

Abstract
Background
Pathogenic mutations in FBN1, encoding the glycoprotein, fibrillin-1, cause Marfan syndrome (MFS) and related connective tissue disorders. In the present study, qualitative and quantitative effects of 16 mutations, identified in FBN1 in MFS patients with systematically described phenotypes, were investigated in vitro.
Methods
Qualitative analysis was performed with reverse transcription-PCR (RT-PCR) and gel electrophoresis, and quantitative analysis to determine the FBN1 mRNA levels in fibroblasts from the 16 patients with MFS was performed with real-time PCR.
Results
Qualitative analysis documented that the mutations c.4817-2delA and c.A4925G led to aberrant FBN1 mRNA splicing leading to in frame deletion of exon 39 and in exon 39, respectively. No difference in the mean FBN1 mRNA level was observed between the entire group of cases and controls, nor between the group of patients with missense mutations and controls. The mean expression levels associated with premature termination codon (PTC) and splice site mutations were significantly lower than the levels in patients with missense mutations. A high level of FBN1 mRNA in the patient with the missense mutation c.G2447T did not segregate with the mutation in three of his first degree relatives. No association was indicated between the FBN1 transcript level and specific phenotypic manifestations.
Conclusions
Abnormal FBN1 transcripts were indicated in fibroblasts from patients with the splice site mutation c.4817-2delA and the missense mutation c.A4925G. While the mean FBN1 mRNA expression level in fibroblasts from patients with splice site and PTC mutations were lower than the mean level in patients with missense mutations and controls, inter-individual variability was high. The observation that high level of FBN1 mRNA in the patient with the missense mutation c.G2447T did not segregate with the mutation in the family suggests that variable expression of the normal FBN1 allele may contribute to explain the variability in FBN1 mRNA level.
Electronic supplementary material
The online version of this article (doi:10.1186/s12881-015-0260-4) contains supplementary material, which is available to authorized users.
Keywords: FBN1, Marfan syndrome, FBN1 mRNA expression
Background
Mutations in FBN1, encoding fibrillin-1 cause Marfan syndrome (MFS; OMIM #154700) and other heritable connective tissue disorders, referred to as fibrillinopathies [1, 2]. The phenotypes caused by FBN1 mutations range from isolated, minor manifestations to a lethal, neonatal form of MFS [3, 4]. MFS is an autosomal dominantly inherited disorder, exhibiting variable clinical expressivity [5]. Major clinical manifestations are found in the cardiovascular (aortic aneurysm with dissection), ocular (ectopia lentis), and skeletal systems [6].
FBN1 (NM 000138.4) contains 65 exons encoding profibrillin-1, a 350 kDa glycoprotein that is processed to fibrillin-1 [7]. Human fibrillin-1 is a main component of 10–12 nm microfibrils located in the extracellular matrix (ECM) of connective tissues [8]. The protein is modular, comprising 47 epidermal growth factor-like (EGF) domains, seven transforming growth factor β (TGF-β) binding protein-like domains, two hybrid domains, and one proline-rich region [9]. Forty-three of the 47 EGF domains are calcium binding domains (cbEGF), of which each is characterised by six cysteine residues, normally forming three disulphide bonds and a calcium binding consensus sequence which is involved in protein structure stabilisation [10].
At the latest update of the UMD-FBN1 mutations database (http://www.umd.be/FBN1/ on 28/08/14), 1847 different mutations and 1096 protein variants have been identified in FBN1 [www.umd.be/FBN1/] in patients with MFS and a spectrum of related fibrillinopathies [2]. Missense mutations are the most frequent (55 %) type of mutations in FBN1, typically affecting cysteins in the highly conserved cbEGF domains [11]. These mutations may be associated with increased proteolytic degradation of fibrillin-1 [12–14]. Twenty-five percent of all known FBN1 mutations are frameshift or nonsense mutations leading to premature termination codons (PTC) [15, 16], potentially generating truncated fibrillin-1 variants that may assemble into the extracellular microfibrils. Truncated transcripts are usually degraded by the nonsense-mediated mRNA decay (NMD) mechanism [17], which would then result in reduced or no expression of truncated fibrillin-1 thus ameliorating negative effects of microfibrils on ECM. Splice site mutations are also frequent in MFS [18, 19]. Infrequently, large genomic deletions involving single or multiple exons of the FBN1 gene, as well as whole FBN1 deletions, have been identified [20–22].
Analyses of fibrillin-1 in cultured dermal fibroblasts from MFS patients have revealed abnormalities in the synthesis, secretion, and deposition of fibrillin-1 in the ECM [23–25]. Further, mutant fibrillin-1 may cause abnormal structure of microfibrils and ECM [26]. Alteration of TGF-β binding protein like domains may play an additional role in the pathogenesis of fibrillinopathies, as increased TGF-β signalling causes deregulation of cytokine function [27, 28]. Two models of MFS pathogenesis have been suggested. According to the haploinsufficiency model, the pathogenesis is based on reduction in the levels of normal fibrillin-1 [22, 29], and according to the dominant-negative model, mutant fibrillin-1 assembles with molecules of the wild-type protein, thereby disrupting the function of ECM [30].
