

Abstract
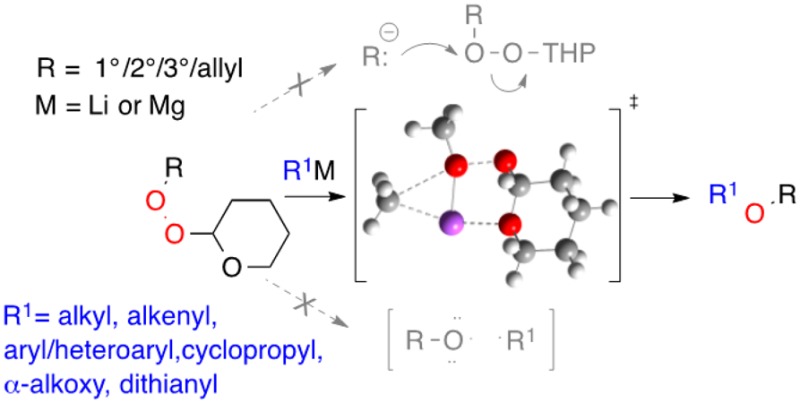
Although transfer of electrophilic alkoxyl (“RO+”) from organic peroxides to organometallics offers a complement to traditional methods for etherification, application has been limited by constraints associated with peroxide reactivity and stability. We now demonstrate that readily prepared tetrahydropyranyl monoperoxyacetals react with sp3 and sp2 organolithium and organomagnesium reagents to furnish moderate to high yields of ethers. The method is successfully applied to the synthesis of alkyl, alkenyl, aryl, heteroaryl, and cyclopropyl ethers, mixed O,O-acetals, and S,S,O-orthoesters. In contrast to reactions of dialkyl and alkyl/silyl peroxides, the displacements of monoperoxyacetals provide no evidence for alkoxy radical intermediates. At the same time, the high yields observed for transfer of primary, secondary, or tertiary alkoxides, the latter involving attack on neopentyl oxygen, are inconsistent with an SN2 mechanism. Theoretical studies suggest a mechanism involving Lewis acid promoted insertion of organometallics into the O–O bond.
Introduction
Methods for ether synthesis are overwhelmingly based upon the attack of nucleophilic oxygen on electrophilic carbon.1−4 Our lab has been interested in expanding the scope of an umpoled strategy based upon reaction of carbanions with electrophilic oxygen.5 The reaction of dialkyl peroxides with carbanions, while long known,6 has largely remained a curiosity applied mainly to transfer of methoxyl and other unhindered alkoxides.7,8 Even a slight increase in steric bulk results in reduced yields,7 and reactions of di-t-butyl peroxide furnish multiple products.9,10 We became interested in a general method for selective transfer of 1°, 2°, or 3° alkoxides from a mixed peroxide to a carbanion. In approaching this problem, we were encouraged by evidence for the strong influence of substituent groups on peroxide reactivity. Bistrimethylsilyl peroxide efficiently transfers OSiMe3 to a variety of organometallic reagents;11 curiously, tBuOOSiMe3 transfers neither OSiMe3 nor tBuO.12 Lithiated peroxides, sometimes described as “oxenoid” in character, transfer LiO to a variety of carbanions,13 and similar reactivity patterns have been observed with other metalloperoxides.14 Bisacyl peroxides even transfer acyloxy groups to enolates and enamines.15 However, transfer of alkoxide is more challenging. The strain within bicyclic peroxides has been harnessed to facilitate reaction of more hindered peroxides; unfortunately, the presence of the scaffold limits the range of potential targets.16 Electronic activation in the form of peresters has been employed to achieve transfer of tertiary alkoxides to Grignard reagents,17 but broad application is limited by the propensity of peresters to undergo C-to-O rearrangement or fragmentation to carbonyls.18,19 We were particularly intrigued by reports describing enhanced reactivity of acyclic peroxyacetals and peroxyaminals with Grignard reagents.20,21 However, the reactions of peroxyaminals displayed variable regioselectivity,20 while reactions of a mixed ethoxy/peroxy acetal required a significant excess of the Grignard reagent.21 In preliminary work, we discovered that tetrahydropyranyl (THP) monoperoxyacetals selectively transfer the attached OR to lithiated 1,3-dithianes.5b We now describe investigations of reactions between carbanions and several classes of organic peroxides, with an emphasis on monoperoxyacetals. (Figure 1).
Figure 1.

Substrate classes.
Results
Table 1 illustrates preparation of substrates. Base-promoted alkylation of hydroperoxides with primary and secondary triflates22 provided good yields of t-butyl/alkyl peroxides (8a–11a), monoperoxyacetals (8c,238b–10b, 12b, 13b(24)), and a 2-methoxyethyl peroxide (9e). Tetrahydropyranyl monoperoxyacetals (14b, 15b) were also prepared through acetalization of hydroperoxides with dihydropyran.25 Ag(I)-promoted hydroperoxide alkylation was used to prepare allyl peroxide 16b.26 Alkyl/silyl peroxides 8d and 10d were prepared through silylation of hydroperoxides.27
Table 1. Substrate Synthesis.


Thermal Stability
Thermal analysis was conducted on one of the dialkyl peroxides (10a) and one of the monoperoxyacetals (10b) to provide some baseline stability data for each class. The results, detailed in Supporting Information, revealed that 10a and 10b are relatively stable, undergoing exothermic decomposition only once heated past 170 and 120 °C, respectively.
Table 2 illustrates the reactivity of t-butyl/alkyl and trialkylsilyl/alkyl peroxides toward sp3 organometallics. t-Butyl peroxide 8a reacted with an excess of RLi over a period of several hours to furnish ethers derived from selective attack on the less hindered oxygen. The corresponding reactions with Grignard reagents were successful, but somewhat slower. The corresponding trialkylsilyl peroxide (8d) reacted slowly with n-BuLi to furnish a large number of products (TLC).
Table 2. Reactions of Dialkyl and Alkyl/Silyl Peroxides.

| substrate | R1M (equiv) | time (h) | product | yield (%) |
|---|---|---|---|---|
| 8a | n-BuLi (2) | 4 | 17a | 70 |
| 8a | hexMgBr (3) | 8 | 17b | 42 |
| 8a | allylMgBr (2) | 0.5 | 17c | 27 |
| 8a | allylLi (2.5)a | 4 | 17c | 70 |
| 8d | n-BuLi (2) | 8 | multiple |
From allylSnBu3 and n-BuLi.
Scheme 1 summarizes reactions of THP monoperoxyacetals with sp3 organometallics. Reactions with RLi proceeded slowly at −78 °C but were complete within minutes at 0 °C. The monoperoxyacetals failed to react with Grignard reagents at −78 °C but were consumed rapidly at 0 °C, providing ethers in yields equal or superior to those obtained with RLi. Transfer of 1°, 2°, or 3° alkoxides occurred with similar yields; in each case; we observed only the products of regioselective displacement of OTHP.
Scheme 1. Reaction of sp3 RM with Peroxyacetals.

(a) RLi (1.1 equiv), −78 °C; (b) hexylMgBr (1.1 equiv), 0 °C; (c) Me3SiCH2MgCl (1.3 equiv), 0 °C.
Reactions with sp2 Organometallics
Monoperoxyacetal 8b reacted readily with PhLi (−78 °C) and PhMgBr (0 °C) to furnish good yields of the phenyl ether (18a, Scheme 2). Reaction with an alkenyl magnesium bromide or vinyl lithium, the latter generated in situ, furnished the corresponding enol ethers; the moderate yields may in part reflect decomposition during purification. Reaction of 2-thienyl lithium with primary monoperoxyacetals furnished a moderate yield of 2-alkoxythiophenes 18d and 18e, representatives of a class of molecules previously explored only to a limited extent.28
Scheme 2. Reaction of Peroxyacetals with sp2 RM.

THF, −78 to rt (RLi) or 0 °C to rt (RMgX).
Metalated Alkynes
Alkynyllithium or alkynylmagnesium reagents failed to react with a dialkyl peroxide (8a) or a monoperoxyacetal (8b); starting materials could be recovered even after prolonged reaction periods (eq 1).
 |
1 |
Reaction with Resonance-Stabilized Nucleophiles
Neither monoperoxyacetals nor a dialkyl peroxide underwent intermolecular C–O bond formation with enolates (eq 2) or azaenolates (eq 3); only slow decomposition was observed. The results were not entirely unexpected given our observations during tandem reactions of enolates with iodoperoxides.5a In a control experiment, reaction of a THF solution of 8b with stoichiometric potassium t-butoxide (eq 4) resulted in slow formation of byproducts indicative of E1CB fragmentation.19,29 The corresponding t-butyl peroxide 8a (not shown) decomposed more slowly to furnish citronellal.
 |
2 |
Lithiated Dithianes
Lithiated 1,3-dithianes have been widely applied as acyl anion equivalents for reaction with carbon electrophiles.30 In the only report of their reactivity toward peroxides, reaction with bistrimethylsilyl peroxide results in preferential transfer of a silyl group.11a We recently demonstrated successful reaction of THP monoperoxyacetals with lithiated dithianes to furnish S,S,O-orthoesters and derived difluoroalkyl ethers.5b Being curious about the influence of the peroxide electrophile on the reactivity toward inductively stabilized anions, we investigated reactions of a monoperoxyacetal, a silyl/alkyl peroxide, and a dialkyl peroxide toward a common lithiated dithiane (Scheme 3). The monoperoxyacetal 10b and alkyl/silyl peroxide 10d gave similar yields of difluoroether 19, isolated following fluorodesulfurization of the initial orthoester products. In contrast, the mixed alkyl/t-butyl peroxide 10a was much less reactive. Reactions were conducted exclusively in THF; previous investigations of reactions of lithiated trithianes5b observed no benefit from the presence of additives (LiCl, MgBr2) or polar cosolvents (HMPA).
