimensions | |||
| a, b, c (Å) | 105.47, 45.66, 67.95 | 105.60, 45.65, 67.82 | 105.70, 45.42, 67.98 |
| α, β, γ (Å) | 90, 118.09, 90 | 90, 118.03, 90 | 90, 118.32, 90 |
| Resolution (Å) | 50-1.78 (1.84-1.78) | 50-1.61 (1.67-1.61) | 50-1.58 (1.64-1.58) |
| Number of unique reflections | 27,367 (2533) | 35,154 (2785) | 38,987 (3797) |
| Rmerge (%)a | 5.7 (46.7) | 3.4 (35.5) | 3.3 (28.9) |
| I/σ | 33.7 (3.0) | 54.1 (3.9) | 55.7 (5.3) |
| Completeness (%) | 99.1 (92.7) | 94.8 (75.9) | 99.7 (96.9) |
| Redundancy | 7.0 (5.6) | 7.2 (5.1) | 6.2 (4.8) |
| Refinement | |||
| Resolution | 50-1.78 (1.89-1.78) | 50-1.62 (1.70-1.62) | 50-1.58 (1.66-1.58) |
| Rwork (%)b | 16.96 (28.63) | 18.17 (27.26) | 18.82 (40.85) |
| Rfree (%)c | 20.20 (31.24) | 19.95 (28.41) | 22.31 (40.07) |
| No. of atoms | |||
| Protein | 2,195 | 2,256 | 2,224 |
| Water | 272 | 396 | 384 |
| Ligands | 27 | 22 | 30 |
| B Factors | |||
| Protein | 28.61 | 29.18 | 26.15 |
| Water | 34.89 | 38.22 | 36.46 |
| Ligands | 42.41 | 41.07 | 35.02 |
| r.m.s.d. | |||
| Bond lengths (Å) | 0.007 | 0.008 | 0.009 |
| Bond angles (°) | 1.086 | 1.052 | 1.175 |
| Ramachandran plot | |||
| Most favored region (%) | 90.3 | 92.6 | 89.9 |
| Additional allowed region (%) | 9.2 | 6.9 | 9.2 |
| Generously allowed region (%) | 0.5 | 0.5 | 0.9 |
| Disallowed region (%) | 0 | 0 | 0 |
a Rmerge = Σ|(I − <I>)|/σ(I), where I is the observed intensity.
b Rwork = Σhkl‖Fo| − |Fc‖/Σhkl |Fo|, calculated from a working dataset.
c Rfree is calculated from ∼1000 reflections randomly chosen and not included in refinement.
The AO7RE·UbcH5B structure was determined by molecular replacement using phenix.automr of the PHENIX program suite (44). The crystal structure of WT UbcH5B (PDB entry 2ESK) (6) was used as the search model. The AO7RE structure including zinc ions coordinated by the AO7 RING domain were built into the electron density map, first using phenix.autobuild and then by manual interpretation. The structure was refined with phenix.refine of PHENIX, and the model building and adjustment was done with COOT (45). About 1000 reflections were randomly selected for cross-validation. Ethylene glycol, di(hydroxyethyl) ether, and oxalate molecules from the crystallization solutions and water molecules were included at the last stage of the refinement on the basis of difference electron density (Fo − Fc, above 3σ) and verified with omit maps. The refined structure was validated using the PROCHECK (46) and WHATIF (47) programs. The AO7REY165A·UbcH5B and AO7REP199A·UbcH5B structures were determined with the difference Fourier method using the AO7RE·UbcH5B structure as the start model, and the structures were refined with the same strategy as the AO7RE·UbcH5B structure. The figures were generated with PyMol (DeLano Scientific LLC). Refinement statistics of the three structures are listed in Table 2.
35S-Labeled E2 Binding Assay
Binding was carried out using equimolar amounts (∼50 picomoles) of each GST fusion protein. In vitro translated [35S]methionine-labeled E2s were generated using the E. coli T7-S30 Extract System (Promega) from pET15b-UbcH5B or pDEST17-Ube2E3. Binding assays using in vitro translated UbcH5B were carried out by incubating purified GST fusion proteins prebound to GS 4B with ∼105 cpm (∼180 fmol) of [35S]methionine-labeled E2 in 25 mm Tris-HCl, pH 7.4, 50 mm NaCl, 5 mm DTT, 0.5% Nonidet P-40 overnight at 4 °C in 250 μl. Beads were washed with the same buffer and resolved by SDS-PAGE under reducing conditions.
In Vitro Ubiquitination
Autoubiquitination assays employed equimolar amounts (∼1 μm) of purified GST fusion proteins that were prebound to GS 4B in 50 μl of ubiquitination assay buffer (50 mm Tris-HCl, pH 7.4, 0.2 mm ATP, 0.5 mm MgCl2, 0.1 mm DTT). Approximately 2 μm UbcH5B and 50–100 nm His-tagged purified mouse ubiquitin-activating enzyme (E1) were used in all reactions. Baculovirus encoding His6-tagged mouse E1 was a gift from Kazuhiro Iwai. For experiments involving UbI44A, purified recombinant WT ubiquitin was used in parallel. Purified bovine WT ubiquitin from Sigma (catalog #U6253) was employed in all other experiments. WT ubiquitin was used at 4.0 μm in all reactions. Samples were incubated at 37 °C for 2 h (unless otherwise indicated) with continuous shaking. Beads were washed and processed as described (22). Autoubiquitination assays using ∼190 fmol of [35S]methionine-labeled in vitro transcribed and translated forms of AO7 were carried out in solution using UbcH5B and E1 in 25 μl of ubiquitination assay buffer. Reactions were quenched with SDS sample buffer and resolved by SDS-PAGE under reducing conditions and assessed by Storm phosphorimaging (GE Healthcare) and quantified using ImageQuant (GE Healthcare).
Single-round Ubiquitin Turnover
Loading of 35S-labeled UbcH5B in the presence of excess ubiquitin (5 μm) using His-tagged purified mouse E1 ubiquitin-activating enzyme was carried out as described (22). To assess discharge of ubiquitin from UbcH5B, E1 activity was quenched with apyrase (Sigma) (23). Then [35S]methionine-labeled ubiquitin-loaded UbcH5B (∼2 nm) was combined with ubiquitin (80 μm) and purified GST fusion proteins (∼1 μm) in 50 μl of 50 mm Tris-HCl, pH 7.4. E2∼Ub was allowed to bind to GST fusions at 4 °C for 10 min before raising the temperature to 25 °C and beginning turnover reactions. Samples were resolved by SDS-PAGE without reducing agent and processed for visualization and quantification as above. Discharge was monitored by measuring the fraction of UbcH5B∼Ub remaining at each time point. Data analysis was carried out with Prism (GraphPad Software). Data shown are the average of three independent experiments; error bars represent S.D.
For discharge using purified UbcH5B, WT or S22R UbcH5B (UbcH5BS22R) were purified as described above, except gel filtration was performed using 50 mm HEPES, pH 7.5, 150 mm NaCl, 5 mm DTT. Purified UbcH5B (5 μm) was loaded with ubiquitin in which all seven lysines were mutated to arginines (ubiquitin K0 (UbK0)) (5 μm) using human E1 (both from R&D Systems) at 30 °C for 20 min in buffer containing 50 mm HEPES, pH 7.5, 100 mm NaCl, 2 mm ATP, 2 mm MgCl2, 100 μm DTT. E1 activity was quenched as described (23). UbcH5B∼Ub (0.5 μm) was combined with 400 μm Lys and 5 μm purified, GST-cleaved AO7RE with amino acids 238–245 deleted (AO7REΔ) in 50 mm HEPES, pH 7.5, 100 mm NaCl in the presence or absence of 160 μm UbK0. Discharge proceeded at 25 °C, and samples were assessed at the indicated time points. Samples were resolved by SDS-PAGE without reducing agent, and UbcH5B was detected by immunoblotting.
Cell Experiments
HEK-293 cells were cultured in DMEM containing 10% (v/v) fetal bovine serum, 2 mm glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin in a humidified incubator at 37 °C. Cells were transfected as described (48) with a 4:1 ratio of plasmids encoding forms of HA-AO7 and GFP. After 36–48 h, cells were treated with 80 μg/ml cycloheximide, 10 μm lactacystin, or 50 μm MG132 (all from Sigma) and lysed and processed as described (49).
Microscale Thermophoresis (MST)
UbcH5B-Green was expressed in E. coli BL21 (DE3) and purified with GS 4B. The immobilized protein was incubated on beads with FlAsH-EDT2 for 20 min at room temperature, thoroughly washed in buffer consisting of 50 mm Tris-HCl, pH 7.5, and 100 mm NaCl, and released from beads by cleavage with PreScission Protease. Microscale Thermophoresis was performed with titration of AO7 RING constructs with labeled UbcH5B in Monolith NT.115 (NanoTemper Technologies GmbH). Measurements were repeated at least twice in triplicates. Data were fitted with the equation for mass action by nonlinear least squares to estimate Kd. For UbcH5B-Green binding to ubiquitin, the apparent Kd was reported, as saturation was not achieved even at the highest concentration of ubiquitin (1 mm).