We aimed to investigate qualitative and quantitative effects of 16 mutations in FBN1 on FBN1 mRNA in cultured fibroblasts from 16 MFS patients, comparing with fibroblasts from individuals with no known connective tissue disorder.
Methods
Patients and cell cultures
In this study we analyzed the FBN1 mRNA levels in fibroblasts from 16 patients with MFS in whom a presumptive disease-causing mutation has been identified (Table 1). All patients have been systematically examined as previously described [31], and satisfied the diagnostic Ghent criteria from 1996 [32]. The study has been approved by the Norwegian Regional Ethics Committee South-East. Fourteen of the presumptive disease-causing mutations have been reported by us [31]. As controls, fibroblast cultures from four individuals with no known connective tissue disorder were established in Department of Medical Genetics, Oslo University Hospital, and two were commercially obtained (BioNordika, Medprobe, Lysaker, Norway). In order to study intrafamilial variability, FBN1 expression levels were determined in three first degree relatives of one of the patients.
Table 1.
FBN1 genotype, predicted effect, fibroblast FBN1 expression level, and patient characteristics
| FBN1 nucleotide change | Protein change | Affected domain | In silico predictiona | Type of mutation | FBN1 mRNA % of controlsb | Clinical phenotypec |
|---|---|---|---|---|---|---|
| c.G629A | p.Cys210Tyr | Hybrid | Probably damaging | Missense | 120 ± 35 | DOsp |
| c.G1027A | p.Gly343Arg | TGF-β1 | Possibly damaging | Missense | 219 ± 35 | Dos |
| c.G2447T | p.Cys816Phe | cbEGF9 | Probably damaging | Missense | 212 ± 52 | DOCsi |
| c.T2848C | p.Cys950Arg | cbEGF10 | Probably damaging | Missense | 100 ± 16 | DOCsi |
| c.T4348G | p.Cys1450Gly | cbEGF21 | Probably | Missense | 169 ± 26 | DOoCcSsi |
| c.T5866C | p.Cys1956Arg | cbEGF29 | damaging | Missense | 141 ± 24 | DOoSi |
| c.G6388A | p.Glu2130Lys | cbEGF32 | Possibly damaging | Missense | 119 ± 27 | DOSsip |
| c.G7094A | p.Cys2365Tyr | TGF-β7 | Possibly damaging | Missense | 109 ± 18 | DOS |
| Probably damaging | ||||||
| c.4269_4270delAC | p.Pro1424Argfs*6 | cbEGF20 | Fs, introducing | PTC (fs) | 88 ± 23 | DCcSi |
| c.5559delT | p.Gln1854Lysfs*39 | cbEGF27 | Fs, introducing | PTC (fs) | 75 ± 19 | DCcs |
| c.T6339Ad | p.Tyr2113X | TGF-β6 | PTC skip of exon 25 | PTC (fs) | 53 ± 8 | Ds |
| c.3083-2A > G | - | Intronic | Skip of exon 25 | Splice site | 51 ± 10 | DOoCcSs |
| c.4211-1G > A | - | Intronic | Skip of exon 34 | Splice site | 59 ± 11 | DOoCcspi |
| c.4817-2delA | p.Ile1607_Asp1648del | Intronic | Skip of exon 39 | Splice site | 80 ± 16 | DOCcsi |
| c.4942 + 2 T > C | - | Intronic | Skip of exon 39 | Splice site | 72 ± 12 | DOoCcSsp |
| c.A4925G | p.Thr1643_Asp1648del | cbEGF23 | CSS | Splice site (CSS) | 91 ± 17 | DOosi |
Exons are numbered according to the reference sequence GenBank NM_000138.4. cb, calcium binding
EGF, epidermal growth factor, fs frame shift, PTC premature termination codon, TGF transforming growth factor, CSS cryptic splice site
a In silico prediction of mutations was obtained using ALAMUT prediction algorithms
bMean FBN1 mRNA expression levels in five parallel wells of fibroblasts from MFS patients compared to controls (n = 6) ± SD. The level in controls was assigned as 100 %
cAffection of organ systems is given according to the original Ghent nosology for the diagnosis MFS. Dura mater: Major criterion: D. Ocular system: Major criterion: O, Minor manifestations: o. Cardiovascular system: Major criteria: C, Minor criteria: c. Skeletal system: Major manifestations: S, minor manifestations: s. Skin and integument: Minor criteria implying involvement: i, Pulmonary system: Minor criteria implying involvement: p. dThe mutation was contained in the UMD-FBN1 database (recurrent mutation)
Biopsies from forearm skin were grown in complete Chang medium (Sigma-Aldrich, St. Louis, MO) from the same batch, supplemented with L-glutamine, penicillin and streptomycin (Invitrogen, Carlsbad, CA), and incubated at standard conditions (37° C, 5 % CO2). Fibroblasts from each individual were cultured in five parallels in a 6-well plate (seeding wells). Cells were harvested at passage number 3–5 in late logarithmic growth phase, assessed by microscopy.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
RNA was extracted from fibroblasts according to the manufacturer’s instruction, using the RNAqueous Small Scale Phenol-Free Total RNA isolation kit (Ambion, Cambridgeshire, UK), and quantified with a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE). cDNA synthesis using 600–1200 ng of RNA was performed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA).
