

Abstract
Hsp104, a protein disaggregase from yeast, can be engineered and potentiated to counter TDP-43, FUS, or α-synuclein misfolding and toxicity implicated in neurodegenerative disease. Here, we reveal that extraordinarily disparate mutations potentiate Hsp104. Remarkably, diverse single missense mutations at 20 different positions interspersed throughout the middle domain (MD) and small domain of nucleotide-binding domain 1 (NBD1) confer a therapeutic gain of Hsp104 function. Moreover, potentiation emerges from deletion of MD helix 3 or 4 or via synergistic missense mutations in the MD distal loop and helix 4. We define the most critical aspect of Hsp104 potentiation as enhanced disaggregase activity in the absence of Hsp70 and Hsp40. We suggest that potentiation likely stems from a loss of a fragilely constrained autoinhibited state that enables precise spatiotemporal regulation of disaggregase activity.
Graphical abstract
Hsp104, a hexameric AAA+ protein from yeast, catalyzes the construction and deconstruction of yeast prions.1–3 Hsp104 is also essential in the reactivation of disordered aggregates that accumulate upon exposure to environmental stress.4–7 Because yeasts have harnessed infectious amyloids (prions) for adaptive purposes,8 these tight amyloid-regulatory pathways could potentially be reformulated to counter protein misfolding implicated in neurodegenerative disease in humans.9–11 Protein misfolding is implicated in numerous neurodegenerative disorders including amyotrophic lateral sclerosis (ALS) and Parkinson’s disease (PD).12,13 In ALS, certain subsets of patients display accumulations of misfolded TDP-43 or FUS.14 In PD patients, α-synuclein (α-syn) forms highly toxic prefibrillar oligomers and amyloid fibrils that accumulate in Lewy bodies.12 Treatments for nearly all protein-misfolding disorders remain palliative, and therapeutics that target the underlying causes of these diseases are needed.9 Accumulations of α-syn amyloid fibrils, as well as TDP-43 and FUS aggregates, are widely considered intractable. No therapeutics are available to eliminate these structures or their precursors. The genes encoding TDP-43 and FUS expression are essential, and thus therapies that reactivate these proteins to their native fold rather than degrading or downregulating them are needed.15 Due to the complexities of protein misfolding, it remains unclear if small molecule therapeutics can effectively target any of these disorders. Thus, it is crucial to explore alternative therapeutic strategies, such as the rewiring of proteostasis networks by engineering protein disaggregases with enhanced activity.9
Hsp104 has only limited ability to recover natively folded TDP-43, FUS, or α-syn from aggregates, preamyloid oligomers, and amyloid fibers, substrates that it does not ordinarily encounter.9,16,17 Thus, we engineered potentiated Hsp104 variants to counter TDP-43, FUS, and α-syn misfolding.17–20 Using random mutagenesis, we generated large libraries of Hsp104 variants that we screened for suppression of proteotoxicity in yeast.17,18 We isolated a series of potentiated Hsp104 variants that suppress TDP-43, FUS, and α-syn aggregation and toxicity, while also restoring nuclear localization of TDP-43 and plasma membrane localization of α-syn in yeast.17 Potentiated Hsp104 variants are the first disaggregases, or even chaperones, engineered to optimize proteostasis against neurodegenerative disease. These variants also suppress the toxicity and aggregation associated with mutant versions of TDP-43, FUS, and α-syn linked to more severe disease phenotypes.21 We have also demonstrated that potentiated Hsp104 variants suppress dopaminergic neurodegeneration in a C. elegans model of PD.17 Surprisingly, at certain positions, missense mutation to nearly any amino acid potentiates Hsp104.17 To the best of our knowledge, generic mutation of specific residues to any amino acid leading to a therapeutic gain-of-function is unprecedented.9,17 Thus, we sought to better understand the extent to which the Hsp104 middle domain (MD) can be modulated to confer potentiation.
Here, we have employed a modified yeast-based screening assay to isolate a broad series of potentiated Hsp104 variants. We have identified many new variants, and the scope of potentiating mutations is broad and unanticipated. We have genetically mapped the positions at which missense mutations confer Hsp104 potentiation and find that they comprise at least ~14% of the residues in the MD. However, potentiation is not conferred by any mutation in the MD, as we also report a similar number of missense mutations that do not confer potentiation. Using a series of Hsp104 MD deletion constructs, we have also elucidated the structural requirements for Hsp104 potentiation. To fine-tune Hsp104 variants and enhance their substrate or conformer specificity, it is crucial to accurately understand the mechanistic and structural basis for Hsp104 potentiation. Our studies provide new insight into the basis for Hsp104 potentiation and raise new questions as to why Hsp104 might have evolved to maintain a fragilely constrained autoinhibited state.
RESULTS AND DISCUSSION
Missense Mutations throughout the Middle Domain Potentiate Hsp104
We have previously reported the isolation of a series of potentiated Hsp104 MD variants that suppress the toxicity of TDP-43, FUS, and α-syn.9,17,21 These mutations were located in MD helix 1, the distal loop between MD helix 1 and 2, and helix 3, as well as the small domain of NBD1 (Supporting Information Table 1).9,17,21 Using pure-protein biochemistry, we determined that these variants were all potentiated by similar mechanistic principles and were typically enhanced in a nonspecific manner, suppressing the aggregation and toxicity of TDP-43, FUS, and α-syn.17 The potentiating mutations are seemingly dissimilar, and we could determine no unifying features to link them. We hypothesized that additional potentiating mutations likely exist and sought to more comprehensively identify potentiated Hsp104 variants.
