

Abstract
Chronic alcohol consumption induces hepatic oxidative stress resulting in production of highly reactive electrophilic α/β-unsaturated aldehydes that have the potential to modify proteins. A primary mechanism of reactive aldehyde detoxification by hepatocytes is through GSTA4-driven enzymatic conjugation with GSH. Given reports that oxidative stress initiates GSTA4 translocation to the mitochondria, we hypothesized that increased hepatocellular damage in ethanol (EtOH)-fed GSTA4−/− mice is due to enhanced mitochondrial protein modification by reactive aldehydes. Chronic ingestion of EtOH increased hepatic protein carbonylation in GSTA4−/− mice as evidenced by increased 4-HNE and MDA immunostaining in the hepatic periportal region. Using mass spectrometric analysis of biotin hydrazide conjugated carbonylated proteins, a total of 829 proteins were identified in microsomal, cytosolic and mitochondrial fractions. Of these, 417 were novel to EtOH models. Focusing on mitochondrial fractions, 1.61-fold more carbonylated proteins were identified in EtOH-fed GSTA4−/− mice compared to their respective WT mice ingesting EtOH. Bioinformatic KEGG pathway analysis of carbonylated proteins from the mitochondrial fractions revealed an increased propensity for modification of proteins regulating oxidative phosphorylation, glucose, fatty acid, glutathione and amino acid metabolic processes in GSTA4−/− mice. Additional analysis revealed sites of reactive aldehyde protein modification on 26 novel peptides/proteins isolated from either SV/GSTA4−/− PF or EtOH fed mice. Among the peptides/proteins identified, ACSL, ACOX2, MTP, and THIKB contribute to regulation of fatty acid metabolism and ARG1, ARLY, and OAT, which regulate nitrogen and ammonia metabolism having direct relevance to ethanol-induced liver injury. These data define a role for GSTA4-4 in buffering hepatic oxidative stress associated with chronic alcohol consumption and that this GST isoform plays an important role in protecting against carbonylation of mitochondrial proteins.
Abbreviations: ADPH, adipophilin; ALD, alcoholic liver disease; ALT, alanine aminotransferase; CID, collision-induced dissociation; Cyp2E1, Cytochrome P4502E1; ETD, electron transfer dissociation; EtOH, ethanol; GSTA4, glutathione S-transferase isoform A4; 4-HHE, 4-hydroxy-2-hexenal; 4-HNE, 4-hydroxy-2-nonenal; MDA, malondialdehyde; 4-ONE, 4-oxononenal; PF, Pair-fed
Keywords: Ethanol, Lipid peroxidation, GSTA4, Protein carbonylation, Liver, Oxidative stress, Mitochondria
Graphical abstract
Highlights
-
•
We demonstrate increased mitochondrial carbonylation in GSTA4-4 KO mice chronically fed EtOH.
-
•
Using LC-MS we identify 829 total carbonylated proteins (417 novel to murine ALD).
-
•
Pathway analysis revealed a propensity for adduction of fatty acid metabolic and electron transport proteins.
-
•
Using MS/MS, 26 novel adducted peptides were identified.
-
•
Reactive aldehyde modification of proteins contributes to pathogenesis of ALD.
1. Introduction
Alcoholic liver disease (ALD) is a major contributor of liver failure in the United States today. A common phenotype of ALD is a hepatocellular environment characterized by pronounced lipid accumulation with enhanced oxidative stress [1], [2]. In this pro-oxidant environment, increased lipid peroxidation occurs resulting in the accumulation of reactive aldehydes including 4-hydroxynonenal (4-HNE), 4-oxononenal (4-ONE), acrolein, and malondialdehyde (MDA) [3], [4]. Following their production, the aforementioned reactive aldehydes react with DNA as well as Cys, Lys, and His residues within proteins [4].
In chronic ETOH models, we have identified numerous proteins including the lipid phosphatase PTEN, protein kinase AMPKα and molecular chaperone Grp78 as targets of electrophilic carbonylation by reactive aldehydes in vivo [5], [6], [7]. Concurrently, using mass spectrometry and a global proteomic approach, we have characterized the “lipid peroxidome” formed during chronic ETOH administration [8]. From those seminal reports, it was determined that increased lipid peroxidation due to ethanol consumption results in increased carbonylation of key proteins involved in cellular metabolic pathways involved in fatty acid metabolism, drug metabolism, oxidative phosphorylation, and the TCA cycle. Interestingly, all of these hepatic pathways are reportedly impaired during ALD [9], [10], [11], [12], [13], [14].
An important mechanism for removal of toxic lipid aldehydes is via conjugation with glutathione [15], [16] by Glutathione S-transferase A4-4 (GSTA4) [17]. GSTA4 is a phase 2 detoxifying enzyme whose expression is increased in response to oxidative stress. For instance, the exposure of GSTA4 transfected HepG2 cells to lipid aldehydes, including 4-HNE results in cellular resistance to toxicity [18]. In chow-fed SV 129 mice, deletion of GSTA4 results in an age-dependent increased propensity for obesity and increased lipid peroxidation, a phenotype not evident in GSTA4−/− C57BL/6J mice [17]. Furthermore, consumption of a high fat diet increased adipocyte oxidative stress and mitochondrial damage. Additionally, deletion of GSTA4-4 significantly enhances protein carbonylation and hepatocellular damage following carbon tetrachloride (CCl4) administration, further highlighting the importance of GSTA4 in removal of lipid aldehydes [19].
In this study, we used the GSTA4−/− mouse model treated chronically with ethanol to identify and validate hepatic proteins that are modified by electrophilic products of lipid peroxidation as well as evaluate their involvement in the early stages of ALD. The enhanced mass spectrometric techniques used in this study have identified novel protein targets of reactive aldehydes associated with chronic EtOH. Most importantly, the use of GSTA4−/− mice provide insight into hepatocellular mitochondrial systems impacted by deranged detoxification of electrophilic aldehydes generated by oxidative stress.
2. Experimental methods
2.1. EtOH Administration
SV 129 WT (n=7/group) and GSTA4−/− (n=6/group) male mice were fed Lieber-DeCarli EtOH liquid diets containing EtOH, or Lieber-DeCarli control liquid diet for 6 weeks. In the EtOH groups, EtOH calories replaced carbohydrate calories. This group of animals was acclimated to the diet by increasing the percentage of EtOH slowly to a maximum of 28% EtOH-derived calories as described elsewhere [20]. At sacrifice, livers were weighed and subcellular fractions (cytosolic, mitochondrial, microsomal) prepared as previously described [2], [6]. Pieces of freshly isolated hepatic tissue were formalin-fixed for further analysis. Blood alcohol concentrations were determined using blood isolated during the dark phase of housing as previously described [21].
2.2. Histological evaluation
Formalin fixed slides were immunohistochemically evaluated for 4-HNE, adipophilin (ADPH) (Fitzgerald, Ind. Acton, MA), or MDA as previously described [6].