Scheme 3. Reactivity towards Lithiated Dithianes.

Attempted Synthesis of Trihaloethers
Trifluoromethyl ethers are of interest as degradation-resistant structural components in agrochemicals and drug molecules.31 However, methods for their introduction remain limited.32 We recently reported an indirect approach to introduction of OCF3 groups via reaction of a monoperoxyacetal with a lithiated trithiane, followed by fluorodesulfurization of the resulting S,S,O-orthocarbonate,5b and we became interested in a more direct approach based upon reaction of a peroxide with a trifluoromethyl anion. This approach requires the targeted C–O bond formation to occur more rapidly than α-elimination of the nucleophile.33 We began our investigations with the Ruppert-Prakash reagent, CF3SiMe3.32,33 The trifluoromethyl ether was not observed despite application of different activators, solvents (DMF, THF), or reaction temperatures (Scheme 4). We also failed to detect ether formation for base-promoted reaction with trifluoroacetaldehyde hydrate,34 or with KCF3 generated via low temperature deprotonation of fluoroform.35 Formation of aldehydes derived from Kornblum fragmentation of the peroxide (see eq 4) was observed in some of these reactions. Interested in whether the displacements might be more successful in the presence of a lithium cation, we investigated reactions with lithiated trihalomethanes but saw no reaction at temperatures (−110 °C for LiCCl3, −50 °C for LiCBr3) where the reagents were stable.
Scheme 4. Attempted Synthesis of Trihalomethyl Ethers.

Reaction with α-Alkoxylithium Reagents
Lithiated ethers (α-alkoxylithiums) have been employed as synthons for acyl anions and hydroxymethyl anions.36 As illustrated in Table 3, the alkoxylithium reagent generated from a tributylstannyl MOM ether (20) reacted with primary alkyl/t-butyl peroxides or a monoperoxyacetal to give moderate yields of the mixed O,O-acetals.
Table 3. Synthesis of O,O-Acetals.
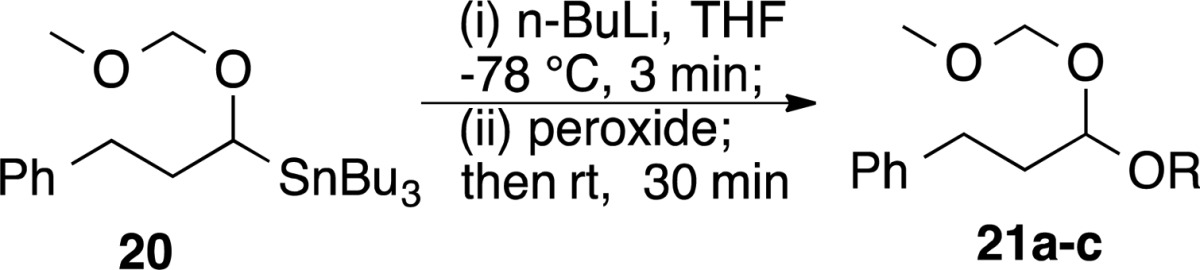
| peroxide | acetal | yield (%) |
|---|---|---|
| C8H17OOtBu (11a) | 21a: R = octyl | 57 |
| Ph(CH2)3OOtBu (9a) | 21b: R = (CH2)3Ph | 55 |
| C10H21OOTHP (12b) | 21c: R = decyl | 55 |
The same reaction, when repeated with methoxypropyl peroxyacetal 8c, did not proceed to completion. The mixed acetal (21d) was isolated in low yield and accompanied by the tertiary alcohol (22) derived from trapping of acetone generated from the cleaved acetal (eq 5).
 |
5 |
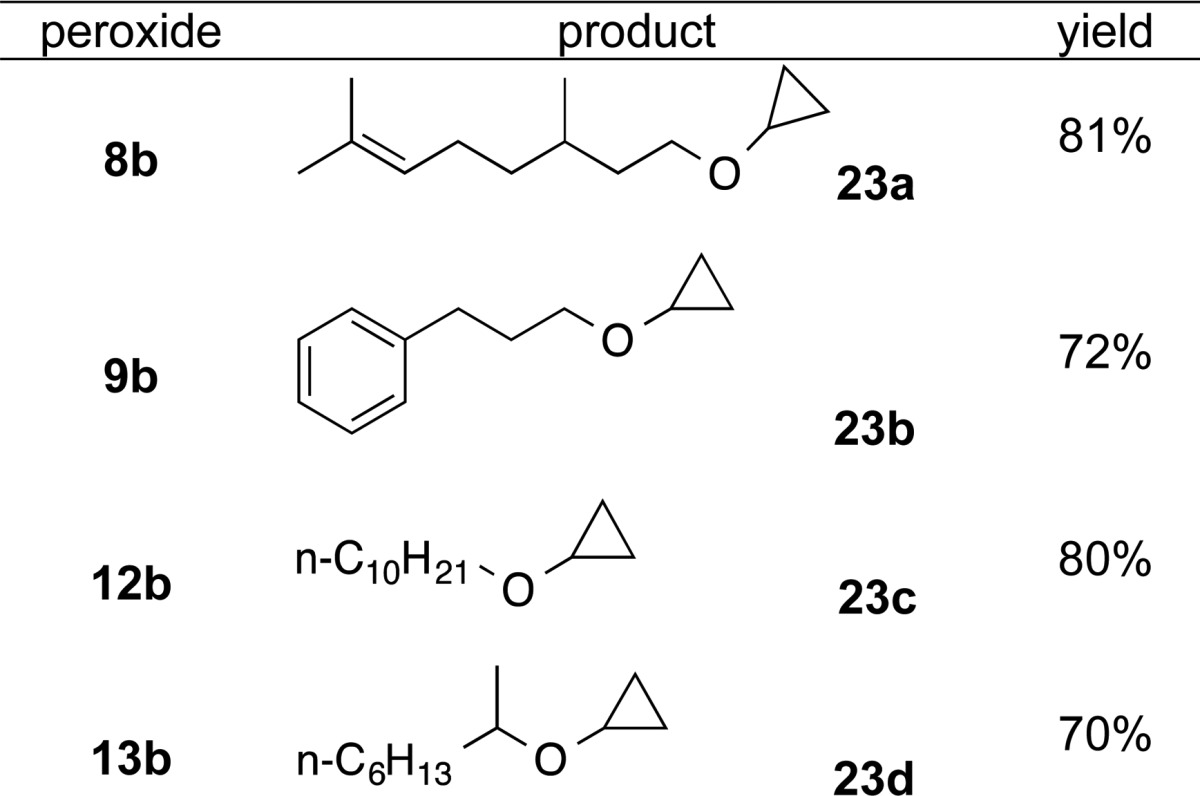
Application to Cyclopropyl Ethers
We were interested in application of the reaction of monoperoxyacetals and organometallics as an approach to cyclopropyl ethers, a relatively challenging class of targets for which multiple approaches have been reported.37 In approaching this class of reactions, we were encouraged by the reported reaction of t-butyl perbenzoates with cyclopropyl magnesium bromide.38 We found that the reaction of monoperoxyacetals with cyclopropyl magnesium bromide furnishes an efficient approach to primary and secondary cyclopropyl alkyl ethers (Table 4).
Table 4. Synthesis of Cyclopropyl Ethers.
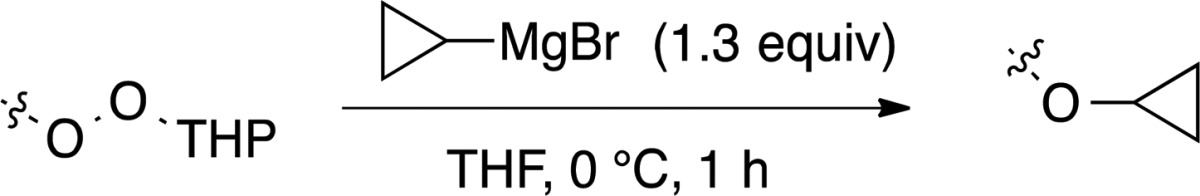
Mechanistic Investigations
In an effort to gain information about the possible role of single electron transfer in the C–O bond-forming step, we investigated reactions of phenylmethylpropyl peroxides; the derived alkoxy radical undergoes cleavage to acetone and benzyl radical at ∼108/s (Scheme 5).39 Dialkyl peroxide 24a was prepared through a known procedure.40 Silyl peroxide 24d was prepared through Co(II)-mediated peroxidation of methallyl benzene with oxygen and triethylsilane.41 THP acetal 24b was prepared by deprotection of the silyl peroxide and acetalization of the intermediate hydroperoxide.25
Scheme 5. Preparation of Radical Probes.