NMR Spectroscopy
NMR samples were prepared in 50 mm Tris-HCl, pH 7.5, 2 mm tris(2-carboxyethyl)phosphine; experiments were performed at 25 °C. For titration experiments, 15N-labeled UbcH5B samples were concentrated up to 0.3 mm, whereas the concentration of AO7 217–258 ranged from 0.15 to 1.5 mm. NMR spectra were acquired on 600-MHz Bruker spectrometers equipped with cryoprobes. Titration experiments were monitored by recording two-dimensional 1H,15N correlated spectra at various ratios of UbcH5B to AO7 217–258. All NMR data were processed using NMRPipe. The assignments and analysis were derived using software SPARKY (50). Titration curves were fitted to 1:1 model by software written in MATLAB (kindly provided by David Fushman) (51).
Results
A Region Beyond the AO7 RING Finger Is Required for E2 Binding
Analogous to mouse AO7 (3), a RING-containing fragment of human AO7 that extends 15 amino acids beyond the last predicted Zn2+-coordinating Cys (AO7R) (see Fig. 1A for a schematic) was insufficient to bind UbcH5B, as assessed using GST fusion proteins and GS pulldown (Fig. 1B). However, an extension of 57 amino acids beyond the last RING Cys (AO7R extended; AO7RE), terminating at residue 258, was adequate for stable E2 binding. This difference was validated by MST where AO7RE showed an ∼80-fold greater affinity for UbcH5B relative to AO7R (180 nm versus 14 μm; Fig. 1C and Table 1).
TABLE 1.
Kd values measured by MST
| E2 | Interacting protein | Kd |
|---|---|---|
| UbcH5B | AO7R | 14 ± 1 μm |
| UbcH5B | AO7RE | 180 ± 32 nm |
| UbcH5B | AO7REY165A | 141 ± 13 nm |
| UbcH5B | AO7REP199A | 62 ± 15 nm |
| UbcH5B | AO7REI137A | 318 ± 24 nm |
| UbcH5B | Ubiquitin | 215 ± 80 μm |
| UBE2G2a | G2BR of gp78 | 28 ± 2 nm |
a Kd = 21 ± 4 nm based on isothermal calorimetry titration (22).
AO7 Forms a Clamp-like Structure around UbcH5B
Attempts were first undertaken to crystallize AO7RE alone. The large scale purification of AO7RE or variants, including AO7R, necessary for crystallography or NMR, resulted in non-refoldable precipitated protein. To circumvent this difficulty, AO7RE and UbcH5B were co-expressed (see “Experimental Procedures”). This AO7RE·UbcH5B complex was crystallized and diffracted to 1.8 Å (Table 2). There is one AO7RE·UbcH5B complex in the asymmetric unit, which includes two Zn2+ ions (Fig. 2A).
FIGURE 2.

Crystal structure of the AO7RE·UbcH5B complex. A, ribbon diagram of the structure (residue 1 of UbcH5B and residues 126–128, 178–194, and 256–258 of AO7 are not observed and are presumably disordered). UbcH5B is shown in cyan. AO7RE is shown in yellow except for the Linker, in red. Helices, strands, and loops are illustrated as spirals, arrows, and tubes, respectively. The N and C termini and secondary structure elements are labeled in black and red for UbcH5B and AO7, respectively. The disordered peptide (residues 178–194 after the α1 helix) of AO7RE is represented with a dashed curve. Two zinc ions in AO7 RING are illustrated. B, superposition of AO7 RING with other RING and U-box structures (PDB entries 3FL2, 1Z6U, 1FBV, 3HCT, 1LDJ, 2CKL, 2C2V, 3EB6, and 3LRQ). The AO7 RING is shown in yellow, the central α-helix of AO7 RING is labeled, and the disordered peptide in loop 2 of the AO7 RING is indicated with a dashed curve. C–E, the interactions of UbcH5B with AO7 RING (C), Linker (D), and U5BR (E, viewed from two directions). UbcH5B is shown in cyan, RING and U5BR are in yellow, and Linker is in red. The residues involved in electrostatic (cutoff distance = 3.5 Å) and van der Waals (cutoff distance = 4.0 Å) interactions at the interface are shown as sticks. Dashed lines in black indicate salt bridges and hydrogen bonds. The residues involved in the interactions are labeled in black and red for UbcH5B and AO7, respectively.
In comparison to other published structures, UbcH5B in the AO7RE·UbcH5B complex features a typical E2 fold (for review, see Ref. 52) and does not change significantly upon binding AO7RE. The root mean square deviations between UbcH5B in the AO7RE·UbcH5B complex and several free UbcH5B structures (PDB entries 2ESK, 2ESO, 2ESP, and 2ESQ) are ∼0.5 Å. The number and property of residues involved in RING binding as well as those surrounding the active site Cys, including the highly conserved Asn-77 (53, 54), are similar among all structures.
In the complex, AO7RE is clamp-shaped and surrounds UbcH5B (Fig. 2A; for linear representation see Fig. 1A). The buried surface between UbcH5B and AO7RE is ∼2950 Å2. The AO7RE fragment contains three structural motifs: a highly atypical N-terminal RING domain (residues 129–216), a secondary UbcH5B-binding region (residues 228–255, U5BR), and an extended linker region connecting the RING to the U5BR domain (residues 217–227, Linker) (Fig. 2A). The RING contains a β-hairpin, a central α-helix (α1), and two loops that contribute, as predicted, four of the eight coordinating residues of the C3H2C3 RING to Zn2+ 1 and Zn2+ 2 (3). This core of the RING is followed by a region that is unique to AO7, which contains a second α-helix (α2) that stacks “underneath” the core and interacts extensively through hydrophobic interactions with the structural elements of the core of this RING (Fig. 2A). This region connects directly to the Linker. The AO7 RING is also unique among canonical RING fingers in that the α1 helix has two additional turns at its C-terminal end and a 17-residue disordered Gln-Glu-rich region in loop 2 (residues 178–194; Fig. 2B, for sequence see Fig. 1A).
All three components of the AO7RE clamp interact with UbcH5B. The RING of AO7 covers loops β3β4 and β4α2 of UbcH5B and also interacts with its α1 helix (Fig. 2A). Residues from both the AO7 α1 helix and residues in proximity to the Zn2+-coordinating cysteines of the two loops are involved in the interaction (Fig. 3A). Hydrophobic interactions dominate the RING-UbcH5B interface. The Val-136, Ile-137, Tyr-165, and Pro-199 side chains of AO7 face the hydrophobic surface composed of Pro-61, Phe-62, Pro-95, and Ala-96 from loops β3β4 and β4α2 of UbcH5B (Fig. 2C). Leu-139 and Tyr-140 stack with the hydrophobic portions of side chains of Arg-5, Lys-8, Glu-9, and Asp-12 from the UbcH5B α1 helix (Fig. 2C). Several hydrogen bonds are formed across the RING-UbcH5B interface, including those between AO7 Val-136 and Ile-137 and UbcH5B Arg-5 and between AO7 Pro-199 and UbcH5B Ser-94 (Figs. 2C and 3A). This interaction pattern is conserved among RING and U-box complexes with E2s (39, 55–57).
FIGURE 3.

Interactions between UbcH5B and the U5BR. A–C, summary of intramolecular interactions between WT (A) and mutant (B and C) AO7 RING fingers and UbcH5B. Solid lines indicate van der Waals interactions (cutoff distance = 4.0 Å). Dashed lines indicate hydrogen bonds or salt bridges (cutoff distance = 3.5 Å). The secondary structure assignments of the residues are illustrated. Residues in red were mutated to Ala, and co-crystallization was undertaken or attempted as described in the “Results” section. D, trajectories of two 15N-labeled UbcH5B residues that shift upon titration with AO7 fragment 217–258 (Linker and U5BR) used to calculate Kd. E, chemical shift perturbations in the UbcH5B upon interaction with the AO7 fragment (residues 217–258). Unlabeled AO7 was titrated onto 15N-labeled UbcH5B. Upon titration of 15N-labeled UbcH5B, residues on the UbcH5B backside β-sheet are perturbed significantly in areas corresponding to U5BR binding. In contrast, the Linker does not perturb the UbcH5B significantly. The perturbation (Δδ) is defined as the difference in peak frequencies of the free and AO7-saturated states. Δδ is calculated by the formula Δδ = [(δNf − δNs)2/5 + (δHf − δHs)2]1/2, where δHf, δHs and δNf, δNs are the proton and nitrogen frequencies in free and saturated state, respectively. UbcH5B residues contacting AO7 in the crystal structure are shown as circles on the data. Black circles represent UbcH5B contacts with the AO7 U5BR in the crystal structure, whereas gray-filled circles represent UbcH5B contacts with the AO7 Linker.
