

Abstract
The observation that Cadmium (Cd2+) inhibits Msh2-Msh6, which is responsible for identifying base pair mismatches and other discrepancies in DNA, has led to the proposal that selective targeting of this protein and consequent suppression of DNA repair or apoptosis promote the carcinogenic effects of the heavy metal toxin. It has been suggested that Cd2+ binding to specific sites on Msh2-Msh6 blocks its DNA binding and ATPase activities. To investigate the mechanism of inhibition, we measured Cd2+ binding to Msh2-Msh6, directly and by monitoring changes in protein structure and enzymatic activity. Global fitting of the data to a multiligand binding model revealed that binding of about 100 Cd2+ ions per Msh2-Msh6 results in its inactivation. This finding indicates that the inhibitory effect of Cd2+ occurs via a nonspecific mechanism. Cd2+ and Msh2-Msh6 interactions involve cysteine sulfhydryl groups, and the high Cd2+:Msh2-Msh6 ratio implicates other ligands such as histidine, aspartate, glutamate, and the peptide back bone as well. Our study also shows that cadmium inactivates several unrelated enzymes similarly, consistent with a nonspecific mechanism of inhibition. Targeting of a variety of proteins, including Msh2-Msh6, in this generic manner would explain the marked broad-spectrum impact of Cd2+ on biological processes. We propose that the presence of multiple nonspecific Cd2+ binding sites on proteins and their propensity to change conformation on interaction with Cd2+ are critical determinants of the susceptibility of corresponding biological systems to cadmium toxicity.
Humans can be exposed to toxic levels of cadmium (Cd2+) from cigarette smoke and industrial pollution associated with the manufacture and use of materials such as nickel–cadmium batteries, plastics, pigments, metal alloys, and coatings. Cd2+ poses a severe and intractable health hazard because it can accumulate in and damage several tissues, including the liver, kidneys and lungs, pancreas, testis, and bone, and because it is a potent carcinogen (the biological half-life of Cd2+ is 15–30 years in humans). Cd2+ has been reported to alter cell growth, differentiation, and apoptosis by disrupting a variety of cellular processes, including signal transduction, gene regulation, responses to oxidative stress, and DNA processing and repair. For these reasons, the targets and mechanisms by which this heavy metal toxin effects cellular changes are subject to intense investigation (1–3).
The mutagenic effects of Cd2+ have been attributed, at least in part, to inhibition of DNA mismatch repair (MMR)1 (4). This evolutionarily conserved DNA system is responsible for correcting base pair mismatches and insertion/deletion loops (IDL) formed during DNA replication and inducing cell cycle arrest and apoptosis in response to extensive DNA damage. Thus, inhibition of MMR increases genomic instability and increases susceptibility to cancer (5, 6). Recent studies of the effects of Cd2+ on MMR in eukaryotes suggest that the metal ion targets Msh2-Msh6, an essential protein in the system, and it has been proposed that Cd2+ binding to a specific site on Msh2-Msh6 blocks its activities (7, 8). The DNA repair reaction sequence involves (a) recognition of an error in a DNA strand, (b) incision of the strand in the vicinity of the error, (c) excision of the strand past the error, followed by (d) strand resynthesis. Msh2 and Msh6, eukaryotic homologues of MutS protein, form a heterodimer that primarily recognizes base pair mismatches and short IDLs (1–2 bases). Eukaryotic homologues of MutL, another conserved MMR protein (e.g., Saccharomyces cerevisiae Mlh1-Pms1 or Homo sapiens Mlh1-Pms2), have an endonuclease activity that preferentially acts on a DNA strand containing a nick and may create sites for initiation of excision in the vicinity of the mismatch (9). ExoI exonuclease catalyzes strand excision, and RPA single-stranded DNA binding protein, DNA polymerase δ, PCNA sliding clamp, and RFC clamp loader are responsible for DNA resynthesis. Msh2-Msh6 also recognizes mismatches containing damaged bases such as O6-methylguanine, and in the case of the apoptosis reaction sequence, debate continues as to whether the recognition event signals the cell cycle arrest and apoptosis machinery directly or perhaps indirectly through toxic DNA intermediates created by futile repair attempts (5, 6).
The mechanistic basis for suppression of DNA MMR by Cd2+ has yet to be resolved. The MMR pathway clearly has several potential protein targets for Cd2+-mediated inactivation, and alteration of DNA structure and dynamics by Cd2+ binding may be a contributing factor as well (10). Thus far, the focus of investigation has been on Msh2-Msh6, the best characterized of the core MMR proteins. The common model for prokaryotic MutS and eukaryotic Msh2-Msh6 actions in MMR is as follows: (a) MutS/Msh2-Msh6, likely in an ADP-bound state (11, 12), slides on DNA, interrogating base pair structure and/or dynamics to find mismatches (13); (b) conserved phenylalanine and glutamate residues help MutS/Msh2-Msh6 make contacts with base pairs and recognize a mismatch (14–17); (c) mismatch binding triggers replacement of ADP with adenosine 5′-triphosphate (ATP) (18) and stabilization of MutS/Msh2-Msh6 in an ATP-bound state (11, 12, 19); and, (d) ATP-bound MutS/Msh2-Msh6 activates MutL/Mlh1-Pms1 and initiates the next steps in MMR (18, 20, 21).
Recent studies of S. cerevisiae Msh2-Msh6, which is related closely to the human Msh2-Msh6 protein, indicate that its mismatched DNA binding and ATPase activities are suppressed in the presence of Cd2+, with one report suggesting that the ATPase activity is impacted more severely than mismatch binding (7, 8). The mechanism by which Cd2+ inhibits Msh2-Msh6 function is not known, although it has been speculated that selective binding of Cd2+ to a site on Msh2-Msh6, such as a hypothetical Zn2+ finger domain, could disrupt its interactions with ATP and DNA, either directly or through allostery.
To understand the mechanism of Msh2-Msh6 inactivation by Cd2+, which could in turn facilitate efforts to mitigate its toxicity, we undertook a quantitative analysis of the impact of Cd2+ on S. cerevisiae Msh2-Msh6. The specific aim was to identify Cd2+ binding site(s) on Msh2-Msh6 and determine how the interaction triggers changes in its DNA binding and ATP binding/hydrolysis activities. Surprisingly, our data revealed that Msh2-Msh6 has a very high capacity for binding Cd2+ ions in a nonspecific manner, and these interactions lead to alteration of protein structure and loss of function. Cd2+ blocks the activities of several other unrelated proteins in a similar fashion, consistent with the nonspecific nature of the inhibition mechanism. On the basis of these findings, we propose that interactions between Cd2+ and multiple binding sites on a protein that lead to changes in its conformation constitute a generic mechanism of Cd2+-mediated inhibition of function.
Experimental Procedures
Proteins, DNA, Nucleotides, and Other Reagents. S. cerevisiae Msh2-Msh6 (22), Thermus aquaticus MutS (11), S. cerevisiae RFC (22), and Escherichia coli phosphate binding protein (PBP) (23) were overexpressed and purified from E. coli, and PBP was labeled with N-[2(1-maleimidyl)ethyl]-7-(diethylamino)coumarin-3-carboxamide (MDCC) as described previously. E. coli γ complex and overexpression clones for E. coli MutS and MutL were gifts from Michael O'Donnell (The Rockefeller University). Synthetic DNAs (37 bp) were purchased from Integrated DNA Technologies, purified by denaturing polyacrylamide gel electrophoresis, and annealed to prepare duplex DNA substrates, as described (12). Radioactive nucleotides were purchased from Perkin-Elmer Life Sciences, and nonradioactive nucleotides and CdCl2 were purchased from Sigma-Aldrich. Fluorescein-5-maleimide, SYBR Green, and Measure-iT cadmium assay kit were purchased from Invitrogen. PEI-Cellulose F thin-layer chromatography (TLC) plates were purchased from EM Science.