We generated an Hsp104 MD library using domain-specific error-prone PCR.17 We coexpressed this library of variants with TDP-43 in Δhsp104 yeast and isolated toxicity suppressors by plating the yeast on galactose-supplemented media. Here, a galactose-inducible promoter controls TDP-43 and Hsp104 expression. Expression was performed at 34 °C, which we predicted would disfavor selection of the previously isolated variants due to their mild toxicity at temperatures of 34 °C and above.17,21 We uncovered numerous novel potentiating mutations in MD helix 2, as well as several in helix 4, two regions in which we had not identified potentiating mutations before17 (Figure 1A, Supporting Information Table 1). We confirmed that the potentiated Hsp104 variants do not alter the expression of TDP-43 and that the Hsp104 variants are expressed at similar or lower levels than Hsp104WT (Figure 1B).
Figure 1.

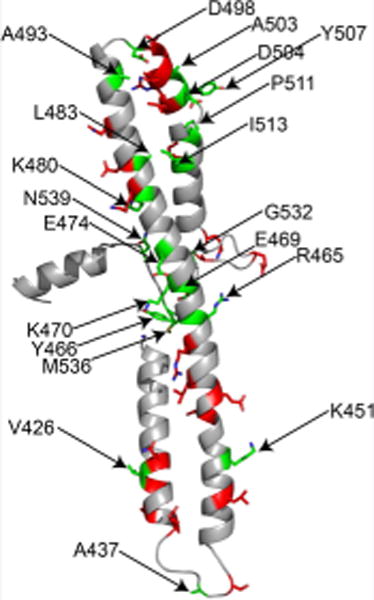
Hsp104 potentiation. Diverse missense mutations in disparate positions throughout the MD and small domain of NBD1 potentiate Hsp104. (A) Δhsp104 yeasts integrated with genes encoding TDP-43, FUS, or α-synuclein were transformed with the indicated Hsp104 missense mutants or control. Strains were serially diluted 5-fold and spotted on glucose (off) or galactose (on) media. (B) Selected strains from A were induced for 5 h, lysed, and immunoblotted. 3-Phosphoglycerate kinase (PGK) serves as a loading control. (C) Potentiating mutations are distributed throughout the MD of Hsp104. The MD (green, residues 411–538) is comprised of four helices and is inserted into nucleotide-binding domain 1 (NBD1, blue). Potentiating mutations shown in this figure are further described in this paper and our earlier work.17 (D) Homology model of the MD and a portion of the small domain of NBD1 of Hsp104, where side chains studied are shown as sticks. Residues where the tested mutations potentiate Hsp104 are shown in green and are numbered. Residues where the tested mutations do not potentiate Hsp104 are shown in red.
We also tested if the toxicity suppression by potentiated Hsp104 variants involves Sse1 or Sse2, the yeast homologues of Hsp110, which functions as a mammalian protein disaggregase.22 To eliminate this possibility, we confirmed that several variants retain their ability to suppress TDP-43 toxicity in Δsse1 and Δsse2 yeast strains (Figure S1). Thus, rescue of TDP-43 toxicity in yeast by potentiated Hsp104 variants does not require Sse1 in the same way that Hsp104 requires Sse1 to propagate [PSI+] and [URE3] in yeast.23–25
As with the majority of previously isolated potentiated Hsp104 variants,17 each of the novel potentiating variants also suppressed the toxicity of FUS and α-syn (Figure 1A). With the identification of these new mutations, we have now identified numerous missense mutations that confer potentiation in every helix of the MD as well as in the small domain of NBD1 (Figure 1C,D). The disparate locations and diversity of mutations that confer a therapeutic gain of Hsp104 function is remarkable and unanticipated.
We also isolated several colonies that harbored more than one Hsp104 mutation. Individual yeast cells can harbor multiple plasmids with the same selection marker. When multiple mutations are identified in a single colony, it is unclear which of the mutations are potentiating. Thus, we constructed each of the mutations as single missense mutants and retested them for suppression of TDP-43 toxicity. For nearly all of the colonies, we identified at least one potentiating mutation (Figure 1A). Many colonies harboring multiple point mutations only harbored a single activating mutation while the other comaintained mutations did not modify TDP-43, FUS, or α-syn toxicity (Supporting Information Table 2). Thus, it is important to note that Hsp104 is not potentiated by any mutation in the MD. Indeed, Hsp104 MD missense mutations can be isolated that are neutral or inhibitory.26
Curiously, we identified K470Q as an activating variant and K470R as a variant that does not modify toxicity (Figure 1A, Supporting Information Table 2). This finding was surprising because we previously found that conservative mutations such as alanine to glycine or valine potentiate Hsp104.17 Thus, at some MD positions (e.g., K470), potentiation requires a higher degree of side-chain modification, while at other positions (e.g., A503) virtually any side-chain alteration confers potentiation.