Qualitative analysis of FBN1 mRNA
In order to analyse FBN1 mRNA from patients and control fibroblast cultures, 10 overlapping cDNA amplicons covering the whole FBN1 mRNA was PCR amplified using cDNA specific primers (Additional file 1). Primers were designed using Primer 3 (www.primer3.sourceforge.net). PCR fragments were resolved by gel electrophoresis using ethidium bromide-containing 2 % NuSieve GTG Agarose gel (Cambrex BioScience, Rockland, ME), which was run at 70 V for 6 h. PCR products were purified using AMPure beads (Beckman Coulter Inc, Brea, CA) and sequenced directly in both directions by ABI PRISM 3730 Genetic Analyzer (Applied Biosystems).
Quantitative FBN1 mRNA analysis
FBN1 mRNA levels were determined by the 7900 HT Fast Real-Time PCR System (Applied Biosystems) using TaqMan Gene Expression Assays for FBN1 (Hs009731199_m1, Applied Biosystems) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Hs99999905_m1, Applied Biosystems). Real-time PCR was performed in triplicates in a 384-well plate and run according to the manufacturer’s recommendation. Negative controls included water and no cDNA template.
Amplification levels of FBN1 were calculated according to the 2-ddCT method [33] including normalization to the mRNA levels of the house keeping gene GADPH, and to the FBN1 mRNA levels in the six controls.
Statistical analysis
All results were tested for statistical significance with the two-tailed, unpaired T- test. P-values <0.05 were considered statistically significant.
Computer analysis of mutational effect
Consequences of the investigated mutations were predicted using ALAMUT software (www.interactive-biosoftware.com/alamut/).
Results
Qualitative FBN1 mRNA analysis
Sequence analysis of cDNA from fibroblasts verified the presence of the eight heterozygous missense mutations, c.G629A, c.G1027A, c.G2447T, c.T2848C, c.T4348G, c.T5866C, c.G6388A, and c.G7094A, previously identified in genomic DNA [31]. Ten overlapping amplicons covering the FBN1 transcript (Table 1) were analyzed by gel electrophoresis and appeared normal in these eight patients (data not shown). Fragment analysis in fibroblasts from patients with premature termination codons (PTCs) (c.4269_4270delAC, c.5559delT, and c.T6339A) did not reveal any aberrant FBN1 transcripts, and sequencing of the amplicons did not indicate any defects in the mRNA sequence (data not shown). Fragment analysis of FBN1 mRNA in the patients with splice site mutations (c.3083-2A > G, c.4211-1G > A, c.4817-2delA, and c.4942 + 2 T > C) demonstrated an aberrant transcript in the patient with the c.4817-2delA mutation (Fig. 1a, lane 3) only. Furthermore, the predicted missense mutation, c.A4925G, was shown by fragment analysis to result in an aberrant FBN1 transcript (Fig. 1a, lane 4). Sanger sequencing of the cDNA amplicon of exons 36–44 from the patients with the c.4817-2delA and c.A4925G mutations showed two in frame deletions, of c.4925-c.4942 (Fig. 1b) and a skip of exon 39 (Fig. 1c), respectively. This suggests that the c.A4925G mutation introduced a cryptic splice site (CSS), resulting in loss of the terminal 18 nucleotides of exon 39 (Fig. 1d).
Fig. 1.
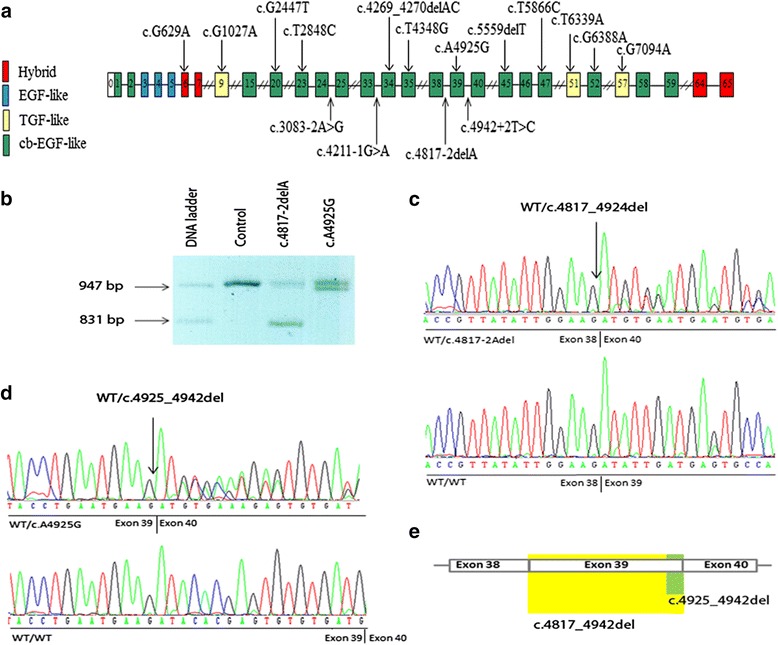
Localization of mutations in FBN1 and results from qualitative analysis (a) Schematic presentation of FBN1 with the localization of the 16 mutations investigated in this study indicated (b) Analysis of cDNA fragments containing FBN1 exons 36–44 from the patient with c.4817-2delA (lane 3), c.A4925G (lane 4), and control (lane 2). The fragment size marker is in lane 1. Lanes 3 and 4 show fragments corresponding to the normal (955 bp) and truncated cDNA fragments of 829 bp and 937 bp from patients with c. 4817-2delA and c. A4925G, respectively. c Fragment of FBN1 cDNA sequence in patient with 4817 + 2 T > C. d Fragment of FBN1 cDNA sequence in patient with A4925G. e Aberrant splicing resulting from the mutations, c.4817-2delA and c.A4925G lead to two distinct in frame deletions affecting exon 39 (schematic presentation). Skip of exon 39 (c.4817-4942del) (yellow), was confirmed in the patient with the c.4817-2delA splice site variant, whereas deletion of c.4925-4942 (green) was identified in the patient with the c.A4925G variant, which likely introduced a cryptic splice site