2.3. Biotin hydrazide purification
Total carbonylated proteins were purified from 500 μg cytosolic extracts (5PF/5EtOH SV/GSTA4-4−/−), mitochondrial extracts (5PF/5EtOH SV/GSTA4-4−/−) and microsomal extracts (4PF/4EtOH SV, 3PF/3EtOH GSTA4-4−/−) following biotin hydrazide derivatization (5 mM/2 h/RT/dark) and NaBH4 reduction (10 mM/100 mM NaOH 1hr/dark). Biotin hydrazide linked carbonylated proteins were incubated along with an untreated pooled samples (negative control/background) overnight using Streptavidin HiTrap columns (GE Biosciences) [8]. Columns were washed 5X in PBS/2 M Urea and eluted with 0.1 M NH4OH. Elutions were dried down using a roto-evaporator, resuspended in 6X SDS PAGE loading buffer and loaded on 10% SDS PAGE gels. Gels were run for 20 min at or until the dye front migrated approximately 1 cm into the resolving gel. Gels were stained with Coomassie blue overnight and stained 1 cm bands excised, destained, and digested with trypsin as previously described [8]. Peptides were extracted, dried down, and resuspended in 0.1% formic acid in ddH2O before loading on the mass spectrometer.
2.4. LC-MS/MS analysis
For LC-MS/MS analysis, 8μl of each peptide mixture in 0.1 M formic acid was loaded on a Bruker Amazon Speed LC-MS/MS (Bruker Daltronics, Billerica, MA). The instrument was operated using data-dependent alternating collision-induced dissociation (CID) MS/MS and electron transfer dissociation (ETD) with a threshold of 10,000 total ion current (TIC). Data analysis was performed using ProteinScape V3.1.2 (Bruker Daltonics Inc. Billerica, MA) and Mascot (v2.1.04, Matrixscience). First a global search for proteins was conducted with a Mascot cutoff score of 15 with the following variable modifications of carbamidomethly (C) and oxidized (M) (Protein identification confidence value of 99%, peptide identification confidence value of 95%, Peptide tolerance: 0.5 Da, MS/MS tolerance: 0.5 Da with 2 missed cleavages). After the initial search a second search was conducted for post-translational modification by reactive aldehydes using the masses shown in Table S1. For this 2nd iteration, peptide significance required a Mascot score higher than 22 and each spectra was verified manually for false positive identification.
2.5. Bioinformatics
UniProt IDs from proteins identified were initially converted into gene symbols using the UniProt website (UniProt Release 2015_08) [22]. Gene symbols functionally annotated for KEGG 2015 pathways [23] using EnrichR [24]. Initially, pathways were examined for over-representation in 6 different lists of proteins/gene symbols derived from the mitochondrial fraction: (1) in 3 (50% of samples) or more samples from the PF/GSTA4-4−/− group and not in background; (2) in 3 (50% of samples) or more samples from the EtOH/GSTA4-4−/− group and not in background; (3) in 3 (50% of samples) or more samples from the PF/SV group and not in background; 4) in 3 (50% of samples) or more samples from the EtOH/SV group and not in background; (5) in 3 (50% of samples) or more samples from all four groups (PF/EtOH SV/GSTA4-4−/−) and not in background and 6) in background sample. To further examine the pathways that were over-represented by proteins/gene symbols that were identified in the GSTA4-4−/− mice and not the SV mice in the mitochondrial fraction, 3 additional lists were constructed: (1) in 3 or more samples from the EtOH/GSTA4-4−/− group but less than 3 samples in the other 3 groups and not in background; (2) in 3 or more samples from the PF/GSTA4-4−/− group but less than 3 samples in the other 3 groups and not in background; and 3) in 3 or more samples from the EtOH/GSTA4-4−/− group and 3 or more samples from the PF/GSTA4-4−/− group but less than 3 samples in the 2 SV groups and not in background. Pathways that were nominally significant (unadjusted p-Value<0.01 in the initial comparison of 6 lists; p-Value<0.05 in the comparison of the 3 GSTA4-4−/− exclusive lists) in at least one of the lists were retained. When no proteins from a list were found in the pathway, a p-Value of 1 was assumed. Hierarchical clustering was used to describe the relationships among the enriched pathways with respect to their significance in the compared lists, i.e., pathways that are enriched in the same groups clustered together. Hierarchical clustering was done using Euclidean distance between binary indicators of significance (p<0.05 vs. p>=0.05). The hierarchical clustering analysis and the heatmap were generated using R Statistical Software (version 3.2.1).
2.6. Statistical analysis
The data are presented as means±SE. Comparisons between genotype and diet were accomplished by two-way ANOVA, followed by Student Newman-Keuls post hoc analysis. Comparisons between two groups was accomplished by Student's T-test. Statistical significance was set at P<0.05. Prism 5 for Windows (GraphPad Software, San Diego, CA) was used to perform all statistical tests.
3. Results
As we previously reported, this 6 week model of chronic ethanol consumption, by GSTA4−/− mice resulted in a significant increase in hepatocellular steatosis and liver triglycerides [23]. Whereas the mean ALT value was 2-fold higher in ethanol-fed GSTA4-4−/− mice (74.8±15.1), as compared to the respective pair-fed controls (35.5±14.8), a large variation in responses of individual animals resulted in a trend towards higher levels of necrosis (p<0.06) but did not achieve statistical significance [20]. In the present study, we have extended these previously published results by exploring the effects EtOH in combination with GSTA4−/− on protein carbonylation. To further expand our previous observations, lipid accumulation was examined by immunohistochemical analysis of adipophilin (ADPH, perilipin 2, PLIN2), a validated marker hepatocyte lipid accumulation as well as unstimulated stellate cells which contain retinoic acid rich vesicles [25]. Thus, ADPH is an ideal marker for determining both steatosis as well as the transition of hepatic stellate cells to an active state, which is signaled by retinoic acid release. As shown in Fig. 1 (Panels A–D), liver sections prepared from SV PF or ETOH-fed mice show only ADPH positive staining in stellate cells with no evidence of hepatocellular lipid accumulation. Interestingly, pair-fed (PF), GSTA4−/− mice exhibited obvious hepatocellular lipid accumulation with no indication of retinoic acid loss from stellate cells. However, the chronic ingestion of EtOH by GSTA4−/− mice markedly increased ADPH staining, reflecting increased lipid accumulation within hepatocytes. To further explore the effects of 6-weeks consumption of EtOH on hepatocellular pathology in GSTA4−/− mice, fibrosis was evaluated by picrosirius red staining and αSMA immunohistochemistry. Based on the images in Fig. 1(Panels E–M), there is no apparent difference in Sirius red staining in either group. Nor was there a significant increase in αSMA staining evident after this relatively short period of EtOH exposure (Data not shown). These data suggest that following short term administration of EtOH, deletion of GSTA4 contributes primarily to hepatocellular lipid accumulation without detectable stellate cell activation.
Fig. 1.

Effects of chronic EtOH on hepatic lipid accumulation and fibrosis in SV and GSTA4−/− mice. Paraffin embedded formalin fixed tissue sections were analyzed immunohistochemically using polyclonal antibodies directed against ADPH (Panels A–D) or stained with Sirius red (Panels E–M). Top panels (A–D); adipophilin staining of tissue sections (Original magnification 400X). Middle panels (E–H); Picrosirius red staining of liver sections (original magnification 200X). Bottom panels (I–M); Polarized light exposure of picrosirius red staining. Figures are representative of three PF/EtOH groups of SV and GSTA4−/− mice respectively.