Missense Mutations in the MD Distal Loop and Helix 4 Synergize to Potentiate Hsp104
For each colony identified as a hit that harbored multiple missense mutations, we constructed each of the mutations individually and assessed them for TDP-43 toxicity suppression. With just one exception, each colony harboring multiple mutations contained at least one mutation that conferred potentiation. The exception was one colony that harbored three mutations: A430V, K514E, and N534I. None of these missense mutations conferred potentiation individually (Figure 2A, Supporting Information Tables 1 and 2). Therefore, we constructed and tested all four combinations of the three mutations to determine if multiple mutations were required in combination to confer potentiation. Surprisingly, Hsp104A430V–K514E and Hsp104A430V–N534I displayed potentiation, and Hsp104A430V–K514E–N534I displayed even greater activity, whereas the single mutations were inactive (Figure 2A,B). While these mutations do not potentiate Hsp104 independently, in combination they display robust synergy. We confirmed that the Hsp104 variants do not alter expression of TDP-43 and that the Hsp104 variants are expressed at similar or lower levels than Hsp104WT (Figure 2C). We hypothesize that each of these individual mutations might subtly perturb the MD, but insufficiently to relieve autoinhibition. However, in combination, these mutations relieve autoinhibition to potentiate Hsp104 and robustly suppress TDP-43 toxicity.
Figure 2.
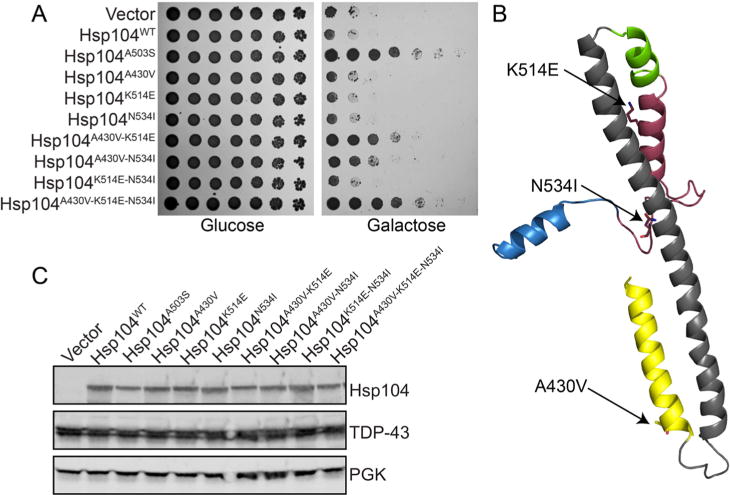
Synergy between mutations in the MD distal loop and helix 4. This synergy yields potentiated Hsp104 variants. (A) Δhsp104 yeasts integrated with pAG303GAL-TDP-43 were transformed with each of the indicated pAG416GAL-Hsp104 variants or vector control. Strains were serially diluted 5-fold and spotted on glucose (off) or galactose (on) media. (B) Homology model of the MD and a portion of the small domain of NBD1 of Hsp104. Side chains of the key residues are shown as sticks. (C) Selected strains from A were induced for 5 h, lysed, and immunoblotted. 3-Phosphoglycerate kinase (PGK) serves as a loading control.
Second Generation Hsp104 MD Variants Display Reduced Temperature Sensitivity
We predicted that by screening the Hsp104 library at elevated temperatures, this selective pressure might promote isolation of variants with a diminished temperature-sensitive growth phenotype. Hsp104 is not required for yeast growth at 37 °C (Figure 3).21,27 We also observed no noticeable difference in growth between Δhsp104 yeast overexpressing Hsp104 from an exogenous plasmid or vector alone at 30 or 37 °C (Figure 3).17,21 However, overexpression of potentiated Hsp104A503V results in a growth defect at 37 °C, likely due to promiscuous unfolding of modestly destabilized substrates at this temperature.17,21 Thus, we tested the novel variants for temperature-sensitivity by expressing them in Δhsp104 yeast at 30 and 37 °C. As a control, we compared growth to that of yeast expressing Hsp104A503S, a potentiated variant identified in earlier work as one of the least temperature-sensitive.17 When overexpressed at 30 °C, none of the variants, including Hsp104A503S, display a growth defect (Figure 3A). When overexpressed at 37 °C, Hsp104A503S displays a moderate growth defect (Figure 3A).17 Promisingly, many of the newly isolated variants display a diminished temperature-sensitive phenotype as compared to Hsp104A503S (Figure 3A). Hsp104R465G, Hsp104E469D, Hsp104K470Q, Hsp104E474V, and Hsp104G532S display substantially more robust growth than Hsp104A503S at 37 °C, though they still display mild temperature sensitivity relative to the controls (Figure 3A). These variants also maintain a robust level of toxicity suppression of the disease-associated substrates (Figure 1A). We also tested the synergistic mutations and found that Hsp104A430V–N534I displayed nearly no temperature-sensitive growth phenotype, growing at levels similar to the vector and Hsp104WT controls (Figure 3B). Hsp104A430V–N534I suppresses TDP-43 toxicity (Figure 2B), but less so than Hsp104A503S or the other potentiated synergistic variants. This finding suggests that diminished temperature-sensitivity incurs a cost of diminished overall protective activity against neurodegenerative disease proteins. Nonetheless, Hsp104A430V–N534I does buffer TDP-43 toxicity, indicating that potentiation is not always linked with a temperature-sensitive growth phenotype.