DNA Binding Assays
DNA binding to Msh2-Msh6 was measured by gel mobility shift assays. In 10 μL reactions containing buffer A (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, and 5 mM MgCl2), Msh2-Msh6 (0.2 μM) was incubated with DNA (0.16 μM) and CdCl2 (0–400 μM) for 20 min at 25 °C, followed by electrophoresis at 4 °C on a native 4.5% TBE-acrylamide gel. The gels were stained with SYBR Green according to the manufacturer's protocol, and the DNA was visualized and quantified on a PhosphorImager (GE Healthcare).
ATPase Assays
The ATPase activity was measured under steady-state conditions at 30 °C with Msh2-Msh6 (0.03–2 μM) and 500 μM α32P-ATP in buffer A with varying amounts of CdCl2 (0–500 μM). PEI cellulose TLC (0.6 M potassium phosphate, pH 3.4) was used to resolve α32P-ADP from α32P-ATP, and kcat was calculated from initial ADP production rates for each Msh2-Msh6 and CdCl2 concentration (experiments with DTT were performed in the presence of 5 mM freshly dissolved DTT). Steady-state ATPase activities of other proteins (0.5 μM) were measured similarly; T. aquaticus MutS (40 °C; 50 mM Hepes-NaOH, pH 7.8, 100 mM KCl, and 5 mM MgCl2); S. cerevisiae RFC [25 °C; 30 mM Hepes-NaOH, pH 7.5, 10 mM Mg(OAc)2, and 5% glycerol]; E. coli γ complex (25 °C; 20 mM Tris-HCl, pH 7.5, 50 mM NaCl, and 8 mM MgCl2); and, E. coli MutS and MutL (25 °C; 20 mM Tris-HCl, pH 8.0, 100 mM NaCl, and 5 mM MgCl2).
The ATPase activity was measured under presteadystate conditions using a stopped-flow instrument (KinTek Corp., Austin TX) and MDCC-labeled PBP to detect phosphate (Pi) release (11). Forty microliters of Msh2-Msh6 (2 μM) and MDCC-PBP (20 μM) was mixed with 40 μL of ATP (1 mM) and CdCl2 (0–1 mM) in buffer A at 30 °C, and the change in MDCC-PBP fluorescence on binding Pi was measured by excitation at 425 nm and emission at >450 nm (final concentrations: 1 μM Msh2-Msh6, 10 μM MDCC-PBP, 500 μM ATP, and 0–500 μM CdCl2). The kinetic traces were fit to a burst equation ([Pi] = (A0 e−kt + Vt + F0)/mPi) where [Pi] is the Pi concentration, A0 is the amplitude, k is the observed burst rate constant, V is the velocity of the linear phase, F0 is the initial fluorescence intensity, and mPi is the slope of the Pi standard curve measured under the same conditions.
Protein Structure Assays
The change in Msh2-Msh6 (0.05–2 μM) intrinsic fluorescence on titration with CdCl2 (0–500 μM) was measured using a FluoroMax-3 spectrofluorometer (Horiba Jobin Yvon). Msh2-Msh6 in buffer A was mixed with increasing CdCl2 for 1 min at 25 °C, followed by excitation at 290 nm and emission measured at 340 nm. Control experiments with buffer replacing CdCl2 were performed concurrently to correct for photobleaching effects. The Cd2+ effect on cysteine labeling was measured by incubating Msh2-Msh6 (0.5 μM) with fluorescein-5-maleimide (2.5 μM) and 0–300 μM CdCl2 in 20 μL of 50 mM potassium phosphate, pH 7.2, and 100 mM NaCl for 5 min at 25 °C. The reactions were quenched by adding SDS and DTT and analyzed by SDS-PAGE, and the fluorescein-labeled protein was quantified on a PhosphorImager.
Cadmium Binding Assays
Cadmium binding was assayed by incubating Msh2-Msh6 (1 μM) or MutS (1 μM) in 10 μL of buffer A with CdCl2 (0–200 μM) for 10minat 25 °C and measuring the free Cd2+ remaining in solution by the fluorescence-based Measure-iT cadmium assay (Invitrogen), according to the manufacturer's protocol (lower detection limit, 1 pmol or 0.1 μM Cd + in 10 μL of solution). Free Cd2+ concentrations were determined from linear standard plots generated concurrently with each experiment, using both freshly prepared and the assay kit CdCl2 stock solutions and corresponding bound Cd2+ concentrations plotted vs total CdCl2.
Results
Exposure to Cadmium Changes Msh2-Msh6 Conformation and Suppresses Its Mismatch Binding and ATPase Activities
The effect of cadmium on S. cerevisiae Msh2-Msh6 was assessed by measuring the mismatched DNA binding, ATP binding, and hydrolysis activities of the protein, as well as monitoring its conformation via intrinsic fluorescence in the presence of CdCl2. Gel mobility shift analysis (GMSA) revealed loss of Msh2-Msh6 binding to G:T mismatch-containing DNA [37 nucleotide (nt)] on titration with CdCl2 (Figure 1A), and the Msh2-Msh6 · DNA complex was essentially undetectable at about 200 μM CdCl2; a similar inhibition profile was observed for Msh2-Msh6 binding to fully matched DNA (data not shown). The saturating concentration of CdCl2 is very high relative to that of Msh2-Msh6 (0.2 μM). However, Cd2+ ions may also bind DNA in the reaction via contacts with guanine-N7, the A:T base pair, and backbone groups, as dissociation constants for these interactions have been reported in the low micromolar range (10,24). Thus, it is possible that the GMSA reports Cd2+ binding to both protein and DNA, as altering the properties of either macromolecule could disrupt Msh2-Msh6·DNA complex.
Figure 1.

Cadmium changes Msh2-Msh6 conformation and inhibits its mismatch binding and ATPase activities. (A) The gel mobility shift analysis shows a decrease in the Msh2-Msh6·G:T DNA complex with increasing CdCl2 in the reaction. (B) The steady-state Msh2-Msh6 ATPase activity decreases with increasing CdCl2 (kcat = 0.17 to 0.02 s−1), and the data fit to a hyperbola yield K1/2 = 9 μM. (C) The presteady state Pi release assays show a burst of Msh2-Msh6-catalyzed ATP hydrolysis at 1.3 s−1 rate and 1.1 μM amplitude. Increasing CdCl2 inhibits both the burst and the steady-state ATPase rates. (D) Intrinsic Msh2-Msh6 fluorescence decreases with increasing CdCl2, with K1/2 = 6 μM.
The ATPase activity of Msh2-Msh6 was measured in the absence of DNA, under steady-state conditions, and it was found to decrease from a kcat value of 0.17 s−1 to about 0.02 s−1 with increasing CdCl2 (Figure 1B). A plot of the kcat values vs CdCl2 concentration fit to a hyperbola yields an apparent inhibition constant K1/2 of 9.2 ± 2 μM. We also measured Msh2-Msh6 ATPase activity under presteady-state conditions to determine which steps in the reaction are affected by Cd2+ (ATP binding to Msh2-Msh6 appears not to be disrupted significantly by Cd2+, according to data from nitrocellulose membrane filtration experiments; Supporting Information, Figure S1A). A series of stopped-flow experiments was performed in which the change in MDCC-labeled PBP fluorescence on binding Pi was used as a reporter of ATP hydrolysis and Pi release by Msh2-Msh6 (Figure 1C). As reported previously, in the absence of DNA, Msh2-Msh6 catalyzes a burst of ATP hydrolysis and Pi release (1.3 s−1) in the first catalytic turnover, followed by the slower, linear steady-state phase (0.3 s−1) (12). The amplitude of the burst phase decreases with increasing CdCl2 in the reaction, as does the ATPase rate (Figure 1C). Rapid-quench experiments, which directly measure α32P-ATP hydrolysis, yielded similar results (data not shown; ref 12). These data indicate that Cd2+ suppresses the ATP hydrolysis step in the reaction. Curiously, a substantially higher concentration of CdCl2 is required to inhibit Msh2-Msh6 ATPase activity in presteady state experiments (Figure 1C) as compared with steady-state experiments (Figure 1B), which differ only in the concentration of Msh2-Msh6 in the reactions, i.e., 1 vs 0.03 μM, respectively; the reason for this discrepancy is clarified in the next section. In any case, the results all confirm that cadmium has a severe inhibitory effect on both the DNA binding and the ATPase activities of Msh2-Msh6.