We previously reported that ingestion of EtOH results in an increase in hepatic 4-hydroxynonenal staining and serum levels of auto-antibodies directed against MDA-adducted proteins levels in GSTA4−/− mice [20]. To determine if increases hepatocellular protein modification by MDA occurs in tandem with increased serum levels or MDA antibodies, liver sections prepared from PF and EtOH-fed SV/GSTA4−/− were examined for post-translational modification by either MDA or 4-HNE (Fig. 2). In the GSTA4−/− mice, sections prepared from both PF and EtOH mice displayed significantly more 4-HNE staining compared to their SV counterparts. Consistent with our previous report of increased anti-MDA antibody concentrations following chronic ethanol treatment, EtOH ingestion markedly increased MDA staining in the GSTA4−/− genotype with ethanol-consuming mice displaying the most intense staining.
Fig. 2.

Effects of chronic EtOH on hepatic protein carbonylation in SV and GSTA4−/− mice. Paraffin embedded formalin fixed tissue sections were analyzed immunohistochemically using polyclonal antibodies directed against 4-HNE and MDA. Figures are representative of three PF/EtOH groups of SV and GSTA4−/− mice respectively.
We recently employed biotin hydrazide derivatization followed by global LC-MS/MS analysis to identify carbonylated proteins in cellular fractions (cytosolic, mitochondrial and microsomal) isolated from PF/EtOH fed C57BL/6J mice [8]. In that study, individual samples collected from PF and EtOH-fed animals were pooled within their respective treatment groups for analysis. In the present study, however, we examined hepatocellular extracts for protein carbonylation in fractions prepared from individual mice to evaluate animal-to-animal replication of treatment-specific hepatic protein modifications. In addition, for negative controls, untreated whole cell extracts were pooled, purified by streptavidin and processed. As shown in Table 1 and Table S2, a total of 829 carbonylated proteins, collectively, were identified in cytosolic, mitochondrial and microsomal samples prepared from PF and EtOH-fed SV and GSTA4−/− mice. Of these, 326 proteins were previously identified in C57BL6J mice subjected to the same EtOH feeding regimen but 417 have not been previously identified in murine EtOH models [8]. Surprisingly, 47 proteins were identified in our untreated samples. Data presented in Table 1 demonstrate that when compared to PF SV mice, the number of carbonylated proteins identified in PF/EtOH fed GSTA4−/− mice is markedly higher (1.6–1.8 fold) in the mitochondrial fraction but the same increase is not evident in the cytosolic or microsomal fractions.
Table 1.
Total number of proteins identified to be carbonylated in each fraction. 500 μg of cytosolic (cyto), mitochondrial (mito) and microsomal (micro) extracts were treated with biotin hydrazide and subjected to LC-MS/MS analysis as described in methods.
| Table 1 | Fraction | Number of animals used | # Identified proteins |
|---|---|---|---|
| All fractions (PF/EtOH) | All | 20 | 829 |
| SV PF | mito | 5 | 214 |
| SV EtOH | mito | 5 | 230 |
| GSTA4−/− PF | mito | 5 | 389 (+1.81-fold) |
| GSTA4−/− EtOH | mito | 5 | 371 (+1.61-fold) |
| SV PF | cyto | 5 | 238 |
| SV EtOH | cyto | 5 | 244 |
| GSTA4−/− PF | cyto | 5 | 224 (−0.06-fold) |
| GSTA4−/− EtOH | cyto | 5 | 195 (−0.13-fold)) |
| SV PF | micro | 4 | 209 |
| SV EtOH | micro | 4 | 191 |
| GSTA4−/− PF | micro | 3 | 116 (−0.45-fold) |
| GSTA4−/− EtOH | micro | 3 | 133(−0.31-fold) |
3.1. Bioinformatic analysis of mitochondrial carbonylated proteins in PF/EtOH fed SV/GSTA4−/−mice.
To identify proteins preferentially targeted by carbonylation in GSTA4−/− mice a threshold was established that required proteins to be identified in at least 50% of animals (mitochondrial, cytosolic and microsomal proteins). As shown in Table S3, 342 proteins were identified in at least 50% of the animals in any group. From the Venn diagram presented in Fig. S1, 119 proteins were common to all 4 groups. Examining the effect of EtOH, approximately 2-fold more proteins were unique in the SV EtOH group when compared to SV PF controls. In comparison, the GSTA4−/− PF displayed a 3-fold increase of unique proteins when compared to respective SV controls indicating that protein carbonylation is increased in GSTA4−/− mice fed diets high I polyunsaturated fats, even in the absence of EtOH consumption. Examining the effects of EtOH and GSTA4−/−, EtOH induced carbonylation of 22 unique proteins.
In murine models, chronic EtOH reportedly impairs mitochondrial function contributing to hepatocyte death [13]. During conditions of increased oxidative stress, GSTA4 reportedly localizes to mitochondria [26], [27]. In adipose tissue, deletion of GSTA4 induces mitochondrial dysfunction [28]. As evidenced by the carbonylated proteins identified, in the liver GSTA4 deletion increases mitochondrial protein carbonylation up to 1.8-fold when compared to respective SV PF/EtOH groups (Table 1). Due to the increase in the number of mitochondrial carbonylated proteins in GSTA4−/− mice, subsequent analyses’ were focused on 235 mitochondrial carbonylated proteins identified in at least 50% of mice in each group. From the Venn diagram presented in Fig. 3 and Table S4, 63 proteins were common to all 4 groups. Examining the distribution of proteins unique to each genotype, there were significantly more mitochondrial proteins identified in either GSTA4−/− PF (43 GSTA4−/− PF/2SVPF), GSTA4−/− EtOH fed (23 GSTA4−/− EtOH/1SV EtOH) and both GSTA4−/−PF/EtOH (49 GSTA4−/− PF/EtOH vs 1SVPF/EtOH) mice as compared to the respective SV groups. This difference strongly supports the contribution of GSTA4 in removal of mitochondrial reactive aldehydes. Furthermore, both GSTA4−/− PF and GSTA4−/− EtOH exhibited clear differences in patterns of protein carbonylation suggesting influences from both the PF diet and by EtOH.
Fig. 3.

Venn diagram showing the distribution of mitochondrial proteins in SV/GSTA4−/− PF and EtOH fed mice. Diagram was prepared as described in Methods using mitochondrial proteins identified as carbonylated in at least 50% of mice in each group.
To further determine pathway specific effects of increased mitochondrial protein carbonylation, KEGG analysis was performed on proteins identified in the mitochondrial fractions. As a negative control, analysis included proteins identified as background from our untreated samples. From the pathway analysis using the KEGG database, in all groups, carbonylation of proteins implicated in fatty acid, xenobiotic, butanoate and amino acid metabolism (Fig. 4, Table S5). As shown, deletion of GSTA4 contributed to increased carbonylation of proteins regulating urea, bile acid, glycolytic, oxidative phosphorylation, amino acid and glutathione metabolism.
Fig. 4.