Residues equivalent to AO7 Arg-202, which immediately follows the last Zn2+-coordinating Cys, have been implicated in binding to thioester-linked ubiquitin in RING-type E3·E2-Ub structures7 (8, 9, 11, 15, 16) and potentially serve as “allosteric linchpins” in the activation of E2∼Ub by RING-type domains (9). Interestingly, unlike most RING-type E3s in complex with E2s, Arg-202 exhibited static disorder with two conformations in a 0.56/0.44 ratio (Fig. 2C, see also Fig. 6C). The major conformation allows for the conserved hydrogen bonding with UbcH5B Gln-92 or the equivalent in other E2s (e.g. Refs. 39, 56, and 57; for review, see Ref. 1), whereas the minor conformation, which is not generally observed, led to a hydrogen bond with the AO7 Gln-197 side chain and a van der Waal interaction with UbcH5B Ser-94. The AO7RE·UbcH5B structure was, of necessity, determined in the absence of thioester-linked ubiquitin because of the need for co-expression.
FIGURE 6.

Assessment of potential for ubiquitin backside binding in cells. A, HEK-293 cells were transfected as indicated with plasmids encoding HA-tagged AO7 amino acids 2–439 (HA-AO7) or HA-AO7 lacking amino acids 238–245 (HA-AO7Δ). Each transfection was divided into four before carrying out a cycloheximide chase. B and C, HEK-293 cells were transfected as in Fig. 5A and treated with either MG132 (50 μm) or lactacystin (LCN; 10 μm). D, HEK-293 cells were transfected with plasmid encoding HA-AO7Δ or HA-AO7Δ in which Tyr-165 in the central α-helix of the RING was mutated to Ala. Cells were then divided for cycloheximide (CHX) chase as in Fig. 6A. GFP served as a transfection efficiency control in all experiments.
The AO7RE Linker covers the outer surface of the UbcH5B α1 helix (Fig. 2D). On one side of the Linker, Met-222, Leu-224, and Tyr-225 of AO7 contact Leu-3, His-7, and Leu-10 of UbcH5B, respectively, and on the other side, salt bridges and hydrogen bonds are formed between the proteins.
The U5BR of AO7RE contains a long α-helix (α3) and a short C-terminal loop that interacts extensively with the backside of UbcH5B (Fig. 2E). From the middle of the α3 helix to the C terminus of AO7RE, several hydrophobic side chains (Leu-241, Tyr-242, Ile-251, Ile-252, and Leu-254) interact with UbcH5B. AO7 Tyr-242 forms an edge-to-face interaction with UbcH5B Phe-51, whereas the AO7 Ile-252 side chain penetrates deeply into a hydrophobic pocket formed by side chains from the UbcH5B α4α2 loop (Fig. 2E). A number of hydrogen bonds and salt bridges surround the hydrophobic interface. The guanidinium groups of AO7 Arg-244 and Arg-248 each form a salt bridge with the C-terminal (Met-147) carboxyl group of UbcH5B. In addition, the AO7 Arg-238 and Gln-245 side chains form hydrogen bonds with side chains from the UbcH5B β-sheet, whereas the main chain groups of AO7 Gly-249, Gly-250, and Ile-252 are hydrogen-bonded to several UbcH5B side chains (Fig. 2E). The U5BR is hook-shaped in general, anchoring on the backside of UbcH5B on the opposite side of the E2 from the site of covalent binding of UbcH5B to Ub and from the closed conformation of UbcH5B∼Ub (for review, see Ref. 1). By NMR, the U5BR together with the Linker exhibited a dissociation constant (Kd) of 173 ± 40 μm for UbcH5B (Fig. 3D). Assessment of binding of this region by NMR using 15N-labeled UbcH5B demonstrated that, in solution, as in the crystal structure, most contacts with UbcH5B distal to the RING finger are in the U5BR (Fig. 3E). Notably, the site of U5BR binding to UbcH5B overlaps the site of non-covalent UbB binding to the UbcH5 family of E2s (Kd > 200 μm; Table 1) (17, 20).
Requirements for AO7-UbcH5B Binding and Ubiquitination
To begin to assess the significance of the U5BR, we carried out a structure-based mutational analysis. Mutation of two U5BR residues that contact the β-sheet core of UbcH5B (Fig. 2E), Y242A and Q245A (AO7REYQ-AA), significantly diminished binding (Fig. 4A). Deletions within the U5BR of either amino acids 238–245, referred to hereafter as AO7REΔ, or amino acids 246–252 resulted in loss of high-affinity UbcH5B binding, as did deletion of amino acids 217–222 of the Linker (Fig. 4A).
FIGURE 4.

Effects of disruption of the U5BR. A, indicated GST fusions of mutations and deletions in AO7RE were assessed for binding to UbcH5B as in Fig. 1B. AO7REYQ-AA refers to Ala mutations of Tyr-242 and Gln-245. B, GST fusions prebound to GS were subject to autoubiquitination reactions and, after washing of beads, eluted material was immunoblotted (IB) for ubiquitin. C, 35S-labeled in vitro transcribed and translated GST fusions were incubated in solution in the presence of E1, UbcH5B, and ubiquitin for the indicated times and ubiquitination assessed. D, quantification of results from C. E, in vitro transcribed and translated 35S-labeled UbcH5B was loaded with ubiquitin. After inactivation of E1, the rate of loss of covalently-bound ubiquitin was measured in the presence of GST-AO7RE or GST-AO7REΔ and ∼80 μm free ubiquitin. Results were normalized to those obtained with GST. Concentrations of GST fusions and E2 were ∼1 μm and 2 nm, respectively. Data represent the average and S.D. of three independent experiments. F, representative experiment used in E.
A GST fusion of AO7RE was next compared with AO7R and AO7REΔ, both of which lack an intact U5BR, in autoubiquitination assays with UbcH5B (Fig. 4B). In these assays, except for free ubiquitin, essentially all of the ubiquitin immunoreactivity is found on the AO7 fusion proteins (data not shown). Compared with AO7RE, both AO7R and AO7REΔ exhibited a substantial increase in ubiquitination, including a marked increase in higher molecular weight forms (Fig. 4B). This suggests increased activity with loss of the U5BR. As results with AO7REΔ and AO7R were indistinguishable, most experiments that follow were carried out comparing AO7RE to AO7REΔ.
Immunoblotting for ubiquitin was corroborated by directly assessing ubiquitination of 35S-labeled AO7 (Fig. 4, C and D). A higher proportion of AO7REΔ molecules was modified at each time accompanied by a greater upward shift in migration compared with AO7RE, even after extended incubation. Single-round discharge experiments where UbcH5B∼Ub was first preincubated with an ∼500-fold excess of AO7RE or AO7REΔ confirmed that AO7REΔ, which lacks an intact U5BR, had greater activity than AO7RE (Fig. 4, E and F). These demonstrate that the differences observed in autoubiquitination assays are due to activity and are not accounted for by differences in the rate of exchange of UbcH5B for UbcH5B∼Ub (58, 59). Thus, loss of the U5BR enhances AO7 activity despite its decreased affinity for UbcH5B.
AO7REΔ Reveals a Role for Ubiquitin Backside Binding
One explanation for the unexpected increase in reactivity with disruption of the U5BR could lie in the increased accessibility of UbB to UbcH5B when the U5BR is not intact. To address this we employed an UbcH5B mutant that cannot bind UbB (UbcH5BS22R) (17). This mutant does not significantly alter UbcH5B binding to AO7RE (Fig. 5A) and has relatively minor effects on AO7RE autoubiquitination (Fig. 5B, compare lanes 1, 7, and 13 to lanes 2, 8, and 14). In sharp contrast, when either AO7REΔ or AO7R was evaluated with UbcH5BS22R, there was a dramatic decrease in ubiquitination, with low molecular weight ubiquitin-modified forms of the E3 only detectable at later time points (Fig. 5B).
FIGURE 5.

Role of backside binding in AO7 ubiquitination. A, binding assays were carried out as in Fig. 1B, percent of input bound is indicated. B, time course of ubiquitination was carried out as in Fig. 4B. C, loss of E2∼Ub was assessed as in Fig. 4E. Data are from three independent experiments. D, ubiquitination was carried out as in Fig. 4B. E, binding assay was carried out as in Fig. 1B. IB, immunoblot. F, ubiquitination assay was carried out as in Fig. 4B for 90 min using UbcM2 as the E2. G, purified, bacterially expressed WT or S22R UbcH5B was loaded with equimolar amounts of ubiquitin lacking Lys (UbK0), and the release of UbK0 was assessed during incubation in the presence or absence of added UbK0 (160 μm) and with AO7REΔ from which GST had been cleaved. UbcH5B was detected by immunoblotting. Note the ratio of E2∼Ub to E2 over the time course. Efficiency of loading is less than in experiments using 35S-labeled E2 and WT ubiquitin (e.g. Fig. 4F), as the amount of UbK0 used was kept low during loading to maximize the effect of UbK0 added during the discharge phase.