Figure 3.

Temperature sensitivity. Novel potentiated Hsp104 variants can display a diminished temperature-sensitive phenotype. (A) W303Δhsp104 yeasts were transformed with the indicated 416GAL-Hsp104 plasmid or empty vector control. Yeasts were grown to saturation in synthetic raffinose medium, serially diluted, and spotted onto SD-Ura or SGal-Ura media and incubated at 30 or 37 °C. Plates were analyzed after 2–3 days of growth. (B) Experiments were carried out as in A to test the putative synergistic variants.
Deletion of MD Helix 3 or 4 Confers Hsp104 Potentiation
We have isolated numerous potentiated Hsp104 variants with missense mutations dispersed throughout all four helices of the MD as well as the small domain of NBD1. It is perplexing that mutation at such disparate positions should activate the protein so profoundly. Thus, we were curious if deletion of entire helices or motifs, or perhaps even deletion of the entire MD, might also potentiate Hsp104. In the E. coli homologue of Hsp104, ClpB, deletion of motif 1 (helix 1 and a portion of helix 2), motif 2 (a portion of helix 2, helix 3, and helix 4), or the entire MD confers a hyperactive state, which is highly toxic to E. coli at regular growth temperatures.28,29 While Hsp104 and ClpB are often assumed to function by the same mechanism,30 we have established several key mechanistic differences between Hsp104 and ClpB.16,26 Thus, we constructed a comprehensive series of deletion constructs to elucidate the MD requirements for Hsp104 potentiation. We constructed Hsp104ΔMotif 1, Hsp104ΔMotif 2, and Hsp104ΔMD, which eliminate motif 1 (Δ412–459), motif 2 (Δ460–524), or the entire MD (Δ412–524), respectively (Figure 4A, right). For comparison, we also constructed Hsp104Δ411–531, which has been characterized in vitro.31 Plasmids harboring the Hsp104 variants were cotransformed in Δhsp104 yeast integrated with the TDP-43 gene. Hsp104ΔMotif 2 suppresses TDP-43 toxicity, while neither Hsp104ΔMotif 1, Hsp104ΔMD, nor Hsp104Δ411–531 suppresses TDP-43 toxicity (Figure 4A, left). This finding is in striking contrast to ClpB, in which deletion of either motif or the entire MD confers a hyperactive state.28,29,32 Thus, the requirements for Hsp104 potentiation are strikingly different from the requirements for generating hyperactive ClpB variants, indicating profound differences between Hsp104 and ClpB.
Figure 4.
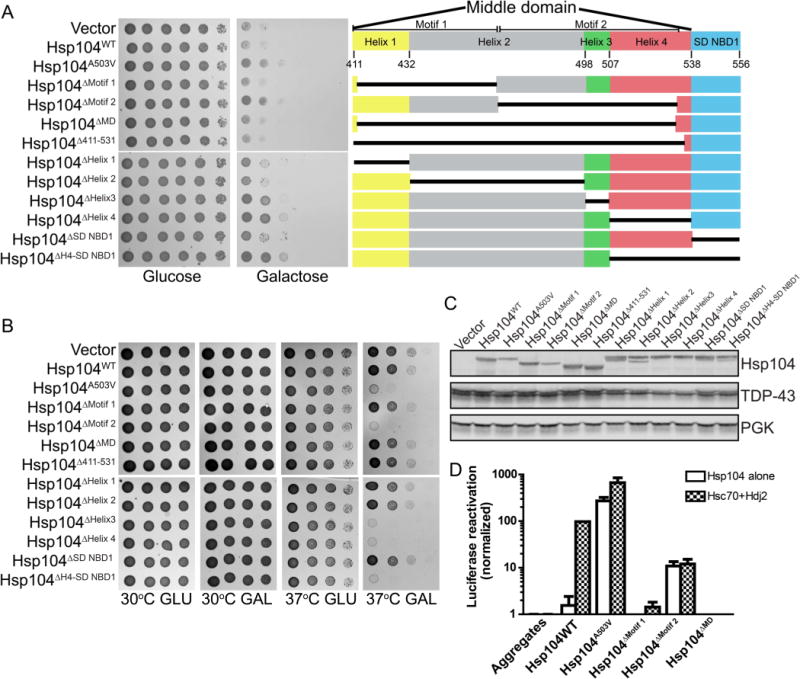
Deletion of Hsp104 MD motifs and helices. Deletion of MD motif 2, helix 3, or helix 4 potentiates Hsp104. (A) W303Δhsp104 yeasts integrated with pAG303GAL-TDP-43 were transformed with the indicated 416GAL-Hsp104 deletion construct, empty plasmid, Hsp104WT, or Hsp104A503V. Strains were serially diluted 5-fold and spotted on glucose (off) or galactose (on) media (left). Organization of the middle domain and small domain of NBD1 (right). (B) W303Δhsp104 yeasts were transformed with the indicated 416GAL-Hsp104 plasmid or empty vector control. Yeasts were grown to saturation in synthetic raffinose medium, serially diluted, and spotted onto SD-Ura or SGal-Ura media and incubated at 30 or 37 °C. Plates were analyzed after 2–3 days of growth. (C) Selected strains from A were induced for 5 h, lysed, and immunoblotted. 3-Phosphoglycerate kinase (PGK) serves as a loading control. (D) Luciferase aggregates were incubated with the purified Hsp104 variants (0.167 μM hexamer) plus (checkered bars) or minus (clear bars) Hsc70 (0.167 μM) and Hdj2 (0.167 μM). Values represent means ± SEM (n = 3).