KEGG Pathway Bioinformatic analysis of mitochondrial proteins. For enrichment analysis, six groups of proteins were identified and enrichment of pathways for each group are represented in each column. The SV-PF column represents pathways enriched for proteins identified in at least 50% of the pair-fed 129 mice, but not in the background sample. The GSTKO-PF column represents pathways enriched for proteins identified in at least 50% of the pair-fed GSTA4−/− mice, but not in the background sample. The SV-EtOH column represents pathways enriched for proteins identified in at least 50% of the ethanol-fed 129 mice, but not in the background sample. The GSTKO-EtOH column represents pathways enriched for proteins identified in at least 50% of the ethanol-fed GSTA4−/− mice, but not in the background sample. The column labeled ‘all 4 groups’ represents pathways enriched for proteins identified in at least 50% of samples in each of the four groups (PF/EtOH SV/GSTA4−/−), but not in the background sample. The background column represents pathways enriched for proteins identified in the background sample. Proteins functionally annotated using KEGG pathways [23] were examined for enrichment using EnrichR [24]. KEGG pathways that were nominally significant (p<0.01) in at least one of the 6 protein lists are included in the graphic. The colors of the heatmap range from white (unadjusted p-Value>0.05) to bright red based on the log base 10 transformation of the unadjusted p-value. A p-Value of 1 was used when the KEGG pathway was not represented by any proteins in the list. KEGG pathways (rows) are ordered based on hierarchical clustering using the Euclidean distance and a binary indicator of significance (p<0.05 vs. p>=0.05).
To further focus our bioinformatic analyses’, an additional KEGG analysis was performed using only mitochondrial proteins that were identified as unique to GSTA4−/− PF/EtOH (49 proteins) or PF (43 proteins) or EtOH groups (23 proteins). From Fig. 5 and Table S6, of the proteins unique to GSTA4 groups, EtOH consumption induced a propensity for carbonylation of glyoxylate cycle and carboxylate cycle proteins whereas PF induced carbonylation glutathione, bile acid synthesis and taurine metabolic pathways. Examining proteins unique to both groups, increased carbonylation was evident in fatty acid, amino acid and xenobiotic metabolic pathways.
Fig. 5.

KEGG Bioinformatic analysis of mitochondrial carbonylated proteins unique to GSTA4−/− PF, GSTA4−/− EtOH or PF/EtOH groups. For enrichment analysis, three groups of proteins were identified and enrichment of pathways for each group are represented in each column. The GSTKO-PF only column represents pathways enriched for proteins identified in at least 50% of the pair-fed GSTA4−/− mice, but less than 50% of samples in all three of the other groups and not identified in the background sample. The GSTKO-EtOH only column represents pathways enriched for proteins identified in at least 50% of the ethanol-fed GSTA4−/− mice, but less than 50% of samples in all three of the other groups and not identified in the background sample. The GSTKO-EtOH and PF only column represents pathways enriched for proteins identified in at least 50% of the ethanol-fed GSTA4−/− mice and at least 50% of pair-fed GSTA4−/− mice, but less than 50% of samples in the SV groups and not identified in the background sample. Proteins functionally annotated using KEGG pathways [23] were examined for enrichment using EnrichR [24]. KEGG pathways that were nominally significant (p<0.05) in at least one of the 3 protein lists are included in the graphic. The colors of the heatmap range from white (unadjusted p-Value>0.05) to bright red based on the log base 10 transformation of the unadjusted p-value. A p-value of 1 was used when the KEGG pathway was not represented by any proteins in the list. KEGG pathways (rows) are ordered based on hierarchical clustering using the Euclidean distance and a binary indicator of significance (p<0.05 vs. p>=0.05).
To determine if there are common proteins modified in each animal representing each treatment group, carbonylated proteins were examined that were present in all of the 5 animals in all of the respective treatment groups (Table S7, S8). A significant number of proteins (19) were identified in all samples evaluated. These include aldehyde dehydrogenase isoform 2 (ALDH2), heat shock protein 71 kDa (HSP7C, HSPA8), mitochondrial glutathione S-transferase (MGST1), and protein disulfide isomerase isoform A6 (PDIA6) all of which have been identified as targets of protein carbonylation [8]. In the SV PF and EtOH alone or PF/EtOH combined groups only 7 proteins were carbonylated in all animals. Of the proteins identified in GSTA4−/− mice, 19 proteins were identified in all 5 GSTA4−/− PF mice, 13 protein targets in all 5 GSTA4−/− EtOH mice and a total 21 proteins found in all mice in both GSTA4−/− PF/EtOH groups. These data reinforce the contribution of GSTA4 in removal of reactive aldehydes generated within the mitochondria. Of interest, a predominance of cytochrome P450 isozymes and fatty acid metabolic enzymes were identified as carbonylated in the GSTA4−/− groups, indicating the possibility for defects in hepatocellular detoxification and lipid accumulation. This phenomenon further increased with the addition of EtOH in GSTA4−/− mice as evident by carbonylation of cellular detoxification enzymes (glutathione S-transferase isoform P1 (GSTπ), cytochrome P450 1A2 (CYP1A2), cytochrome P450 2C50 (CYP2C50) and fatty acid metabolic proteins (3-ketoacyl-CoA thiolase A (ACAA1A, THIKA), long chain specific Acyl-CoA dehydrogenase (ACADL). Carbonylation of these proteins could contribute to increased lipid accumulation as well as altered GST activity during chronic EtOH consumption [1], [2], [8].
Using tissue extracts from animal models, identification of in vivo sites of carbonylation has proven to be a difficult task. Confounding factors that contribute to the paucity of identification of sites of carbonylation from proteins collected from animal tissue include the relative abundance of total peptides compared to carbonylated peptides recovered and identified, aldehyde adduct stability as well as instrument sensitivity. In our previous work using collision induced dissociation LC-MS/MS, pooled samples and post-translational modification by 4-HNE, 4-ONE, acrolein and MDA, we identified 8 novel peptides that were carbonylated in PF/EtOH fed C57BL/6J mice [8]. In the present study, protein isolated from individual (PF/EtOH-fed, SV/GSTA4−/−, (cytosolic, microsomal and mitochondrial fractions) animals were combined with a Bruker Amazon Speed CID/ETD mass spectrometer to examine post-translational modification of Cysteine, Lysine or Histidine residues by acrolein, MDA, 4-HNE, 4-ONE, 4-HHE, crotonaldehyde, 2-pentenal, 2-hexenal, 2-heptenal, 2-octenal and 2-nonenal [29]. To enhance peptide statistical stringency, Protein Prospector false positive peptides were discarded regardless of MASCOT score and each spectra was manually validated. As depicted in Fig. 6 and Table S9, 26 novel peptides were determined to possess aldehyde modification of on Cys, Lys or His residues. Of these, carbonylated proteins were identified in all fractions, both diets and all genotypes. Examining amino acid preference, no obvious differences were detected between specific amino acids modified and type of aldehyde modification. Importantly, deletion of GSTA4 resulted in increased carbonylated peptide identification when compared to respective SV groups. Of particular relevance to ALD are peptides derived from ACSL1, peroxisomal acyl-coenzyme A oxidase 2 (ACOX2), MTP, and THIKB, all of which assist in regulating hepatocellular fatty acid homeostasis. In addition, arginase (ARG1), argininosuccinate lyase (ARLY) and ornithine aminotransferase (OAT) are central for regulation of hepatic ammonia detoxification while aldehyde dehydrogenase isoform 2 (ALDH2) plays an important role in the detoxification of ethanol-derived acetaldehyde. HSP7C (HSP8) is a house-keeping chaperone assisting in prevention of protein aggregation [30] while fructose-bisphosphate aldolase B (ALDOB), ATP synthase subunit alpha (ATPA), Pyruvate carboxylase (PYC), Succinate dehydrogenase (SDHA) regulate glucose and ATP metabolism. The tandem mass spectra for these peptides are shown in Fig. 6 and Supplemental Fig. 1.