To understand the mechanistic basis for this difference, we once again carried out single round turnover experiments (Fig. 5C). In the presence of AO7RE, the rate of ubiquitin transfer from UbcH5BS22R was comparable with WT UbcH5B. In contrast, using AO7REΔ, ubiquitin transfer from UbcH5BS22R was dramatically diminished relative to the rate from WT UbcH5B. This strongly supports the idea that UbB binding plays a critical role in ubiquitination mediated by AO7REΔ but not by AO7RE, where this binding is blocked by the high-affinity interaction with the U5BR.
To complement our findings with S22R, we wished to assess the impact of mutating Ile-44 of ubiquitin (UbI44A). Ile-44 is central to, and essential for, the interaction of UbB with the backside of UbcH5B (17). Unfortunately, mutating Ile-44 disrupts E2 function, as exemplified in studies using the E2, Cdc34, for which UbB binding is not implicated in function (12). This disruption is due, at least in part, to loss of a critical interaction of the E2 with ubiquitin covalently bound to its active site (E2∼Ub) (8, 9, 11). However, in Cdc34, a complementary mutation restores function (12). Similarly, for AO7RE, where UbB binding to UbcH5B was blocked by the U5BR, UbI44A resulted in a loss of function, but a mutation of UbcH5B analogous to that made in Cdc34 (UbcH5BS108L) resulted in a significant restoration of activity (Fig. 5D, lanes 1–4). The restoration of function with UbcH5BS108L allowed us to assess the role of disrupting UbB binding using UbI44A and AO7REΔ. Unlike the significant restoration of activity seen with AO7RE, this combination failed to restore function with AO7REΔ (Fig. 5D, lanes 5–8). Together with the results obtained with UbcH5BS22R, these findings demonstrated the stimulatory role of UbB binding to UbcH5B when the U5BR is disrupted. An additional important finding that arises from these experiments is that, when UbB binding is precluded, an intact U5BR is required for significant ubiquitination (Fig. 5, B–D).
To further evaluate the positive role of the U5BR when UbB binding is lost or diminished, we turned to another E2, UbcM2/Ube2e3/UbcH9, which tightly binds AO7 (3, 60). This binding is also U5BR-dependent (Fig. 5E). In contrast to UbcH5, UbcM2 shows very little UbB binding and exhibits markedly decreased activity with AO7 relative to UbcH5B (60). When ubiquitination was assessed, activity was seen with AO7RE (Fig. 5F). However, ubiquitination was essentially undetectable with UbcM2 when the U5BR was interrupted (AO7REΔ, AO7R), reinforcing the importance of the U5BR in facilitating ubiquitination in the absence of significant UbB binding.
The stimulatory effect of UbB binding was originally postulated to be due to this ubiquitin itself being linked to an E2 (E2∼UbB), resulting in concatemerization of E2∼Ub molecules through backside binding. This would then facilitate chain formation, with backside-bound ubiquitin serving as the acceptor ubiquitin (17, 18). To evaluate whether this is indeed a requirement, a purified protein system was used to assess ubiquitin transfer from E2, with AO7REΔ as the E3. UbcH5B was loaded in the presence of an equimolar amount of UbK0, which does not function as an ubiquitin acceptor, and then allowed to discharge in the presence of excess free Lys8 (400 μm) (61, 62). When UbK0 was added during the discharge assay at a significantly higher concentration than at the loading step (160 versus 5 μm), the rate of discharge increased with WT UbcH5B (Fig. 5G, top two panels, compare the relative amounts of E2 and E2∼UbK0 as a function of time). In contrast, excess UbK0 had no effect on discharge when ubiquitin backside binding was disrupted by the S22R mutation (Fig. 5G, bottom two panels). This suggests that, although UbB binding is critical for ubiquitin transfer by UbcH5B, UbB does not necessarily serve as an acceptor. This is consistent with recent results using the RING E3 RNF38 (20).
Ubiquitin Backside Binding Can Be Functionally Important in Cells
In cells some E3s undergo autoubiquitination, often resulting in ubiquitin chains that target them for proteasomal degradation (for review, see Ref. 4). If this is true for AO7 and UbB binding is important in cells, which has not been previously assessed, then disruption of the U5BR should destabilize AO7 by enhancing autoubiquitination. Indeed, HA-AO7 (amino acids 2–439) lacking U5BR amino acids 238–245 (HA-AO7Δ), is short-lived relative to a version containing an intact U5BR (HA-AO7; Fig. 6A) and is stabilized by inhibitors of the proteasome (Fig. 6, B and C). Importantly, degradation of HA-AO7Δ is markedly diminished when its own ligase activity is compromised by mutation of a conserved residue in the RING-E2 interface (Y165Α; Fig. 6D, see also below), demonstrating that the accelerated loss of HA-AO7Δ is not due to misfolding. These findings are highly suggestive of the cellular relevance of non-covalent UbB binding.
Structures of AO7 RING Finger Mutations in Complex with UbcH5B
For other RING-type E3s, specific E3s residues at the canonical interface with E2 have been shown to affect the ability of RINGs to bind to E2 or activate E2∼Ub (Refs. 8, 9, 16, 36–39, 63, and 64; for review, see Ref. 1). However, parsing the effects of these mutations on low-affinity RING-E2 binding versus ligase function is difficult. Included among conserved interface residues shown to be important are bulky hydrophobic residues in loop 1 between the first two Zn2+-coordinating cysteines (usually an Ile) (36) and at the beginning of the central α-helix (37, 38) as well as a highly-conserved Pro in loop 2 (39), also between the Zn2+-coordinating residues (for review, see Ref. 1). Importantly, the effects of these mutations on the RING-E2 interface have not been assessed at a detailed structural level. We took advantage of the AO7 clamp and its high affinity for UbcH5B to evaluate mutations in each of these positions by co-crystallization (Zn2+ 1 loop, I137A; central α-helix, Y165A; Zn2+ 2 loop, P199A). AO7REY165A·UbcH5B and AO7REP199A·UbcH5B were successfully crystallized, but crystals for AO7REI137A·UbcH5B were not obtained. Neither the Y165A nor the P199A mutation caused significant overall conformational changes in the proteins (Fig. 7A). The root mean square deviation values for the three structures are ∼0.5 Å. All three mutated AO7 RING residues are located at the hydrophobic interface between AO7 and UbcH5B (Fig. 2A). The Ile-137 side chain faces the hydrophobic pocket surrounded by Pro-61, Phe-62, and Pro-95 of UbcH5B, contributing most to the hydrophobic interactions between AO7 RING and UbcH5B, providing a possible explanation for the inability to obtain a crystal structure of this mutant with E2.
FIGURE 7.
