

Abstract
Background
Campylobacter species are the most prevalent bacterial pathogen causing acute enteritis worldwide. In contrast to Campylobacter jejuni, about 5 % of Campylobacter coli strains exhibit susceptibility to restriction endonuclease digestion by DpnI cutting specifically 5’-GmATC-3’ motifs. This indicates significant differences in DNA methylation between both microbial species.
The goal of the study was to analyze the methylome of a C. coli strain susceptible to DpnI digestion, to identify its methylation motifs and restriction modification systems (RM-systems), and compare them to related organisms like C. jejuni and Helicobacter pylori.
Results
Using one SMRT cell and the PacBio RS sequencing technology followed by PacBio Modification and Motif Analysis the complete genome of the DpnI susceptible strain C. coli BfR-CA-9557 was sequenced to 500-fold coverage and assembled into a single contig of 1.7 Mbp. The genome contains a CJIE1-like element prophage and is phylogenetically closer to C. coli clade 1 isolates than clade 3. 45,881 6-methylated adenines (ca. 2.7 % of genome positions) that are predominantly arranged in eight different methylation motifs and 1,788 4-methylated cytosines (ca. 0.1 %) have been detected. Only two of these motifs correspond to known restriction modification motifs. Characteristic for this methylome was the very high fraction of methylation of motifs with mostly above 99 %.
Conclusions
Only five dominant methylation motifs have been identified in C. jejuni, which have been associated with known RM-systems. C. coli BFR-CA-9557 shares one (RAATTY) of these, but four ORFs could be assigned to putative Type I RM-systems, seven ORFs to Type II RM-systems and three ORFs to Type IV RM-systems. In accordance with DpnI prescreening RM-system IIP, methylation of GATC motifs was detected in C. coli BfR-CA-9557. A homologous IIP RM-system has been described for H. pylori. The remaining methylation motifs are specific for C. coli BfR-CA-9557 and have been neither detected in C. jejuni nor in H. pylori.
The results of this study give us new insights into epigenetics of Campylobacteraceae and provide the groundwork to resolve the function of RM-systems in C. coli.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2317-3) contains supplementary material, which is available to authorized users.
Keywords: Campylobacter coli, Genome, Methylation, Motifs, Methylome, Restriction modification systems, Isoschizomer digestion assay, SMRT sequencing, PacBio
Background
Campylobacteriosis is the most prevalent form of bacterial acute enteritis worldwide. In symptomatic cases it is characterized by a prodromal phase with fever, vomiting, and headaches followed by watery or bloody diarrhea and abdominal cramps [1, 2]. In consequence of acute enteritis, extraintestinal post-infectious sequelae, namely, the Guillain-Barré syndrome, inflammatory bowel disease, and reactive arthritis may occur [3, 4]. The average incidence reported in the European Union was 64.8 per 100,000 population in 2013 [5], in the USA 14.3 cases per 100,000 population in 2012, and in China 161 cases per 100,000 population in urban areas compared to 37 cases per 100,000 population in rural areas [6]. In Europe, 80.6 % were reported to have been caused by Campylobacter jejuni and 7.1 % by Campylobacter coli [5].
C. coli is phylogenetically subdivided into three clades [7, 8]: clade 1 isolates commonly colonize swine but can also be isolated from poultry and humans, although less frequently. Clades 2 and 3 are typically isolated from environmental waters [8, 9].
At the moment, seven completed C. coli chromosomal genome sequences [10–13], several scaffold genomes, and various contigs have been deposited in the NCBI Genome database [14–17]. The completed genome sequences, range from 1.685 to 1.872 Mb, have a G + C content of about 31 to 32 %, and contain 1715 – 1970 predicted genes including 1642 – 1861 protein coding ORFs [10–13].
One of the major epigenetic mechanisms in prokaryotes is DNA methylation [18]. DNA methylation patterns influence gene expression [19], through silencing of transcription [20, 21] as well as DNA replication initiation [22, 23] and mismatch repair [24]. DNA methylation also serves as a protection of the host genome against extraneous DNA [18] through restriction-modification systems (RM-systems). RM-systems consist of two components: (i) a restriction endonuclease that recognizes a specific DNA motif and (ii) a cognate DNA methyltransferase that methylates the same DNA, preventing its cleavage by the restriction endonuclease [25]. The majority of RM-systems can be categorized into four types [25–29]:
Type I RM-systems typically consist of three types of subunits: two restriction endonuclease subunits (R), which facilitate DNA cleavage, one specificity subunit (S) for recognition of specific DNA sequence motifs, and two DNA methylase subunits (M) that catalyse N6 adenine methylation [30, 31]. This composition enables Type I RM-systems to digest unmethylated DNA, whereas hemimethylated DNA is further methylated and fully methylated DNA is insusceptible to restriction [32].
Type II RM-systems are mostly composed of two homodimeric R subunits and a separated M subunit. The R and M subunits recognize the same DNA motif, which is typically a 4–8 bp palindrome [33].
Type III RM-systems are comprised of two modification (Mod) subunits and two restriction (R) subunits. Type III RM-systems must bind to two inversely oriented copies of its 5–6 bp asymmetric recognition motif. Cleavage of unmethylated DNA typically occurs 25–27 bp away from the binding sites [34].
Type IV RM-systems consist of two separate R subunits cleaving DNA that contains methylated, hydroxymethylated or glucosyl-hydroxymethylated cytosines. Cleavage typically occurs 30 bp away from one of the binding sites [35]. Furthermore, there exist many orphan DNA methylases that are not part of a RM-system e.g. DNA adenine methylase (Dam) and cell cycle-regulated DNA methyltransferase (CcrM) [29].
It must be considered that the genes encoding for particular components of a RM-system are diverse within microbial species i.e. C. jejuni and C. coli [36–38]. In C. jejuni the putative Type I RM-system locus (genes cj1549–cj1553; hsd locus) reveals significant diversity regarding gene order, chromosomal location, intervening ORFs and gene sequence leading to the classification of the C. jejuni hsd loci into at least three families, namely the IAB, IC, and IF family. Especially sequence variations in the hsdS gene suggest at least 30 different target sequence specificities and therewith differences in DNA methylation [39]. Holt and coworkers demonstrated that cj1051c (cjeI) encodes an active restriction-modification Type IIG enzyme in C. jejuni that significantly decreases transformation efficiency with plasmids [40]. Additionally it was shown that Type IIS restriction modification enzyme Cj0030/Cj0031 is subject to phase variable gene expression due to mutations in polyC/G tracts [41–43]. Additionally cj0139/cj0140 encode a putative 5-methylcytosine-restriction system, cj0690c a Type II RM-system and cj0979c a nuclease that could be part of a RM-system [41, 42].
Furthermore it was shown for C. jejuni that gene product of cj1461 is a N6-adenine-specific DNA methyltransferase that is not a Dam homologue and not part of a RM-system. Knockout of cj1461 affects flagellar appearance, motility, adherence, and invasion indicating its role for epigenetic control of proteins involved in these processes [44]. Further non-ubiquitous modification/methyltransferase gene loci have been observed in C. jejuni ST-677 isolates, namely fixL (cjj5070_14950c) that showed homology with DNA adenine/modification methylases in Campylobacter rectus, a homolog of iceA1/nlaIII (cjj5070_14940c), cjj5070_14910c predicted as ulcer associated adenine-specific DNA methyltransferase, which is an ortholog of the CATG-specific methyltransferase hpyIM of H. pylori, and the orphan DNA methyltransferase in ORF cjj5070_08940 [45]. Three strain-specific RM-systems have been reported for C. jejuni ST403 complex: R. HinPI restriction endonuclease (cje135_03870), Modification methylase Hhal (cje135_03865), and R.Pab1 restriction endonuclease (cje135_02348) [46].
In contrast to C. coli, Type III RM-systems have been reported for C. jejuni subsp. jejuni, C. jejuni subsp. doylei, Campylobacter lari, and Campylobacter upsaliensis [47].