We next deleted each helix of the Hsp104 MD and tested for suppression of TDP-43 toxicity. Supporting our findings with deletion of entire motifs, deletion of helices 1, 2, or the small domain of NBD1 does not potentiate Hsp104, while deletion of helices 3 or 4 confers potentiation (Figure 4A). Similar results were obtained for FUS and α-synuclein (Figure S2). We also tested the variants for temperature sensitivity and, consistent with our results in suppression of TDP-43 toxicity, we found that deletion of motif 2, helix 3, or helix 4 confers a temperature-sensitive growth phenotype (Figure 4B). We confirmed that expression of the Hsp104 deletion constructs does not alter TDP-43 expression levels and found that the deletion constructs were expressed at similar or lower levels than Hsp104WT (Figure 4C).
To further assess the activity of the deletion constructs, we purified Hsp104ΔMotif 1, Hsp104ΔMotif 2, and Hsp104ΔMD and tested the variants for reactivation of chemically denatured luciferase in vitro. We found that deletion of motif 1 or the entire middle domain inactivates Hsp104, while deletion of just motif 2 preserves Hsp104 activity (Figure 4D). However, Hsp104ΔMotif 2 retains only ~15% activity in comparison to Hsp104 in the presence of Hsc70 and Hdj2 (Figure 4D). However, Hsp104ΔMotif 2 displays similar activity in the presence or absence of Hsc70 and Hdj2 (Figure 4D), whereas Hsp104 is inactive without Hsc70 and Hdj2. Thus, our results reveal a key feature governing Hsp104 potentiation. Potentiation depends strongly on obviation of the requirement for Hsp70 and Hsp40 for disaggregase activity, while total activity is not as important. Collectively, these data establish that, surprisingly, the MD of Hsp104 regulates Hsp104 activity differently than the MD of ClpB regulates ClpB activity.26,28,29,32
We have demonstrated that Hsp104, an AAA+ protein disaggregase from yeast, can be mutated at disparate positions to relieve autoinhibition and confer Hsp104 potentiation. Potentiated Hsp104 variants suppress the toxicity of diverse substrates implicated in ALS and PD, whereas Hsp104 is ineffective. We have comprehensively illustrated the sequence-specific requirements for Hsp104 potentiation via mutations in the MD and have uncovered a diverse array of potentiating mutations. Perplexingly, mutation at any of 20 different residues located throughout the MD and small domain of NBD1 (a 145 amino acid stretch) confer Hsp104 potentiation (Supporting Information Table 1). Additionally, at many of these positions, mutation to diverse classes of residues, including both conservative and nonconservative substitutions, confers poten-tiation.17 We are aware of no other examples in which mutation at such disparate positions to both conservative and non-conservative substitutions leads to a therapeutic gain-of-function.9,17 It is perplexing that mutation of at least ~14% of residues in the MD can confer this effect. However, Hsp104 autoinhibition is not relieved by simply mutating any residue.26 We have identified an additional 25 residues where mutation does not confer potentiation (Supporting Information Table 2, Figure 1D). We suggest that Hsp104 has evolved to maintain an autoinhibited state, in which it is restrained from generically unfolding all substrates. However, this autoinhibited state is fragile, as seemingly subtle but diverse changes to side chains can transform Hsp104 into its potentiated form.
Using a series of MD deletion constructs, we have further elucidated the structural requirements for Hsp104 potentiation. Deletion of the MD hyperactivates ClpB.28,29,32 However, in the Hsp104 backbone, the requirements for potentiation are more complex and deletion of the entire MD does not confer potentiation. Rather, deletion of helix 3, helix 4, or both confers potentiation. These findings suggest that the requirements for Hsp104 potentiation are strikingly different from the requirements for hyperactivation of ClpB. In the future, it will be important to develop a complete map of potentiating mutations covering every position in the MD and small domain of NBD1, as we currently have mapped only ~30% of this region. Our studies provide new insights into the basis for Hsp104 potentiation and provide clues as to how Hsp104 might be fine-tuned to enhance specificity. We suggest that potentiating mutations in the MD relieve an autoinhibitory function of this domain or mimic an allosteric event that enhances disaggregase activity in the absence of Hsp70 and Hsp40. Independence from Hsp70 and Hsp40 might be particularly important if the functionality of Hsp70 and Hsp40 has been compromised by excessive sequestration in protein aggregates.33 It will be interesting to more thoroughly elucidate the structural requirements that dictate which MD residues yield potentiated states via missense mutation and which cannot. We hypothesize that potentiation likely stems from structural perturbation and partial loss of the coiled-coil structure of the MD, because the diverse range of potentiating mutations would be highly unlikely to all introduce a similar increased structural stability or specific structural feature. The idea that structural destabilization is the underlying basis for potentiation is supported by the lower expression levels of potentiating versus nonpotentiating variants (Figure 4C).