Monoperoxyacetal 24b underwent rapid reaction with n-BuLi to furnish mainly the butyl ether (25a), accompanied by smaller amounts of the tertiary alcohol 26 and recovered starting material (Table 5). The corresponding reaction with excess hexylMgBr furnished only the hexyl ether (25b) resulting from displacement of OTHP. In contrast, reaction of di-t-alkyl peroxide 24a with n-BuLi generated multiple products and only trace amounts of the ether. The reaction of silyl peroxide 24d with n-BuLi provided the most convincing evidence for the intermediacy of radicals in the form of a significant amount of pentylbenzene product of radical–radical coupling.
Table 5. RM Reactions with Radical Probesa.
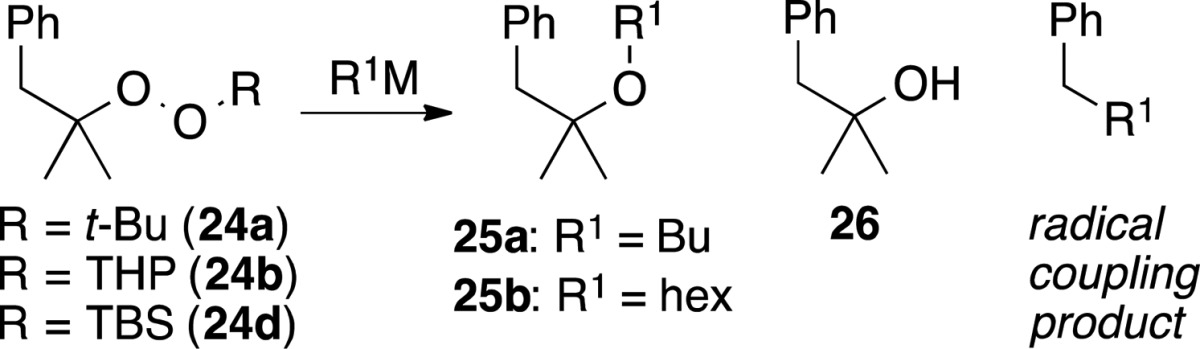
| subs | R1M | conv. (%) | ether | alcohol | coupling |
|---|---|---|---|---|---|
| 24b | n-BuLi | 93 | R1 = Bu (55%) | 15% | |
| 24b | hexMgBr | 100 | R1 = hex (75%) | ||
| 24a | n-BuLi | 55 | 32% | PhC5H11 (3%) | |
| 24d | n-BuLi | 59 | 11%b | PhC5H11 (10%) |
Conditions: n-BuLi (1 equiv), THF, −78 °C, allow to warm to rt; hexylMgBr (2 equiv), 0 °C, allow to warm to rt.
Et3SiOH isolated in 24% yield.
Competition between a Dialkyl Peroxide and a Monoperoxyacetal
A competition between a dialkyl peroxide and a monoperoxyacetal for capture of a limiting quantity of n-BuLi generated only the ether (27) derived from the monoperoxyacetal (eq 6).
 |
6 |
sp3 vs sp2 RLi
A competition between n-BuLi and PhLi for a limiting quantity of a monoperoxyacetal revealed a strong preference for reaction with the sp3 reagent (eq 7).
 |
7 |
Importance of Chelation vs Acetal Structure
In an effort to probe the basis for the enhanced reactivity of the monoperoxyacetals, we investigated C–O bond formation with 2-methoxyethyl peroxide 9e. This substrate, which possesses the potential for metal chelation but lacks a peroxyacetal, provided the corresponding either in lower yields than had been observed even with simple t-butyl peroxides (eq 8).
 |
8 |
Theoretical Investigations
In an effort to gain better understanding of the reactions described above and the influence of reaction conditions on product formation, we conducted B3LYP/6-31+G(d,p) calculations for the simple model reaction of the transfer of methoxide from a methyl peroxide or peroxyacetal to a methyl anion (Scheme 6). Our calculations focused on the impact of changing the metal from Li to Na and of changing the leaving group from Ot-Bu to OTHP.
Scheme 6. Predicted Relative Gas-Phase Energies and Solution-Phase Enthalpies of Intermediates and Transition States.

B3LYP/6-31+G(d,p), 0 K; units in kcal/mol.
THF at 298 K with the SMD continuum solvent model; units in kcal/mol and enthalpies in parentheses.
All four sets of conditions appear to favor the same mechanism: first, formation of a metal-peroxy complex (II), second, passage through a four-centered transition state (TS III), third, formation of a complex of the incipient ether with the metal alkoxide (IV), and, finally, cleavage to final products (V). (Early calculations found the same mechanism for the addition of alkyl lithium reagents to carbon–carbon and carbon–oxygen multiple bonds.)42,43 The calculations predict that the lithiated system is significantly more reactive than the sodiated system. This suggests that the metal ion’s interaction with the peroxy moiety is primarily electrostatic;42 the greater charge density on the smaller Li+ leads to greater stabilization. The presence of the THP group is found to stabilize the initial organometallic-peroxide complex (II) and to dramatically lower the energy for insertion into the peroxide bond. An approximate treatment of THF solvation using the SMD continuum model predicts the relative enthalpies of most intermediates and transition states to be several kcal/mol higher in energy than the gas-phase calculations. However, the solvation calculations predict the same trends in reactivity: the Li+ ion and the THP substituent both significantly stabilize both the reactive complex and the transition state for insertion.
Discussion
Whereas reactions of dialkyl peroxides are largely limited to transfer of methoxide or primary alkoxide,6−8,10 monoperoxyacetals have significantly enhanced reactivity toward unstabilized carbanions, transferring primary, secondary, or tertiary alkoxides through highly regioselective attack on the nonacetal oxygen of the peroxide. In contrast to peresters, monoperoxyacetals appear to promote alkoxide transfer without significantly destabilizing the peroxide.44
Reaction is observed between monoperoxyacetals and sp3 and sp2 RLi and RMgX reagents at −78 °C (organolithium reagents) or 0 °C (Grignard reagents). Both acyclic (2-methoxyprop-2-yl) and cyclic (THP) peroxyacetals offer good reactivity; however, the acyclic peoxyacetals sometime generate side products derived from liberation of a reactive carbonyl group in the presence of the organometallic. The monoperoxyacetals do not undergo intermolecular reactions with enolates or similar stabilized carbanions, paralleling earlier observations from our lab.5a The monoperoxyacetals also fail to react with metalated alkynes, an interesting outcome given the reported reactivity of lithiated hydroperoxides toward carbanions,13,45 as well as our observation of successful reaction of peroxyacetals with inductively stabilized anions such as lithiated dithianes and lithiated ethers. However, the lack of reactivity toward sp carbanions is consistent with results observed with bistrimethylsilyl peroxide, which undergoes mainly silyl exchange with lithiated alkynes.12
Several mechanisms have been discussed with regard to the reaction of dialkyl peroxides with unstabilized organometallics. While early results with unhindered peroxides were consistent with SN2-type attack on the O–O bond,7 reactions of di-t-alkyl peroxides have been reported to proceed via intermediate alkoxy radicals.10,46 Our work found no evidence for radical intermediates in attack of organolithium reagents on THP peroxyacetals.
If the reactions involve neither simple displacement nor cleavage to alkoxy radicals, then what? Lithiated peroxides are often thought to react with nucleophiles through oxenoid-type insertion into the C–Li bond; attack on alkenyl lithium and cyclopropyl lithium appears to occur without loss of stereochemistry.13 However, whereas the transfer of LiO from lithiated hydroperoxides to organolithium reagents has been calculated to involve in-line arrangement of the carbanion, the lithiated oxygen, and the departing oxygen,13b our calculations suggest side-on insertion of the RLi into the Lewis acid activated O–O bond. This theoretical result is supported by the similar yields obtained for transfer of primary, secondary, and tertiary alkoxides, a result which would seem to rule out an SN2-type attack on the backside of the breaking O–O bond. Calculations predict, and experiments confirm, that the presence of the acetal group significantly lowers the energy for insertion of unstabilized organometallics and directs the reaction toward exclusive transfer of the nonanomeric oxygen. The bidentate coordination available to the organometallic/Lewis acid is not sufficient to explain the results, as evidenced by the poor reactivity of the methoxyethyl peroxide. However, the interaction of Lewis acids with peroxyacetals and ozonides (1,2,4-trioxolanes) has been previously demonstrated to activate both C–O and O–O bonds.47
In conclusion, we have shown that monoperoxyacetals, readily prepared and easily handled derivatives of hydroperoxides, enable highly efficient intermolecular transfer of alkoxide to unstabilized organolithium and organomagnesium reagents along with some inductively stabilized organolithium species. The peroxyacetal activating group promotes highly selective transfer of the nonanomeric OR, regardless of steric bulk. The bond-forming process, which appears to involve metal-promoted insertion into the O–O linkage, appears to be mechanistically distinct from similar reactions of alkyl or alkyl silyl peroxides. The new methodology offers a new tool with which to approach ethers, including some poorly accessible through existing methods.