Until to date the methylomes of C. jejuni subsp. jejuni (3 isolates) and of the related microbial species Helicobacter pylori (2 isolates) have been analyzed [48–50], but so far no C. coli methylome has been examined.
Studies performing isoschizomer digestion assays indicated differences in methylation at GATC sites in genomic DNA of C. coli isolates, which suggested host-associated DNA modification systems [51, 52].
In this study we analyzed the first methylome of C. coli using SMRT DNA sequencing. This approach facilitates analysis of methylation motifs as well as RM-system gene loci in parallel. For SMRT sequencing a C. coli isolate was selected, which was tested positive for GATC site methylation isoschizomer digestion assay screening.
Results and discussion
SMRT sequencing and annotation
Screening of 50 C. coli isolates performing 5’-G(m)ATC-3’-specific isoschizomer digestion assays revealed six bacterial isolates showing Dam activity. Of these six isolates, the isolate BfR-CA-9557 was selected for SMRT sequencing, since it reproducibly tested positive in five biological independent analyses. Furthermore, its origin from broiler hearts sampled at a retail store in Berlin, Germany, indicates its relevance for food hygiene and thus for the infection of humans.
MLST typing revealed sequence type ST-1589 and clonal complex CC-828. Accordingly, it is a potentially human pathogenic isolate of clade 1.
Susceptibility testing indicated a quinolone resistant phenotype (zone diameter 0 mm in disc diffusion; minimal inhibition concentration (MIC) measured by broth microdilution: 16 μg/ml ciprofloxacin, 32 μg/ml nalidixic acid). In contrast, the isolate was tested susceptible for macrolides, tetracyclines and aminoglycosides (MIC values: 2 μg/ml erythromycin, <=0,5 μg/ml tetracycline, 1 μg/ml gentamicin, 2 μg/ml streptomycin).
Sequencing of the C. coli isolate using one Pacific Bioscience SMRT cell resulted in 74,742 continuous long reads (CLR) with an average (total) length of 14,514 (1.08°109) base pairs (Additional file 1: Figure S1). 142,135 subreads (i.e. individual fragments) of high quality and an average length of 7,602 bp could be extracted from the CLRs.
Utilizing the Pacific Bioscience high-quality de novo genome assembly algorithm (HGAP.2), we obtained a single polished contig of 1,720,506 base pairs with an average 500-fold coverage and a confidence score of >99.99 % (Additional file 2: Figure S2). The G + C content of the contig was 31.4 % and the most closely related genome sequence available in the NCBI genome database was that of C. coli 15–537360 (taxonomy ID 1358410, 87 % coverage). No plasmids were detected in this isolate.
Application of the NCBI/RAST/Prodigal annotation pipelines resulted in 1637/1769/1797 predicted coding sequences and 275/366/453 (16.8/20.7/25.2 %) predicted hypothetical proteins. (GenBank ID: CP011777, Additional files 3 and 4). Furthermore, 54 RNA genes (44 tRNA/9 rRNA/1 ncRNA) were predicted by NCBI, 52 RNAs (43 tRNA/9 rRNA) by RAST, and 54 RNAs (44 tRNA/9 rRNA/1 tmRNA) by Prodigal.
The RAST subsystem coverage was 57 % (1003 genes), of which amino acid metabolism (296 of 1557 associated terms, 19.0 %), protein metabolism (216 terms, 13.9 %), and cofactors, vitamins, prosthetic groups, and pigments (144, 9.2 %) represented the largest groups (Fig. 1).
Fig. 1.
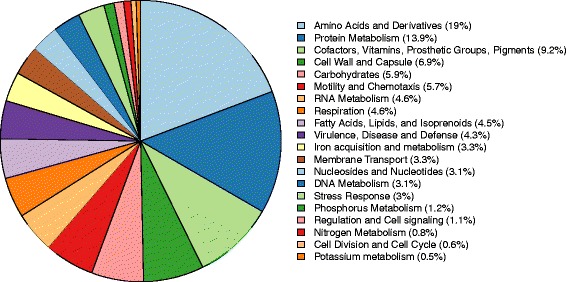
Pie chart representing RAST subsystems identified in the BFR-CA-9557 genome. The 20 most abundant subsystems on the “category” level as identified by RAST are represented by a particular color indicated at the right-hand side of the figure
Figure 2a shows a comparison of the BfR-CA-9557 genome sequence to other C. coli genomes (76339, clade 3 and RM4661, clade 1) using the Artemis tool. Here, homologous regions as identified by BLAST are indicated by red (aligned in the same direction) or blue line segments (opposite direction). It can be seen that C. coli 76339 mainly shows three stretches of inverse homologous regions to BfR-CA-9557 and lacks similarity in the region of the identified CJIE1 (green box, see also below). RM4661 displays a long stretch of close homology with minor gaps, but also inverse homologous segments within the plasmid and the beginning and end of the chromosome. A similar pattern can be observed for the comparison of BFR-CA-9557 to C. jejuni NCTC 11168 (Fig. 2b). This indicates that C. coli BfR-CA-9557 is phylogenetically closer to clade 1 and therewith to C. jejuni islolates than to clade 3 C. coli isolates.
Fig. 2.

Comparison of C. coli BFR-CA-9557 genome to those of C. coli RM4661 and 76339 a as well as C. jejuni NCTC 11168 b using the Artemis Comparison Tool (ACT). Comparison of the BFR-CA-9577 genome to those of C. coli clade 1 strain RM4661 and C. coli clade 3 strain 76339 as well as to the genome of C. jejuni NCTC 11168 indicates that the clade 1 strain BFR-CA-9577 genome organization is more related to C. coli clade 1 and C. jejuni strains than to clade 3 strains. The CJIE1-homologue prophage in the BFR-CA-9557 genome is indicated in green
Poly-G/C tracts and the capsular polysaccharid gene locus
Analysis of poly-G/C tracts within the contig of BfR-CA-9557 revealed 17 locations with homopolymeric stretches of at least eight G or C nucleotides (Tables 1 & 2). Most of these tracts occur in the vicinity of hypothetical proteins, however, two motifs could be identified within the capsular polysaccharid gene locus (cps) close to ORFs identified as glycosyltransferases (EC 2.4.99.-) by RAST. Other ORFs with neighboring poly-G/C motifs comprise e.g. transferases and ligases.
Table 1.
Positions of poly-G tracts in the BFR-CA-9557 genome
| Poly G on forward strand | Pos. rel. to | ORF location | |||||
|---|---|---|---|---|---|---|---|
| Position | Length | RAST annotation of closest ORF | ATG | STOP | Start | Stop | Strand |
| 46933 | 9 | hypothetical protein | −52 | −2292 | 46985 | 49225 | + |
| 258726 | 10 | FIG 00470070: hypothetical protein | −15 | −1924 | 258741 | 260650 | + |
| 689956 | 10 | UDP-N-acetylmuramoylalanine--D-glutamate ligase (EC 6.3.2.9) | −6 | −1232 | 691179 | 689971 | - |
| 851085 | 10 | FIG 00470965: hypothetical protein | 556 | −671 | 850529 | 851756 | + |
| 1562460 | 10 | Putative transferase | −48 | −590 | 1562508 | 1563050 | + |
| 1565336 | 9 | FIG 00469667: hypothetical protein | 251 | −423 | 1565085 | 1565759 | + |
| 1575332 | 10 | Motility accessory factor | 167 | −1780 | 1575165 | 1577112 | + |
| 1645063 | 9 | Ferrous iron transport protein B | 1576 | −269 | 1643487 | 1645332 | + |
Poly-G/C tracts were searched in the BFR-CA-9557 genome using regular expression describing at least 8 consecutive G/C. The first two columns denote the genome location and length of the expression found. Column 3 shows the RAST annotation of the ORF closest to the homopolymeric stretch. Column 4 and 5 represent the relative position of the stretch to the ORF’s start (4) and Stop codon (5), with negative numbers representing upstream locations. Columns 6 to 8 denote the location and orientation of the ORF
Table 2.