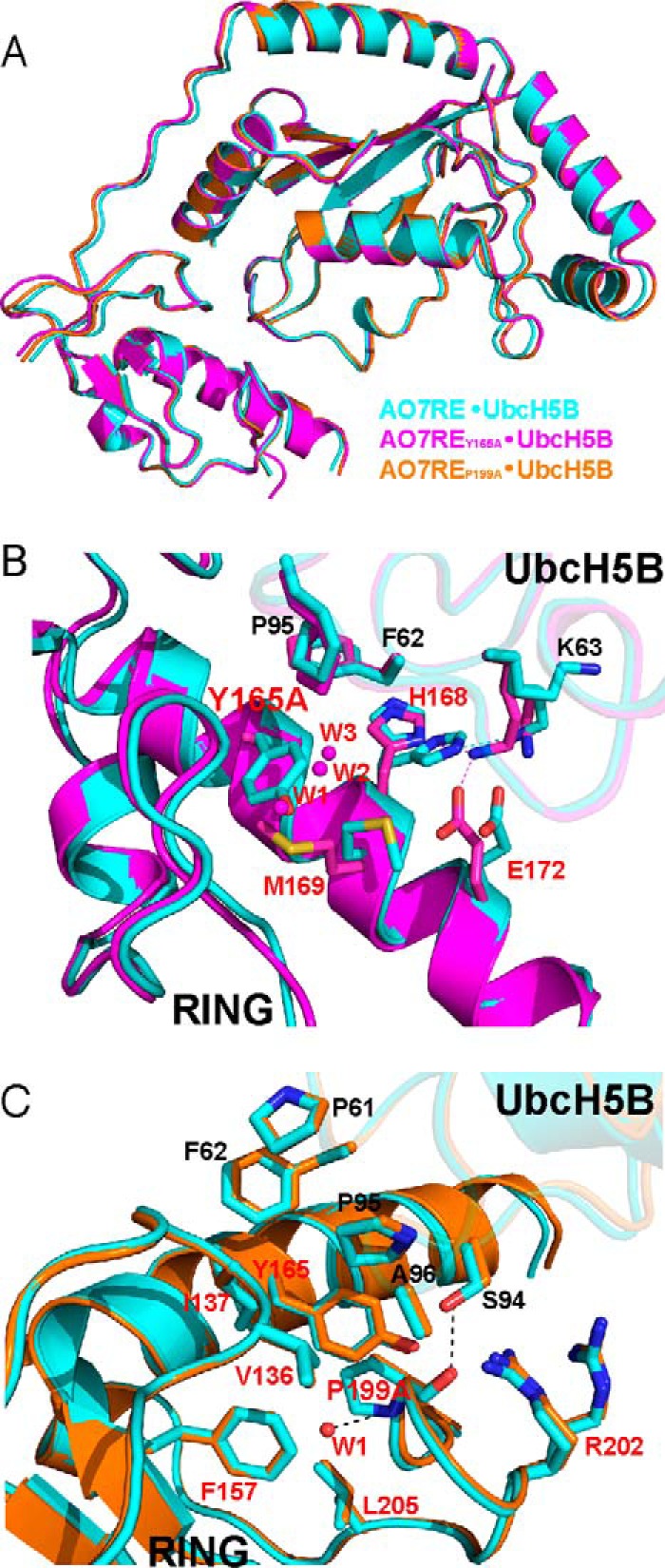
Structures of UbcH5B in complex with AO7RE mutants. A, superimposition of the AO7RE·UbcH5B (in cyan), AO7REY165A·UbcH5B (in magenta), and AO7REP199A·UbcH5B (in orange) complexes. B, the Y165A mutation resulted in a change in the AO7 Met-169 side-chain orientation inasmuch as the Met-169 side chain partially occupies the space created by the Y165A mutation, causing the AO7 Glu-172 side chain to bend toward the Met-169 side chain such that Glu-172 forms a new salt bridge with Lys-63 in the β3β4 loop of UbcH5B. The His-168 side chain, which is disordered in the WT, is stabilized in the mutant. Such conformational changes reshape the local E2-binding surface of the RING. Furthermore, several water molecules (W1, W2, and W3) invade into the cavity created by the Y165A mutation. The mutant and WT structures are shown in magenta and cyan, respectively. Residues at the RING-E2 interface are shown as sticks. The water molecules are shown as spheres. C, the P199A mutation brings a water molecule (W1) into the hydrophobic core of AO7 RING, which is hydrogen-bonded to the main chain amide of the Ala-199 residue. This does not disturb the conformation of nearby side chains. The residues on the interface or surrounding the hydrophobic core and the water molecule are illustrated. The mutant and WT structures are shown in orange and cyan, respectively.
Although the Y165A and P199A mutations of AO7 do not bring significant changes to the overall structure of the AO7RE-UbcH5B complex, they each cause localized changes to the hydrophobic environment at the RING-UbcH5B interface. In AO7REY165A·UbcH5B, the mutation induced the rearrangement of three side chains on the E2 binding surface of the RING central α-helix and invasion of several water molecules (Fig. 7B). Consequently, the landscape of the E2 binding surface of the RING is reshaped in the localized area of the mutation, and the hydrophobicity in the center of the RING-UbcH5B interface is changed. This leads to an alteration of several interactions, including the formation of two new RING interactions with Lys-63 of UbcH5B (Fig. 3B, compare with Fig. 3A). In AO7REP199A·UbcH5B, there is no notable conformational change in other interface residues, but the mutation brings a water molecule (W1) into the hydrophobic core of AO7RE (Fig. 7C and Fig. 3C compare with Fig. 3A). We observed no significant alterations in E2 residues in the α2 helix and the 310 (η1) helix with the Y165A and P199A mutations. These are regions that recent RING-type E3·UbcH5-Ub structures have implicated as being involved in RING-dependent ubiquitin binding and in formation of the activated closed conformation of E2∼Ub (for review, see Ref. 1). Additionally, AO7 Arg-202 remained disordered (Fig. 7C). The critical Asp-87 of UbcH5B was similarly disordered as it is in the WT complex. Asn-77 (53, 54) also did not change with mutations in the RING-E2 interface.
Consequence of RING Finger Mutations
The crystallographic analyses demonstrate that structural changes seen with AO7REY165A and AO7REP199A, as compared with WT AO7RE, are largely limited to local changes in the AO7RE-UbcH5B binding landscape, without substantial decreases in interactions at the RING-E2 interface. This is surprising considering their consequences for other RINGs. Consistent with limited alteration of interactions, binding of UbcH5B as assessed by GS pulldown showed no difference between AO7RE WT, Y165A, and P199A after extensive washing, whereas I137A demonstrated diminished binding (Fig. 8A). The affinity of Tyr-165 and Pro-199 mutants of AO7RE for UbcH5B were found to be greater than (Y165A) or similar to (P199A) that of AO7RE, whereas the affinity of I137A was marginally less than AO7RE (<1.5-fold) (Table 1 and Fig. 8B).
FIGURE 8.
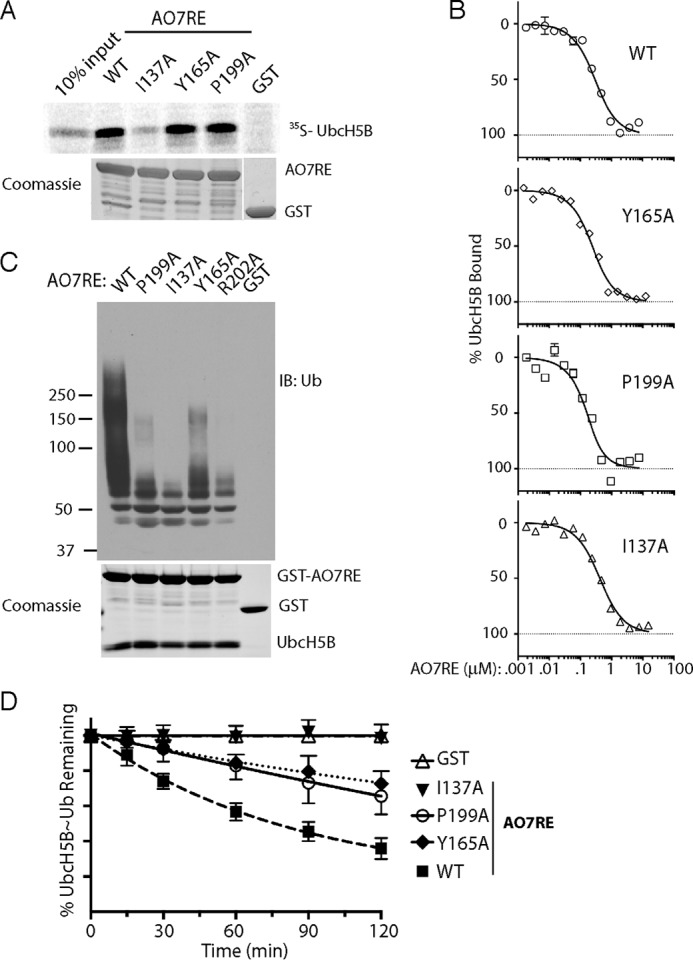
Consequences of RING finger mutations. A, 35S-labeled UbcH5B was incubated with the indicated GST fusions as in Fig. 1B. B, data were from MST as in Fig. 1C, including the same data set for WT AO7RE. See Table 1 for calculated the Kd values. C, equal amounts of the indicated AO7RE mutations were purified in complex with UbcH5B by washing co-expressed complexes only once, and ubiquitination was assessed after the addition of an equal amount of purified UbcH5B. Twice as much E2 was added in the GST control reaction. A Coomassie stain of the purified complexes is shown below. D, single round turnover experiments were carried out as in Fig. 4E. Data are graphed as the fraction of E2∼Ub remaining at each time point and are the average of three independent experiments.
In contrast, autoubiquitination with the Y165A and P199A mutants was markedly reduced compared with AO7RE (Fig. 8C). The I137A mutation (36) dramatically decreased ubiquitination. Similarly, mutation of Arg-202, whose equivalent in other RING type E3s has been shown, using E2-Ub, to be essential to maintain the closed conformation necessary for ubiquitin transfer (8, 9, 11, 15, 16), severely inhibits ubiquitination. The functional effects of the I137A, Y165A, and P199A RING mutations were confirmed in single turnover experiments (Fig. 8D). All of these mutations reduce the rate of ubiquitin transfer. In the context of the AO7 clamp, this cannot be explained by a simple effect on affinity for E2. This is particularly the case for Y165A and P199A, where the RING-E2 interface is largely unaltered and there are no substantial changes in E2 residues identified as being important for binding ubiquitin (8, 9, 11, 15, 16, 20). Accordingly, both Tyr-165 and Pro-199 play substantial roles in AO7 function without having significant effects on the E2-RING interface.