It is perplexing that a poorly conserved region of Hsp104, the MD, plays a crucial role in regulating Hsp104 activity. Poor sequence conservation is often thought to reflect the unimportance of that region. For Hsp104, the opposite is true. It has been postulated that poor sequence conservation of the MD might have allowed ancestral forms of Hsp104 to rapidly acquire new functions.27 Indeed, the mutability of the MD may have provided the capacity for ancestral forms of Hsp104 to rapidly evolve and promote yeast survival through environmental stress and simultaneously regulate the inheritance of diverse prion-based, bet-hedging devices that can be beneficial in some environments.8,11,34,35 These diverse activities, encompassing disordered aggregate reactivation, prion fragmentation, and prion dissolution place different physical demands on Hsp104, which has resulted in the evolution of a disaggregase with increased operational plasticity in comparison to ClpB, which functions primarily in stress tolerance and disordered aggregate reactivation.16
Why is Hsp104 not naturally potentiated? Potentiated Hsp104 variants can display defects in the propagation of potentially beneficial prions.36 Moreover, it seems probable that the temperature-sensitive growth defect has likely selected against naturally potentiated variants, even though they can antagonize the excessive aggregation and toxicity caused by the over-expression of a single protein in yeast, such as TDP-43, FUS, or α-syn.17,21 Thus, it would seem that elevated levels of a single, aggregation-prone, toxic protein is an unusual stress for yeast, which has not been a significant selection pressure governing Hsp104 primary sequence and activity. Hsp104 appears to have evolved to exist in a naturally and fragilely constrained autoinhibited state, which likely enables precise spatiotemporal regulation of disaggregase activity. Thus, disaggregase activity would only be elicited when or where it is needed, perhaps by the Hsp70 chaperone system37 or by natural prion substrates.1,16 It is curious that Hsp104 has not acquired secondary mutations to make accessing the potentiated state improbable. Thus, the MD likely serves as a switch that maintains Hsp104 in the autoinhibited state but allows for the nonrepressed state to be rapidly accessed. However, this regulation appears too tight to enable Hsp104 to antagonize the excessive aggregation and toxicity caused by the overexpression of a single protein in yeast, such as TDP-43, FUS, or α-syn.9,17,21 It remains possible that the proteomes and environmental challenges faced by other species might have created selective pressures that enabled evolution of naturally potentiated Hsp104 variants. Thus, it will be fascinating to compare the activity of Hsp104 orthologues from different species with disparate proteomes that inhabit diverse environments. Indeed, a naturally occurring Hsp104 variant could display enhanced activity that eradicates the aggregation of human neurodegenerative disease proteins. Regardless, our work suggests that optimization of the MD may prove critical to engender activity and specificity against noncognate substrates connected with human disease.
MATERIALS AND METHODS
Yeast Strains, Media, and Plasmids
All yeasts were W303aΔhsp104 (MATa, can1–100, his3–11,15, leu2–3,112, trp1–1, ura3–1, ade2–1).7 Yeasts were grown in a rich medium (YPD) or in synthetic media lacking the appropriate amino acids. Media were supplemented with 2% glucose, raffinose, or galactose. Vectors encoding TDP-43, FUS, and α-syn (pAG303GAL-TDP-43, pAG303GAL-FUS, pAG303GAL-α-syn-YFP, and pAG304Gal-α-syn-YFP) were from A. Gitler and M. Duennwald.38–40 pRS416GAL-Hsp104 variants have been described previously.17 All mutations were constructed using QuikChange site-directed mutagenesis (Agilent) and confirmed by DNA sequencing. To assess the importance of Hsp110 for suppression of toxicity, we used BY4741Δsse1 and Δsse2 yeast strains from B. Johnson.
Yeast Transformation and Spotting Assays
Yeasts were transformed according to standard protocols using polyethylene glycol and lithium acetate.41 The Hsp104 MD library generated by error-prone PCR has been described previously.17 This library was screened to isolate suppressors of TDP-43 toxicity as described,17,18 but in this work selection was carried out at 34 °C. Single colonies were selected, grown, and screened to confirm toxicity suppression was Hsp104 dependent.17,18 For each verified hit, the missense mutations were constructed using QuikChange site-directed mutagenesis (Agilent) and confirmed by DNA sequencing.
For the spotting assays, yeasts were grown to saturation overnight in raffinose-supplemented dropout media at 30 °C. Cultures were serially diluted and spotted in duplicate onto synthetic dropout media containing glucose or galactose. Plates were analyzed after growth for 2–3 days at 30 °C.
Assessing Toxicity of Hsp104 Variants
W303Δhsp104 yeasts were transformed with the indicated 416GAL-Hsp104 plasmid. Yeasts were diluted and grown to saturation overnight in raffinose-supplemented dropout media at 30 °C. Cultures were serially diluted and spotted in duplicate onto SD-Ura or SGal-Ura media and incubated at 30 or 37 °C. Plates were analyzed after 48–72 h of growth.