Quantitative FBN1 mRNA analysis
Relative FBN1 mRNA levels in fibroblasts from the 16 patients compared to the level in six controls were determined by qRT-PCR. The mean FBN1 transcript level in fibroblasts from all 16 MFS patients were not significantly different from controls (Fig. 2). Furthermore, there was no significant difference in the mean FBN1 transcript level in the group of eight patients with missense mutations, the group of three patients with PTC, or in the group of five patients with splice site mutations compared to controls (Fig. 2a). However, the mean FBN1 transcript level in the two groups of patients with PTC and splice site mutations were both significantly lower than in the group of patients with missense mutations (Fig. 2a). Individually, the patients with the missense mutations c.G1027A and c.G244T showed significantly higher FBN1 transcript levels than controls (Additional file 1). To investigate if the high level in the latter patient could be due to the c.G2447T mutation, we investigated if the high FBN1 mRNA level segregated with the mutation in three first degree relatives; two with the heterozygous c.G2447T and one without. The results showed that all four had higher FBN1 mRNA levels than the mean level in controls, with the highest level in the index patient and his unaffected sister, who did not have the heterozygous c.G2447T mutation (Fig. 2b).
Fig. 2.
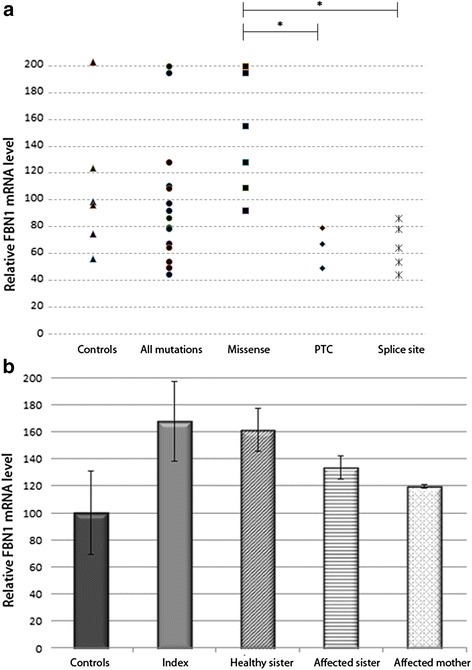
Results from quantitative analysis of FBN1 mRNA (a) FBN1 transcript levels in fibroblasts from controls (n = 6) and MFS patients with missense (n = 8), PTC (n = 3), and splice site mutations (n = 5), respectively. Significant differences (p < 0.05) were observed between mean FBN1 transcript levels in patients with missense mutations and patients with PTC mutations and between the levels in patients with missense mutations and patients with splice site mutations. b FBN1 transcript levels in fibroblasts from the patient with the missense mutation, c.G2447T, his affected mother and sister, both having the c.G2447T, and his unaffected sister, who tested negative for the mutation, compared to the level in controls (n = 6). The results are presented as the mean of two experiments with five seeding wells ± SD. The level in controls was assigned as 100 %
Discussion
Qualitative analysis
Sequencing of cDNA from fibroblasts from the patient with the splice site mutation, c.4942 + 2 T > C did not reveal any abnormal FBN1 transcript, indicating that the transcript affected by this mutation was efficiently eliminated by NMD. The c.4817-2delA caused a skip of exon 39 in the FBN1 transcript demonstrated by gel electrophoresis and Sanger sequencing. The fact that the patient had a high FBN1 mRNA level suggests that most of the FBN1 transcripts were rescued from NMD. This result is consistent with results from other studies, which have indicated that splice site mutations maintaining the reading frame were not degraded by NMD and therefore did not cause decreased FBN1 expression [20, 34].
Sequencing of cDNA isolated from the patient with the mutation c.A4925G showed that the mutation predicted to cause the missense p.Asp1642Gly in fact caused the deletion of 18 nucleotides. This finding is explained by introduction of a CSS, which may cause splicing at a position of a transcript where it is usually not spliced [35]. Introduction of CSS has previously been associated with human diseases, including MFS [36, 37].