Discussion
Backside Binding to E2s
A central issue in RING-type E3 function is to understand how ubiquitination efficiently occurs despite generally low-affinity interactions between these E3s and E2s. As this study and others clearly demonstrate, non-covalent binding of ubiquitin to the backside of a subset of E2s (UbB) is one means by which ubiquitination is stimulated. This binding occurs between the Ile-44 “hydrophobic patch” on ubiquitin and a specific region on the backside of these E2s (17–20). The U5BR, which is a primarily helical, secondary E2 binding domain that forms the upper part of the AO7 clamp, traverses the UbB binding site on UbcH5B and precludes UbB binding (Fig. 9A).
FIGURE 9.

Backside binding to E2s. A, comparison of the AO7RE U5BR·UbcH5B (in cyan) and non-covalently associated ubiquitin (UbB)·UbcH5B (in orange) complexes. The superposition is based on the UbcH5B Cα positions. The Ser-22 side chain of UbcH5B is shown as sticks, and the Ile-44 hydrophobic patch of ubiquitin is circled in red. The U5BR helix crosses the loop connecting the β3 and β4 strands of ubiquitin. Ser-22 of UbcH5B is also covered by the overlapping region of the U5BR and ubiquitin. B, comparison of the AO7RE U5BR·UbcH5B (in cyan), gp78 G2BR·Ube2g2 (in magenta) and Rad18 R6BD·Rad6B (in red) complexes. The superposition of the complexes is based on the E2 Cα positions. Ser-22 in UbcH5B and the corresponding residues in Ube2G2 and Rad6B (Val-25 and Ser-25, respectively) are shown as sticks. G2BR contains only a straight helix and lies on the backside of E2; R6BD is a twisted helix and spans the β-sheet surface of Rad6b. The U5BR has an extra C-terminal loop in addition to the long helix, anchors to a hydrophobic valley on the UbcH5B surface, and thus provides more extensive interaction with E2. The indicated N and C termini are of ubiquitin, U5BR, G2BR and R6BD.
Several other RING-type E3s also contain secondary E2 binding domains, most of which are characterized by a prominent α-helix that crosses the UbB binding site, although some contain additional N- or C-terminal features that enhance interactions with E2. For the mammalian endoplasmic reticulum-associated degradation (ERAD) E3 gp78 and the yeast CUE1 protein, which functions with the ERAD E3s HRD1 and DOA10, these E2 binding helices cross the UbB binding site at a markedly different orientation then that of the U5BR when aligned based on the conserved E2 fold (Fig. 9B) (16, 21–23). Unlike the U5BR, these regions all allosterically stimulate ubiquitination with ERAD E2s (16, 22, 23). Single helices that cross the UbB binding site are also found in the RING-type E3s Rad18 and Bre1, which function with the E2 Rad6. These exhibit yet a different orientation (Fig. 9B) and limit ubiquitination by blocking UbB binding, favoring monoubiquitination (18, 19). More complex E2 backside binding is provided by the binding of the “winged-helix B” domain of the cullin-like subunit of the anaphase promoting complex/cyclosome to UbcH10. In this case the first of two involved helices exhibits an orientation that resembles a severely truncated U5BR (24).
In all cases, these secondary E2 binding domains are either separated from the RING by >100 amino acids or are on distinct polypeptides (16, 18, 19, 21–24). The AO7 U5BR represents the first known example of an E2 backside binding region that is contiguous with a RING-type domain, the two being separated only by the short Linker necessary to traverse the N-terminal α-helix of UbcH5B. These contiguous domains result in a clamp-like structure that binds to UbcH5B with high affinity. Despite the tight binding conferred by the AO7 clamp, the rate of transfer of ubiquitin from E2∼Ub is slower when the clamp is intact. This is due to blocking of UbB binding by the U5BR region of AO7. However, in the absence of the potential for UbB binding (e.g. when an E2 with limited UbB binding is used or potentially when the local concentration of ubiquitin is limited), the high-affinity binding of the intact clamp increases activity relative to a disrupted clamp. Thus, the AO7 clamp uniquely has context-dependent effects that either decrease or increase catalysis of E2∼Ub.
The question arises as to why there is a more substantial stimulation of catalysis by UbB binding and a disrupted AO7 clamp (AO7R or AO7REΔ) compared with the high-affinity intact clamp, where UbB binding is blocked (see Figs. 4E and 5C). A definitive answer cannot be discerned from the structures presented herein. There are hints that arise from recent elegant work of Buetow et al. (20), where the authors provide evidence that UbB increases the affinity of UbcH5B-Ub for the RNF38 RING. This is marginally greater (∼2-fold) than the affinity between AO7RE and UbcH5B. Importantly, they also find alterations in the structure of the α1 helix and the α1β1 loop of UbcH5B in the context of RING·UbcH5B-Ub and, with UbB binding, changes in dynamics of this region are noted. This promotes conformations of UbcH5B∼Ub that facilitate catalysis (20). A possibility, therefore, is that combinatorial differences in these factors (affinity, structure, and dynamics) account for the differential effect of UbB relative to the U5BR. Interestingly, the three AO7RE·UbcH5B structures in this study also exhibit conformational changes in UbcH5B in the α1 helix and the α1β1 loop (compared with free UbcH5B, PDB entries 2ESK, 2ESP, 2ESO, and 2ESQ). However, these changes are not identical with, or directly comparable with those observed by Buetow et al. (20); regardless, they are all within the conformational space of this region. Definitively assessing the effects of the AO7 clamp versus UbB on E2∼Ub structure and dynamics will require additional extensive structural and biochemical studies.
It now becomes important to understand whether UbB binding is relevant in vivo. The fact that binding of UbcH5B-Ub to RING increases the affinity of UbB from 200 to 14 μm in the case of RNF38 (20) makes cellular levels of ubiquitin (20–80 μm; Refs. 65 and 66) sufficient to have an effect. Our cellular data showing proteasomal degradation of AO7 when the clamp is disrupted and the site of UbB binding is accessible provide in vivo data consistent with its importance. Whether this apparent in vivo UbB binding effect is due to the free ubiquitin pool, polyubiquitin chains, ubiquitin already attached to substrates, E2∼Ub, or a combination thereof, and whether ubiquitin-like domains also contribute are interesting questions that remain to be addressed.
Complexity of the E2-RING Interface
The significance of specific conserved RING residues at the E2 interface is generally poorly understood. Taking advantage of the AO7 clamp we have, for the first time, explored the importance of mutations of critical residues in the central α1 helix and the second Zn2+-coordinating loop (Y165A and P199A, respectively) at a structural level. Interestingly, we find that, for both mutants, there is (i) no significant decrease in RING affinity for UbcH5B, (ii) only minimal changes in the RING-E2 interface, and (iii) compared with AO7RE, no difference in the positions of residues implicated in the allosteric RING-mediated activation of E2∼Ub (Refs. 6, 8, 9, 11, 14–16, and 20; for review, see Ref. 1). Despite this, mutation of either Tyr-165 or Pro-199 resulted in a marked decrease in activity with UbcH5B. How this occurs is not clear, but there are at least two possibilities. First, although neither of these residues is predicted to have direct interactions with ubiquitin covalently linked to E2 (E2∼Ub), it is possible that through local changes they indirectly affect its positioning. However, there is no evidence to support this based on other structural studies in which the positioning of ubiquitin in E2-Ub was examined (Refs. 8, 9, 11, 14–16, and 20; for review, see Ref. 1). A second, attractive, possibility is that one or both of these residues are involved in transmitting allosteric changes from the RING to the active site of UbcH5B and thereby affect ubiquitin transfer. Understanding this will require analysis of the effect of these mutations on structure and dynamics using E2-Ub with either AO7 or other RING E3s. Regardless, our data strongly suggest that the full impact of specific residues at the RING-E2 interface is yet to be discerned. Although the physiological function of AO7 awaits determination, the unique clamp-like structure that we describe and the functional consequences of this clamp provide valuable tools for further analysis of RING-type E3 function.
Author Contributions
S. L., Y. C. T., X. J., and A. M. W. conceptualized the study. S. L., Y-H. L., J. M., M. B. M., D. K. S., and Y. C. T. carried out experiments as well as generated and purified reagents. V. A. H. and J. L. generated and purified critical reagents. Y-H. L. and X. J. solved and interpreted structures and generated corresponding figures. P. A. R. helped design and analyze affinity studies. S. L., J. M., M. B. M., D. K. S., Y. C. T., and A. M. W. analyzed biochemical and cell biological studies. Y. C. T., X. J., and A. M. W. oversaw the study. S. L., Y-H. L., J. M., M. B. M., D. K. S., P. A. R., Y. C. T., X. J., and A. M. W. all contributed to preparing figures and/or writing the manuscript.