Experimental Procedures
General Experimental
Reagents and solvents were used as supplied commercially, except for DCM and THF, which were distilled from CaH2 and Na/Ph2CO, respectively. Reagents supplied as solutions were dispensed based upon manufacturer-supplied concentrations. Reactions were conducted under an atmosphere of N2 except where noted. Thin-layer chromatography (TLC) was performed on 0.25 mm hard-layer silica G plates visualized with a UV lamp or by staining: 1% ceric sulfate and 10% ammonium molybdate in 10% H2SO4 (general stain, after heating); 3% vanillin in 3% H2SO4 in EtOH (general stain, after heating); or a 1% N,N′-dimethyl-p-phenylenediamine in 1:20:100 acetic acid/water/methanol (specific for peroxides).48 Unless otherwise noted, NMR spectra were acquired in CDCl3; 1H spectra are reported as chemical shift (multiplicity, J couplings in Hz, number of protons). IR spectra were recorded as neat films on a ZrSe crystal; selected absorbances are reported in cm–1. Abbreviations: hexane = Hex; EA = ethyl acetate; THF = tetrahydrofuran; TBDMS (or TBS) = tert-butyldimethylsilyl; THP = tetrahydropyranyl.
Calculations
Optimized geometries, harmonic vibrational frequencies, and electronic energies of all structures in Scheme 6 were obtained with the B3LYP level of density functional theory and the 6-31+G(d,p) basis set.49,50–502 All minima reported (structures I, II, IV, and V) contain all real frequencies, and all transition states (structures TS III) contain one imaginary frequency. Intrinsic reaction coordinate calculations confirmed that each transition state connected a metal-peroxy complex II and the corresponding metal alkoxide-ether complex IV. The relative electronic energies were corrected by the differences in zero-point vibrational energy scaled by 0.9806.51 Solution-phase enthalpies were determined as follows: The thermal corrections to the electronic energies were calculated based on the B3LYP/6-31+G(d,p) geometries and harmonic frequencies. Solvation corrections were based on single-point energy calculations using the continuum SMD model of Truhlar and co-workers.52
t-Butyl Hydroperoxide (1)
[75-91-2], 5.5 M in decane, was used as purchased.
2-Tetrahydropyranyl (THP) Hydroperoxide (2)
[4676-84-0] was synthesized using a modified version of a known procedure.24 To a 0 °C solution of 50 wt % aq. H2O2 (6.66 mL, ∼118 mmol) was added 10% aq. H2SO4 (0.1 mL) The mixture was stirred for 10 min, after which 3,4-dihydro-2H-pyran (4.94 g, 58.8 mmol) was added over a period of 3 min. The reaction was stirred for 1 h at 0 °C and then diluted with sat. aq. NH4Cl (15 mL). The solution was extracted with ether (150 mL), and the organic layer was washed with aq. sat. (NH4)2SO4 (5 × 10 mL). The resulting solution was dried over Na2SO4 and concentrated on a rotary evaporator. The residue obtained was purified by silica gel flash chromatography using a gradient of 4–25% EA/Hex to afford the hydroperoxide as a thick and colorless oil (4.60 g, 66%). Spectral data were identical to those previously reported.19
2-Methoxyprop-2-yl Hydroperoxide (3)
[10027-74-4] was prepared as a colorless oil (82%, 878 mg) from 2,3-dimethyl-2-butene (10.0 mmol, 841 mg) using a known procedure.23,55Rf = 0.3 (15% EA:Hex).
8-Hydroperoxy-2,6-dimethyloct-2-ene (4)
[123369-58-4] was prepared by alkylation
of a 1,1-dihydroperoxycyclodecane,
followed by hydrolysis of the resulting bisperoxyacetal.22
A solution of 3,7-dimethyloct-6-en-1-ol (0.936 g, 6 mmol) in dry DCM (18 mL) was cooled to 0 °C, and triflic anhydride (1.4 equiv) and 2,6-lutidine (1.5 equiv) were sequentially added via syringe. The reaction was stirred for 15 min at 0 °C and then diluted with ice-cold hexane (∼40 mL). The resulting solution was washed with ice cold 0.1 M aq. KHSO4 (40 mL), and the separated aqueous layer was extracted with another portion of cold Hex (25 mL). The combined organic layers were dried over Na2SO4 and concentrated on a rotary evaporator (bath temperature 10 °C). The residue was subjected to 0.5 mmHg for approximately 5 min to generate nearly pure 3,7-dimethyloct-6-en-1-yl trifluoromethanesulfonate (1.693 g, 98%) as a light brown oil, which was placed in a −20 °C freezer and used within an hour:56Rf = 0.59 (10% EA/Hex); 1H NMR: 5.11–5.07 (m, 1H), 4.61–4.57 (m, 2H), 2.06–1.96 (m, 2H), 1.93–1.85 (m, 1H), 1.72–1.68 (m, 1H), 1.70 (s, 3H), 1.66–1.64 (m, 1H), 1.62 (s, 3H), 1.42–1.32 (m, 1H), 1.29–1.20 (m, 1H), 0.96 (d, J = 6.3 Hz, 3H); 13C NMR: 131.8, 123.9, 118.5 (q, J = 323 Hz), 76.2, 36.6, 36.0, 28.6, 25.6, 25.1, 19.0, 17.6.
To a 0 °C solution of cyclododecanone-1,1-dihydroperoxide (2.196 g, 9.469 mmol, synthesized as previously described)57 in dry THF (50 mL) was added KOtBu (2 equiv), followed by the triflate described above (6.0 g, 20.8 mmol). The reaction mixture was stirred until the starting material was no longer visible (TLC, ∼20 min). The reaction mixture was then quenched with water (25 mL) and extracted with EA (50 mL × 2). The combined organic layers were dried over Na2SO4 and concentrated on a rotary evaporator. The residue was purified by silica chromatography using 1% EA/hex to furnish 1,1-bis-(3,7 dimethyl-6-octenylperoxy) cyclododecane (2.84 g, 59%) as a colorless oil: Rf = 0.75 (10% EA:Hex); 1H NMR: 5.10 (m, 2H), 4.17–3.99 (m, 4H), 2.07–1.91 (m, 4H), 1.72–1.64 (m, 9H), 1.69 (s, 3H), 1.61 (s, 3H), 1.58–1.53 (m, 7H), 1.48–1.28 (m, 20H), 1.24–1.15 (m, 2H), 0.92 (d, J = 6.5, 6H); 13C NMR: 131.2, 124.6, 113.1, 73.4, 37.1, 34.6, 29.7, 27.0, 26.1, 25.7, 25.4, 22.3, 21.9, 19.6, 19.4, 17.6; HRMS (TOFMS-ES): Calcd for C32H60O4Na (M + Na)+ 531.4389 found: 531.4373; IR: 2926, 2850, 1445, 1052.
To a room temperature solution of the bisperoxyacetal (1.01 g, 2 mmol) in THF (20 mL) was added a solution of 50% aq. H2SO4 (1.8 mmol, 6 equiv), and the reaction mixture was stirred at 55 °C until starting material was nearly consumed (TLC, 1–3 h). The reaction was then quenched with saturated aq. Na2CO3 (25 mL), and the resulting mixture was extracted with EA (30 mL × 2). The combined organic layers were dried over Na2SO4 and concentrated on a rotary evaporator. The crude product was purified by silica chromatography using 1–3% EA/Hex to furnish hydroperoxide 4 as a colorless oil (0.364 mg, 53%). The molecule has previously been reported without characterization:58Rf = 0.4 (10% EA/Hex); 1H NMR: 8.53 (s, 1H), 5.09 (m, 1H), 4.10–4.00 (m, 2H), 2.06–1.93 (m, 2H), 1.73–1.63 (m, 1H), 1.68 (s, 3H), 1.60 (s, 3H), 1.54–1.35 (m, 2H), 1.33–1.14 (m, 2H), 0.91 (d, J = 6.5, 3H); 13C NMR: 131.3, 124.6, 75.5, 37.1, 34.3, 29.5, 25.7, 25.4, 19.5, 17.6. IR: 3617–3101 (broad peak), 2915, 1455, 1377, 1053 (mass spec not attempted due to volatility).
1-Hydroperoxy-1-methylcyclohexane (5)
[4952-03-8] was prepared through a two-step procedure.