The active sites for DNA binding and ATP binding/hydrolysis are positioned at opposite ends of Msh2-Msh6, across a distance of approximately 50 Å, according to the structure of the human protein (14). If Cd2+ binding to Msh2-Msh6 underlies the inhibitory effect, it is likely that the interaction induces changes in protein conformation that affect both active sites. To test this hypothesis, we assessed Msh2-Msh6 conformation in the presence of CdCl2 by measuring its intrinsic fluorescence (Figure 1D). Titration of Msh2-Msh6 (0.05 μM) with CdCl2 causes a decrease in protein fluorescence, and the data fit to a hyperbola yield a K1/2 value of 5.5 ± 1 μM, which is comparable to the apparent inhibition constant determined from steady-state ATPase assays (Figure 1B, K1/2 = 9.2 μM). Together, these data support the hypothesis that Cd2+ binding triggers conformational changes in Msh2-Msh6 that cause loss of both its ATPase and DNA binding activities.
Stoichiometry of Cadmium Binding to Msh2-Msh6 That Results in Loss of Function
According to the steady-state ATPase and protein fluorescence data, cadmium inhibits Msh2-Msh6 activities with an apparent inhibition constant in the 5–10 μM range. To determine the affinity and stoichiometry of the interaction between Cd2+ and Msh2-Msh6, we titrated the protein with increasing amounts of CdCl2 and measured the concentration of free Cd2+ remaining in solution using a fluorescence-based assay. Surprisingly, cadmium binding saturates at a ratio of about 170–180 Cd2+ ions per Msh2-Msh6 (Figure 2A, 1 μM Msh2-Msh6 in the reaction). The inset in Figure 2A shows the isotherm at low CdCl2 concentrations, and there is no detectable difference between the first Cd2+ ion and the subsequent Cd2+ ions binding to Msh2-Msh6. Thus, the interactions between Cd2+ and Msh2-Msh6 appear nonspecific, with no particular binding site displaying significantly higher affinity for Cd2+ than any other site on the protein. In a parallel experiment, we also measured Cd2+ binding to T. aquaticus MutS, a prokaryotic homologue of Msh2-Msh6, to compare and contrast Cd2+ effects on these evolutionarily related proteins. MutS also binds a large number of Cd2+ nonspecifically, although in this case binding saturates at a ratio of about 50 Cd2+ ions per MutS dimer (Figure 2A). One distinction between these two proteins is that the Msh2-Msh6 dimer has 27 cysteines while MutS dimer has two cysteines. Free thiol groups are known as good ligands of Cd2+ ions, and the difference in cysteine count between Msh2-Msh6 and MutS may be linked to differences in their interactions with and response to cadmium. This hypothesis is considered further in a later section.
Figure 2.
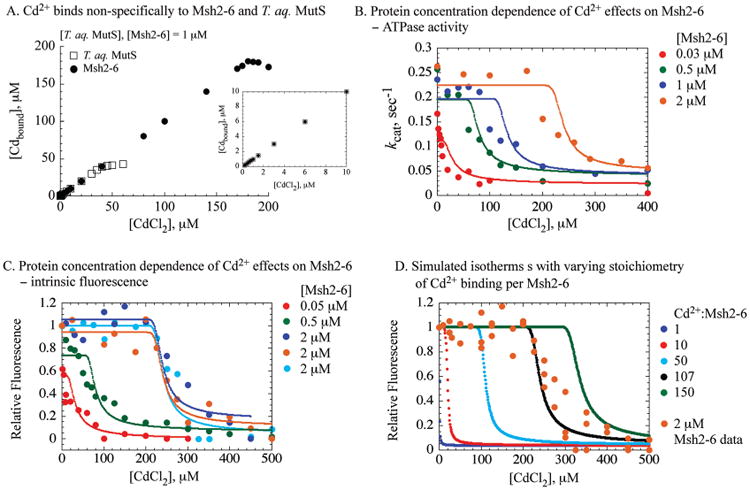
High stoichiometry of Cd2+ binding is required for inhibition of Msh2-Msh6 function. (A) Titration of Msh2-Msh6 and T. aquaticus MutS with CdCl2 results in saturation of Cd2+ binding at ∼180 ions per Msh2-Msh6 and ∼50 ions per MutS dimer. The inset shows the binding isotherms at low CdCl2 concentrations. (B) The ATPase activity of 0.03 (red), 0.5 (green), 1.0 (blue), and 2.0 μM (brown) Msh2-Msh6 and (C) the intrinsic fluorescenceof0.05 (red), 0.5 (green), and 2.0 μM (blue, brown, and cyan) Msh2-Msh6 in the presence of increasing CdCl2. Experimental data and corresponding fits to amultisite binding model are shown. The best global fit of the data yielded an apparent dissociation constant KD = 0.5 ± 0.14 μM for the Cd2+ binding sites, an equilibrium constant KC = 4.0 ± 1.1 for protein conformation change, and the number of Cd2+ binding sites S= 107.9 ± 3.1. Simulations using these parameters are shown as solid lines of the same color as the corresponding data. (D) Protein fluorescence data for 2 μM Msh2-Msh6 are shown as circles, and a corresponding simulation using the best fit parameters is shown as a black line. Other lines show simulations performed with the same parameters except with the number of binding sites, S, set to 1(blue), 10(red), 50(cyan), and 150 (green).
The high Cd2+:Msh2-Msh6 binding ratio offers an explanation for our observation that far greater amounts of CdCl2 appear necessary to block the ATPase activity of 1 vs 0.03 μM Msh2-Msh6 under otherwise identical reaction conditions (Figure 1). To explicitly determine the stoichiometry of Cd2+ ion binding required to inhibit Msh2-Msh6 function, we performed a series of experiments with varying protein concentrations and measured the effects of Cd2+ on both Msh2-Msh6 conformation and ATPase activity. The resulting titration curves showed an increasingly pronounced sigmoidal shape with higher Msh2-Msh6 concentrations (Figure 2B, C, 0.5, 1.0, and 2.0 μM). This protein concentration-dependent sigmoidicity can be explained by the fact that the changes in fluorescence and ATPase activity occur only after a threshold of Cd2+ ions bound to Msh2-Msh6 is reached.
The data were fit to the Hill equation, which is used commonly to analyze sigmoidal titration curves and estimate the number of ligands binding to a macromolecule. The fits yielded Hill coefficients ranging from 1.5 to 6.8 and apparent equilibrium dissociation constants from 58 to 290 μM for the data sets with 0.5 to 2 μM protein, respectively (note: Msh2-Msh6 is a stable dimer within this concentration range). These parameters have no physical meaning, although the Hill coefficient may provide a reliable estimate of the number of ligand binding sites in the case of extreme positive cooperativity (25). There is, however, no evidence to justify an assumption of cooperativity in Cd2+ binding to Msh2-Msh6. Therefore, we developed a simple model in which multiple, independent binding sites of equal affinity on Msh2-Msh6 are occupied by cadmium, after which the protein undergoes conformational change that alters its intrinsic fluorescence and renders it inactive.