Fig. 6.

Representative tandem mass spectra of in vivo adducted peptides isolated from proteins regulating fatty acid and nitrogen metabolism. (A) ACSL (B) ACLY.
4. Discussion
Chronic ethanol consumption causes liver damage through dysregulation of a number of complex hepatocellular pathways. A major contributor to alcohol-induced liver injury is enhanced oxidative stress and the resulting increase in lipid peroxidation. Previous studies from our laboratory using murine models have demonstrated that chronic ethanol consumption results in increased hepatic protein carbonylation by reactive aldehydes including 4-HNE, 4-ONE, MDA, and acrolein [5], [6], [8]. In those studies, protein carbonylation resulted in significant enzyme inhibition potentially contributing to ALD. In our early work, protein identification was performed using 2-dimensional electrophoresis followed by spot excision and subsequent identification by mass spectrometry. Due to technical difficulties, including antibody sensitivity and protein abundance, early attempts identified only a few proteins. Most recently, we have employed a global biotin hydrazide derivatization/streptavidin purification of cellular fractions to identify significantly more proteins that are carbonylated in ALD [8], [31]. In the present report, we further extend our studies on protein carbonylation using GSTA4−/− mice, which are more susceptible to protein carbonylation, as well as the technical advances using fast-switching CID/ETD fragmentation. In addition, the LC-MS/MS search database has been expanded to include masses for Michael addition products of 4-HHE, 2-pentenal, 2-hexenal, 2-heptenal, 2-octenal and 2-nonenal resulting in significantly more peptides identified. Furthermore, carbonylated proteins were isolated from livers of individual mice and processed separately, whereas our previous study utilized pooled fractions [8]. Using these experimental approaches, a total 829 carbonylated proteins are identified in the mouse models used in this study. Of these, 417 have not been previously reported in chronic EtOH models.
Mitochondria are proposed to be a primary target organelle impacted by chronic ethanol ingestion. This proposition is consistent with reports of suppressed ATP production, increased acetate and increased protein acetylation animal models of ALD [13], [32], [33]. In a related study, EtOH also decreased glycolytic function. Research has demonstrated that following 4-weeks consumption of EtOH, increased oxidation of mitochondrial DNA is evident. Using both global approaches and immunoprecipitation techniques, mitochondrial proteins are primary targets of lipid peroxidation supporting the hypothesis that increased mitochondrial oxidative stress and lipid peroxidation/protein carbonylation contribute to mitochondrial dysfunction [8], [34]. Andringa et al. determined that 4-HNE modification of electron-transfer-flavoprotein, alpha polypeptide (ETFα) and dimethylglycine dehydrogenase (DMDH), enoyl CoA hydratase, and cytochrome c increased with ethanol [34]. In that study, only modification by 4-HNE was considered whereas in the present study our searches incorporated proteins modified by a spectrum of reactive aldehydes that have been previously reported to contribute to cellular damage [29], [35], [36], [37]. In the present study, deletion of GSTA4 enhanced mitochondrial carbonylation by 1.8-fold when compared to SV groups reflecting the contribution of this GSH-conjugating enzyme in general detoxification of mitochondrial lipid aldehydes [19]. Of interest, from bioinformatic approaches, increased carbonylation of the oxidative phosphorylation pathway was evident (Fig. 4, Fig. 5 and Table S5). Protein modification of electron transport chains would result in decreased ATP production contributing not only to mitochondrial dysfunction but hepatocellular dysfunction following EtOH consumption. Concurrently increased carbonylation of fatty acid, urea and detoxification pathways is increased which may contribute to increased lipid accumulation and oxidative damage evident in both PF and EtOH-fed GSTA4−/− mice.
A hallmark of chronic alcohol consumption is the reduction of hepatocellular GSH resulting in decreased redox capacity and increased protein carbonylation [8]. Dysregulation of hepatocellular detoxification and oxidative stress pathways clearly contributes to cellular damage [2]. Previously, increased carbonylation of HSP72, HSP90, glucose regulated protein 78 (GRP78) and protein disulfide isomerase (PDI) have been demonstrated in ALD [7], [38], [39], [40], [41]. In the present study, we find that carbonylation of GSTπ, PDIA3, UDP-glucuronyl transferases UGT1A1 and UGT2B1 is a consequence of GSTA4 deletion. Of these 4 proteins, polymorphisms of GSTπ are associated with increased human susceptibility to alcoholic cirrhosis and the involvement of PDIA3 in protein folding is dysregulated in ALD [42], [43]. Therefore, carbonylation of these proteins could impair their function exacerbating the hepatocellular damage resulting from chronic ethanol ingestion.
Hepatic lipid homeostasis is regulated transcriptionally and post-translationally by changing levels of β-oxidation, lipid synthesis, and lipid transport. Defects in all three of these pathways have been described to contribute to increases in lipid accumulation in ALD [9], [44], [45]. Protein carbonylation can alter activity of enzymes in β-oxidation and increased carbonylation of AMPKα contributes to dysregulation of activation in β-oxidation following EtOH consumption in C57BL/6J mice [5]. In the present study, we have identified nearly all of the proteins regulating β-oxidation as targets of reactive aldehydes. Furthermore, site specific adducts are identified in Acyl-CoA Synthetase Long-Chain Family Member 1 (ACSL1C242MDA), 3-ketoacyl-CoA thiolase B (ACAA1, THIKBK293MDA), microsomal triglyceride transfer protein (MTPK414MDA) and Acyl-CoA Oxidase 2 (ACOX2K556ONE). Aldehyde modification of any of these proteins could inhibit enzymatic activity and promote lipid accumulation. These results provide a potential mechanistic basis for the reports that chronic alcohol abuse significantly impairs hepatic β-oxidative processes [10], [14]. Future studies are necessary to determine the molecular outcome of carbonylation on overall ACSL1, THIKB, MTP, and ACOX2 activity. Collectively, the data presented here demonstrate reactive aldehyde modification of numerous enzymes regulating fatty acid oxidation suggesting this as an important mechanism underlying hepatic lipid accumulation in ALD.
Surprisingly, in our previous report, we identified apolipoprotein E (ApoE), non-specific lipid transfer protein (NLTP), MTP and Ras-related protein Rab-1 as putative lipid droplet associating proteins that are carbonylated. In this study, all of the aforementioned proteins were identified as well as ApoA, ApoB and numerous additional Rab GTPases. Furthermore, hepatocellular damage and steatosis is increased in GSTA4−/− mice compared to our previous C57BL/6J model. Increased steatosis would provide additional lipid substrates for peroxidation and contribute to the observed increased in protein modification. Future studies will be necessary to determine the ramification of carbonylation of lipid droplet associated proteins on defects in lipid transport during chronic EtOH consumption.
The identification of protein adducts in PF control mice is intriguing. Increased lipid peroxidation and protein carbonylation are also contributing factors in the pathogenesis of nonalcoholic steatohepatitis (NASH). In a recent publication, increased serum lipid peroxidation strongly correlated with NASH severity in human patients [46], [47], [48]. Furthermore, in a murine model of NASH (MCD-methionine-choline deficient), increased oxidative stress contributed to stimulation of both the adaptive and humoral immune response [49]. Examining the effects of the PF diet, GSTA4−/− mice presented with increased carbonylation compared to the PF-SV group supporting the hypothesis that deletion of GSTA4-4 contributes to enhancement of oxidative stress-induced protein carbonylation.