Triethyl(1-methylcyclohexylperoxy)silane
(CAS 634600-89-8) was prepared through a modification of a reported procedure.59 1-Methyl cyclohexene (0.96 g, 10 mmol) and Et3SiH (2.32 g., 20 mmol) were sequentially added to a solution of Co(acac)2 (1 mmol) in ethanol (35 mL). The reaction mixture was placed under an atmosphere of O2 (balloon) until no starting material was observed (TLC, ∼14 h). The residue obtained upon concentration in vacuo was purified by silica gel chromatography using 1–8% EA:Hex to furnish the 1-methyl-1-cyclohexyl triethylsilyl peroxide as a as colorless oil (0.844 g, 34%): Rf = 0.7 (10% EA/Hex). Spectral data matched those previously reported.60
To a solution of the triethylsilylperoxide (0.78 g, 3.19 mmol) in THF (10 mL) was added tetra n-butyl ammonium fluoride (1 M solution in THF, 1.2 equiv).47 The reaction was stirred at room temperature until no starting material could be observed (TLC, ∼5 min). The residue obtained upon concentration in vacuo was diluted with hexane (30 mL) and washed with water (10 mL). The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by short-column (3″) flash chromatography with 5% EA/Hex (two iterations were required to remove silanol/siloxane byproducts) to furnish the hydroperoxide 5 as a colorless oil (0.266 g, 64%). Although the hydroperoxide is commercially available, NMR data are difficult to access and are provided here for convenience: Rf = 0.4 (10% EA:Hex); 1H NMR: 7.34 (s, 1H), 1.80–1.74 (m, 2H), 1.63–1.53 (m, 2H), 1.51–1.38 (m, 5H), 1.34–1.29 (m, 1H), 1.25 (s, 3H); 13C NMR: 81.7, 34.4, 25.6, 23.8, 22.2, 6.55, 5.76.
3-Phenylpropyl Hydroperoxide (6)
[60956-33-4] was prepared using a route analogous to that employed for hydroperoxide 4.
3-Phenylpropyl trifluoromethanesulfonate [66950-73-0] was prepared (1.34 g, quant.) as a light brown oil from 3-phenylpropanol (0.681 g, 5 mmol) using the procedure described above: Rf = 0.5 (10% EA/Hex). Other spectral data matched those in a previous report.5b
Using a similar procedure as described earlier, cyclododecanone 1,1-dihydroperoxide (1.05 g, 4.545 mmol) was reacted with the 3-phenylpropyl triflate (2.6 g, 10 mmol) to furnish the bisperoxyacetal (1.4381 g, 67%) as a colorless oil: Rf = 0.6 (15% EA/Hex); 1H NMR: 7.32–7.29 (m, 4H), 7.23–7.21 (m, 6H), 4.11 (t, J = 6.4, 4H), 2.73 (t, J = 7.5, 4H), 2.02–1.95 (m, 4H), 1.74–1.70 (m, 4H), 1.56–1.46 (m, 4H), 1.45–1.31 (m, 14H); 13C NMR: 141.7, 128.4, 128.3, 125.8, 113.3, 74.2, 32.3, 29.5, 27.1, 26.1, 22.3, 22.0, 19.4; IR: 2914, 2845, 1462, 1053
Hydrolysis of the bisperoxyacetal (1.35 g, 2.88 mmol) by a similar procedure as described above furnished hydroperoxide 6 as a colorless oil (0.725 g, 82%): Rf = 0.57 (20% EA/Hex). Spectral data matched those previously reported.61
10-Phenyldecane Hydroperoxide (7)
The title compound was prepared by an analogous strategy as described for hydroperoxides 4 and 6.
10-phenyldecyl trifluoromethanesulfonate (1.098 g, quant.) was prepared from 10-phenyldecanol (0.70 g, 3 mmol) as a light brown oil: Rf = 0.65 (10% EA/Hex); 1H NMR: 7.33–7.28 (m, 2H), 7.22–7.18 (m, 3H), 4.56 (t, J = 6.5 Hz, 2H), 2.63 (t, J = 7.7 Hz, 2H), 1.85 (m, 2H), 1.69–1.59 (m, 2H), 1.46–1.33 (m, 12H); 13C NMR: 142, 128.4, 128.2, 125.5, 118.6 (q, J = 320 Hz), 77.7, 35.9, 31.5, 29.4, 29.37, 29.30, 29.27, 29.22, 28.8, 25.0. HRMS (ESI): Calcd for C17H25F3NaO3 S (M + Na) 389.1374, found 389.1369.
Using a similar procedure as described above, reaction of 1,1-dihydroperoxycyclododecanone (0.293 g, 1.26 mmol) and the 10-phenyl decyl triflate (1.02 g, 2.787 mmol) was used to prepare 1,1-bis-(10-phenyldecyldioxy)cyclodecanone: (0.834 g, 99%) as a colorless oil: Rf = 0.74 (15% EA/Hex); 1H NMR: 7.32–7.29 (m, 4H), 7.21–7.19 (m, 6H), 4.09 (t, J = 6.6, 4H), 2.62 (t, J = 7.7, 4H), 1.72–1.59 (m, 12H), 1.37–1.30 (m, 42H); 13C NMR: 142.9, 128.4, 128.2, 125.5, 113.1, 75.0, 36.0, 31.5, 29.57, 29.53, 29.48, 29.36, 27.8, 27.0, 26.2, 26.1, 22.3, 21.9, 19.4. HRMS (TOF-MS-ES+): Calcd for C44H72O4Na (M + Na)+ 687.5328 found 687.5300; IR: 2914, 2845, 1462, 1053.
Using a procedure similar to that described above, hydrolysis of the bis-(10-phenyldecyl)peroxyacetal (0.755 g, 1.14 mmol) furnished 10-phenyldecyl hydroperoxide as a colorless oil (0.243 g, 64%): Rf = 0.55 (15% EA/Hex); 1H NMR: 8.04 (t, J = 6.5, 1H), 7.33–7.28 (m, 2H), 7.22–7.20 (m, 3H), 4.04 (t, J = 6.6, 2H), 2.63 (t, J = 7.7, 2H), 1.68–1.61 (m, 4H), 1.32 (m, 12H); 13C NMR: 142.9, 128.4, 128.2, 125.5, 76.6, 36.0, 31.5, 29.5, 29.4, 29.3, 27.5, 25.9; HRMS (TOF-MS-EI+): Calcd for C16H25O (M – OH)+ 233.1905; found: 233.1920; IR: 3390, 2922, 2852, 1453, 696.
Synthesis of Peroxides via Alkylation of Hydroperoxides with Triflates (Method A)
Synthesis of dialkyl peroxides from alkyl triflates was based upon reported procedures.22 The hydroperoxide (10 mmol in a minimum amount of THF) was added under nitrogen to a 0 °C solution of KOtBu (10 mmol) in dry THF (50 mL). The reaction mixture was stirred for 3 min, after which was added preformed alkyl triflate (10 mmol, neat from syringe, remaining triflate rinsed into the reaction using 1 mL of THF). The reaction was stirred for 15 min at the same temperature and then quenched with water (20 mL). The mixture was extracted with 10% EA/Hex (50 mL × 2), and the organic layer was dried over Na2SO4. The residue obtained upon concentration was purified by flash silica chromatography using 1% EA/Hex.
Peroxides via Acetalization (Method B)
Acetalization of alkyl hydroperoxides to form tetrahydropyranyl (THP) monoperoxyacetals employed a modification of a reported procedure.25 A mixture of alkyl hydroperoxide (1.0 mmol) and 2,3-dihydropyran (1.0 mmol) was cooled to 0 °C, after which was added 0.05 mL of a solution of 10% H2SO4 in THF. The stirred reaction mixture was allowed to warm to rt over a period of 45 min. The reaction was diluted with 10% ether/Hex (15 mL), and the resulting solution was washed with water (2 mL). The separated organic layer was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel chromatography using 4% EA/Hex.
Ag-Promoted Alkylation (Method C)
Dialkyl peroxide synthesis using silver oxide was based upon a modification of reported procedures.26b,26c The alkyl hydroperoxide (1.0 mmol) was added to a suspension (EA, 10 mL) of freshly prepared silver oxide (1.2 mmol). Alkyl halide (1.0 mmol) was added, and the reaction was stirred at rt until no starting material could be observed (TLC, approximately 4–10 h). The reaction mixture was filtered through a small pad of Celite, and the residue was washed with EA (10–20 mL). The combined filtrates were concentrated, and the residue was purified by silica gel chromatography using 1–3% EA/Hex.
Trialkylsilylation of Hydroperoxides (Method D)
Trialkylsilylation was performed based upon a reported procedure, except for a different order of addition and the use of water rather than aqueous acid for the wash.27 To a room temperature solution of alkyl hydroperoxide (1.0 mmol) in DMF (2 mL) was added tert-butyl dimethylsilyl chloride (1.2 mmol), followed by imidazole (1.4 mmol). The reaction was stirred for 1 h and then diluted with water (10 mL). The hexane extracts (1 × 50, 1 × 10 mL) were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel chromatography (4.0 in. tall × 0.5 in. column) using 1% ether/Hex.