Positions of poly-C tracts in the BFR-CA-9557 genome
| Poly C on forward strand | pos. rel. to | ORF location | |||||
|---|---|---|---|---|---|---|---|
| Position | Length | RAST annotation of closest ORF | ATG | STOP | Start | Stop | Strand |
| 442261 | 11 | Putative lipoprotein of ferric iron transporter system | 139 | −3 | 442254 | 442132 | - |
| 1099189 | 10 | hypothetical protein | 739 | −7 | 1099187 | 1098459 | - |
| 1257487 | 9 | Phosphoglycerol transferase | 1728 | −256 | 1257735 | 1255767 | - |
| 1427910 | 9 | Filamentous haemagglutinin domain protein | 1554 | −9 | 1427911 | 1426364 | - |
| 1547848 | 9 | FIG 00469527: hypothetical protein | 637 | −596 | 1548436 | 1547219 | - |
| 1587049 | 8 | hypothetical protein | 679 | −575 | 1587617 | 1586377 | - |
| 1607917 | 9 | FIG 00470049: hypothetical protein | 2305 | 34 | 1607875 | 1605620 | - |
| 1660758 | 9 | CMP-N-acetylneuraminate-beta-galactosamide-alpha-2,3-sialyltransferase (EC 2.4.99.-) | 64 | −620 | 1661370 | 1660702 | - |
| 1668862 | 8 | CMP-N-acetylneuraminate-beta-galactosamide-alpha-2,3-sialyltransferase (EC 2.4.99.-) | 40 | −1587 | 1670442 | 1668829 | - |
Poly-G/C tracts were searched in the BFR-CA-9557 genome using regular expression describing at least 8 consecutive G/C. The first two columns denote the genome location and length of the expression found. Column 3 shows the RAST annotation of the ORF closest to the homopolymeric stretch. Column 4 and 5 represent the relative position of the stretch to the ORF’s start (4) and Stop codon (5), with negative numbers representing upstream locations. Columns 6 to 8 denote the location and orientation of the ORF
The cps locus of BfR-CA-9557 ranges from 1,656,271 to 1,691,672 (1,660,702-1,686,508 excluding flanking kps regions). Spanning 35,401 bp and encoding 29 ORFs (25,806 bp; 21 ORFs; excluding flanking kps regions) it is of comparable size to the largest known other Campylobacter cps cluster, which measures 38 kb excluding flanking kps regions (C. jejuni strain X) [53]. The cps loci of different C. coli and C. jejuni strains are very variable in size and gene content and there are only three genes that are comparatively conserved: the capsular polysaccharide export system periplasmic protein gene KpsD, the GDP-mannose 4,6-dehydratase gene and the capsular polysaccharide biosynthesis/export periplasmic protein wcbA/kpsC. The highest sequence similarities exist to the cps clusters of C. coli strains RM4661 (query coverage 71 %, identity 99 %) and FB1 (query coverage 54 %, identity 99 %). Compared to the cps locus of strain X, query coverage of 28 % (identity 87 %) was observed. The genes for kpsC, hddC, gmhA2, hddA, dmhA, fcl-1, fcl-2, hypothetical protein x.25 and kpsF are present in both cps clusters.
N-linked flagellar glycosylation locus
The N-linked flagellar glycosylation locus extends from 1,319,815-1,332,144 (12,329 bp). It consist of 10 ORFs and is 99 % identical to the corresponding locus in other C. coli genomes e.g. RM4661 (clade 1) or 76339 (clade 3). In comparison to other C. jejuni strains (e.g. M1 and 81116), insertion of the lipid carrier UDP-N-acetylgalactosaminyltransferase gene and an α-1,4-N-acetylgalactosamine transferase PglH gene was observed.
Directly upstream of the N-linked flagellar glycosylation locus the lipooligosaccharide biosynthesis locus (LOS locus) is located. The LOS locus extends from the UDP-glucose 4-epimerase gene galE to the D-glycero-D-manno-heptose 1,7-bisphosphate phosphatase gene rfaD. The LOS locus is one of the more variable regions in Campylobacter genomes. LOS loci containing 8 (RM2095) to 19 (81116) ORFs have been described and depending on gene content and organization 19 different LOS classes (A-S) have been defined [54, 55]. The LOS locus of BfR-CA-9557 ranges from position 1,332,138 to 1,349,328 (17,190 bp) and contains 17 ORFs. No sialyltransferase cstII/III homologue genes and no N-acetylgalactosaminyltransferase neuABC homologue genes are present in this cluster and therefore BfR-CA-9557 expresses an unsialylated LOS. It does not exactly belong to one of the LOS classes described for C. jejuni but it is closer related to the LOS classes E, P, O, and H. Remarkably, the waaM and waaV homologue genes are immediate neighbours, therefore the established sequencing approach would have been difficult due to problems resolving repetitive genomic regions [54].
Virulence-associated genes
Neither dimethylsulfoxide (DMSO) reductase systems nor gamma glutamyltranspeptidase (ggt) gene could be detected in the BfR-CA-9557 genome, which is typical for a clade 1 C. coli isolates, but three genes homologous to the iron transport protein TonB were present in the BfR-CA-9557 genome, which has been reported as typical for clade 2 and 3 C. coli isolates [13]. The cytolethal distending toxin (cdt) operon contains all three subunits and is therewith complete.
Campylobacter jejuni integrated element 1-like element & CRISPR elements
At position 1,427,993-1,467,476 we could identify a 39,483 bp sized Campylobacter jejuni integrated element 1 (CJIE1)-homologue prophage that shows 96 % identity (at 86 % query coverage) to CJIE1 described in C. jejuni RM1221 [45]. No CRISPR elements were found by any method.
Methylation motifs and RM-systems in the genome of C. coli BfR-CA-9557
In addition to reconstruction of the genome sequence SMRT sequencing allows determination of base modification by analysis of the sequencing kinetics. Using the SMRT Analysis Modification and Motif detection, we could identify 45,881 putatively N-6-methylated adenines (m6A, ~2.7 % of genome), 1788 4-methylated cytosines (m4C, ~0.1 %) and further 53,350 rather unspecific “modified bases” where the type of modification was not recognized by the software (Additional file 5: Figure S3).
Methylated bases were arranged within eight different dominant methylation motifs (Table 3, Fig. 3). All motifs are recognized by N-6 adenine-specific methyltransferases. A small fraction (2.8 %) of m6A bases were not clustered into any of the motifs, and no consensus motif could be identified for either m4C-methylated bases or the majority of other unspecific modified bases.
Table 3.
Methylation motifs of C. coli BFR-CA-9557
| No. | Motif | Modified Position | Modification Type | % Motifs Detected | # of Motifs Detected | # of Motifs in Genome | Mean Modi-fication QV1 | Mean Motif Coverage | Partner Motif |
|---|---|---|---|---|---|---|---|---|---|
| A | RAATTY | 3 | m6A | 98.67 | 27795 | 28170 | 309.40 | 235.73 | RAATTY |
| B | GATC | 2 | m6A | 99.66 | 7512 | 7538 | 344.25 | 257.15 | GATC |
| C | RCATC | 3 | m6A | 99.43 | 4381 | 4406 | 274.48 | 256.54 | |
| D | CAAGAA | 6 | m6A | 99.71 | 2069 | 2075 | 290.35 | 249.45 | |
| E | GGGTDA | 6 | m6A | 99.44 | 1607 | 1616 | 327.70 | 254.15 | |
| F | DACATTGB | 4 | m6A | 69.69 | 223 | 320 | 80.60 | 255.05 | |
| G1 | TAAANNNNNGTG | 3 | m6A | 99.75 | 392 | 393 | 271.96 | 259.91 | CACNNNNNTTTA |
| G2 | CACNNNNNTTTA | 2 | m6A | 99.75 | 392 | 393 | 331.81 | 263.02 | TAAANNNNNGTG |
| H1 | CAAYNNNNNNNTTYG | 3 | m6A | 99.58 | 237 | 238 | 316.89 | 246.49 | CRAANNNNNNNRTTG |
| H2 | CRAANNNNNNNRTTG | 4 | m6A | 99.58 | 237 | 238 | 276.97 | 247.15 | CAAYNNNNNNNTTYG |
1 QV = quality value
Methylation motifs were identified using the PacBio SMRT Analysis software (see Methods). Column 2 shows the sequence consensus of the motif, whereby non-uniform positions are represented by IUPAC ambiguity codes. Column 3 and 4 denote the position of the modified base within the motif and the type of methylation. Column 5 represents the fraction of a motif’s occurrences in the genome (column 7) for which a methylation has been detected (column 6). Column 8 and 9 denote the average modification quality (in Phred Q-scores) and average coverage of motifs detected as modified. The last column shows the partner motif, i.e. the reverse complement of the motif
Fig. 3.