In binge and chronic EtOH models, hepatic ammonia concentrations are significantly elevated indicative of dysregulation of ammonia detoxification [41], [50], [51]. Using a long-term EtOH administration model, urea synthesis was significantly decreased but activity of ornithine transcarbamoylase (OTC) and carbamyl phosphate synthetase (CPS) were not significantly different between PF and EtOH fed groups [52]. We previously reported that ammonia metabolism/urea biosynthesis pathways are significant targets of reactive aldehydes during chronic EtOH consumption [8]. In that study, all 5 proteins of the urea cycle (ARG1, ASSY, ARLY, CPS1, OTC) were identified as targets of carbonylation. In the present study, we have extended these findings by showing the same proteins are carbonylated in ethanol-treated GSTA4−/− but now have identified site-specific aldehyde adducts on argininosuccinate lyase, ornithine aminotransferase and arginase. Inhibition of ArlyK288HNE and Arg1K211acrolein could contribute to increased ammonia concentrations found in ALD. However, inhibition of OatK374acrolein could be beneficial. Clearly the functional outcomes of carbonylation of these proteins in this complex pathway of ammonia processing in the etiology of ALD remain to be elucidated.
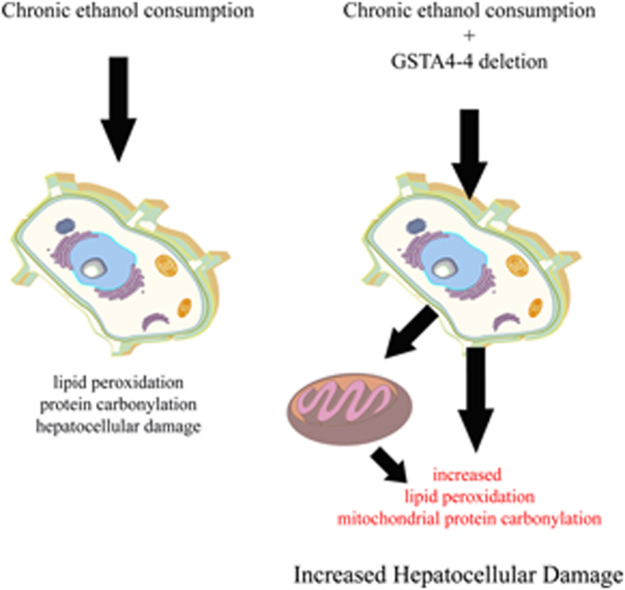
Fig. 7, presents an overall scheme of the results reported here which directly depict the contribution of GSTA4 in mitochondrial detoxification of reactive aldehydes produced as a consequence of oxidative stress and lipid peroxidation. Collectively, the data presented here reinforce the importance of lipid peroxidation and protein carbonylation in ethanol-induced changes in hepatocellular lipid accumulation, dysregulation of urea metabolism and antioxidant responses. We stress however, that each protein identified in this study will require individual analysis to determine the specific effects of carbonylation. Concurrently, although we present our negative control data (Table S2 far right column), many of the targets that are identified in the untreated samples have previously been identified as targets of carbonylation using alternative methods such as 2-dimensional electrophoresis and 4-HNE antibodies (actin, aldehyde dehydrogenases, heat shock proteins) [41], [53], [54]. This indicates that there are proteins that are carbonylated but will also bind nonspecifically to streptavidin columns. Given these ramifications each protein that is identified will need to be further examined using multiple techniques to determine the effects of carbonylation. Therefore these proteins have not been removed from our overall list. Further, we stress that the effects of each carbonylated peptide will also require additional study. These studies do validate the benefits of collection and analyses of liver from individual mice and well as the use of CID/ETD to identify novel adducted peptides and their specific sites of modification providing new mechanistic information concerning the involvement of reactive aldehydes in ALD.
Fig. 7.

Summary. In hepatic mitochondria, by its ability to conjugate reactive aldehydes to GSH, GSTA4 contributes to the removal of reactive aldehydes. When compared to SV wild type control animals, mice possessing a deletion of GSTA4 exhibit increased mitochondrial protein carbonylation.
Conflict of interest
The authors have no conflicts of interest to report.
Author contributions
CTS: Assisted in preparing and processing samples-mass spectrometry, histology, data analysis, prepared all final figures and wrote the initial manuscript preparation.
KSF: Assisted in preparing samples for lc-ms/ms analysis, data analysis and manuscript review.
AHS: Assisted in data analysis-Bioinformatics.
LMS: Assisted in data analysis-Bioinformatics
KEM: Assisted in sample procurement.
JJG: Assisted in sample preparation-lc-ms/ms.
BE: Assisted in sample procurement.
DJO: Assisted in data analysis and figure preparation-Histology.
PZ: Developed GSTA4-/- mice.
MJR: Assisted in experimental design, concepts and manuscript review.
DRP: Assisted in experimental design, concepts and manuscript review.
Acknowledgements
This work was funded by a grant from the NIH/NIAAA 5R37 AA009300-18 (D.R.P.). The authors would like to acknowledge Joe Gomez (Skaggs School of Pharmacy and Pharmaceutical Sciences Mass Spectrometry Core Facility) for carbonylation sample analysis and E. Erin Smith, HTL(ASCP)CMQIHC of the University of Colorado Denver Cancer Center Research Histology Core for assistance in preparing histology slides. The UCDCCRHC is supported in part by NIH/NCRR Colorado CTSI Grant number UL1 RR025780 and the University of Colorado Cancer Center Grant (P30 CA046934).
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2015.11.013.