8-(tert-Butylperoxy)-2,6-dimethyloct-2-ene (8a)
[1592933-34-0]: Using method A (above), t-butyl hydroperoxide (5.5 M in decane, 0.588 mL, 3.24 mmol) was reacted with 3,7-dimethyl-6-octenyl triflate (described above in relation to preparation of peroxide 4, 1.025 g, 3.559 mmol) to furnish 8a as a colorless oil (0.511 g, 69%); Rf = 0.6 (10% EA/Hex). Spectral data matched those in literature reports.5a
2-(3,7-Dimethyloct-6-ene-1-yl)peroxy)-tetrahydro-2H-pyran (8b)
Using method A, THP hydroperoxide 1 (0.63 g, 5.34 mmol) was reacted with 3,7-dimethyl-6-octenyl triflate (1.69 g, 5.87 mmol) to furnish peroxide 9a as a colorless oil (0.854 g, yield: 62%) Rf = 0.42 (10% EA/Hex); 1H NMR: 5.15 (t, J = 3.5 Hz, 1H), 5.10 (m, 1H), 4.19–4.00 (m, 2H), 4.02 (m, 1H), 3.66–3.61 (m, 1H), 1.98 (m, 2H), 1.78–1.71 (m, 2H), 1.70–1.65 (m, 1H), 1.68 (s, 3H), 1.64–1.53 (m, 5H), 1.60 (s, 3H), 1.49–1.42 (m, 1H), 1.41–1.32 (m, 1H), 1.23–1.1 (m, 1H), 0.92 (d, J = 6.6 Hz, 3H); 13C NMR: δ: 131.2, 124.7, 100.8, 73.7, 62.5, 37.1, 34.6, 29.6, 27.96, 25.7, 25.4, 25.1, 19.7, 19.5, 17.6. HRMS (ESI+ TOF) calcd for C15H28NaO3 (M + Na): 279.1931; found: 279.1934; IR: 2923, 2861, 1440, 1040.
8-((2-Methoxypropan-2-yl)peroxy)-2,6-dimethyloct-2-ene (8c)
Using method A, hydroperoxide 3 (0.674 g, 6.35 mmol, 1.5 equiv) was reacted with 3,7-dimethyl-6-octenyl triflate (1.21 g, 4.2 mmol) to furnish 8c as a colorless oil (0.627 g, yield: 61%); Rf = 0.4 (10% EA/Hex); 1H NMR: 5.1 (m, 1H), 4.11–4.01 (m, 2H), 3.33 (s, 3H), 2.06–1.91 (m, 2H), 1.71–1.64 (m, 1H), 1.69 (s, 3H), 1.61 (s, 3H), 1.62–1.54 (m, 1H), 1.47–1.31 (m, 2H), 1.40 (s, 6H), 1.24–1.10 (m, 1H), 0.91 (d, J = 6.5 Hz, 3H); 13C NMR131.2, 124.7, 104.5, 73.4, 49.2, 37.1, 34.6, 29.6, 25.7, 25.4, 22.76, 22.74, 19.5, 17.6; HRMS (TOF-MS-ES+) calcd for C14H28NaO3 (M + Na): 267.1931; found: 267.1926; IR: 2954, 2874, 1457, 836.
t-Butyldimethylsilyl 3,7-Dimethyl-6-octenyl Peroxide (8d)
Using method D, alkyl hydroperoxide 4 (0.22 g, 1.279 mmol) was silylated with t-butyldimethylsilyl chloride (TBS-Cl) to furnish 8d as a colorless oil (0.212 g, 58%): Rf = 0.75 (10% EA/Hex); 1H NMR: 5.12 (m, 1H), 4.06–3.97 (2H), 2.06–1.91 (m, 2H), 1.69 (s, 3H), 1.67–1.59 (m, 1H), 1.61 (s, 3H), 1.59–1.51 (m, 1H), 1.46–1.30 (m, 2H), 1.22–1.15 (m, 1H), 0.95 (s, 9H), 0.91 (d, J = 6.6), 0.17 (s, 6H); 13C NMR: 131.2, 124.6, 75.3, 37.1, 34.5, 29.6, 26.1, 25.7, 25.4, 19.6, 18.1, 17.6, −5.9; HRMS (TOF-MS-EI+) calcd for C16H34NaO2Si (M + Na): 309.2220; found: 309.2218; IR: 2928, 2857, 1461, 834.
3-tert-Butylperoxypropyl Benzene (9a)
[419568-76-6]: Using method A, tert-butyl hydroperoxide (1.8 mL, 5.0–6.0 M in decane, 10 mmol) was reacted with 3-phenyl propyl triflate (2.68 g, 10 mmol) to furnish 9a as a colorless liquid (1.5151 g. 73%). Rf (10% EA/Hex) = 0.65. Spectral data matched those in a literature report.62
2-(3-Phenylpropylperoxy)tetrahydro-2H-pyran (9b)
[1630792-49-2]: Using method A, THP hydroperoxide 1 (1.18 g, 10 mmol) was reacted with 3-phenylpropyl triflate (2.68 g, 10 mmol) to furnish peroxide 9b as a colorless oil (1.937 g, 82%); Rf = 0.66 (15% EA/Hex). Spectral properties matched those in a literature report.5b
3-(2-Methoxyethylperoxy)propyl Benzene (9e)
The title compound was prepared in two steps.
2-Methoxyethyl trifluoromethanesulfonate [112981-50-7] was prepared (2.08 g, ∼quant) from 2-methoxyethanol (760 mg, 10.0 mmol) using the procedure described above and used without purification. Spectral data matched those reported.63Rf = 0.5 (30% EA/hex).
Using method A, alkyl hydroperoxide 6 (0.70 g, 4.6 mmol) was reacted with 2-methoxyethyl trifluoromethanesulfonate (0.956 g, 4.6 mmol) to furnish 9e as a colorless oil (0.723 g, 74%): Rf = 0.62 (20% EA/Hex); 1H NMR: 7.32–7.27 (2H), 7.22–7.18 (3H), 4.15 (m, 2H), 4.05 (t, J = 6.4, 2H), 3.62 (m, 2H), 3.40 (s, 3H), 2.71 (t, J = 7.8, 2H), 2.01–1.93 (2H); 13C NMR: 141.6, 128.46, 128.39, 125.9, 73.59, 73.54, 69.8, 59.1, 32.2, 29.4; HRMS (TOF-MS-CI+) calcd for C12H18O3Na [M + Na]+: 233.1154; found: 233.1152; IR: 2924, 1496, 1453, 745.
10-(tert-Butylperoxy)decylbenzene (10a)
Using method A, t-butyl hydroperoxide (1.41 mL, 5.0–6.0 M in decane, 7.76 mmol) was reacted with 10-phenyldecyl triflate (3.125 g, 8.54 mmol) to furnish 10a as a colorless oil (2.42 g, 92%). Rf = 0.73 (10% EA/Hex); 1H NMR: 7.33–7.20 (m, 2H), 7.22–7.17 (m, 3H), 3.96 (t, J = 6.7 Hz, 2H), 2.62 (t, J = 7.8 Hz, 2H), 1.69–1.56 (m, 4H), 1.32–1.28 (m, 12H), 1.27 (s, 9H); 13C NMR: δ: 142.9, 128.4, 128.2, 125.5, 80.0, 75.1, 36.0, 31.5, 29.5, 29.3, 27.8, 26.3, 26.2. HRMS (ESI+ TOF): Calcd for C20H34NaO2 (M + Na): 329.2451; found: 329.2470; IR: 2924.8, 2853.72, 1361.5, 697.
2-(10-Phenyldecylperoxy)tetrahydro-2H-pyran (10b)
[1630792-52-7]: Using method A, THP hydroperoxide 2 (0.20 g, 1.77 mmol) was reacted with 10-phenyldecyl triflate (0.65 g, 1.775 mmol) to furnish 10b as a colorless oil (0.448 g, 75%); Rf = 0.42 (10% EA/Hex). Spectral properties matched those in a literature report.5b
t-Butyldimethylsilyl 10-Phenyldecyl Peroxide (10d)
Using method D, alkyl hydroperoxide 7 (0.22 g, 0.89 mmol) was converted into the corresponding silyl peroxide 10d, which was obtained as a colorless oil (0.267 g, 82%): Rf = 0.8 (10% EA/Hex); 1H NMR: 7.32–7.28 (m, 2H), 7.22–7.19 (m, 3H), 3.99 (t, J = 6.6 Hz, 2H), 2.63 (t, J = 7.7 Hz, 2H), 1.66–1.56 (m, 4H), 1.32 (m, 12H), 0.97 (s, 9H), 0.19 (s, 6H); 13C NMR: 142.9, 128.4, 128.2, 125.5, 76.6, 36.0, 31.5, 29.5, 29.3, 27.7, 26.2, 26.1, 18.1, −5.8; HRMS (ESI+ TOF) calcd for C22H40NaO2Si (M + Na): 387.2690; found: 387.2677; IR: 2925, 2854, 1462, 1248.
tert-Butylperoxy Octane (11a)
[38375-34-7]: 1-Octyl trifluoromethanesulfonate [71091-89-9] was prepared (3.08 g, 98%) as a light brown oil from 1-octanol (1.56 g, 12 mmol) using the general procedure described above. The 1H NMR spectrum matched data previously reported:64Rf = 0.52 (10% EA/Hex); 1H NMR: 4.55 (t, J = 6.54, 2H), 1.84 (m, 2H), 1.46–1.40 (m, 2H), 1.36–1.29 (m, 8H), 0.90 (t, J = 6.9, 3H); 13C NMR: 118.2 (q, J = 320.1 Hz), 76.7, 31.6, 29.2, 28.9, 28.8, 25.0, 22.5, 14.0.
t-Butyl hydroperoxide (1.81 mL, 5.5 M in decane, 10 mmol) was reacted with octyl triflate (2.88 g, 11 mmol) to furnish 11a as a colorless oil (1.78 g, 88%): Rf = 0.79 (10% EA/Hex). The molecule has been previously reported without spectral characterization:651H NMR: 3.93 (t, J = 6.7 Hz, 2H), 1.63–1.55 (m, 2H), 1.34–1.26 (m, 10H), 1.25 (s, 9H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR: 79.9, 75.1, 31.8, 29.4, 29.2, 27.8, 26.3, 26.2, 22.6, 14.0; HRMS (ESI-TOF-MS) Calcd for C12H26NaO2 (M + Na): 225.1825; found: 225.1840; IR: 2924, 2856, 1361, 1197.