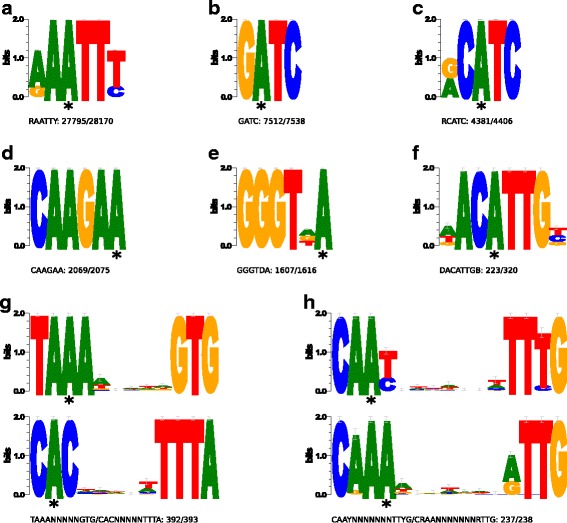
Sequence logos of eight methylation motifs. The consensus sequences of all eight motifs are depicted as sequence logo as obtained by the WebLogo 3 server (weblogo.threeplusone.com/create.cgi). The height of each stack indicates the degree of conservation (bits). The height of the letters represents the relative frequency of the base. The asterisk under a particular letter indicates the modified/methylated base. The two motifs in G and in H are partner motifs that are methylated at both strands. All motifs are recognized by N-6 adenine-specific methyltransferases
The motifs G1 & G2 and H1 & H2 (Table 3) are partner motifs and motifs A and B represent palindromic sequences, all of these partners containing methylated bases on both strands. In contrast, bases of motifs C, D, E, and F were methylated on only one strand. The major methylation motifs A, B, C, E, G, and H were almost completely methylated across their occurrences within the genome. The percentage of methylation ranges from 98.67 % to 99.75 %. In contrast, only 69.69 % of the m6A methylation motif F sequences were methylated.
Using the recognition sequence search feature within the Restriction Enzyme Database – REBASE [56], only two out of the eight motifs (GATC, RAATTY) could be matched to existing recognition sequences of restriction systems (both Type II; http://rebase.neb.com/cgi-bin/pacbioget?17032). The remaining motifs represent yet unknown recognition sequences with the longer motifs (G and H) most likely being specific to C. coli or to this particular strain.
Using the REBASE sequence search feature and the NCBI, G2L and Prodigal/Prokka annotations four ORFs encoding subunits of Type I, seven ORFs encoding subunits of Type II, and three ORFs encoding subunits of Type IV RM-systems have been identified (Table 4). Only two of the REBASE predicted recognition sequences correspond to a specific motif detected by SMRT sequencing, namely: GAATTC/RAATTY (motif A) recognized by the DNA modification methylase (Adenine-specific methyltransferase) FokIM_2 (ORF #02605) and GATC (motif B) recognized by the DNA modification methyltransferase DpnA (ORF #2895). Additionally REBASE predicts a second candidate for a GATC-specific DNA modification methyltransferase in ORF #8910. In contrast, RAST and Prodigal predict a 16S rRNA (guanine(966)-N(2))-methyltransferase for this ORF.
Table 4.
Putative C. coli BFR-CA-9557 restriction modification systems
| ORF #1 | Strand | Position in genome | Description | Type/subunit | Predicted rec. seq. |
|---|---|---|---|---|---|
| 465 | + | 86828-89146 | hsdR, Type I restriction-modification system2C restriction subunit R (EC 3.1.21.3) | I/R | - |
| 485 | + | 92205-93437 | hsdS, Type I restriction-modification system2C specificity subunit S (EC 3.1.21.3), EcoKI specificity protein | I/S | - |
| 495 | + | 94709-96196 | Type I restriction-modification system2C DNA-methyltransferase subunit M (EC 2.1.1.72) | I/M | - |
| 6540 | - | 1246404-1250480 | Type I restriction-modification system2C DNA-methyltransferase subunit M (EC 2.1.1.72) / Type I restriction-modification system2C specificity subunit S (EC 3.1.21.3) | I/MS | (GAGNNNNNGT)3,4 |
| 720 | + | 139123-140043 | fokIM_1, Modification methylase FokI, (EC 2.1.1.72), homologue to ulcer associated adenine specific DNA methyltransferase | II/M | (GGATG)3 |
| 1470 | - | 275768-271959 | Type IIS restriction enzyme Eco57I | II/S | - |
| 2605 | + | 488272-489375 | DNA modification methylase (Adenine-specific methyltransferase), fokIM_2, Modification methylase FokI, (EC 2.1.1.72) | II/M | GAATTC |
| 2895 | + | 541751-542602 | DNA modification methyltransferase, dpnA, Modification methylase DpnIIB (EC 2.1.1.72) | II/M | GATC |
| 6520 | - | 1240865-1243951 | N-6 adenine specific DNA methyltransferase | II/M | - |
| 7730 | + | 1464470-1465285 | DNA adenine methylase, dpnM, Modification methylase DpnIIA, EC 2.1.1.72 | II/M | - |
| 8910 | + | 1701685-1702374 | 16S rRNA (guanine(966)-N(2))-methyltransferase (EC 2.1.1.171) | II/M | GATC5 |
| 2225 | + | 409683-409970 | McrBC 5-methylcytosine-specific restriction endonuclease system2C McrB subunit2C putative | IV/R | - |
| 22302 | + | 409982-411148 | McrBC 5-methylcytosine-specific restriction endonuclease system2C McrB_1 subunit2C putative | IV/R | - |
| 22302 | + | 411208-412158 | McrBC 5-methylcytosine-specific restriction endonuclease system2C McrB_2 subunit2C putative | IV/R | - |
| 2235 | + | 412124-413494 | McrBC 5-methylcytosine-specific restriction enzyme subunit McrC | IV/R | - |
1ORF # according to the NCBI annotation pipeline for RAST and Prodigal ORF # see Additional files 3 and 4
2disrupted ORF
3predicted sequence does not correspond to any motif detected by SMRT sequencing
4According to REBASE ORF #6540 encodes a Type II RM-system
5In contrast to RAST and Prodigal REBASE predicts a second candidate for a GATC-specific DNA modification methyltransferase for this ORF
Restriction modification systems have been identified as outlined in section “Methods”. Column 1 to 3 denote the number, strand direction and genome position of the ORF as identified by the NCBI annotation pipeline. Column 4 contains the description of the ORF in terms of aggregated annotations from NCBI, RAST and Prodigal. The type and predicted recognition sequence of the motif are shown in columns 5 and 6
The two recognition sequences GAGNNNNNGT and GGATG predicted for the Type I restriction-modification system2C DNA-methyltransferase subunit M (ORF #6540) and modification methylase FokI homologue (ORF #720), respectively, do not correspond to any motif detected by SMRT sequencing in the genome of C. coli BfR-CA-9557.
REBASE search hits to the remaining ten RM-system subunit genes have not been associated with a specific recognition sequence. Therewith, REBASE was not able to predict a corresponding RM-system subunit for the motifs C, D, E, F, G, and H. This is most likely due to the fact that besides motifs A (RAATTY) and B (GATC) none of the published C. jejuni and H. pylori methylomes contains one of these motifs [49, 50, 57].