Immunoblotting
Yeasts were grown and induced in galactose containing medium for 5 h. Cultures were normalized to A600 nm = 0.6; 6 mL of cells were harvested and treated in 0.1 M NaOH for 5 min at RT. Cell pellets were resuspended into 100 μL 1 × SDS sample buffer and boiled. Cleared lysates were separated by SDS-PAGE (4–20% gradient, BioRad) and transferred to a PVDF membrane. Membranes were blocked using LI-COR blocking buffer for 1 h at RT. Primary antibody incubations were performed at 4 °C overnight. Antibodies used: anti-TDP-43 polyclonal (Proteintech), anti-Hsp104 polyclonal (Enzo Life Sciences), and anti-PGK monoclonal (Invitrogen). Blots were processed using the LI-COR Odyssey Fc Imaging system.
Protein Purification and Luciferase Reactivation Assays
Potentiated Hsp104 variants were purified as described.17 Hsp104 concentrations refer to the hexamer concentration. Firefly luciferase was purchased from Sigma while Hsc70 and Hdj2 were purchased from Enzo Life Sciences. Luciferase reactivation assays were performed as described16,17 using 50 nM aggregated luciferase with 0.167 μM Hsp104 hexamer in the presence or absence of 0.167 μM Hsc70 and Hdj2.
Supplementary Material
Acknowledgments
We thank A. Gitler, S. Lindquist, B. Johnson, and M. Duennwald for kindly sharing reagents, and K. Mack and M. Torrente for critiques. Our studies were supported by an American Heart Association Post-Doctoral Fellowship and Target ALS Springboard Fellowship (M.E.J); NIH Director’s New Innovator Award DP2OD002177, NIH grants R21HD074510 and R01GM099836, a Muscular Dystrophy Association Research Award (MDA277268), Packard Center for ALS Research at Johns Hopkins University, Target ALS, an Ellison Medical Foundation New Scholar in Aging Award, and The Life Extension Foundation (J.S.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.5b00765.
Supporting Tables 1 and 2, Figures S1 and S2 captions (PDF)
Author Contributions
M.E.J. and J.S. conceived and designed the experiments. M.E.J., A.I.C, and J.S. contributed key reagents. M.E.J., K.Y., and A.T. performed the experiments. M.E.J. and J.S. analyzed the data. M.E.J. and J.S. wrote the paper.
The authors declare no competing financial interest.
References
- 1.Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304:1793–1797. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- 2.Shorter J, Lindquist S. Destruction or potentiation of different prions catalyzed by similar Hsp104 remodeling activities. Mol Cell. 2006;23:425–438. doi: 10.1016/j.molcel.2006.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shorter J, Lindquist S. Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. EMBO J. 2008;27:2712–2724. doi: 10.1038/emboj.2008.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 5.Parsell DA, Sanchez Y, Stitzel JD, Lindquist S. Hsp104 is a highly conserved protein with two essential nucleotide-binding sites. Nature. 1991;353:270–273. doi: 10.1038/353270a0. [DOI] [PubMed] [Google Scholar]
- 6.Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature. 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- 7.Sanchez Y, Lindquist SL. HSP104 required for induced thermotolerance. Science. 1990;248:1112–1115. doi: 10.1126/science.2188365. [DOI] [PubMed] [Google Scholar]
- 8.Newby GA, Lindquist S. Blessings in disguise: biological benefits of prion-like mechanisms. Trends Cell Biol. 2013;23:251–259. doi: 10.1016/j.tcb.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 9.Jackrel ME, Shorter J. Reversing deleterious protein aggregation with re-engineered protein disaggregases. Cell Cycle. 2014;13:1379–1383. doi: 10.4161/cc.28709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vashist S, Cushman M, Shorter J. Applying Hsp104 to protein-misfolding disorders. Biochem Cell Biol. 2010;88:1–13. doi: 10.1139/o09-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shorter J. Hsp104: a weapon to combat diverse neurodegenerative disorders. Neurosignals. 2008;16:63–74. doi: 10.1159/000109760. [DOI] [PubMed] [Google Scholar]
- 12.Cushman M, Johnson BS, King OD, Gitler AD, Shorter J. Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci. 2010;123:1191–1201. doi: 10.1242/jcs.051672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 14.Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- 15.Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19:R46–64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeSantis ME, Leung EH, Sweeny EA, Jackrel ME, Cushman-Nick M, Neuhaus-Follini A, Vashist S, Sochor MA, Knight MN, Shorter J. Operational Plasticity Enables Hsp104 to Disaggregate Diverse Amyloid and Nonamyloid Clients. Cell. 2012;151:778–793. doi: 10.1016/j.cell.2012.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackrel ME, DeSantis ME, Martinez BA, Castellano LM, Stewart RM, Caldwell KA, Caldwell GA, Shorter J. Potentiated Hsp104 Variants Antagonize Diverse Proteotoxic Misfolding Events. Cell. 2014;156:170–182. doi: 10.1016/j.cell.2013.