2-(Decylperoxy)-tetrahydro-2H-pyran (12b)
[1630792-51-6]: Decyl trifluoromethanesulfonate [53059-89-5] was prepared (3.1 g, quant = 2.9 g) as a light brown oil from 1-decanol (1.58 g, 10.0 mmol) using the procedure described above. Spectra closely matched those in previous reports:66Rf = 0.52 (10% EA/Hex); 1H NMR: 4.55 (t, J = 6.5 Hz, 2H), 1.84 (m, 2H), 1.43 (m, 2H), 1.36–1.28 (m, 12H), 0.9 (t, J = 6.8 Hz, 3H); 13C NMR: 118.6 (q, JC–F = 320 Hz), 77.7, 31.8, 29.4, 29.3, 29.2, 28.8, 25.0, 22.6, 14.0.
Using method A, 2-tetrahydropyranyl hydroperoxide 2 (1.18 g, 10 mmol) was reacted with decyl triflate (2.98 g, 10 mmol) to furnish peroxide 12b as a colorless oil (2.05 g, 79%); Rf = 0.56 (15% EA/Hex). Spectral properties matched those in a literature report.5b
2-(2-Octylperoxy)-tetrahydro-2H-pyran (13b)
[1630792-50-5]: 2-Octyl trifluoromethanesulfonate [58864-30-5] was prepared (3.58 g, estimated 91%) as a light brown oil from 2-octanol (2.0 g, 15 mmol) using the procedure described above. Rf = 0.3 (10% EA/Hex). Spectral properties matched those in a previous report.5b
Using method A, hydroperoxide 2 (0.595 g, 5.01 mmol) was reacted with 2-octyl triflate (1.47 g, 5.61 mmol) to furnish 13b as a colorless oil (0.808 g, 70%) Rf = 0.61 (20% EA/Hex). Spectral properties matched those in a literature report.5b
Bis-(tetrahydro-2H-pyranyl) Peroxide (14b)
[685877-38-7]: Using method B, acetalization of 2-tetrahydropyranyl hydroperoxide 2 (0.35 g, 2.97 mmol) with dihydropyran (0.249 g, 2.97 mmol) furnished peroxide 14b as a colorless oil (0.493 g, 84%). This molecule has been previously prepared without NMR characterization:67Rf = 0.50 (10% EA/Hex); 1H NMR: 5.22 (m, 2H), 4.09 (m, 1H), 4.00 (m, 1H), 3.63–3.60 (m, 2H), 1.76–1.74 (m, 4H), 1.66–1.53 (m, 8H); 13C NMR: 101.8, 100.2, 62.5, 62,1, 27.9, 27.7, 25.1, 25.0, 19.6, 19.4.
2-(1-Methylcyclohexylperoxy)-tetrahydro-2H-pyran (15b)
Using method B, hydroperoxide 5 (0.266 g, 2.04 mmol) was reacted with dihydropyran (0.171 g, 2.04 mmol) to furnish 15b as a colorless oil (0.326 g, 74%). Rf = 0.50 (10% EA/Hex); 1H NMR: 5.04 (broad triplet, 1H), 4.02 (m, 1H), 3.60 (m, 1H), 1.75 (m, 5H), 1.68–1.53 (m, 6H), 1.47–1.35 (m, 6H), 1.26 (s, 3H); 13C NMR: 101.0, 81.5, 62.7, 35.0, 27.8, 25.7, 25.1, 24.3, 22.5, 22.3, 20.0; HRMS (ESI+ TOF) calcd for C12H22NaO3 (M + Na): 237.1461; found: 237.1469; IR: 2931, 2852, 1443, 962.
(E,Z)-2-(Undec-2-en-1-yl peroxy)tetrahydro-2H-pyran (16b)
Using a known procedure,68 cross-metathesis of 1-octene (6.72 g, 4.8 mmol) and allyl bromide (1.45 g, 12 mmol) in the presence of the Grubbs II catalyst furnished 1-bromo-2-undecene (1.109 g, 80%) as a colorless oil consisting of a 84:16 E/Z mixture using a known procedure. The portions of the spectra corresponding to the major (E) isomer closely matched a previous report [67952-61-8]:69Rf = 0.8 (hexane); 1H NMR: (both isomers, partial integrals given): 5.82–5.58 (m, 2H), 4.01 (d, J = 8.3 Hz, 0.32 H), 3.96 (d, J = 8.2 Hz, 1.68 H), 2.14 (m, 0.32H), 2.07 (m, 1.68), 1.40–1.35 (m, 2H), 1.34–1.28 (m, 10H),5.82– 0.89 (t, J = 6.8 Hz, 3H); 13C NMR: (major isomer): 136.7, 126.2, 33,6, 32.0, 31.9, 29.4, 29.2, 29.1, 28.8, 22.7, 14.1.
Using method C, hydroperoxide 2 (0.118 mg, 1 mmol) was reacted with an E/Z-mixture of 2-undecenyl bromide (0.232 g, 1.0 mmol) to furnish peroxide 16b as a colorless oil (0.197 mg, 73%) consisting of an inseparable 84:16 E/Z mixture: Rf = 0.42 (10% EA/Hex); 1H NMR: 5.82–5.74 (m, 1H), 5.69–5.55 (m, 1H), 5.16 (m, 1H), 5.64 (d, J = 6.8 Hz, 0.24 H), 4.51 (d, J = 6.8 Hz, 1.76 H), 4.01 (m, 1H), 3.64–3.60 (m, 1H), 2.13–2.02 (m, 2H), 1.73–1.68 (m, 2H), 1.65–1.53 (m, 4H), 1.38–1.35 (m, 2H), 1.31–1.26 (m, 10H), 0.87 (t, J = 6.8 Hz, 3H); 13C NMR: 137.8, 123.7, 100.8, 76.2, 62.4, 32.3, 31.8, 29.4, 29.2, 29.1, 28.8, 27.8, 25.1, 22.6, 19.6, 14.0. HRMS (TOF-MS-ES+) calcd for C16H30NaO3 (M + Na): 293.2087; found: 293.2087; IR: 2923, 2851, 1202, 962.
Etherification of Peroxides with sp3 RM
The alkyllithium (0.55 mmol, 1.1 equiv, typically as a solution in Hex) was added to the solution of peroxide (0.50 mmol) in dry THF (3 mL) at −78 °C, and the reaction was stirred for 15 min. For THP monoperoxyacetals, the reaction was brought to room temperature for 15 min and then quenched with water (2 mL); for other peroxides, the reaction was allowed to stir at room temperature for the time indicated. The mixture was extracted with 20% ether in Hex (1 × 25, 1 × 5 mL), and the combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by silica gel chromatography (0.5 × 4 in. column) using 1% ether in hexane. Note: For volatile ethers, column fractions were carefully concentrated on a rotary evaporator at 20 °C bath temperature and then briefly (1 min) concentrated on high vacuum while held in an ice bath.
For reaction with Grignard reagents, a solution of the alkyl magnesium bromide (0.55 mmol, 1.1 equiv) in diethyl ether was added to a 0 °C solution of the THP monoperoxyacetal (0.5 mmol) in dry THF (3 mL). The reaction was stirred for 15 min and then brought to room temperature. After 15 min, the reaction was quenched with water (2 mL) and extracted with 20% ether/Hex (1 × 25, 1 × 5 mL). The combined organic layers were dried over Na2SO4, and the concentrated residue was purified by silica gel chromatography (0.5 × 4 in. column) using 1% ether/Hex. In the case of volatile ethers, column fractions were concentrated on a rotary evaporator at 20 °C bath temperature and subsequently subjected to high vacuum (<1 mm) for 1 min while placed in an ice bath.
8-Butoxy-2,6-dimethyloct-2-ene (17a)
[71077-30-0]: Using the general procedure described above, THP monoperoxyacetal 8b (128 mg, 0.5 mmol) was reacted with n-BuLi (0.34 mL, 1.6 M in Hex, 0.55 mmol) to furnish ether 17a as a colorless oil (86 mg, 81%). The same procedure, when applied to dialkyl peroxide 8b or 2-methoxypropyl peroxyacetal 8c furnished 17a in 70% and 87% yields, respectively. Spectral data matched those in previous reports.5aRf = 0.6 (5% EA/Hex).