Quantitative analysis of FBN1 mRNA expression
FBN1 mRNA expression in MFS patients and controls
We observed high inter-individual variability in FBN1 expression levels in fibroblasts from individuals with no connective tissue disorder, as well as in MFS patients (Fig. 2a), similar to results reported by other investigators [38, 39]. Some studies have indicated that FBN1expression may be affected by the passage number or different growth conditions for the fibroblasts [26]. In the present study the fibroblasts were cultured under uniform conditions. No trend was observed with respect to expression level compared to age or gender of the skin biopsy donors, the source of the control fibroblasts, or if cells were harvested in different growth phases (data not shown). It has been suggested that the clinical variability in MFS might be explained by varying expression levels of both mutant and normal FBN1 transcripts [38, 40–42]. In line with this, Hutchinson and co-workers demonstrated that the variable reduction of total FBN1 transcript in three related individuals carrying a PTC mutation was due to variation in the expression of the normal FBN1 allele rather than by NMD of mutant RNA [41]. Further, Aubert and co-workers recently carried out differential allelic expression analysis demonstrating reduced FBN1 transcript levels in patients with PTC and further that 90 % of the transcript originates from the wild type allele [38]. In the present study, the similar high levels of FBN1 mRNA in both affected and non-affected relatives of the patient with the mutation, c.G2447T indicated that the high level in these family members was not caused by the mutation (Fig. 2b).
FBN1 expression, type and location of the mutations
In the present study, eight of the 16 mutations were missense mutations. Our finding of normal or high FBN1 mRNA levels (ranging from 94 to 219 % of controls) in patients with missense mutations is consistent with findings in other studies [11, 30, 43]. In spite of the high expression level, the MFS phenotype in the patients with missense mutations and high FBN1 mRNA level could not be considered as mild (Additional file 1). An association between ectopia lentis and missense mutations in the cbEGF domain affecting cysteine residues, in the presence of normal levels of FBN1 mRNA, has been reported [14]. In the present study, five of the eight missense mutations were located in cbEGF domains of fibrillin-1; four of them affecting cysteine. All four patients with a mutation in cbEGF domain affecting cysteine had ectopia lentis, and their mean FBN1 mRNA level was 156 % of controls. To our knowledge, few reports exist on the effects of missense mutations in TGF-β binding protein like domains and hybrid domains on FBN1 mRNA expression. Only one missense mutation in our patients was located in a hybrid domain (also referred to as a TGF-β binding protein like domain); in a moderately affected patient with relatively high FBN1 mRNA level. Two missense mutations were located in TGF-β1 and TGF-β7 domains, respectively, and the clinical phenotypes of the two patients were rather mild. Their FBN1 mRNA levels differed two-fold, being 109 to 219 % of controls (Additional file 1). Missense mutations may cause disease through a dominant-negative effect. In line with this, previous studies have demonstrated that missense mutations in FBN1 caused increased intracellular misfolded fibrillin-1 [11, 30, 43], which is able to cause a severe phenotype in the presence of normal or high FBN1 mRNA level.
In the two MFS patients with the small deletions c.4269_4270delAC and c.5559delT, the mean FBN1 mRNA levels were 75 % and 88 % of the level in controls. Both patients had major affection of the cardiac system (Table 1), and both deletions were predicted to cause frameshifts resulting in truncated proteins if the transcripts do not undergo NMD. Analysis by RT-PCR and gel electrophoresis indicated that only the wild type transcripts were present, indicating NMD. These results are consistent with several studies that have shown that nonsense and frameshift mutations in FBN1 result in efficient NMD [16, 43–45]. However, there are also reports on large out-of-frame deletions of FBN1 that did not cause reduction of the mRNA levels [21, 22]. Our patient with a nonsense mutation in exon 51 [c.T6339A (p.Tyr2113X)] was clinically moderately affected, although the FBN1 mRNA level was much lower (53 % of the level in controls) than in the two cases with PTC mutations discussed above (Table 1). Another nonsense mutation in the same codon has been reported to cause exon skipping and NMD of the mutant FBN1 transcript [15]. This indicates that NMD is activated and is the underlying mechanism causing the low mRNA level in our patient.
The effect of splice site mutations on mRNA is difficult to predict [19, 46]. Four splice site mutations were investigated in this study: the c.4942 + 2 T > C, c.4817-2delA, c.4211-1G > A, and c.3083-2A > G. In silico analysis indicated that a skip of exon 39 was likely in the first two cases, and we demonstrated this for the c.4817-2delA. The mutations, c.4942 + 2 T > C and c.4817-2delA were associated with 72 % and 80 % of FBN1 mRNA expression compared to controls, respectively, and the patients were severely affected with several major, including major cardiovascular, manifestations of MFS (Table 1). The splice site mutations c.3083-2A > G and c.4211-1G > A were associated with 51 % and 59 %, respectively, of FBN1 mRNA expression compared to controls. These patients were also severely affected (Table 1).