As shown in reaction scheme, Cd2+ ions (L) bind to Msh2-Msh6 complex (M) with S binding sites. Once the binding sites are populated, the complex transforms into inactive form (M′LS).
| (1) |
The fraction of altered, Cd2+-bound complex, [M′LS]/[MT], is calculated as described in the Appendix. The value F of the observed property of the reaction (either fluorescence or ATPase kcat) is calculated as
| (2) |
where F0 and F1 are values of the properties of the native protein in the absence of Cd2+ and for the altered Cd2+-bound protein, respectively (in the case of the ATPase activity, F1 = 0).
To test the model and obtain accurate estimates of its parameters, all of the fluorescence and ATPase data were fit simultaneously to the model using gfit, a program designed for global analysis of data from different types of experiments (Supporting Information; 26). The best fit to the data yielded stoichiometry S = 107 ± 3 for Cd2+ binding to Msh2-Msh6 with an apparent dissociation constant KD = 0.5 ± 0.1 μM and a conformational change equilibrium constant KC = 4 ± 1 (Figure 2). The search for alternative good or better fits was performed by random restart method. The fitting procedure was repeated over 300 times with randomly chosen starting parameters. Each procedure produced either a very similar set of parameters or a significantly worse fit. Notably, the analysis provided a relatively narrow confidence interval for Cd2+ stoichiometry. Accordingly, simulations using different numbers of Cd2+ binding sites per Msh2-Msh6 (S = 1…150) show that the shapes of the titration curves are affected strongly by the value of S (Figure 2D, 2 μM protein). Thus, both the intrinsic fluorescence and the ATPase responses of Msh2-Msh6 to cadmium exposure indicate that changes in conformation and loss of function occur as a result of Cd2+ ions binding the protein at a high ratio of about 100:1.
Nonspecific Cadmium Binding Affects Several Proteins in a Similar Manner
The above findings argue against an inhibition mechanism whereby site-specific Cd2+ binding to Msh2-Msh6 directly impacts its ATPase or DNA binding activities. The lack of specificity evidenced by the high stoichiometry of Cd2+ ions necessary for Msh2-Msh6 inhibition implies that the heavy metal toxin may inactivate other proteins, unrelated to Msh2-Msh6, in a similar manner. We tested this hypothesis by assaying S. cerevisiae RFC complex, a clamp loader that catalyzes assembly of PCNA clamps onto DNA during replication and various DNA processing reactions, including DNA MMR. The data in Figure 3A show that CdCl2 effects near identical suppression of both Msh2-Msh6 and RFC ATPase activities (0.03 μM proteins). A survey of yet other ATPases, assayed at higher protein concentrations of 0.5 μM, shows that correspondingly higher concentrations of CdCl2 are required to inactivate S. cerevisiae RFC, E. coli MutS, E. coli MutL, and the E. coli γ complex clamp loader (Figure 3B). Interestingly, the ATPase activity of T. aquaticus MutS, which is a homologue of Msh2-Msh6 and also binds a large number of Cd2+ ions nonspecifically (Figure 2A), is comparatively resistant to inactivation under the same conditions. A possible reason for this difference is explored in the next section.
Figure 3.
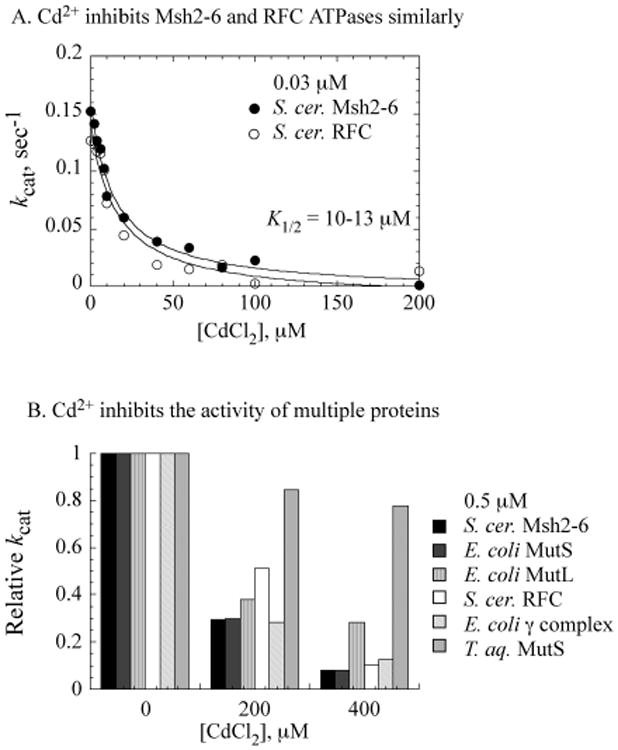
Nonspecific cadmium binding inhibits different proteins in a similar manner. (A) S. cerevisiae Msh2-Msh6 and RFC clamp loader (0.03 μM) exhibit anidentical decrease in ATPase activity with increasing CdCl2. (B) A variety of other proteins (0.5 μM) exhibit the same ATPase inhibition profile with increasing CdCl2, except for T. aquaticus MutS.
Inhibitory Effects of Nonspecific Cadmium Binding Involve Sulfhydryl Groups on Msh2-Msh6
To characterize the Cd2+ interactions that lead to inhibition of Msh2-Msh6 function, we probed further the observed difference between Cd2+ binding to T. aquaticus MutS vs Msh2-Msh6 (Figure 2A). Numerous Cd2+ ions bind nonspecifically to both proteins, although the binding stoichiometry is lower for T. aquaticus MutS. Correspondingly, T. aquaticus MutS is less susceptible to inhibition by Cd2+, as its ATPase activity declines by only about 20% in the presence of 400 μM CdCl2 vs greater than 90% decline for Msh2-Msh6 (Figure 4A, 0.5 μM proteins). We hypothesized that that the difference in the number of cysteines on T. aquaticus MutS relative to Msh2-Msh6 may contribute to differences in their Cd2+ binding stoichiometry and the impact of Cd2+ on their activities (2 vs 27 cysteines, respectively). Evidence supporting participation of multiple cysteine thiols on Msh2-Msh6 in Cd2+ binding was provided by a labeling experiment in which the protein was reacted with fluorescein-5-maleimide. As shown in Figure 4B, addition of increasing concentrations of CdCl2 to the reaction blocks thiol labeling on both Msh2 and Msh6 subunits, consistent with their involvement in coordinating Cd2+ ions (labeling of 0.5 μM Msh2-Msh6 is reduced to <30%, in the presence of 300 μM CdCl2). Furthermore, the ATPase activity of Msh2-Msh6 is resistant to high concentrations of CdCl2 in the presence of DTT (Figure 4A). DTT likely competes effectively against thiol groups and other ligands on Msh2-Msh6, thereby protecting the protein against Cd2+. These results, together with the observation that all of the ATPases tested above have several cysteine residues with the exception T. aquaticus MutS, suggest that participation of thiol groups in nonspecific interactions between proteins and Cd2+ may play a key role in mediating its inhibitory effect on the proteins (S. cerevisiae RFC, 28 cysteines/pentamer; S. cerevisiae Msh2-Msh6, 27/dimer; E. coli γ complex, 27/pentamer; E. coli MutL, 14/dimer; E. coli MutS, 12/dimer; and T. aquaticus MutS, 2/dimer).
Figure 4.