Appendix A. Supplementary material
Supplementary material
Supplementary material
Supplementary material
Supplementary material
References
- 1.Smathers R.L., Galligan J.J., Stewart B.J., Petersen D.R. Overview of lipid peroxidation products and hepatic protein modification in alcoholic liver disease. Chem. Biol. Interact. 2011;192:107–112. doi: 10.1016/j.cbi.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galligan J.J., Smathers R.L., Shearn C.T., Fritz K.S., Backos D.S., Jiang H., Franklin C.C., Orlicky D.J., Maclean K.N., Petersen D.R. Oxidative stress and the ER stress response in a murine model for early-stage alcoholic liver disease. J. Toxicol. 2012:207594. doi: 10.1155/2012/207594. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esterbauer H., Schaur R.J., Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free. Radic. Biol. Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 4.Schaur R.J. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol. Asp. Med. 2003;24:149–159. doi: 10.1016/s0098-2997(03)00009-8. [DOI] [PubMed] [Google Scholar]
- 5.Shearn C.T., Backos D.S., Orlicky D.J., Smathers-McCullough R.L., Petersen D.R. Identification of 5′ AMP-activated kinase as a target of reactive aldehydes during chronic ingestion of high concentrations of ethanol. J. Biol. Chem. 2014;289:15449–15462. doi: 10.1074/jbc.M113.543942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shearn C.T., Smathers R.L., Backos D.S., Reigan P., Orlicky D.J., Petersen D.R. Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free. Radic. Biol. Med. 2013;65:680–692. doi: 10.1016/j.freeradbiomed.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galligan J.J., Fritz K.S., Backos D.S., Shearn C.T., Smathers R.L., Jiang H., MacLean K.N., Reigan P.R., Petersen D.R. Oxidative stress-mediated aldehyde adduction of GRP78 in a mouse model of alcoholic liver disease: functional independence of ATPase activity and chaperone function. Free. Radic. Biol. Med. 2014;73:411–420. doi: 10.1016/j.freeradbiomed.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galligan J.J., Smathers R.L., Fritz K.S., Epperson L.E., Hunter L.E., Petersen D.R. Protein carbonylation in a murine model for early alcoholic liver disease. Chem. Res. Toxicol. 2012;25:1012–1021. doi: 10.1021/tx300002q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.You M., Crabb D.W. Molecular mechanisms of alcoholic fatty liver: role of sterol regulatory element-binding proteins. Alcohol. 2004;34:39–43. doi: 10.1016/j.alcohol.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 10.You M., Matsumoto M., Pacold C.M., Cho W.K., Crabb D.W. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004;127:1798–1808. doi: 10.1053/j.gastro.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 11.You M., Considine R.V., Leone T.C., Kelly D.P., Crabb D.W. Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology. 2005;42:568–577. doi: 10.1002/hep.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baio D.L., Czyz C.N., Van Horn C.G., Ivester P., Cunningham C.C. Effect of chronic ethanol consumption on respiratory and glycolytic activities of rat periportal and perivenous hepatocytes. Arch. Biochem. Biophys. 1998;350:193–200. doi: 10.1006/abbi.1997.0514. [DOI] [PubMed] [Google Scholar]
- 13.Young T.A., Bailey S.M., Van Horn C.G., Cunningham C.C. Chronic ethanol consumption decreases mitochondrial and glycolytic production of ATP in liver. Alcohol Alcohol. 2006;41:254–260. doi: 10.1093/alcalc/agl017. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Villafranca J., Guillen A., Castro J. Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie. 2008;90:460–466. doi: 10.1016/j.biochi.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y., Sharma R., Sharma A., Awasthi S., Awasthi Y.C. Lipid peroxidation and cell cycle signaling: 4-hydroxynonenal, a key molecule in stress mediated signaling. Acta Biochim. Pol. 2003;50:319–336. [PubMed] [Google Scholar]
- 16.Engle M.R., Singh S.P., Czernik P.J., Gaddy D., Montague D.C., Ceci J.D., Yang Y., Awasthi S., Awasthi Y.C., Zimniak P. Physiological role of mGSTA4-4, a glutathione S-transferase metabolizing 4-hydroxynonenal: generation and analysis of mGsta4 null mouse. Toxicol. Appl. Pharmacol. 2004;194:296–308. doi: 10.1016/j.taap.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 17.Singh S.P., Niemczyk M., Saini D., Awasthi Y.C., Zimniak L., Zimniak P. Role of the electrophilic lipid peroxidation product 4-hydroxynonenal in the development and maintenance of obesity in mice. Biochemistry. 2008;47:3900–3911. doi: 10.1021/bi702124u. [DOI] [PubMed] [Google Scholar]
- 18.Gallagher E.P., Huisden C.M., Gardner J.L. Transfection of HepG2 cells with hGSTA4 provides protection against 4-hydroxynonenal-mediated oxidative injury. Toxicol. In Vitro. 2007;21:1365–1372. doi: 10.1016/j.tiv.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dwivedi S., Sharma R., Sharma A., Zimniak P., Ceci J.D., Awasthi Y.C., Boor P.J. The course of CCl4 induced hepatotoxicity is altered in mGSTA4-4 null (−/−) mice. Toxicology. 2006;218:58–66. doi: 10.1016/j.tox.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 20.Ronis M.J., Mercer K.E., Gannon B., Engi B., Zimniak P., Shearn C.T., Orlicky D.J., Albano E., Badger T.M., Petersen D.R. Increased 4-hydroxynonenal protein adducts in male GSTA4-4/PPARalpha double knockout mice enhance injury during early stages of alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2014;308:G403–G415. doi: 10.1152/ajpgi.00154.2014. ajpgi 00154 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smathers R.L., Galligan J.J., Shearn C.T., Fritz K.S., Mercer K., Ronis M., Orlicky D.J., Davidson N.O., Petersen D.R. Susceptibility of L-FABP−/− mice to oxidative stress in early-stage alcoholic liver. J. Lipid Res. 2013;54:1335–1345. doi: 10.1194/jlr.M034892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magrane M., Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database (Oxf.) 2011 doi: 10.1093/database/bar009. 2011:bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinforma. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee T.F., Mak K.M., Rackovsky O., Lin Y.L., Kwong A.J., Loke J.C., Friedman S.L. Downregulation of hepatic stellate cell activation by retinol and palmitate mediated by adipose differentiation-related protein (ADRP) J. Cell. Physiol. 2010;223:648–657. doi: 10.1002/jcp.22063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raza H., Robin M.A., Fang J.K., Avadhani N.G. Multiple isoforms of mitochondrial glutathione S-transferases and their differential induction under oxidative stress. Biochem. J. 2002;366:45–55. doi: 10.1042/BJ20020533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raza H., John A. 4-hydroxynonenal induces mitochondrial oxidative stress, apoptosis and expression of glutathione S-transferase A4-4 and cytochrome P450 2E1 in PC12 cells. Toxicol. Appl. Pharmacol. 2006;216:309–318. doi: 10.1016/j.taap.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 28.Curtis J.M., Grimsrud P.A., Wright W.S., Xu X., Foncea R.E., Graham D.W., Brestoff J.R., Wiczer B.M., Ilkayeva O., Cianflone K., Muoio D.E., Arriaga E.A., Bernlohr D.A. Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes. 2010;59:1132–1142. doi: 10.2337/db09-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ichihashi K., Osawa T., Toyokuni S., Uchida K. Endogenous formation of protein adducts with carcinogenic aldehydes: implications for oxidative stress. J. Biol. Chem. 2001;276:23903–23913. doi: 10.1074/jbc.M101947200. [DOI] [PubMed] [Google Scholar]