Acknowledgments
We thank Marzena Dyba and Sergey Tarasov of the Biophysics Resource, Structural Biophysics Laboratory, Center for Cancer Research for assistance with MST and Mei Yang for providing reagents. Some experiments were carried out with assistance from Erin Marshall and Alex Roumeliotis (both supported by the Werner H. Kristen Student Internship Program). We thank R. Andrew Byrd, Ranabir Das, and Stanley Lipkowitz for invaluable discussions and critical reading of this manuscript. We also thank Ranabir Das and R. Andrew Byrd for NMR analyses and Gary Pauly for FlAsH-EDT2.
This work was supported by the Intramural Research Program of the, National Institutes of Health, National Cancer Institute, Center for Cancer Research. The authors declare that they have no conflict of interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5D1K, 5D1L, and 5D1M) have been deposited in the Protein Data Bank (http://wwpdb.org/).
E2-Ub or UbcH5B-Ub indicates forms of E2 bound to ubiquitin through the active site in which the active site Cys is replaced by a Ser or Lys to allow for E1-mediated charging with ubiquitin and relative resistance to hydrolysis when bound to RING-type domains.
The excess Lys added as an acceptor did not substantively accelerate the rate of discharge in the presence of AO7REΔ.
- Ub
- ubiquitin
- E2∼Ub (UbcH5B∼Ub)
- ubiquitin thioester-linked to E2
- UbB
- ubiquitin that binds to the backside of E2
- RING
- really interesting new gene
- AO7RE
- AO7 RING extended
- E1
- ubiquitin-activating enzyme
- GS
- glutathione-Sepharose
- UbK0
- ubiquitin with all Lys replaced with Arg
- MST
- microscale thermophoresis
- U5BR
- UbcH5B binding region.
References
- 1.Metzger M. B., Pruneda J. N., Klevit R. E., and Weissman A. M. (2014) RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta 1843, 47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deshaies R. J., and Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 [DOI] [PubMed] [Google Scholar]
- 3.Lorick K. L., Jensen J. P., Fang S., Ong A. M., Hatakeyama S., and Weissman A. M. (1999) RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl. Acad. Sci. U.S.A. 96, 11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weissman A. M., Shabek N., and Ciechanover A. (2011) The predator becomes the prey: regulating the ubiquitin system by ubiquitylation and degradation. Nat. Rev. Mol. Cell Biol. 12, 605–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirisako T., Kamei K., Murata S., Kato M., Fukumoto H., Kanie M., Sano S., Tokunaga F., Tanaka K., and Iwai K. (2006) A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 25, 4877–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ozkan E., Yu H., and Deisenhofer J. (2005) Mechanistic insight into the allosteric activation of a ubiquitin-conjugating enzyme by RING-type ubiquitin ligases. Proc. Natl. Acad. Sci. U.S.A. 102, 18890–18895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pruneda J. N., Stoll K. E., Bolton L. J., Brzovic P. S., and Klevit R. E. (2011) Ubiquitin in motion: structural studies of the ubiquitin-conjugating enzyme approximately ubiquitin conjugate. Biochemistry 50, 1624–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dou H., Buetow L., Sibbet G. J., Cameron K., and Huang D. T. (2012) BIRC7-E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat. Struct. Mol. Biol. 19, 876–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pruneda J. N., Littlefield P. J., Soss S. E., Nordquist K. A., Chazin W. J., Brzovic P. S., and Klevit R. E. (2012) Structure of an E3:E2∼Ub complex reveals an allosteric mechanism shared among RING/U-box ligases. Mol. Cell 47, 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soss S. E., Klevit R. E., and Chazin W. J. (2013) Activation of UbcH5c∼Ub is the result of a shift in interdomain motions of the conjugate bound to U-box E3 ligase E4B. Biochemistry 52, 2991–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plechanovová A., Jaffray E. G., Tatham M. H., Naismith J. H., and Hay R. T. (2012) Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature 489, 115–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saha A., Lewis S., Kleiger G., Kuhlman B., and Deshaies R. J. (2011) Essential role for ubiquitin-ubiquitin-conjugating enzyme interaction in ubiquitin discharge from Cdc34 to substrate. Mol. Cell 42, 75–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wickliffe K. E., Lorenz S., Wemmer D. E., Kuriyan J., and Rape M. (2011) The mechanism of linkage-specific ubiquitin chain elongation by a single-subunit E2. Cell 144, 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakatani Y., Kleffmann T., Linke K., Condon S. M., Hinds M. G., and Day C. L. (2013) Regulation of ubiquitin transfer by XIAP, a dimeric RING E3 ligase. Biochem. J. 450, 629–638 [DOI] [PubMed] [Google Scholar]
- 15.Dou H., Buetow L., Sibbet G. J., Cameron K., and Huang D. T. (2013) Essentiality of a non-RING element in priming donor ubiquitin for catalysis by a monomeric E3. Nat. Struct. Mol. Biol. 20, 982–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Das R., Liang Y. H., Mariano J., Li J., Huang T., King A., Tarasov S. G., Weissman A. M., Ji X., and Byrd R. A. (2013) Allosteric regulation of E2:E3 interactions promote a processive ubiquitination machine. EMBO J. 32, 2504–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brzovic P. S., Lissounov A., Christensen D. E., Hoyt D. W., and Klevit R. E. (2006) A UbcH5/ubiquitin noncovalent complex is required for processive BRCA1-directed ubiquitination. Mol. Cell 21, 873–880 [DOI] [PubMed] [Google Scholar]
- 18.Hibbert R. G., Huang A., Boelens R., and Sixma T. K. (2011) E3 ligase Rad18 promotes monoubiquitination rather than ubiquitin chain formation by E2 enzyme Rad6. Proc. Natl. Acad. Sci. U.S.A. 108, 5590–5595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turco E., Gallego L. D., Schneider M., and Köhler A. (2015) Monoubiquitination of histone H2B is intrinsic to the Bre1 RING domain-Rad6 interaction and augmented by a second Rad6-binding site on Bre1. J. Biol. Chem. 290, 5298–5310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buetow L., Gabrielsen M., Anthony N. G., Dou H., Patel A., Aitkenhead H., Sibbet G. J., Smith B. O., and Huang D. T. (2015) Activation of a primed RING E3-E2-ubiquitin complex by non-covalent ubiquitin. Mol. Cell 58, 297–310 [DOI] [PubMed] [Google Scholar]
- 21.Li W., Tu D., Li L., Wollert T., Ghirlando R., Brunger A. T., and Ye Y. (2009) Mechanistic insights into active site-associated polyubiquitination by the ubiquitin-conjugating enzyme Ube2g2. Proc. Natl. Acad. Sci. U.S.A. 106, 3722–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das R., Mariano J., Tsai Y. C., Kalathur R. C., Kostova Z., Li J., Tarasov S. G., McFeeters R. L., Altieri A. S., Ji X., Byrd R. A., and Weissman A. M. (2009) Allosteric activation of E2-RING finger-mediated ubiquitylation by a structurally defined specific E2-binding region of gp78. Mol. Cell 34, 674–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Metzger M. B., Liang Y. H., Das R., Mariano J., Li S., Li J., Kostova Z., Byrd R. A., Ji X., and Weissman A. M. (2013) A structurally unique E2-binding domain activates ubiquitination by the ERAD E2, Ubc7p, through multiple mechanisms. Mol. Cell 50, 516–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown N. G., VanderLinden R., Watson E. R., Qiao R., Grace C. R., Yamaguchi M., Weissmann F., Frye J. J., Dube P., Ei Cho S., Actis M. L., Rodrigues P., Fujii N., Peters J. M., Stark H., and Schulman B. A. (2015) RING E3 mechanism for ubiquitin ligation to a disordered substrate visualized for human anaphase-promoting complex. Proc. Natl. Acad. Sci. U.S.A. 112, 5272–5279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams C., van den Berg M., Panjikar S., Stanley W. A., Distel B., and Wilmanns M. (2012) Insights into ubiquitin-conjugating enzyme/co-activator interactions from the structure of the Pex4p:Pex22p complex. EMBO J. 31, 391–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dowil R. T., Lu X., Saracco S. A., Vierstra R. D., and Downes B. P. (2011) Arabidopsis membrane-anchored ubiquitin-fold (MUB) proteins localize a specific subset of ubiquitin-conjugating (E2) enzymes to the plasma membrane. J. Biol. Chem. 286, 14913–14921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Downes B. P., Saracco S. A., Lee S. S., Crowell D. N., and Vierstra R. D. (2006) MUBs, a family of ubiquitin-fold proteins that are plasma membrane-anchored by prenylation. J. Biol. Chem. 281, 27145–27157 [DOI] [PubMed] [Google Scholar]