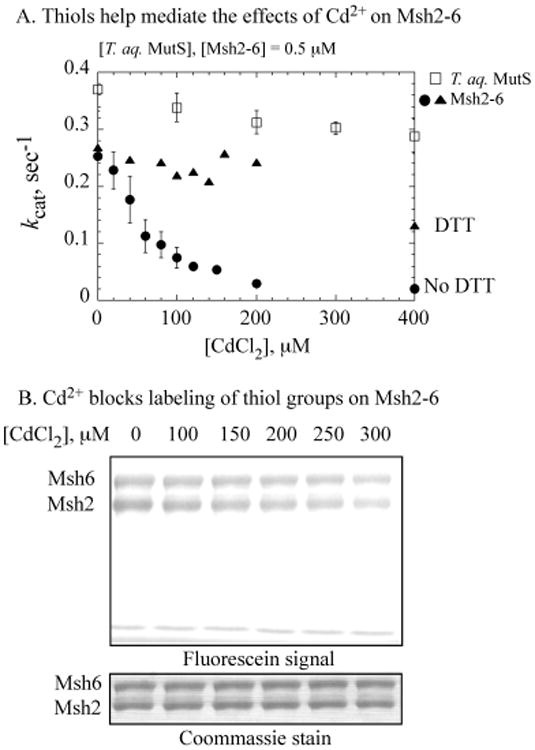
Thiol groups are involved in cadmium-mediated inhibition of Msh2-Msh6. (A) Cd2+ has a relatively minor effect on T. aquaticus MutS (□) as compared with S. cerevisiae Msh2-Msh6 ATPase (●). The addition of reduced DTT to the reaction protects Msh2-Msh6 from Cd2+-mediated inhibition (▴). (B) Cd2+ binding blocks reaction between cysteines on Msh2-Msh6 and fluorescein-5-maleimide. SDS-PAGE analysis shows reduction in fluorescein-labeled Msh2-Msh6 with increasing CdCl2 to <30% labeled protein at 300 μM CdCl2; Coommassie staining of the same gel shows equivalent amounts of protein in each reaction.
Discussion
Multiple Targets of Cadmium-Mediated Toxicity
The potent toxic effects of cadmium, including disruption of DNA metabolic reactions leading to mutations and chromosomal aberrations, make it a group I carcinogen, classified as a hazard to humans by the International Agency for Research on Cancer (1,2). Several targets of Cd2+ have been identified, and various mechanisms have been proposed in the literature to account for Cd2+-mediated toxicity. Cd2+ does not participate directly in redox reactions under physiological conditions, but it induces reactive oxygen species, likely via inhibition of antioxidant enzymes, and can thus promote nucleic acid, lipid, and protein damage (27). In favor of a more direct mechanism, there is evidence for Cd2+ replacing Ca2+ or Zn2+ in metallo-proteins and interfering with Ca2+ transport (28) and Ca2+-dependent signaling (29) or the function of Zn2+-dependent enzymes and Zn2+ finger-containing transcription factors (30). Other examples illustrating the broad spectrum of cadmium toxicity include altered expression of transcription factors (c-fos, c-jun, and c-myc proto-oncogenes) and translation factors (initiation factor 3 and elongation factor 1-δ) (1). Cd2+ can also disrupt extracellular processes such as cell–cell adhesion by interfering with the function of E-cadherin (31). Importantly, Cd2+ is known to disrupt the activities of tumor suppressor p53 (32), which would significantly increase the risk of carcinogenesis.
It has become increasingly apparent in recent years that Cd2+ targets a host of DNA repair mechanisms as well, including base excision repair (BER), nucleotide excision repair (NER), and MMR (3). In BER, for example, Cd2+ suppresses formamidopyrimidine DNA glycosylase (33), which catalyzes removal of damaged bases, as well as 8-oxo-dGTPase (34), which hydrolyzes 8-oxo-dGTP to 8-oxo-dGMP, minimizing its misincorporation into DNA. Other reported BER targets include AP endonuclease (35), DNA polymerase β (36), and DNA ligase (37). In NER, the DNA damage recognition protein XPA is inactivated by Cd2+ (33), and other key NER proteins, including TFIIH and TFIIA, and proteins responsible for repair DNA synthesis, appear to be affected as well (3). More recently, Cd2+ was found to inhibit the MMR system (4). Thus far, only the eukaryotic mismatch recognition protein Msh2-Msh6 has been identified clearly as a target of Cd2+ (7, 8). Together, these studies highlight the marked destructive impact of cadmium on critical processes responsible for DNA damage response and repair and help to explain the high level of mutagenesis, genome instability, and cancer risk associated with exposure to this toxin.
The brief overview of known Cd2+ targets highlights the fact that this metal toxin can interfere with the function of a wide variety of proteins, and ongoing efforts in several laboratories are aimed at identifying the corresponding mechanisms of inhibition. Cd2+ may bind specific site(s) on a protein and disrupt its function, as appears to be the case for Cd2+ binding to the Zn2+ finger motif in XPA (38). However, most of the proteins identified as Cd2+ targets are not canonical metal-binding proteins, and the exact mechanisms by which Cd2+ alters their structure and/or function remain unknown. This is also true in the case of Msh2-Msh6, where it has been speculated that Cd2+ may directly bind a critical residue or compete with Mg2+ in the ATPase site or bind a hypothetical Zn2+ finger motif, thereby disrupting its function in DNA repair (7,8).
Model Mechanism for Nonspecific Cadmium Binding-Mediated Disruption of Protein Structure and Function
In an effort to determine the mechanism by which cadmium inhibits MMR, we quantified Cd2+ binding to S. cerevisiae Msh2-Msh6 and assessed its effects on protein conformation and activity. In parallel, we also examined a prokaryotic homologue, T. aquaticus MutS, with the goal of identifying possible evolutionarily conserved features of the inhibition mechanism. Initial DNA binding and ATPase data indicated that inhibition requires high concentrations of Cd2+ relative to Msh2-Msh6 (Figure 1). Direct measurement of Cd2+ binding showed that the interaction saturates at 170–180 Cd2+ ions per Msh2-Msh6 (Figure 2A). A high ratio of Cd2+: Msh2-Msh6 was also found necessary for alteration of Msh2-Msh6 structure and activity. Isotherms of intrinsic Msh2-Msh6 fluorescence and ATPase activity from titrations with CdCl2 showed increasing sigmoidal character at higher protein concentrations, consistent with Cd2+ ions binding to multiple sites on Msh2-Msh6 (Figure 2B, C). Global analysis of all of the data yielded good fits to a minimal model, in which binding of multiple Cd2+ ions to Msh2-Msh6 is followed by change in protein conformation associated with loss of function. The results show that a strikingly high stoichiometry of about 100 Cd2+ ions (S = 107) is required to bind Msh2-Msh6 with low micromolar affinity to induce loss of function (apparent KD = 0.5 μM).
Nonspecific Interactions between Proteins and Cadmium
These findings raise interesting questions regarding the mechanism of cadmium-mediated inhibition; for example, on the nature of the Cd2+ binding sites on Msh2-Msh6 and, more importantly, the likelihood that an inhibition mechanism that involves Cd2+ binding Msh2-Msh6 at a ratio of about 100:1 is specific to this particular protein.
Several studies have shown that free sulfhydryl groups on cysteine residues are good ligands of Cd2+ ions, since cadmium, a soft acid, interacts strongly with soft donor atoms like sulfur (39). Other known biological ligands of Cd2+ ions include the methylsulfanyl group on methionine, amino, and carboxyl groups, as well as carbonyl groups in the peptide bond (40). We find that Cd2+blocks the reaction between fluorescein-5-maleimide and cysteines on Msh2-Msh6 (Figure 4), consistent with a recent report of thiol involvement in the Cd2+-Msh2-Msh6 interaction; the authors found that addition of free cysteine to the reaction protects Msh2-Msh6 from Cd2+, as does histidine to slightly lesser extent (8). In addition to 27 cysteines, S. cerevisiae Msh2-Msh6 has 57 methionine and 47 histidine residues. Thus, a variety of Cd2+–Msh2-Msh6 complexes are possible, with coordination numbers ranging from 2 to 8. Ligands in solution may also be involved, as illustrated by association of an excessive number of adenosine 5′-3-O-(thio)-triphosphate (ATPγS) molecules with Msh2-Msh6 in the presence of CdCl2 (Supporting Information, Figure S1B). At 200 μM CdCl2, the ratio is 13 ATPγS per Msh2-Msh6, which is remarkable given that the dimer has only two specific binding sites for nucleotides (12). This phenomenon can be explained by the participation of ATPγS molecules in multiple Cd2+ coordination complexes formed on Msh2-Msh6. Given the number of ions bound and the broad variety of ligands involved, binding of Cd2+ to Msh2-Msh6 has to be defined as a nonspecific interaction. Similar nonspecific interactions with Cd2+ have been reported for the metal-binding protein metallothionein (MT), which is best known for chelating seven Cd2+ ions with low nanomolar affinity via multiple sulfur atoms; however, the binding does not saturate even at 20 mol equiv of Cd2+ ions per MT (41).