Motif B (GATC) is present in the methylomes of H. pylori 26695 and J99-R3 [57] but not in any of the publicly available C. jejuni methylomes. In the two H. pylori genomes ORFs hp0092 and jhp0085 encoding a RM-system IIM subunit have been assigned to this motif [58, 59]. The C. coli BfR-CA-9557 homologue of hp0092 (H. pylori 26695) demonstrates a sequence identity of 76 % (553/730; Query coverage: 86 %).
In contrast, the RAATTY motif (A) has not been detected in one of the two H. pylori methylomes, but is was present in C. jejuni F38011, NCTC 11168 as well as 81–176 and in all three isolates a corresponding RM-system subunit has been assigned [50, 57]. The C. coli BfR-CA-9557 homologue of cj0208 (NCTC 11168) demonstrates a sequence identity of 85 % (893/1050; Query coverage: 95 %).
The biological implications of this particular subset of RM-systems are difficult to predict, especially since the majority of methylation motifs and methylases in the genome of C. coli BFR-CA-9557 are quite novel. From the closely related bacterial species H. pylori it has been described in detail that RM-systems play a crucial role in forming strong barriers to prevent uptake of both plasmid and chromosomal DNA [60]. H. pylori bacterial cells are inherently very competent for DNA uptake, but this competence for DNA uptake varies significantly between specific strains. It has been observed that strains showing a very low endogenous RM-system activity demonstrate the highest transformation efficiency. Especially the presence of an RM-system homologous to MboI, which has been used for isoschizomer digestion assays in our study, has been described as major component of the H. pylori transformation barrier [60]. This MboI homologous RM-system named HpyIII is highly conserved among H. pylori strains and mediates protection against MboI digestion.
In MboI sensitive strains the hpyIIIR gene was found absent but a homologue to C. jejuni cj1602, namely hrgA, was detected. HpyIIIR negative but hrgA positive H. pylori strains have been associated with gastric cancer in Asian patients, while the pathogenic mechanism remains unclear [61].
Besides this role for DNA uptake the high intra-species variety in RM-system configuration and therewith the highly diverse methylation status of H. pylori chromosomal DNA was suggested to serve as a new typing system [62]. This DNA methylation based typing system may help to discriminate H. pylori isolates and as well isolates of related bacterial species like C. jejuni or C. coli for epidemiological and clinical purposes.
Conclusions
The objective of the study was to analyze the methylome of a clade 1 C. coli isolate using SMRT sequencing. The C. coli isolate BfR-9557 was tested positive in 5’-GmATC-3’ specific isoschizomer digestion assays and hence chosen for SMRT sequencing. Using one SMRT cell the complete genome of C. coli BfR-CA-9557 was sequenced and assembled into a single contig of 1.7 Mbp. The genome contains a CJIE1-like element prophage. SMRT Analysis Modification and Motif detection have identified eleven different dominant methylation motifs, while 14 RM-system subunits have been identified using REBASE and three different annotation pipelines. Only two of the REBASE predicted recognition sequences correspond to a specific motif detected by SMRT sequencing. The remaining 9 motifs did not correspond to any database record and are therefore characteristic for C. coli or at least for C. coli BfR-CA-9557. However, no comparable motifs have been described in the related microbial species C. jejuni or H. pylori.
Methods
Bacterial isolates, culture conditions, DNA extraction and MLST typing
50 C. coli isolates of different origin were obtained from the Federal Institute for Risk Assessment (BfR) in Berlin, Germany. The bacterial isolates were cultured on Columbia agar base (Merck) supplemented with 5 % sheep blood (BA) and incubated at 42 °C under microaerophilic conditions (5 % O2, 10 % CO2, 85 % N2) for 18 hours prior to genomic DNA extraction. Genomic DNA of all C. coli isolates was extracted using the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s instructions.
Species confirmation was performed using MALDI Biotyper system (Bruker Daltonics, Bremen, Germany). Results with MALDI Biotyper identification score values ≥2.000 were considered correct. Additionally multiplex PCR was used to discriminate between C. jejuni and C. coli [63, 64].
The MLS-type was established using amplification and sequencing primers reported before [65]. The cycling conditions were 94 °C for 1 min, followed by 35 cycles of 94 °C for 120 s, 50 °C for 60 s, 72 °C for 60 s, followed by a final elongation step of 72 °C for 5 min [65]. Amplicons of the seven genes included in the C. jejuni/C. coli MLST scheme were sent for sequencing to Seqlab Sequence Laboratories GmbH (Göttingen, Germany) using 10 pmol of the respective sequencing primer.
5’-GmATC-3’ specific Isoschizomer digestion assay
Screening for a C. coli strain that methylates adenines in G(m)ATC sequences was performed using isoschizomer digestion assays in order to identify a C. coli strain expressing a Dam homologue. Genomic/chromosomal bacterial DNA was digested by the three restriction enzymes MboI, DpnI and Sau3AI. MboI is responsive to Dam methylation. Therefore adenosyl-methylated GATC sequences become refractory to cleavage by MboI but turn susceptible to DpnI cleavage. Additionally the restriction endonuclease Sau3AI that is insensitive to adenosyl-methylation by the Dam system was used as digestion control [51].
Antimicrobial susceptibility testing
C. jejuni BfR-CA-9557 antimicrobial susceptibility to quinolones (ciprofloxacin) macrolides (erythromycin), aminoglycosides (gentamicin, streptomycin) and tetracyclines (tetracycline) was tested using the disc diffusion method according to the standards of the European Committee on Antimicrobial Susceptibility Testing (ESCMID) [66] and the microdilution assay according to the standards of the Clinical and Laboratory Standards Insitute (CLSI) [67], using cut-off values set by EUCAST (www.eucast.org). Antimicrobial test discs (ciprofloxacin, erythromycin, gentamicin, tetracycline) and EUCAMP2 microtiter plates were obtained from Oxoid/ThermoFisher Scientific (Wesel, Germany). For MIC analysis the Sensititre semiautomated system (Trek Diagnostic Systems, Inc, Cleveland, Ohio) was used.
Library Preparation and Sequencing
Genomic DNA of C. coli BfR-CA-9557 (DSM 100395) was extracted using the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s instructions. The DNA libraries have been prepared following the PacBio guidelines and sequenced on a SMRT cell using Pacific Biosciences RS sequencing technology (Pacific Biosciences, Menlo Park, USA) at Functional Genomics Center Zürich (FGCZ, Switzerland). Input genomic DNA concentration was measured using a Qubit Fluorometer dsDNA Broad Range assay (Life Technologies, Carlsbad, USA; p/n 32850). The SMRT bell was produced using the DNA Template Prep Kit 1.0 (Pacific Biosciences; p/n 100-259-100). 10 μg of gDNA were mechanically sheared to an avarage size distribution of 10Kb, using a Covaris gTube (Kbiosciences, Hoddesdon, UK; p/n 520079). A Bioanalyzer 2100 12 K DNA Chip assay (Agilent Technologies, Santa Clara, USA; p/n 5067–1508) was used to assess the fragment size distribution. 5 μg of sheared gDNA were DNA damage repaired and end-repaired using polishing enzymes. A blunt end ligation reaction followed by exonuclease treatment was performed to create the SMRT bell template. A Blue Pippin device (Sage Science, Beverly, USA) was used to size select the SMRT bell template and enrich the big fragments > 8Kbp. The sized selected library was quality inspected and quantified on the Agilent Bioanalyzer 12Kb DNA Chip and on a Qubit Fluorimeter.
A ready-to-sequence SMRT bell-polymerase Complex was created using the P6 DNA/Polymerase binding kit 2.0 (Pacific Biosciences, Menlo Park, USA; p/n 100-236-500) according to the manufacturer instructions.