- 30.Lee Y.H., Chung M.C., Lin Q., Boelsterli U.A. Troglitazone-induced hepatic mitochondrial proteome expression dynamics in heterozygous Sod2(+/−) mice: two-stage oxidative injury. Toxicol. Appl. Pharmacol. 2008;231:43–51. doi: 10.1016/j.taap.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 31.Codreanu S.G., Zhang B., Sobecki S.M., Billheimer D.D., Liebler D.C. Global analysis of protein damage by the lipid electrophile 4-hydroxy-2-nonenal. Mol. Cell. Proteom. 2009;8:670–680. doi: 10.1074/mcp.M800070-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fritz K.S., Galligan J.J., Smathers R.L., Roede J.R., Shearn C.T., Reigan P., Petersen D.R. 4-Hydroxynonenal inhibits SIRT3 via thiol-specific modification. Chem. Res. Toxicol. 2011;24:651–662. doi: 10.1021/tx100355a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fritz K.S., Galligan J.J., Hirschey M.D., Verdin E., Petersen D.R. Mitochondrial acetylome analysis in a mouse model of alcohol-induced liver injury utilizing SIRT3 knockout mice. J. Proteome Res. 2012;11:1633–1643. doi: 10.1021/pr2008384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andringa K.K., Udoh U.S., Landar A., Bailey S.M. Proteomic analysis of 4-hydroxynonenal (4-HNE) modified proteins in liver mitochondria from chronic ethanol-fed rats. Redox Biol. 2014;2C:1038–1047. doi: 10.1016/j.redox.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fontaine F.R., Dunlop R.A., Petersen D.R., Burcham P.C. Oxidative bioactivation of crotyl alcohol to the toxic endogenous aldehyde crotonaldehyde: association of protein carbonylation with toxicity in mouse hepatocytes. Chem. Res. Toxicol. 2002;15:1051–1058. doi: 10.1021/tx0255119. [DOI] [PubMed] [Google Scholar]
- 36.Chavez J.D., Wu J., Bisson W., Maier C.S. Site-specific proteomic analysis of lipoxidation adducts in cardiac mitochondria reveals chemical diversity of 2-alkenal adduction. J. Proteom. 2011;74:2417–2429. doi: 10.1016/j.jprot.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shibata N., Kawaguchi M., Uchida K., Kakita A., Takahashi H., Nakano R., Fujimura H., Sakoda S., Ihara Y., Nobukuni K., Takehisa Y., Kuroda S., Kokubo Y., Kuzuhara S., Honma T., Mochizuki Y., Mizutani T., Yamada S., Toi S., Sasaki S., Iwata M., Hirano A., Yamamoto T., Kato Y., Sawada T., Kobayashi M. Protein-bound crotonaldehyde accumulates in the spinal cord of superoxide dismutase-1 mutation-associated familial amyotrophic lateral sclerosis and its transgenic mouse model. Neuropathology. 2007;27:49–61. doi: 10.1111/j.1440-1789.2006.00746.x. [DOI] [PubMed] [Google Scholar]
- 38.Shearn C.T., Mercer K.E., Orlicky D.J., Hennings L., Smathers-McCullough R.L., Stiles B.L., Ronis M.J., Petersen D.R. Short term feeding of a high fat diet exerts an additive effect on hepatocellular damage and steatosis in liver-specific PTEN knockout mice. PLOS One. 2014;9:e96553. doi: 10.1371/journal.pone.0096553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carbone D.L., Doorn J.A., Kiebler Z., Ickes B.R., Petersen D.R. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J. Pharmacol. Exp. Ther. 2005;315:8–15. doi: 10.1124/jpet.105.088088. [DOI] [PubMed] [Google Scholar]
- 40.Carbone D.L., Doorn J.A., Kiebler Z., Petersen D.R. Cysteine modification by lipid peroxidation products inhibits protein disulfide isomerase. Chem. Res. Toxicol. 2005;18:1324–1331. doi: 10.1021/tx050078z. [DOI] [PubMed] [Google Scholar]
- 41.Carbone D.L., Doorn J.A., Kiebler Z., Sampey B.P., Petersen D.R. Inhibition of Hsp72-mediated protein refolding by 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 2004;17:1459–1467. doi: 10.1021/tx049838g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burim R.V., Canalle R., Martinelli Ade L., Takahashi C.S. Polymorphisms in glutathione S-transferases GSTM1, GSTT1 and GSTP1 and cytochromes P450 CYP2E1 and CYP1A1 and susceptibility to cirrhosis or pancreatitis in alcoholics. Mutagenesis. 2004;19:291–298. doi: 10.1093/mutage/geh034. [DOI] [PubMed] [Google Scholar]
- 43.Aroor A.R., Roy L.J., Restrepo R.J., Mooney B.P., Shukla S.D. A proteomic analysis of liver after ethanol binge in chronically ethanol treated rats. Proteome Sci. 2012;10:29. doi: 10.1186/1477-5956-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang X., Hu M., Rogers C.Q., Shen Z., You M. Role of SIRT1-FoxO1 signaling in dietary saturated fat-dependent upregulation of liver adiponectin receptor 2 in ethanol-administered mice. Antioxid. Redox Signal. 2011;15:425–435. doi: 10.1089/ars.2010.3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.French S.W. Biochemical basis for alcohol-induced liver injury. Clin. Biochem. 1989;22:41–49. doi: 10.1016/s0009-9120(89)80067-0. [DOI] [PubMed] [Google Scholar]
- 46.Stiuso P., Scognamiglio I., Murolo M., Ferranti P., De Simone C., Rizzo M.R., Tuccillo C., Caraglia M., Loguercio C., Federico A. Serum oxidative stress markers and lipidomic profile to detect NASH patients responsive to an antioxidant treatment: a pilot study. Oxid. Med. Cell. Longev. 2014;2014:169216. doi: 10.1155/2014/169216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nobili V., Parola M., Alisi A., Marra F., Piemonte F., Mombello C., Sutti S., Povero D., Maina V., Novo E., Albano E. Oxidative stress parameters in paediatric non-alcoholic fatty liver disease. Int. J. Mol. Med. 2010;26:471–476. doi: 10.3892/ijmm_00000487. [DOI] [PubMed] [Google Scholar]
- 48.Nobili V., Pastore A., Gaeta L.M., Tozzi G., Comparcola D., Sartorelli M.R., Marcellini M., Bertini E., Piemonte F. Glutathione metabolism and antioxidant enzymes in patients affected by nonalcoholic steatohepatitis. Clin. Chim. Acta. 2005;355:105–111. doi: 10.1016/j.cccn.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 49.Sutti S., Jindal A., Locatelli I., Vacchiano M., Gigliotti L., Bozzola C., Albano E. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. 2014;59:886–897. doi: 10.1002/hep.26749. [DOI] [PubMed] [Google Scholar]
- 50.Cascales C., Cascales M., Santos-Ruiz A. Effect of chronic ethanol or acetaldehyde on hepatic alcohol and aldehyde dehydrogenases, aminotransferases and glutamate dehydrogenase. Rev. Esp. Fisiol. 1985;41:19–27. [PubMed] [Google Scholar]
- 51.Leung T.M., Lu Y., Yan W., Moron-Concepcion J.A., Ward S.C., Ge X., Conde de la Rosa L., Nieto N. Argininosuccinate synthase conditions the response to acute and chronic ethanol-induced liver injury in mice. Hepatology. 2012;55:1596–1609. doi: 10.1002/hep.25543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adachi K., Matsuhashi T., Nishizawa Y., Usukura J., Popinigis J., Wakabayashi T. Studies on urea synthesis in the liver of rats treated chronically with ethanol using perfused livers, isolated hepatocytes, and mitochondria. Biochem. Pharmacol. 1995;50:1391–1399. doi: 10.1016/0006-2952(95)02023-3. [DOI] [PubMed] [Google Scholar]
- 53.Aldini G., Orioli M., Carini M. Alpha, beta-unsaturated aldehydes adducts to actin and albumin as potential biomarkers of carbonylation damage. Redox Rep. 2007;12:20–25. doi: 10.1179/135100007X162310. [DOI] [PubMed] [Google Scholar]
- 54.Sampey B.P., Korourian S., Ronis M.J., Badger T.M., Petersen D.R. Immunohistochemical characterization of hepatic malondialdehyde and 4-hydroxynonenal modified proteins during early stages of ethanol-induced liver injury. Alcohol Clin. Exp. Res. 2003;27:1015–1022. doi: 10.1097/01.ALC.0000071928.16732.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Supplementary material
Supplementary material
Supplementary material