Despite their nonspecific nature, the Cd2+–Msh2-Msh6 interactions are characterized by fairly high affinity (apparent KD = 0.5 μM), suggesting that other proteins could be susceptible to this type of Cd2+ inhibition mechanism. Consistent with this hypothesis, higher concentrations of other proteins required correspondingly higher Cd2+ concentrations for inactivation (Figure 3). T. aquaticus MutS, which has only one cysteine per monomer, presented an interesting case as it binds multiple Cd2+ ions, but its ATPase activity is relatively resistant to Cd2+-mediated inactivation (Figures 2A and 4A). It is possible that nonspecific Cd2+ coordination complexes that include one or more sulfur atoms (as terminal or bridging ligands) play a special role in Cd2+-mediated alteration of protein structure and function. Another possible reason for the differential effect of cadmium on T. aquaticus MutS is that it is a thermostable protein and may thus be more resistant to conformational changes on nonspecific Cd2+ binding, as compared with Msh2-Msh6 and the other proteins tested in this study. This hypothesis is consistent with the observation that at very high CdCl2 concentrations, Cd2+ binding-mediated changes in Msh2-Msh6 conformation eventually lead to protein aggregation over time (detected as increasing turbidity at 630 nm), whereas in the case of MutS, no such structural perturbation can be detected even at 2 mM CdCl2 (Supporting Information, Figure S2).
Stoichiometry and affinity constants defining Cd2+ interactions have been reported for specific metal-binding proteins, such as MTs, as well as Cd2+-binding peptides and small molecules, but not for a majority of the proteins reported as targets of Cd2+ toxicity. It is therefore difficult to determine from past in vitro studies whether these proteins are inhibited by a nonspecific mechanism similar toMsh2-Msh6. Moreover, in many cases, the data are generated from reactions containing DTT, which may or may not be in reduced form, further complicating interpretation (7). According to most studies, micromolar concentrations of Cd2+ are sufficient to inactivate the protein in question, although the reported concentration range can vary widely, even for closely related proteins. For example, we can estimate a K1/2 ∼ 5 μM from a study of Cd2+-mediated inhibition of human 8-oxoguanine-DNA glycosylase (estimated hOGG1 concentration = 1 nM) (42), and a K1/2 > 100 μM for mouse 8-oxoguanine-DNA glycosylase (reported mOGG1 concentration = 0.5–2 nM) (43). In case of in vitro studies using cell-free extracts, estimates of stoichiometry or binding constants are further complicated by the presence of Cd2+ binding molecules other than the protein in question. For example, Cd2+ apparently inhibits the cleavage activity of purified N-methylpurine-DNA glycosylase (reported MPG concentration = 0.35 μM) with a K1/2 ∼ 100 μM, whereas in a Hela cell-free extract, which likely has lower amounts of MPG, the K1/2 is ∼ 250 μM (44). Notably, all of these proteins have multiple cysteine, methionine, and histidine residues, and in light of our results with Msh2-Msh6 and other ATPases, the reported range of Cd2+ concentrations is consistent with a nonspecific binding and inhibition mechanism. In the case of in vivo studies, the addition of low micromolar concentrations of Cd2+ to the growth medium can lower the activity of the protein under investigation. However, the concentration of cadmium inside cells can vary widely depending on uptake by transporters and interaction with other ligands (45). Thus, it is difficult to ascertain inhibition constants based on in vivo data, and the nonspecific Cd2+ binding mechanism proposed here could well be applicable, and may even be predominant.
Cadmium can clearly have a disastrous impact on cellular processes by affecting many different proteins. It has been proposed that MMR is a specific target of cadmium toxicity, since exposure to CdCl2 leads to high mutability in S. cerevisiae, while DNA recombination levels and cell viability remain relatively stable (4). Our investigation indicates that Msh2-Msh6 is not affected specifically by Cd2+, but is instead subject to a generic mechanism of inhibition. Results from both in vivo and in vitro analysis are likely influenced by variation in the stoichiometry and affinity of nonspecific Cd2+ binding sites on different proteins, the ability of proteins to adopt “inactive” conformations on Cd2+ binding, the concentration of proteins in the cell or reaction, the status of redundant or compensatory protective mechanisms against DNA damage and cytotoxicity, and the status of protective mechanisms against metal ion toxicity. To unequivocally identify a specific target of cadmium toxicity in vitro, which is distinguished by a selective Cd2+ binding and inhibition mechanism, it is important to assess the Cd2+ binding stoichiometry and affinity of the protein, its amino acid composition, its propensity for conformational changes, as well as the influence of other ligands in the reaction, such as DTT. To identify a specific target of cadmium toxicity in vivo, it is important to assess the extent to which Cd2+ accumulates inside cells and its impact on the cellular redox status, as well as compare its effects on unrelated proteins under the same experimental conditions, particularly if they correlate with protein concentrations. These are all pertinent indicators of the type of mechanism underlying the impact of cadmium on a biological system and, therefore, inform development of strategies to mitigate its toxicity.
Generic Antidotes for a Generic Poison
The primary mechanism of cadmium detoxification in various organisms is chelation by thiol-containing molecules such as glutathione (GSH) and metalloproteins (MT) (46–48). Cd2+ interacts predominantly with the sulfhydryl and amino groups on GSH and can make weak contacts with the glutamyl and glycyl carboxylic acid groups. Dissociation constants for Cd2+-GSH interaction are in the low micromolar range(47,49), which implies that GSH would be ineffective as a competitor against proteins that bind Cd2+ with similar affinity. However, the GSH concentration can range from 1 to 10 mM inside cells, and the ratio of reduced GSH to oxidized GSSG in the cytosol is maintained at 100:1. Thus, high amounts of GSH are available to mop up Cd2+ and protect nonspecific Cd2+-binding proteins from its toxic effects. Moreover, exposure to Cd2+ results in strong stimulation of cysteine and GSH biosynthesis, consistent with the hypothesis that GSH is a key part of the defense mechanism against Cd2+ toxicity (46). MTs are ubiquitous transition metal-binding proteins characterized by high cysteine content and recognized for their role in homeostasis of essential metals and protection against toxic metals (48). Mammalian MTs are reported to bind up to seven Zn2+ or Cd2+ ions in metal–thiolate clusters with dissociation constants in the low nanomolar range (50). Such efficient interaction with Cd2+ would allow MTs to compete effectively with nonspecific Cd +-binding proteins and thus block its toxic effects, at least until excessive cadmium exposure and accumulation overwhelms these defense mechanisms.
In summary, our results confirm that Cd2+ binding inactivates Msh2-Msh6, which in turn would disrupt MMR and lead to a high mutator phenotype and increased risk of carcinogenesis. We have discovered that the mechanism of inhibition does not involve binding of Cd2+ to a particular site on Msh2-Msh6 but rather binding of a large number of Cd2+ ions to multiple sites, which leads to changes in protein conformation and loss of function. Not surprisingly, this inhibition mechanism is not unique to Msh2-Msh6, and Cd2+ inactivates several unrelated proteins in a similar manner. These findings highlight the critical role that GSH and MT play in shielding cellular processes against cadmium toxicity, by competing effectively against nonspecific Cd2+-binding proteins and thus sparing them from catastrophic disruption of structure and function.