Conclusions
Qualitative analysis of FBN1 mRNA from fibroblasts from 16 MFS patients and six controls indicated that the mutations, c.A4925G and c.4817-2delA led to aberrant splicing resulting in frame deletions in exon 39 or deletion of exon 39, respectively. Quantitative mRNA analysis revealed considerable variability in FBN1 mRNA levels in both MFS patients and controls. No difference in the mean FBN1 mRNA level was observed between the entire group of cases and controls, nor between the group of patients with missense mutations and controls, but the mean expression levels associated with PTC and splice site mutations were significantly lower than the levels in controls and patients with missense mutations. In line with evidence from other studies, the mRNA levels in fibroblasts derived from four members of one family suggested that variable expression from the normal FBN1 transcript may contribute to explain the variability in FBN1 mRNA level.
Acknowledgements
The authors have no conflicts of interests to declare. The study was funded by grants from the Norwegian South-Eastern Health Authority, the Department of Cardio-Thoracic Surgery Fund for Cardiac Research at Oslo University Hospital, and the Norwegian National Advisory Unit on Rare Disorders. We thank Bjørg Oda Sandengen for help with establishing fibroblast cultures.
Additional file
Primer sequences used for amplification of FBN1 cDNA fragments Exons are numbered according to the cDNA sequence in GenBank (accession number: NM_000138.4). (DOCX 16 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LT participated in conception and design of the study, established and maintained cell culture, carried out the molecular biology studies, analyzed and interpreted the data, and drafted the manuscript. SSA established and maintained cell culture, carried out the molecular biology studies, analyzed and interpreted the data, and drafted the manuscript. TB analyzed and interpreted the data and revised the manuscript critically for intellectual content. SRH and OG recruited and evaluated subjects in the Norwegian Marfan Syndrome Study at Sunnaas Hospital and Oslo University Hospital, participated in interpretation of the data, and drafted the manuscript. EF participated in the conception and design of the study, analyzed and interpreted the data, and revised the manuscript critically for intellectual content. BP participated in conception and design of the study, analyzed and interpreted the data, and wrote the final manuscript. All authors have given final approval of the version to be published and agree to be accountable for all aspects of the work.
Contributor Information
Lena Tjeldhorn, Email: lena.tjeldhorn@online.no.
Silja Svanstrøm Amundsen, Email: silja.s.amundsen@rr-research.no.
Tuva Barøy, Email: tuva.baroy@medisin.uio.no.
Svend Rand-Hendriksen, Email: svend.rand-hendriksen@medisin.uio.no.
Odd Geiran, Email: odd.geiran@medisin.uio.no.
Eirik Frengen, Email: eirik.frengen@medisin.uio.no.
Benedicte Paus, Email: benedicte.paus@medisin.uio.no.
References
- 1.Dietz HC, Pyeritz RE. Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan syndrome and related disorders. Hum Mol Genet. 1995;4 Spec No:1799–809. doi: 10.1093/hmg/4.suppl_1.1799. [DOI] [PubMed] [Google Scholar]
- 2.Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81:454–66. doi: 10.1086/520125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kainulainen K, Karttunen L, Puhakka L, Sakai L, Peltonen L. Mutations in the fibrillin gene responsible for dominant ectopia lentis and neonatal Marfan syndrome. Nat Genet. 1994;6:64–9. doi: 10.1038/ng0194-64. [DOI] [PubMed] [Google Scholar]
- 4.Milewicz DM, Grossfield J, Cao SN, Kielty C, Covitz W, Jewett T. A mutation in FBN1 disrupts profibrillin processing and results in isolated skeletal features of the Marfan syndrome. J Clin Invest. 1995;95:2373–8. doi: 10.1172/JCI117930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rand-Hendriksen S, Lundby R, Tjeldhorn L, Andersen K, Offstad J, Semb SO, et al. Prevalence data on all Ghent features in a cross-sectional study of 87 adults with proven Marfan syndrome. Eur J Hum Genet. 2009;17:1222–30. doi: 10.1038/ejhg.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collod-Beroud G, Le BS, Ades L, Ala-Kokko L, Booms P, Boxer M, et al. Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum Mutat. 2003;22:199–208. doi: 10.1002/humu.10249. [DOI] [PubMed] [Google Scholar]
- 7.Pereira L, D'Alessio M, Ramirez F, Lynch JR, Sykes B, Pangilinan T, et al. Genomic organization of the sequence coding for fibrillin, the defective product in Marfan syndrome. Hum Molec Genet. 1993;2:961–8. doi: 10.1093/hmg/2.7.961. [DOI] [PubMed] [Google Scholar]