The Pacific Biosciences RS2 instrument was programmed to load and sequence the sample on a single SMRT cell v3.0 (Pacific Biosciences p/n100-171-800), taking one movie of 120 minutes.
The MagBead loading method (PacBio, Menlo Park, USA; p/n 100-133-600) was chosen in order to improve the enrichment the longer fragments.
At the end of the run, a sequencing report was generated for every cell, via the SMRT portal. Thereby, the adapter dimer contamination, the sample loading efficiency, the obtained average read-length and the number of filtered sub-reads have been assessed.
Sequence analysis
Processing of the raw SMRT cell data was performed using the Pacific Biosciences SMRT Analysis System (version 2.3, January 2015; PacBio, Menlo Park, USA).
For de novo assembly of the C. coli BFR-CA-9557 genome the high-quality Hierarchical Genome Assembly Process (RS_HGAP_Assembly.2) was used with standard parameters (for details on algorithms please read https://github.com/PacificBiosciences/Bioinformatics-Training/wiki/HGAP-in-SMRT-Analysis).
The assembled genome was annotated using the rapid annotation using subsystem technology platform (RAST, http://rast.nmpdr.org) [68–70] and the Prodigal/Prokka annotation pipeline [71] implemented at Göttingen Genome Laboratory (G2L).
For identification of methylated bases and modification motifs the RS_Modification_and_Motif_Analysis.1 protocol within the SMRT Analysis System was used with standard parameters on the basis of the previously assembled genome.
Putative restriction modification systems have been identified using the Restriction-ModificationFinder-1.0 server (https://cge.cbs.dtu.dk/services/Restriction-ModificationFinder-1.0/) based on the Restriction Enzyme database (REBASE, http://rebase.neb.com/rebase/rebase.html) [56].
Additionally, homologues of published Campylobacter and Helicobacter restriction modification [46–50] systems have been identified by BLAST search.
Additional checking for clustered regularly interspaced short palindromic repeats (CRISPRs) and CRISPR-associated (cas) genes was performed using CRISPR-finder (http://crispr.u-psud.fr/Server/Advanced.CRISPRfinder.php) [72].
Ethics statement
Ethical clearance for the analysis was obtained from Ethics Committee of the University Medical Center Göttingen, Germany. As the bacterial isolates from human donors were already part of an anonymized strain collection and no evaluation including personal patient data has been performed the Ethics Committee of the University Medical Center Göttingen waived the need for written informed consent from the donor or the next of kin.
Availability of supporting data
The genome is available at NCBI as Campylobacter coli BFR-CA-9557 with AB430 locus tags (BioProject Accession: PRJNA285481 ID: 285481; BioSample: SAMN03754337; GenBank ID: CP011777). Additionally RM-system and methylation motifs can be accessed via the index of the REBASE database (http://tools.neb.com/genomes/) or directly via this link: http://tools.neb.com/genomes/view.php?view_id=35944.
The bacterial isolate Campylobacter coli BFR-CA-9557 was deposited in the strain collection of the Leibniz-Institut DSMZ-Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany (German Collection of Microorganisms and cell cultures); isolate ID: DSM 100395.
Acknowledgements
The authors’ work was supported by the Deutsche Forschungsgemeinschaft (DFG GR906/13-1) and the Forschungsförderungsprogramm of the Universitätsmedizin Göttingen (UMG), Germany. This publication was funded by the Open Access support program of the Deutsche Forschungsgemeinschaft and the publication fund of the Georg August Universität Göttingen. We would like to thank the Functional Genomics Center in Zurich, Switzerland (FGCZ) for processing and sequencing the samples.
Abbreviations
- CC
Clonal complex
- CJIE1
Campylobacter jejuni integrated element 1
- MLST
Multi-locus sequence typing
- RM-system
Restriction modification system
- ST
Sequence type
Additional files
Figure S1. SMRT sequencing of Campylobacter coli BFR-CA-9557. (A) Read length distribution of 74,742 continuous long reads (CLR) obtained from a single SMRT cell after filtering for low quality. The black line depicts the cumulated amount of bases covered by reads of a minimum size as shown on the x-axis. (B) Distribution of read quality values (1 = 100 %) for 74,742 CLRs after filtering. The black line denotes the average length of reads with a quality at least as good as indicated on the x-axis. (C) Subread length distribution of 142,135 subreads (i.e. individual fragments of CLRs). (DOC 74 kb)
Figure S2. Reference Coverage. (A) Reference coverage (number of read base pairs per position) of the polished assembly across the contig comprising 1,720,506 bp. Average reference coverage is 500.8-fold. (B) Histogram of reference coverage across the assembled contig. (DOC 73 kb)
RAST annotation of the C. coli BFR-CA-9557. Annotation of the C. coli BFR-CA-9557 genome using the rapid annotation using subsystem technology platform (RAST, http://rast.nmpdr.org). (XLS 3605 kb)
Prodigal annotation of the C. coli BFR-CA-9557. Annotation of the C. coli BFR-CA-9557 genome using the Prodigal/Prokka annotation pipeline. (XLS 727 kb)
Figure S3. Methylated bases in C. coli BFR-CA-9557. (A) Scatter plot of modification quality values and per-strand coverage of 101,019 bases detected as methylated in the C. coli BFR-CA-9557 genome. (B) Histogram of modification quality values for all bases. (DOC 115 kb)
Footnotes
Competing interests
All authors declare no competing interests.
Authors’ contributions
AEZ and KS conceived the study idea, organized the bacterial isolate collection, and drafted the manuscript. AMG, RL and OB performed bacterial culture, DNA isolation, isoschizomer digestion assays and MLST-PCR. TL, GSL, AT, JS and coordinated DNA sequencing and conducted bioinformatics analysis. TL, AT and JS performed annotation of the genome. UG participated in the study design and helped in drafting the manuscript. All authors assisted in drafting the manuscript and all authors read and approved the final manuscript.
References
- 1.Dasti JI, Tareen AM, Lugert R, Zautner AE, Gross U. Campylobacter jejuni: a brief overview on pathogenicity-associated factors and disease-mediating mechanisms. Int J Med Microbiol. 2010;300(4):205–211. doi: 10.1016/j.ijmm.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Zautner AE, Herrmann S, Groß U. Campylobacter jejuni - The Search for virulence-associated factors. Arch Lebensmittelhyg. 2010;61(3):91–101. [Google Scholar]
- 3.Allos BM. Association between Campylobacter infection and Guillain-Barre syndrome. J Infect Dis. 1997;176(Suppl 2):S125–128. doi: 10.1086/513783. [DOI] [PubMed] [Google Scholar]
- 4.Zautner AE, Johann C, Strubel A, Busse C, Tareen AM, Masanta WO, et al. Seroprevalence of campylobacteriosis and relevant post-infectious sequelae. Eur J Clin Microbiol Infect Dis. 2014;33(6):1019–1027. doi: 10.1007/s10096-013-2040-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Authority EFS. The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2013. EFSA J. 2015;13(1):3991. doi: 10.2903/j.efsa.2018.5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.KR LL, AR S: Campylobacteriosis. http://www.cfsph.iastate.edu/Factsheets/pdfs/campylobacteriosis.pdf 2013.