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackrel ME, Tariq A, Yee K, Weitzman R, Shorter J. Isolating Potentiated Hsp104 Variants Using Yeast Proteinopathy Models. J Visualized Exp. 2014:e52089. doi: 10.3791/52089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sweeny EA, Jackrel ME, Go MS, Sochor MA, Razzo BM, DeSantis ME, Gupta K, Shorter J. The hsp104 N-terminal domain enables disaggregase plasticity and potentiation. Mol Cell. 2015;57:836–849. doi: 10.1016/j.molcel.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackrel ME, Shorter J. Engineering Enhanced Protein Disaggregases for Neurodegenerative Disease. Prion. 2015;9:90–109. doi: 10.1080/19336896.2015.1020277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackrel ME, Shorter J. Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease-linked proteins. Dis Models & Mech. 2014;7:1175–1184. doi: 10.1242/dmm.016113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shorter J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One. 2011;6:e26319. doi: 10.1371/journal.pone.0026319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan Q, Park KW, Du Z, Morano KA, Li L. The role of Sse1 in the de novo formation and variant determination of the [PSI+] prion. Genetics. 2007;177:1583–1593. doi: 10.1534/genetics.107.077982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kryndushkin D, Wickner RB. Nucleotide exchange factors for Hsp70s are required for [URE3] prion propagation in Saccharomyces cerevisiae. Mol Biol Cell. 2007;18:2149–2154. doi: 10.1091/mbc.E07-02-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadlish H, Rampelt H, Shorter J, Wegrzyn RD, Andreasson C, Lindquist S, Bukau B. Hsp110 chaperones regulate prion formation and propagation in S. cerevisiae by two discrete activities. PLoS One. 2008;3:e1763. doi: 10.1371/journal.pone.0001763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeSantis ME, Sweeny EA, Snead D, Leung EH, Go MS, Gupta K, Wendler P, Shorter J. Conserved distal loop residues in the Hsp104 and ClpB middle domain contact nucleotide-binding domain 2 and enable Hsp70-dependent protein disaggregation. J Biol Chem. 2014;289:848–867. doi: 10.1074/jbc.M113.520759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schirmer EC, Homann OR, Kowal AS, Lindquist S. Dominant Gain-of-Function Mutations in Hsp104p Reveal Crucial Roles for the Middle Region. Mol Biol Cell. 2004;15:2061–2072. doi: 10.1091/mbc.E02-08-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oguchi Y, Kummer E, Seyffer F, Berynskyy M, Anstett B, Zahn R, Wade RC, Mogk A, Bukau B. A tightly regulated molecular toggle controls AAA+ disaggregase. Nat Struct Mol Biol. 2012;19:1338–1346. doi: 10.1038/nsmb.2441. [DOI] [PubMed] [Google Scholar]
- 29.Carroni M, Kummer E, Oguchi Y, Wendler P, Clare DK, Sinning I, Kopp J, Mogk A, Bukau B, Saibil HR. Head-to-tail interactions of the coiled-coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation. eLife. 2014;3:e02481. doi: 10.7554/eLife.02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doyle SM, Wickner S. Hsp104 and ClpB: protein disaggregating machines. Trends Biochem Sci. 2009;34:40–48. doi: 10.1016/j.tibs.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 31.Sielaff B, Tsai FT. The M-domain controls Hsp104 protein remodeling activity in an Hsp70/Hsp40-dependent manner. J Mol Biol. 2010;402:30–37. doi: 10.1016/j.jmb.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seyffer F, Kummer E, Oguchi Y, Winkler J, Kumar M, Zahn R, Sourjik V, Bukau B, Mogk A. Hsp70 proteins bind Hsp100 regulatory M domains to activate AAA+ disaggregase at aggregate surfaces. Nat Struct Mol Biol. 2012;19:1347–1355. doi: 10.1038/nsmb.2442. [DOI] [PubMed] [Google Scholar]
- 33.Yu A, Shibata Y, Shah B, Calamini B, Lo DC, Morimoto RI. Protein aggregation can inhibit clathrin-mediated endocytosis by chaperone competition. Proc Natl Acad Sci U S A. 2014;111:E1481–1490. doi: 10.1073/pnas.1321811111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137:146–158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halfmann R, Alberti S, Lindquist S. Prions, protein homeostasis, and phenotypic diversity. Trends Cell Biol. 2010;20:125–133. doi: 10.1016/j.tcb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi A, Hara H, Kurahashi H, Nakamura Y. A systematic evaluation of the function of the protein-remodeling factor Hsp104 in [PSI+] prion propagation in S. cerevisiae by comprehensive chromosomal mutations. Prion. 2007;1:69–77. doi: 10.4161/pri.1.1.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee J, Kim JH, Biter AB, Sielaff B, Lee S, Tsai FT. Heat shock protein (Hsp) 70 is an activator of the Hsp104 motor. Proc Natl Acad Sci U S A. 2013;110:8513–8518. doi: 10.1073/pnas.1217988110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson BS, McCaffery JM, Lindquist S, Gitler AD. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2008;105:6439–6444. doi: 10.1073/pnas.0802082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284:20329–20339. doi: 10.1074/jbc.M109.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, Gitler AD. Molecular Determinants and Genetic Modifiers of Aggregation and Toxicity for the ALS Disease Protein FUS/TLS. PLoS Biol. 2011;9:e1000614. doi: 10.1371/journal.pbio.1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.