- 8.Sakai LY, Keene DR, Engvall E. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol. 1986;103:2499–509. doi: 10.1083/jcb.103.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corson GM, Chalberg SC, Dietz HC, Charbonneau NL, Sakai LY. Fibrillin binds calcium and is coded by cDNAs that reveal a multidomain structure and alternatively spliced exons at the 5' end. Genomics. 1993;17:476–84. doi: 10.1006/geno.1993.1350. [DOI] [PubMed] [Google Scholar]
- 10.Downing AK, Knott V, Werner JM, Cardy CM, Campbell ID, Handford PA. Solution structure of a pair of calcium-binding epidermal growth factor-like domains: implications for the Marfan syndrome and other genetic disorders. Cell. 1996;85:597–605. doi: 10.1016/S0092-8674(00)81259-3. [DOI] [PubMed] [Google Scholar]
- 11.Schrijver I, Liu W, Brenn T, Furthmayr H, Francke U. Cysteine substitutions in epidermal growth factor-like domains of fibrillin-1: distinct effects on biochemical and clinical phenotypes. Am J Hum Genet. 1999;65:1007–20. doi: 10.1086/302582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirschner R, Hubmacher D, Iyengar G, Kaur J, Fagotto-Kaufmann C, Bromme D, et al. Classical and neonatal Marfan syndrome mutations in fibrillin-1 cause differential protease susceptibilities and protein function. J Biol Chem. 2011;286(37):32810–23. doi: 10.1074/jbc.M111.221804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reinhardt DP, Ono RN, Notbohm H, Muller PK, Bachinger HP, Sakai LY. Mutations in calcium-binding epidermal growth factor modules render fibrillin-1 susceptible to proteolysis. A potential disease-causing mechanism in Marfan syndrome. J Biol Chem. 2000;275:12339–45. doi: 10.1074/jbc.275.16.12339. [DOI] [PubMed] [Google Scholar]
- 14.Vollbrandt T, Tiedemann K, El-Hallous E, Lin G, Brinckmann J, John H, et al. Consequences of cysteine mutations in calcium-binding epidermal growth factor modules of fibrillin-1. J Biol Chem. 2004;279:32924–31. doi: 10.1074/jbc.M405239200. [DOI] [PubMed] [Google Scholar]
- 15.Caputi M, Kendzior RJ, Jr, Beemon KL. A nonsense mutation in the fibrillin-1 gene of a Marfan syndrome patient induces NMD and disrupts an exonic splicing enhancer. Genes Dev. 2002;16:1754–9. doi: 10.1101/gad.997502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schrijver I, Liu W, Odom R, Brenn T, Oefner P, Furthmayr H, et al. Premature termination mutations in FBN1: distinct effects on differential allelic expression and on protein and clinical phenotypes. Am J Hum Genet. 2002;71:223–37. doi: 10.1086/341581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng J, Maquat LE. Nonsense codons can reduce the abundance of nuclear mRNA without affecting the abundance of pre-mRNA or the half-life of cytoplasmic mRNA. Mol Cell Biol. 1993;13:1892–902. doi: 10.1128/MCB.13.3.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chao SC, Chen JS, Tsai CH, Lin JM, Lin YJ, Sun HS. Novel exon nucleotide substitution at the splice junction causes a neonatal Marfan syndrome. Clin Genet. 2010;77:453–63. doi: 10.1111/j.1399-0004.2009.01337.x. [DOI] [PubMed] [Google Scholar]
- 19.Liu W, Qian C, Comeau K, Brenn T, Furthmayr H, Francke U. Mutant fibrillin-1 monomers lacking EGF-like domains disrupt microfibril assembly and cause severe marfan syndrome. Hum Mol Genet. 1996;5:1581–7. doi: 10.1093/hmg/5.10.1581. [DOI] [PubMed] [Google Scholar]
- 20.Liu W, Schrijver I, Brenn T, Furthmayr H, Francke U. Multi-exon deletions of the FBN1 gene. BMC Med Genet. 2001;2:11. doi: 10.1186/1471-2350-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh KK, Elligsen D, Liersch R, Schubert S, Pabst B, Arslan-Kirchner M, et al. Multi-exon out of frame deletion of the FBN1 gene leading to a severe juvenile onset cardiovascular phenotype in Marfan syndrome. J Mol Cell Cardiol. 2007;42:352–6. doi: 10.1016/j.yjmcc.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 22.Matyas G, Alonso S, Patrignani A, Marti M, Arnold E, Magyar I, et al. Large genomic fibrillin-1 (FBN1) gene deletions provide evidence for true haploinsufficiency in Marfan syndrome. Hum Genet. 2007;122:23–32. doi: 10.1007/s00439-007-0371-x. [DOI] [PubMed] [Google Scholar]
- 23.Aoyama T, Tynan K, Dietz HC, Francke U, Furthmayr H. Missense mutations impair intracellular processing of fibrillin and microfibril assembly in Marfan syndrome. Hum Mol Genet. 1993;2:2135–40. doi: 10.1093/hmg/2.12.2135. [DOI] [PubMed] [Google Scholar]
- 24.Milewicz DM, Pyeritz RE, Crawford ES, Byers PH. Marfan syndrome: defective synthesis, secretion, and extracellular matrix formation of fibrillin by cultured dermal fibroblasts. J Clin Invest. 1992;89:79–86. doi: 10.1172/JCI115589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaefer GB, Godfrey M. Quantitation of fibrillin immunofluorescence in fibroblast cultures in the Marfan syndrome. Clin Genet. 1995;47:144–9. doi: 10.1111/j.1399-0004.1995.tb03947.x. [DOI] [PubMed] [Google Scholar]