- 7.Sheppard SK, McCarthy ND, Falush D, Maiden MC. Convergence of Campylobacter species: implications for bacterial evolution. Science. 2008;320(5873):237–239. doi: 10.1126/science.1155532. [DOI] [PubMed] [Google Scholar]
- 8.Sheppard SK, Dallas JF, Wilson DJ, Strachan NJ, McCarthy ND, Jolley KA, et al. Evolution of an agriculture-associated disease causing Campylobacter coli clade: evidence from national surveillance data in Scotland. PloS One. 2010;5(12):e15708. doi: 10.1371/journal.pone.0015708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Litrup E, Torpdahl M, Nielsen EM. Multilocus sequence typing performed on Campylobacter coli isolates from humans, broilers, pigs and cattle originating in Denmark. J Appl Microbiol. 2007;103(1):210–218. doi: 10.1111/j.1365-2672.2006.03214.x. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Mukherjee S, Hoffmann M, Kotewicz ML, Young S, Abbott J, et al. Whole-genome sequencing of gentamicin-resistant Campylobacter coli isolated from U.S. retail meats reveals novel plasmid-mediated aminoglycoside resistance genes. Antimicrob Agents Chemother. 2013;57(11):5398–5405. doi: 10.1128/AAC.00669-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pearson BM, Rokney A, Crossman LC, Miller WG, Wain J, van Vliet AH. Complete genome sequence of the Campylobacter coli clinical isolate 15–537360. Genome Announc. 2013;1:6. doi: 10.1128/genomeA.01056-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tatusova T, Ciufo S, Fedorov B, O'Neill K, Tolstoy I. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 2014;42(Database issue):D553–559. doi: 10.1093/nar/gkt1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skarp-de Haan CP, Culebro A, Schott T, Revez J, Schweda EK, Hanninen ML, et al. Comparative genomics of unintrogressed Campylobacter coli clades 2 and 3. BMC Genomics. 2014;15:129. doi: 10.1186/1471-2164-15-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fouts DE, Mongodin EF, Mandrell RE, Miller WG, Rasko DA, Ravel J, et al. Major structural differences and novel potential virulence mechanisms from the genomes of multiple Campylobacter species. Plos Biol. 2005;3(1):e15. doi: 10.1371/journal.pbio.0030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards VP, Lefebure T, Pavinski Bitar PD, Stanhope MJ. Comparative characterization of the virulence gene clusters (lipooligosaccharide [LOS] and capsular polysaccharide [CPS]) for Campylobacter coli, Campylobacter jejuni subsp. jejuni and related Campylobacter species. Infect Genet Evol. 2013;14:200–213. doi: 10.1016/j.meegid.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheppard SK, Didelot X, Jolley KA, Darling AE, Pascoe B, Meric G, et al. Progressive genome-wide introgression in agricultural Campylobacter coli. Mol Ecol. 2013;22(4):1051–1064. doi: 10.1111/mec.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Criscuolo A, de la Blanchardiere A, Coeuret S, Passet V, Saguet-Rysanek V, Vergnaud M, et al. Draft genome sequence of campylobacter coli Strain IPSID-1 isolated from a patient with immunoproliferative small intestinal disease. Genome Announc. 2014;2:2. doi: 10.1128/genomeA.00079-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar R, Rao DN. Role of DNA methyltransferases in epigenetic regulation in bacteria. Subcell Biochem. 2013;61:81–102. doi: 10.1007/978-94-007-4525-4_4. [DOI] [PubMed] [Google Scholar]
- 19.Srikhanta YN, Maguire TL, Stacey KJ, Grimmond SM, Jennings MP. The phasevarion: a genetic system controlling coordinated, random switching of expression of multiple genes. Proc Natl Acad Sci U S A. 2005;102(15):5547–5551. doi: 10.1073/pnas.0501169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reisenauer A, Kahng LS, McCollum S, Shapiro L. Bacterial DNA methylation: a cell cycle regulator? J Bacteriol. 1999;181(17):5135–5139. doi: 10.1128/jb.181.17.5135-5139.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reisenauer A, Shapiro L. DNA methylation affects the cell cycle transcription of the CtrA global regulator in Caulobacter. EMBO J. 2002;21(18):4969–4977. doi: 10.1093/emboj/cdf490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braun RE, O'Day K, Wright A. Autoregulation of the DNA replication gene dnaA in E. coli K-12. Cell. 1985;40(1):159–169. doi: 10.1016/0092-8674(85)90319-8. [DOI] [PubMed] [Google Scholar]
- 23.Yamaki H, Ohtsubo E, Nagai K, Maeda Y. The oriC unwinding by dam methylation in Escherichia coli. Nucleic Acids Res. 1988;16(11):5067–5073. doi: 10.1093/nar/16.11.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marinus MG, Casadesus J. Roles of DNA adenine methylation in host-pathogen interactions: mismatch repair, transcriptional regulation, and more. FEMS Microbiol Rev. 2009;33(3):488–503. doi: 10.1111/j.1574-6976.2008.00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tock MR, Dryden DT. The biology of restriction and anti-restriction. Curr Opin Microbiol. 2005;8(4):466–472. doi: 10.1016/j.mib.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 26.Wilson GG, Murray NE. Restriction and modification systems. Annu Rev Genet. 1991;25:585–627. doi: 10.1146/annurev.ge.25.120191.003101. [DOI] [PubMed] [Google Scholar]
- 27.Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003;31(7):1805–1812. doi: 10.1093/nar/gkg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE--enzymes and genes for DNA restriction and modification. Nucleic Acids Res. 2007;35(Database issue):D269–270. doi: 10.1093/nar/gkl891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casadesus J, Low D. Epigenetic gene regulation in the bacterial world. Microbiol Mol Biol Rev. 2006;70(3):830–856. doi: 10.1128/MMBR.00016-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murray NE. Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle) Microbiol Mol Biol Rev. 2000;64(2):412–434. doi: 10.1128/MMBR.64.2.412-434.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dryden DT, Murray NE, Rao DN. Nucleoside triphosphate-dependent restriction enzymes. Nucleic Acids Res. 2001;29(18):3728–3741. doi: 10.1093/nar/29.18.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vovis GF, Horiuchi K, Zinder ND. Kinetics of methylation of DNA by a restriction endonuclease from Escherichia coli B. Proc Natl Acad Sci U S A. 1974;71(10):3810–3813. doi: 10.1073/pnas.71.10.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pingoud A, Jeltsch A. Structure and function of Type II restriction endonucleases. Nucleic Acids Res. 2001;29(18):3705–3727. doi: 10.1093/nar/29.18.3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meisel A, Bickle TA, Kruger DH, Schroeder C. Type III restriction enzymes need two inversely oriented recognition sites for DNA cleavage. Nature. 1992;355(6359):467–469. doi: 10.1038/355467a0. [DOI] [PubMed] [Google Scholar]
- 35.Sutherland E, Coe L, Raleigh EA. McrBC: a multisubunit GTP-dependent restriction endonuclease. J Mol Biol. 1992;225(2):327–348. doi: 10.1016/0022-2836(92)90925-A. [DOI] [PubMed] [Google Scholar]
- 36.Ahmed IH, Manning G, Wassenaar TM, Cawthraw S, Newell DG. Identification of genetic differences between two Campylobacter jejuni strains with different colonization potentials. Microbiol. 2002;148(Pt 4):1203–1212. doi: 10.1099/00221287-148-4-1203. [DOI] [PubMed] [Google Scholar]
- 37.Poly F, Threadgill D, Stintzi A. Identification of Campylobacter jejuni ATCC 43431-specific genes by whole microbial genome comparisons. J Bacteriol. 2004;186(14):4781–4795. doi: 10.1128/JB.186.14.4781-4795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poly F, Threadgill D, Stintzi A. Genomic diversity in Campylobacter jejuni: identification of C. jejuni 81-176-specific genes. J Clin Microbiol. 2005;43(5):2330–2338. doi: 10.1128/JCM.43.5.2330-2338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller WG, Pearson BM, Wells JM, Parker CT, Kapitonov VV, Mandrell RE. Diversity within the Campylobacter jejuni Type I restriction-modification loci. Microbiol. 2005;151(Pt 2):337–351. doi: 10.1099/mic.0.27327-0. [DOI] [PubMed] [Google Scholar]