Supplementary Material
Appendix
The model of Msh2-Msh6 inhibition by cadmium uses the following assumptions:
– The Msh2-Msh6 complex has S binding sites for Cd2+ with equal affinity KA.
– Binding of Cd2+ to each site can occur independently.
– After all S sites are occupied, unfolding can occur with the equilibrium constant KC.
– Unfolding occurs in one step, which renders the Msh2-Msh6 complex enzymatically inactive and changes its intrinsic tryptophan fluorescence.
– Properties of folded species are not affected by the number of bound Cd2+.
The fraction of unfolded Msh2-Msh6 complex [M′LS]/MT is calculated using concentrations of all intermediate complexes. An intermediate with i Cd2+ bound is formed in the reaction:
| (S1) |
Because multiple empty sites may be present in the left-hand-side complex and the ligand can bind to any one of them, the apparent equilibrium constant for complex formation is directly proportional to S−i + 1, the number of unoccupied sites. The reverse reaction can occur by dissociation of any of i ligands from the right-hand-side complex. Therefore, at equilibrium:
| (S2) |
Concentrations of intermediate complexes:
| (S3) |
where binomial coefficient
Total concentration of the complex:
| (S4) |
Total concentration of ligand:
| (S5) |
Combining eqs S4 and S5 produces an expression for ligand binding density, from which the variable [M] can be eliminated.
| (S6) |
Equation S6 has one unknown variable, [L], and can be solved numerically. However, to avoid computational overflow errors, substitute
| (S7) |
and
| (S8) |
Equation S8 was numerically solved for x using the fzero MATLAB function. The fraction of unfolded complex was calculated as
| (S9) |
The model, data and results of the analysis are available at http://gfit.sourceforge.net.
Footnotes
This work was supported by a grant from the NSF (MCB 0448379). F.N.B. received support from the Barry M. Goldwater Scholarship and Excellence in Education Foundation.
Supporting Information Available: Data on ATPγS binding to Msh2-Msh6 and protein aggregation. This material is available free of charge via the Internet at http://pubs.acs.org.
Abbreviations: MMR, mismatch repair; Msh, Mut S homologue; ATP, adenosine 5′-triphosphate; ATPγS, adenosine 5′-3-O-(thio)-triphosphate; nt, nucleotide; MDCC, N-[2(1-maleimidyl) ethyl]-7-(diethylamino)coumarin-3-carboxamide; PBP, phosphate binding protein; Pi, phosphate; GSH, glutathione; MT, metallothionein.
References
- 1.Beyersmann D, Hartwig A. Carcinogenic metal compounds: Recent insight into molecular and cellular mechanisms. Arch Toxicol. 2008;82:493–512. doi: 10.1007/s00204-008-0313-y. [DOI] [PubMed] [Google Scholar]
- 2.Waalkes MP. Cadmium carcinogenesis. Mutat Res. 2003;533:107–120. doi: 10.1016/j.mrfmmm.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 3.Giaginis C, Gatzidou E, Theocharis S. DNA repair systems as targets of cadmium toxicity. Toxicol Appl Pharmacol. 2006;213:282–290. doi: 10.1016/j.taap.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 4.Jin YH, Clark AB, Slebos RJ, Al-Refai H, Taylor JA, Kunkel TA, Resnick MA, Gordenin DA. Cadmium is a mutagen that acts by inhibiting mismatch repair. Nat Genet. 2003;34:326–329. doi: 10.1038/ng1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsieh P, Yamane K. DNA mismatch repair: Molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129:391–407. doi: 10.1016/j.mad.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: Functions and mechanisms. Chem Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 7.Clark AB, Kunkel TA. Cadmium inhibits the functions of eukaryotic MutS complexes. J Biol Chem. 2004;279:53903–53906. doi: 10.1074/jbc.C400495200. [DOI] [PubMed] [Google Scholar]
- 8.Banerjee S, Flores-Rozas H. Cadmium inhibits mismatch repair by blocking the ATPase activity of the MSH2-MSH6 complex. Nucleic Acids Res. 2005;33:1410–1419. doi: 10.1093/nar/gki291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kadyrov FA, Holmes SF, Arana ME, Lukianova OA, O'Donnell M, Kunkel TA, Modrich P. Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. J Biol Chem. 2007;282:37181–37190. doi: 10.1074/jbc.M707617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Xia YL, Jiang Y, Yan XP. Extracting stoichiometry, thermodynamics, and kinetics for the interaction of DNA with cadmium ion by capillary electrophoresis on-line coupled with electrothermal atomic absorption spectrometry. Electrophoresis. 2008;29:1173–1179. doi: 10.1002/elps.200700674. [DOI] [PubMed] [Google Scholar]
- 11.Antony E, Hingorani MM. Asymmetric ATP binding and hydrolysis activity of the Thermus aquaticus MutS dimer is key to modulation of its interactions with mismatched DNA. Biochemistry. 2004;43:13115–13128. doi: 10.1021/bi049010t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antony E, Hingorani MM. Mismatch recognition-coupled stabilization of Msh2-Msh6 in an ATP-bound state at the initiation of DNA repair. Biochemistry. 2003;42:7682–7693. doi: 10.1021/bi034602h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorman J, Chowdhury A, Surtees JA, Shimada J, Reichman DR, Alani E, Greene EC. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol Cell. 2007;28:359–370. doi: 10.1016/j.molcel.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS. Structure of the human MutSalpha DNA lesion recognition complex. Mol Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 15.Obmolova G, Ban C, Hsieh P, Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature (London) 2000;407:703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 16.Lamers MH, Perrakis A, Enzlin JH, Winterwerp HH, de Wind N, Sixma TK. The crystal structure of DNA mismatch repair protein MutS binding to a G × T mismatch. Nature (London) 2000;407:711–717. doi: 10.1038/35037523. [DOI] [PubMed] [Google Scholar]
- 17.Tessmer I, Yang Y, Zhai J, Du C, Hsieh P, Hingorani MM, Erie DA. Mechanism of MutS searching for DNA mismatches and signaling repair. J Biol Chem. 2008;283:36646–36654. doi: 10.1074/jbc.M805712200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Acharya S, Foster PL, Brooks P, Fishel R. The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol Cell. 2003;12:233–246. doi: 10.1016/s1097-2765(03)00219-3. [DOI] [PubMed] [Google Scholar]
- 19.Mazur DJ, Mendillo ML, Kolodner RD. Inhibition of Msh6 ATPase activity by mispaired DNA induces a Msh2 (ATP)-Msh6(ATP) state capable of hydrolysis-independent movement along DNA. Mol Cell. 2006;22:39–49. doi: 10.1016/j.molcel.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 20.Mendillo ML, Mazur DJ, Kolodner RD. Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end-blocking system. J Biol Chem. 2005;280:22245–22257. doi: 10.1074/jbc.M407545200. [DOI] [PubMed] [Google Scholar]
- 21.Selmane T, Schofield MJ, Nayak S, Du C, Hsieh P. Formation of a DNA mismatch repair complex mediated by ATP. J Mol Biol. 2003;334:949–965. doi: 10.1016/j.jmb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Finkelstein J, Antony E, Hingorani MM, O'Donnell M. Overproduction and analysis of eukaryotic multiproteincomplexes in Escherichia coli using a dual-vector strategy. Anal Biochem. 2003;319:78–87. doi: 10.1016/s0003-2697(03)00273-2. [DOI] [PubMed] [Google Scholar]