- 28.Reverter D., and Lima C. D. (2005) Insights into E3 ligase activity revealed by a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature 435, 687–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Capili A. D., and Lima C. D. (2007) Structure and analysis of a complex between SUMO and Ubc9 illustrates features of a conserved E2-Ubl interaction. J. Mol. Biol. 369, 608–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duda D. M., van Waardenburg R. C., Borg L. A., McGarity S., Nourse A., Waddell M. B., Bjornsti M. A., and Schulman B. A. (2007) Structure of a SUMO-binding-motif mimic bound to Smt3p-Ubc9p: conservation of a non-covalent ubiquitin-like protein-E2 complex as a platform for selective interactions within a SUMO pathway. J. Mol. Biol. 369, 619–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knipscheer P., van Dijk W. J., Olsen J. V., Mann M., and Sixma T. K. (2007) Noncovalent interaction between Ubc9 and SUMO promotes SUMO chain formation. EMBO J. 26, 2797–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saitoh H., Pu R., Cavenagh M., and Dasso M. (1997) RanBP2 associates with Ubc9p and a modified form of RanGAP1. Proc. Natl. Acad. Sci. U.S.A. 94, 3736–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prudden J., Perry J. J., Arvai A. S., Tainer J. A., and Boddy M. N. (2009) Molecular mimicry of SUMO promotes DNA repair. Nat. Struct. Mol. Biol. 16, 509–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sekiyama N., Arita K., Ikeda Y., Hashiguchi K., Ariyoshi M., Tochio H., Saitoh H., and Shirakawa M. (2010) Structural basis for regulation of poly-SUMO chain by a SUMO-like domain of Nip45. Proteins 78, 1491–1502 [DOI] [PubMed] [Google Scholar]
- 35.Jensen J. P., Bates P. W., Yang M., Vierstra R. D., and Weissman A. M. (1995) Identification of a family of closely related human ubiquitin conjugating enzymes. J. Biol. Chem. 270, 30408–30414 [DOI] [PubMed] [Google Scholar]
- 36.Brzovic P. S., Keeffe J. R., Nishikawa H., Miyamoto K., Fox D. 3rd, Fukuda M., Ohta T., and Klevit R. (2003) Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc. Natl. Acad. Sci. U.S.A. 100, 5646–5651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Albert T. K., Hanzawa H., Legtenberg Y. I., de Ruwe M. J., van den Heuvel F. A., Collart M. A., Boelens R., and Timmers H. T. (2002) Identification of a ubiquitin-protein ligase subunit within the CCR4-NOT transcription repressor complex. EMBO J. 21, 355–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joazeiro C. A., Wing S. S., Huang H., Leverson J. D., Hunter T., and Liu Y. C. (1999) The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science 286, 309–312 [DOI] [PubMed] [Google Scholar]
- 39.Mace P. D., Linke K., Feltham R., Schumacher F. R., Smith C. A., Vaux D. L., Silke J., and Day C. L. (2008) Structures of the cIAP2 RING domain reveal conformational changes associated with ubiquitin-conjugating enzyme (E2) recruitment. J. Biol. Chem. 283, 31633–31640 [DOI] [PubMed] [Google Scholar]
- 40.Cenciarelli C., Wilhelm K. G. Jr., Guo A., and Weissman A. M. (1996) T cell antigen receptor ubiquitination is a consequence of receptor-mediated tyrosine kinase activation. J. Biol. Chem. 271, 8709–8713 [DOI] [PubMed] [Google Scholar]
- 41.Plafker S. M., Plafker K. S., Weissman A. M., and Macara I. G. (2004) Ubiquitin charging of human class III ubiquitin-conjugating enzymes triggers their nuclear import. J. Cell Biol. 167, 649–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin J., Li X., Gygi S. P., and Harper J. W. (2007) Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature 447, 1135–1138 [DOI] [PubMed] [Google Scholar]
- 43.Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution: from diffraction images to an initial model in minutes. Acta Crystallogr. D. Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 44.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 46.Laskowski R. A., MacArthur M. W., Moss D. S., and Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 47.Hooft R. W., Vriend G., Sander C., and Abola E. E. (1996) Errors in protein structures. Nature 381, 272. [DOI] [PubMed] [Google Scholar]
- 48.Leboucher G. P., Tsai Y. C., Yang M., Shaw K. C., Zhou M., Veenstra T. D., Glickman M. H., and Weissman A. M. (2012) Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell 47, 547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsai Y. C., Leichner G. S., Pearce M. M., Wilson G. L., Wojcikiewicz R. J., Roitelman J., and Weissman A. M. (2012) Differential regulation of HMG-CoA reductase and Insig-1 by enzymes of the ubiquitin-proteasome system. Mol. Biol. Cell 23, 4484–4494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee W., Tonelli M., and Markley J. L. (2015) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haririnia A., D'Onofrio M., and Fushman D. (2007) Mapping the interactions between Lys-48 and Lys-63-linked di-ubiquitins and a ubiquitin-interacting motif of S5a. J. Mol. Biol. 368, 753–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wenzel D. M., Stoll K. E., and Klevit R. E. (2011) E2s: structurally economical and functionally replete. Biochem. J. 433, 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu P. Y., Hanlon M., Eddins M., Tsui C., Rogers R. S., Jensen J. P., Matunis M. J., Weissman A. M., Weisman A. M., Weissman A. M., Wolberger C., Wolberger C. P., and Pickart C. M. (2003) A conserved catalytic residue in the ubiquitin-conjugating enzyme family. EMBO J. 22, 5241–5250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berndsen C. E., Wiener R., Yu I. W., Ringel A. E., and Wolberger C. (2013) A conserved asparagine has a structural role in ubiquitin-conjugating enzymes. Nat. Chem. Biol. 9, 154–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng N., Wang P., Jeffrey P. D., and Pavletich N. P. (2000) Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin- protein ligases. Cell 102, 533–539 [DOI] [PubMed] [Google Scholar]
- 56.Zhang M., Windheim M., Roe S. M., Peggie M., Cohen P., Prodromou C., and Pearl L. H. (2005) Chaperoned ubiquitylation: crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol. Cell 20, 525–538 [DOI] [PubMed] [Google Scholar]
- 57.Xu Z., Kohli E., Devlin K. I., Bold M., Nix J. C., and Misra S. (2008) Interactions between the quality control ubiquitin ligase CHIP and ubiquitin conjugating enzymes. BMC Struct. Biol. 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eletr Z. M., Huang D. T., Duda D. M., Schulman B. A., and Kuhlman B. (2005) E2 conjugating enzymes must disengage from their E1 enzymes before E3-dependent ubiquitin and ubiquitin-like transfer. Nat. Struct. Mol. Biol. 12, 933–934 [DOI] [PubMed] [Google Scholar]
- 59.Huang D. T., Paydar A., Zhuang M., Waddell M. B., Holton J. M., and Schulman B. A. (2005) Structural basis for recruitment of Ubc12 by an E2 binding domain in NEDD8's E1. Mol. Cell 17, 341–350 [DOI] [PubMed] [Google Scholar]
- 60.Nguyen L., Plafker K. S., Starnes A., Cook M., Klevit R. E., and Plafker S. M. (2014) The ubiquitin-conjugating enzyme, UbcM2, is restricted to monoubiquitylation by a two-fold mechanism that involves backside residues of E2 and Lys-48 of ubiquitin. Biochemistry 53, 4004–4014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pickart C. M., and Rose I. A. (1985) Functional heterogeneity of ubiquitin carrier proteins. J. Biol. Chem. 260, 1573–1581 [PubMed] [Google Scholar]
- 62.Wenzel D. M., Lissounov A., Brzovic P. S., and Klevit R. E. (2011) UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474, 105–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Linke K., Mace P. D., Smith C. A., Vaux D. L., Silke J., and Day C. L. (2008) Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 15, 841–848 [DOI] [PubMed] [Google Scholar]
- 64.Plechanovová A., Jaffray E. G., McMahon S. A., Johnson K. A., Navrátilová I., Naismith J. H., and Hay R. T. (2011) Mechanism of ubiquitylation by dimeric RING ligase RNF4. Nat. Struct. Mol. Biol. 18, 1052–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haas A. L., and Bright P. M. (1985) The immunochemical detection and quantitation of intracellular ubiquitin-protein conjugates. J. Biol. Chem. 260, 12464–12473 [PubMed] [Google Scholar]
- 66.Kaiser S. E., Riley B. E., Shaler T. A., Trevino R. S., Becker C. H., Schulman H., and Kopito R. R. (2011) Protein standard absolute quantification (PSAQ) method for the measurement of cellular ubiquitin pools. Nat. Methods 8, 691–696 [DOI] [PMC free article] [PubMed] [Google Scholar]