- 26.Kielty CM, Shuttleworth CA. Abnormal fibrillin assembly by dermal fibroblasts from two patients with Marfan syndrome. J Cell Biol. 1994;124:997–1004. doi: 10.1083/jcb.124.6.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, et al. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–92. doi: 10.1172/JCI200422715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson PN, Arteaga-Solis E, Baldock C, Collod-Beroud G, Booms P, De PA, et al. The molecular genetics of Marfan syndrome and related disorders. J Med Genet. 2006;43:769–87. doi: 10.1136/jmg.2005.039669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114:172–81. doi: 10.1172/JCI200420641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whiteman P, Handford PA. Defective secretion of recombinant fragments of fibrillin-1: implications of protein misfolding for the pathogenesis of Marfan syndrome and related disorders. Hum Mol Genet. 2003;12:727–37. doi: 10.1093/hmg/ddg081. [DOI] [PubMed] [Google Scholar]
- 31.Rand-Hendriksen S, Tjeldhorn L, Lundby R, Semb SO, Offstad J, Andersen K, et al. Search for correlations between FBN1 genotype and complete Ghent phenotype in 44 unrelated Norwegian patients with Marfan syndrome. Am J Med Genet A. 2007;143A:1968–77. doi: 10.1002/ajmg.a.31759. [DOI] [PubMed] [Google Scholar]
- 32.Paepe D, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet. 1996;62:417–26. doi: 10.1002/(SICI)1096-8628(19960424)62:4<417::AID-AJMG15>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 33.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 34.Eldadah ZA, Brenn T, Furthmayr H, Dietz HC. Expression of a mutant human fibrillin allele upon a normal human or murine genetic background recapitulates a Marfan cellular phenotype. J Clin Invest. 1995;95:874–80. doi: 10.1172/JCI117737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kapustin Y, Chan E, Sarkar R, Wong F, Vorechovsky I, Winston RM, et al. Cryptic splice sites and split genes. Nucleic Acids Res. 2011;39:5837–44. doi: 10.1093/nar/gkr203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gibson MA, Ellis SL, Ades LC, Haan E, Cleary EG. Preferential pre-mRNA utilisation of an upstream cryptic 5' splice site created by a single base deletion mutation in exon 37 of the FBN-1 gene. Eur J Biochem. 1998;256:221–8. doi: 10.1046/j.1432-1327.1998.2560221.x. [DOI] [PubMed] [Google Scholar]
- 37.Robinson DO, Lin F, Lyon M, Raponi M, Cross E, White HE, et al. Systematic screening of FBN1 gene unclassified missense variants for splice abnormalities. Clin Genet. 2012;82:223–31. doi: 10.1111/j.1399-0004.2011.01781.x. [DOI] [PubMed] [Google Scholar]
- 38.Aubart M, Gross MS, Hanna N, Zabot MT, Sznajder M, Detaint D, et al. The clinical presentation of Marfan syndrome is modulated by expression of wildtype FBN1 allele. Hum Mol Genet. 2015; 4. [Epub ahead of print]. [DOI] [PubMed]
- 39.Yao Z, Jaeger JC, Ruzzo WL, Morale CZ, Emond M, Francke U, et al. A Marfan syndrome gene expression phenotype in cultured skin fibroblasts. BMC Genomics. 2007;8:319. doi: 10.1186/1471-2164-8-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo D, Tan FK, Cantu A, Plon SE, Milewicz DM. FBN1 exon 2 splicing error in a patient with Marfan syndrome. Am J Med Genet. 2001;101:130–4. doi: 10.1002/1096-8628(20010615)101:2<130::AID-AJMG1333>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 41.Hutchinson S, Furger A, Halliday D, Judge DP, Jefferson A, Dietz HC, et al. Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Hum Mol Genet. 2003;12:2269–76. doi: 10.1093/hmg/ddg241. [DOI] [PubMed] [Google Scholar]
- 42.Montgomery RA, Geraghty MT, Bull E, Gelb BD, Johnson M, McIntosh I, et al. Multiple molecular mechanisms underlying subdiagnostic variants of Marfan syndrome. Am J Hum Genet. 1998;63:1703–11. doi: 10.1086/302144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Halliday D, Hutchinson S, Kettle S, Firth H, Wordsworth P, Handford PA. Molecular analysis of eight mutations in FBN1. Hum Genet. 1999;105:587–97. doi: 10.1007/s004399900190. [DOI] [PubMed] [Google Scholar]
- 44.Dietz HC, McIntosh I, Sakai LY, Corson GM, Chalberg SC, Pyeritz RE, et al. Four novel FBN1 mutations: significance for mutant transcript level and EGF-like domain calcium binding in the pathogenesis of Marfan syndrome. Genomics. 1993;17:468–75. doi: 10.1006/geno.1993.1349. [DOI] [PubMed] [Google Scholar]
- 45.Hewett DR, Lynch JR, Child A, Sykes BC. A new missense mutation of fibrillin in a patient with Marfan syndrome. J Med Genet. 1994;31:338–9. doi: 10.1136/jmg.31.4.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buratti E, Chivers M, Hwang G, Vorechovsky I. DBASS3 and DBASS5: databases of aberrant 3'- and 5'-splice sites. Nucleic Acids Res. 2011;39:D86–91. doi: 10.1093/nar/gkq887. [DOI] [PMC free article] [PubMed] [Google Scholar]