- 40.Holt JP, Grant AJ, Coward C, Maskell DJ, Quinlan JJ. Identification of Cj1051c as a major determinant for the restriction barrier of Campylobacter jejuni strain NCTC11168. Appl Environ Microbiol. 2012;78(22):7841–7848. doi: 10.1128/AEM.01799-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, et al. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature. 2000;403(6770):665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- 42.Gundogdu O, Bentley SD, Holden MT, Parkhill J, Dorrell N, Wren BW. Re-annotation and re-analysis of the Campylobacter jejuni NCTC11168 genome sequence. BMC Genomics. 2007;8:162. doi: 10.1186/1471-2164-8-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bayliss CD, Bidmos FA, Anjum A, Manchev VT, Richards RL, Grossier JP, et al. Phase variable genes of Campylobacter jejuni exhibit high mutation rates and specific mutational patterns but mutability is not the major determinant of population structure during host colonization. Nucleic Acids Res. 2012;40(13):5876–5889. doi: 10.1093/nar/gks246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim JS, Li J, Barnes IH, Baltzegar DA, Pajaniappan M, Cullen TW, et al. Role of the Campylobacter jejuni Cj1461 DNA methyltransferase in regulating virulence characteristics. J Bacteriol. 2008;190(19):6524–6529. doi: 10.1128/JB.00765-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kivisto RI, Kovanen S, Skarp-de Haan A, Schott T, Rahkio M, Rossi M, et al. Evolution and comparative genomics of Campylobacter jejuni ST-677 clonal complex. Genome Biol Evol. 2014;6(9):2424–2438. doi: 10.1093/gbe/evu194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morley L, McNally A, Paszkiewicz K, Corander J, Meric G, Sheppard SK, et al. Gene loss and lineage-specific restriction-modification systems associated with niche differentiation in the Campylobacter jejuni sequence type 403 clonal complex. Appl Environ Microbiol. 2015;81(11):3641–3647. doi: 10.1128/AEM.00546-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakajima T, Ono K, Tazumi A, Misawa N, Moore JE, Millar BC, et al. Molecular characterisation of a Type III restriction-modification system in Campylobacter upsaliensis. Br J Biomed Sci. 2014;71(2):66–72. doi: 10.1080/09674845.2014.11669967. [DOI] [PubMed] [Google Scholar]
- 48.Yun HC, Kreft RE, Castillo MA, Ehrlich GD, Guymon CH, Crouch HK, et al. Comparison of PCR/electron spray ionization-time-of-flight-mass spectrometry versus traditional clinical microbiology for active surveillance of organisms contaminating high-use surfaces in a burn intensive care unit, an orthopedic ward and healthcare workers. BMC Infect Dis. 2012;12:252. doi: 10.1186/1471-2334-12-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krebes J, Morgan RD, Bunk B, Sproer C, Luong K, Parusel R, et al. The complex methylome of the human gastric pathogen Helicobacter pylori. Nucleic Acids Res. 2014;42(4):2415–2432. doi: 10.1093/nar/gkt1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Loughlin JL, Eucker TP, Chavez JD, Samuelson DR, Neal-McKinney J, Gourley CR, et al. Analysis of the Campylobacter jejuni genome by SMRT DNA sequencing identifies restriction-modification motifs. PloS One. 2015;10(2):e0118533. doi: 10.1371/journal.pone.0118533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edmonds P, Hall BM, Edwards WR, Hartline KM. Presence of methylated adenine in GATC sequences in chromosomal DNAs from Campylobacter species. J Bacteriol. 1992;174(24):8156–8157. doi: 10.1128/jb.174.24.8156-8157.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wright S, Wilson S, Miller WG, Mandrell RE, Siletzky RM, Kathariou S. Differences in methylation at GATC sites in genomic DNA of Campylobacter coli from turkeys and swine. Appl Environ Microbiol. 2010;76(21):7314–7317. doi: 10.1128/AEM.00934-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karlyshev AV, Quail MA, Parkhill J, Wren BW. Unusual features in organisation of capsular polysaccharide-related genes of C. jejuni strain X. Gene. 2013;522(1):37–45. doi: 10.1016/j.gene.2013.03.087. [DOI] [PubMed] [Google Scholar]
- 54.Parker CT, Horn ST, Gilbert M, Miller WG, Woodward DL, Mandrell RE. Comparison of Campylobacter jejuni lipooligosaccharide biosynthesis loci from a variety of sources. J Clin Microbiol. 2005;43(6):2771–2781. doi: 10.1128/JCM.43.6.2771-2781.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parker CT, Gilbert M, Yuki N, Endtz HP, Mandrell RE. Characterization of lipooligosaccharide-biosynthetic loci of Campylobacter jejuni reveals new lipooligosaccharide classes: evidence of mosaic organizations. J Bacteriol. 2008;190(16):5681–5689. doi: 10.1128/JB.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE--a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015;43(Database issue):D298–299. doi: 10.1093/nar/gku1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K, et al. The methylomes of six bacteria. Nucleic Acids Res. 2012;40(22):11450–11462. doi: 10.1093/nar/gks891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kong H, Lin LF, Porter N, Stickel S, Byrd D, Posfai J, et al. Functional analysis of putative restriction-modification system genes in the Helicobacter pylori J99 genome. Nucleic Acids Res. 2000;28(17):3216–3223. doi: 10.1093/nar/28.17.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin LF, Posfai J, Roberts RJ, Kong H. Comparative genomics of the restriction-modification systems in Helicobacter pylori. Proc Natl Acad Sci U S A. 2001;98(5):2740–2745. doi: 10.1073/pnas.051612298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ando T, Xu Q, Torres M, Kusugami K, Israel DA, Blaser MJ. Restriction-modification system differences in Helicobacter pylori are a barrier to interstrain plasmid transfer. Mol Microbiol. 2000;37(5):1052–1065. doi: 10.1046/j.1365-2958.2000.02049.x. [DOI] [PubMed] [Google Scholar]
- 61.Ando T, Wassenaar TM, Peek RM, Jr, Aras RA, Tschumi AI, van Doorn LJ, et al. A Helicobacter pylori restriction endonuclease-replacing gene, hrgA, is associated with gastric cancer in Asian strains. Cancer Res. 2002;62(8):2385–2389. [PubMed] [Google Scholar]
- 62.Xu Q, Morgan RD, Roberts RJ, Blaser MJ. Identification of type II restriction and modification systems in Helicobacter pylori reveals their substantial diversity among strains. Proc Natl Acad Sci U S A. 2000;97(17):9671–9676. doi: 10.1073/pnas.97.17.9671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vandamme P, Van Doorn LJ, Al Rashid ST, Quint WG, van der Plas J, Chan VL, et al. Campylobacter hyoilei Alderton et al. 1995 and Campylobacter coli Veron and Chatelain 1973 are subjective synonyms. Int J Syst Bacteriol. 1997;47(4):1055–1060. doi: 10.1099/00207713-47-4-1055. [DOI] [PubMed] [Google Scholar]
- 64.Wang G, Clark CG, Taylor TM, Pucknell C, Barton C, Price L, et al. Colony multiplex PCR assay for identification and differentiation of Campylobacter jejuni, C. coli, C. lari, C. upsaliensis, and C. fetus subsp. fetus. J Clin Microbiol. 2002;40(12):4744–4747. doi: 10.1128/JCM.40.12.4744-4747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dingle KE, Colles FM, Wareing DR, Ure R, Fox AJ, Bolton FE, et al. Multilocus sequence typing system for Campylobacter jejuni. J Clin Microbiol. 2001;39(1):14–23. doi: 10.1128/JCM.39.1.14-23.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ge B, Wang F, Sjolund-Karlsson M, McDermott PF. Antimicrobial resistance in Campylobacter: susceptibility testing methods and resistance trends. J Microbiol Methods. 2013;95(1):57–67. doi: 10.1016/j.mimet.2013.06.021. [DOI] [PubMed] [Google Scholar]
- 67.Jorgensen JH, Hindler JF. New consensus guidelines from the clinical and laboratory Standards Institute for antimicrobial susceptibility testing of infrequently isolated or fastidious bacteria. Clin Infect Dis. 2007;44(2):280–286. doi: 10.1086/510431. [DOI] [PubMed] [Google Scholar]
- 68.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST) Nucleic Acids Res. 2014;42(Database issue):D206–214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, et al. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep. 2015;5:8365. doi: 10.1038/srep08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 72.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35(Web Server issue):W52–57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]