- 23.Brune M, Hunter JL, Howell SA, Martin SR, Hazlett TL, Corrie JE, Webb MR. Mechanism of inorganic phosphate interaction with phosphate binding protein from Escherichia coli. Biochemistry. 1998;37:10370–10380. doi: 10.1021/bi9804277. [DOI] [PubMed] [Google Scholar]
- 24.Duguid JG, Bloomfield VA, Benevides JM, Thomas GJ., Jr Raman spectroscopy of DNA-metal complexes, II. The thermal denaturation of DNA in the presence of Sr2+, Ba2 +, Mg2+, Ca2+, Mn2+, Co2+, Ni2+, and Cd2+ Biophys J. 1995;69:2623–2641. doi: 10.1016/S0006-3495(95)80133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weiss JN. The Hill equation revisited: Uses and misuses. FASEB J. 1997;11:835–841. [PubMed] [Google Scholar]
- 26.Levin MK, Hingorani MH, Holmes RM, Patel SS, Carson JH. Model-based global analysis of heterogeneous experimental data using gfit. In: Maly IV, editor. Methods in Molecular Biology/Systems Biology. Humana Press, Inc; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 28.Bressler JP, Olivi L, Cheong JH, Kim Y, Maerten A, Bannon D. Metal transporters in intestine and brain: Their involvement in metal-associated neurotoxicities. Hum Exp Toxicol. 2007;26:221–229. doi: 10.1177/0960327107070573. [DOI] [PubMed] [Google Scholar]
- 29.Misra UK, Gawdi G, Akabani G, Pizzo SV. Cadmium-induced DNA synthesis and cell proliferation in macrophages: The role of intracellular calcium and signal transduction mechanisms. Cell Signalling. 2002;14:327–340. doi: 10.1016/s0898-6568(01)00268-6. [DOI] [PubMed] [Google Scholar]
- 30.Witkiewicz-Kucharczyk A, Bal W. Damage of zinc fingers in DNA repair proteins, a novel molecular mechanism in carcinogenesis. Toxicol Lett. 2006;162:29–42. doi: 10.1016/j.toxlet.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 31.Prozialeck WC, Lamar PC, Lynch SM. Cadmium alters the localization of N-cadherin, E-cadherin, and beta-catenin in the proximal tubule epithelium. Toxicol Appl Pharmacol. 2003;189:180–195. doi: 10.1016/s0041-008x(03)00130-3. [DOI] [PubMed] [Google Scholar]
- 32.Meplan C, Mann K, Hainaut P. Cadmium induces conformational modifications of wild-type p53 and suppresses p53 response to DNA damage in cultured cells. J Biol Chem. 1999;274:31663–31670. doi: 10.1074/jbc.274.44.31663. [DOI] [PubMed] [Google Scholar]
- 33.Asmuss M, Mullenders LH, Eker A, Hartwig A. Differential effects of toxic metal compounds on the activities of Fpg and XPA, two zinc finger proteins involved in DNA repair. Carcinogenesis. 2000;21:2097–2104. doi: 10.1093/carcin/21.11.2097. [DOI] [PubMed] [Google Scholar]
- 34.Kasprzak KS, Bialkowski K. Inhibition of antimutagenic enzymes, 8-oxo-dGTPases, by carcinogenic metals. Recent developments J Inorg Biochem. 2000;79:231–236. doi: 10.1016/s0162-0134(99)00240-8. [DOI] [PubMed] [Google Scholar]
- 35.McNeill DR, Narayana A, Wong HK, Wilson DM., 3rd Inhibition of Ape1 nuclease activity by lead, iron, andcadmium. Environ Health Perspect. 2004;112:799–804. doi: 10.1289/ehp.7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Popenoe EA, Schmaeler MA. Interaction of human DNA polymerase beta with ions of copper, lead, and cadmium. Arch Biochem Biophys. 1979;196:109–120. doi: 10.1016/0003-9861(79)90557-5. [DOI] [PubMed] [Google Scholar]
- 37.Yang SW, Becker FF, Chan JY. Inhibition of human DNA ligase I activity by zinc and cadmium and the fidelity of ligation. Environ Mol Mutagen. 1996;28:19–25. doi: 10.1002/(SICI)1098-2280(1996)28:1<19::AID-EM5>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 38.Mustra DJ, Warren AJ, Wilcox DE, Hamilton JW. Preferential binding of human XPA to the mitomycin C-DNA interstrand crosslink and modulation by arsenic and cadmium. Chem Biol Interact. 2007;168:159–168. doi: 10.1016/j.cbi.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 39.Andersen O. Chelation of cadmium. Environ Health Perspect. 1984;54:249–266. doi: 10.1289/ehp.8454249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castagnetto JM, Hennessy SW, Roberts VA, Getzoff ED, Tainer JA, Pique ME. MDB: The Metalloprotein Database and Browser at The Scripps Research Institute. Nucleic Acids Res. 2002;30:379–382. doi: 10.1093/nar/30.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cols N, Romero-Isart N, Capdevila M, Oliva B, Gonzalez-Duarte P, Gonzalez-Duarte R, Atrian S. Binding of excess cadmium(II) to Cd7-metallothionein from recombinant mouse Zn7-metallothionein 1. UV-VIS absorption and circular dichroism studies and theoretical location approach by surface accessibility analysis. J Inorg Biochem. 1997;68:157–166. doi: 10.1016/s0162-0134(97)00085-8. [DOI] [PubMed] [Google Scholar]
- 42.Bravard A, Vacher M, Gouget B, Coutant A, de Boisferon FH, Marsin S, Chevillard S, Radicella JP. Redox regulation of human OGG1 activity in response to cellular oxidative stress. Mol Cell Biol. 2006;26:7430–7436. doi: 10.1128/MCB.00624-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zharkov DO, Rosenquist TA. Inactivation of mammalian 8-oxoguanine-DNA glycosylase by cadmium(II): Implications for cadmium genotoxicity. DNA Repair (Amsterdam) 2002;1:661–670. doi: 10.1016/s1568-7864(02)00074-5. [DOI] [PubMed] [Google Scholar]
- 44.Wang P, Guliaev AB, Hang B. Metal inhibition of human N-methylpurine-DNA glycosylase activity in base excision repair. Toxicol Lett. 2006;166:237–247. doi: 10.1016/j.toxlet.2006.06.647. [DOI] [PubMed] [Google Scholar]
- 45.Bridges CC, Zalups RK. Molecular and ionic mimicry and the transport of toxic metals. Toxicol Appl Pharmacol. 2005;204:274–308. doi: 10.1016/j.taap.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vido K, Spector D, Lagniel G, Lopez S, Toledano MB, Labarre J. A proteome analysis of the cadmium response in Saccharomyces cerevisiae. J Biol Chem. 2001;276:8469–8474. doi: 10.1074/jbc.M008708200. [DOI] [PubMed] [Google Scholar]
- 47.Leverrier P, Montigny C, Garrigos M, Champeil P. Metal binding to ligands: cadmium complexes with glutathione revisited. Anal Biochem. 2007;371:215–228. doi: 10.1016/j.ab.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 48.Miles AT, Hawksworth GM, Beattie JH, Rodilla V. Induction, regulation, degradation, and biological significance of mammalian metallothioneins. Crit Rev Biochem Mol Biol. 2000;35:35–70. doi: 10.1080/10409230091169168. [DOI] [PubMed] [Google Scholar]
- 49.Kadima W, Rabenstein DL. Nuclear magnetic resonance studies of the solution chemistry of metal complexes. 26. Mixed ligand complexes of cadmium, nitrilotriacetic acid, glutathione, and related ligands. J Inorg Biochem. 1990;38:277–288. doi: 10.1016/0162-0134(90)80003-g. [DOI] [PubMed] [Google Scholar]
- 50.Erk M, Raspor B. Evaluation of cadmium-metallothionein stability constants based on voltammetric measurements. Anal Chim Acta. 1998;360:189–194. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.