8-Hexyloxy-2,6-dimethyl-2-octene (17b)
Using the general procedure for Grignard-promoted etherification described above, monoperoxacetal 8b (128 mg, 0.5 mmol) was reacted with n-hexyl magnesium bromide (0.27 mL, 2 M in diethyl ether, 0.55 mmol) to furnish hexyl ether 17b as a colorless oil (106 mg, 88%). The same product could be obtained from dialkyl peroxide 8a except in 42% yield: Rf = 0.70 (10% EA/Hex); 1H NMR: 5.11 (m, 1H), 3.47–3.38 (m, 4H), 2.08–1.94 (m, 2H), 1.69 (s, 3H), 1.65–1.53 (m, 4H), 1.61 (s, 3H), 1.45–1.31 (m, 8H), 1.23–1.11 (m, 1H), 0.91–0.88 (m, 6H); 13C NMR: 131.1, 124.8, 71.0, 69.1, 37.2, 36.7, 31.7, 29.7, 29.6, 25.9, 25.7, 25.4, 22.6, 19.6, 17.6, and 14.0. HRMS (TOF-MS-EI+) calcd for C16H32O: 240.2453; found: 240.2453; IR: 2954, 2927, 2858, 1455, 1376, 1112.
8-Allyloxy-2,6-dimethyloct-2-ene (17c)
[139694-24-9]: To a 0.2 M solution of peroxide 8a (229 mg, 1.0 mmol) in THF was added allyltributyl tin (0.62 mL, 2.0 mmol). The solution was cooled to −78 °C, and n-BuLi (2.5 mmol, 1.0 mL, 2.5 M in Hex) was added dropwise. After 30 min, the reaction was quenched with 10 mL of water and extracted with ether. The combined organic layer was dried with Na2SO, and concentrated under reduced pressure. The residue was then purified by column chromatography (2.5% EA/Hex) to yield 137 mg (70%) of 17c as a colorless oil. The 1H NMR spectrum matched the listing in a previous report.70Rf = 0.25 (5% EA/Hex); 1H NMR: 0.92 (d, 3H, J = 6.7), 1.18 (m, 1H), 1.39 (m, 1H), 1.52–1.68 (s at 1.62 overlapping unresolved signal, 6H), 1.70 (s, 3H), 2.00 (m, 2H), 3.48 (m, 2H), 3.98 (dt, 2H, J = 5.6, 1.4), 5.14–5.08 (m 1H), 5.19 (app. dq, Hcis, J = 10.6, 1.3, 1H), 5.29 (app. dq, Htrans, J = 17.2, 1.6, 1H), 5.9 (m, 1H); 13C NMR: 17.6, 19.6, 25.5, 25.7, 29.6, 36.7, 37.2, 68.7, 71.8, 116.7, 124.8, 131.2, 135.1.
8-tert-Butoxy-2,6-dimethyl-2-octene (17d)
[436141-44-5]: Using the general procedure for etherification described above, peroxyacetal 8b (128 mg, 0.5 mmol) was reacted with t-BuLi (0.32 mL 1.7 M in pentane, 0.55 mmol) to furnish 17d as a colorless oil (55 mg, 51%). Spectral data were nearly identical to those previously described:71Rf = 0.6 (5% EA/Hex); 1H NMR: 5.11 (m, 1H), 3.42–3.31 (m, 2H), 2.06–1.91 (m, 2H), 1.69 (s, 3H), 1.61 (m, 3H), 1.59–1.51 (m, 2H), 1.39–1.27 (m, 2H), 1.19 (s, 9H), 1.17–1.10 (m, 1H), 0.90 (d, J = 6.5 Hz, 3H); 13C NMR: 131.0, 124.9, 72.3, 59.7, 37.7, 37.2, 29.6, 27.5, 25.7, 25.4, 19.6, 17.6.
3-Phenylpropyl Trimethylsilylmethyl Ether (17e)
Using the general procedure for etherification described above, peroxide 9b (118 mg, 0.50 mmol) was reacted with 2-trimethylsilyl methyl magnesium chloride (0.65 mL, 1 M in ether, 0.65 mmol) to furnish ether 17e as a colorless oil (84 mg, 75%): Rf = 0.8 (10% EA/Hex); 1H NMR: 7.31–7.29 (m, 2H), 7.22–7.19 (m, 3H), 3.42 (t, J = 6.3 Hz, 2H), 3.12 (s, 2H), 2.70 (t, J = 7.7 Hz, 2H), 1.91–1.87 (m, 2H), 0.09 (s, 9H); 13C NMR: 142.3, 128.5, 128.2, 125.6, 74.2, 64.7, 32.3, 31.2, −2.98; HRMS(TOF-MS-EI+) calcd for C13H22OSi (M)+: 222.1440; found: 222.1442; IR: 2955, 2845, 1246, 839.
2-Butoxy Octane (17f)
[110458-41-8]: Using the general procedure for etherification described above, peroxide 13b (115 mg, 0.5 mmol) was reacted with n-BuLi (0.34 mL 1.6 M in Hex, 0.55 mmol) to furnish ether 17f as a colorless oil (73 mg, 78%). The 1H NMR data matched those previously reported:72Rf = 0.6 (5% EA/Hex); 13C NMR: 75.3, 68.1, 36.7, 32.3, 31.8, 29.4, 25.6, 22.6, 19.7, 19.4, 14.0, 13.9.
2-Hexyloxy Octane (17g)
[51182-98-0]: Using the general procedure for etherification using a Grignard reagent, peroxide 13b (115 mg, 0.50 mmol) was reacted with n-hexyl magnesium bromide (0.27 mL, 2 M in diethyl ether, 0.54 mmol) to furnish hexyl ether 17g as a colorless oil (95 mg, 88%). The product has been partially characterized:72Rf = 0.6 (5% EA/Hex); 1H NMR: 3.51–3.29 (m, 1H), 3.40–3.29 (m, 2H), 1.57–1.53 (m, 3H), 1.41–1.29 (m, 15H), 1.12 (d, J = 6.1 Hz, 3H), 0.9 (m, 6H); 13C NMR: 75.3, 68.4, 36.7, 31.8, 31.7, 30.1, 29.4, 25.9, 25.6, 22.63, 22.62, 19.7, 14.05, 14.02.
2-Butoxy-tetrahydro-2H-pyran (17h)
[1927-68-0]: Using the general procedure for etherification described above, peroxide 14b (101 mg, 0.50 mmol) was reacted with n-BuLi (0.31 mL 1.6 M in hexane, 0.5 mmol) to furnish ether 17h (12 mg, 15%) accompanied by 16 mg (15%) of recovered 14b. Partial spectral characterization has been reported.73Rf = 0.5 (15% EA/Hex); 1H NMR: 4.59 (m, 1H), 3.89 (m, 1H), 3.76 (m, 1H), 3.52 (m, 1H), 3.40 (m, 1H), 1.84 (m, 1H), 1.72 (m, 1H), 1.65–1.49 (m, 6H), 1.45–1.35 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR: 98.8, 67.3, 62.3, 31.8, 30.8, 25.5, 19.7, 1.4, 13.9.
2-Hexyloxy-tetrahydro-2H-pyran (17i)
[1927-63-5] Using the general procedure for etherification with alkyl magnesium bromide described above, peroxide 14b (107 mg, 0.5 mmol) was reacted with n-hexyl magnesium bromide (0.25 mL, 2 M in diethyl ether, 0.50 mmol) to furnish hexyl ether 17i as a colorless oil (57 mg, 61%). The 1H NMR data matched those in a previous report.74Rf = 0.5 (15% EA/Hex); 1H NMR: 4.57 (m, 1H), 3.90–3.84 (m, 1H), 3.76–3.70 (m, 1H), 3.52–3.47 (m, 1H), 3.41–3.35 (m, 1H), 1.87–1.79 (m, 1H), 1.74–1.68 (m, 1H), 1.62–1.50 (m, 6H), 1.42–1.26 (m, 6H), 0.89 (t, J = 6.8 Hz, 3H); 13C NMR: 98.8, 67.6, 62.3, 31.7, 30.8, 29.7, 25.9, 25.5, 22.6, 19.6, 14.0.
1-Butoxy-1-methyl Cyclohexane (17j)
Using the general procedure for etherification described above, peroxide 15b (107 mg, 0.50 mmol) was reacted with n-BuLi (0.34 mL, 1.6 M in Hex, 0.55 mmol) to furnish ether 17j as a colorless oil (61 mg, 71%): Rf = 0.70 (10% EA/Hex); 1H NMR: 3.32 (t, J = 6.4 Hz, 2H), 1.71–1.64 (m, 2H), 1.61–1.45 (m, 5H), 1.43–1.19 (m, 7H), 1.10 (s, 3H), 0.93 (t, J = 7.20 Hz, 3H); 13C NMR: 72.8, 59.9, 36.5, 32.8, 25.86, 24.6, 22.2, 19.6, 13.99. HRMS (TOF-MS-EI+) calcd for C11H22O: 170.1671; found: 170.1671; IR: 2962, 2925, 2862, 1447, 1081.
1-Hexyloxy 1-Methyl Cyclohexane (17k)
Using the general procedure for etherification described above, peroxide 15b (107 mg, 0.50 mmol) was reacted with n-hexyl magnesium bromide (0.27 mL, 2 M in ether, 0.54 mmol) to furnish hexyl ether 17k as a colorless oil (64 mg, 65%): Rf = 0.65 (5% EA/Hex); 1H NMR: 3.29 (t, J = 6.7 Hz, 2H), 1.71–1.66 (m, 2H), 1.64–1.47 (m, 5H), 1.44–1.27 (m, 11H), 1.11 (s, 3H), 0.90 (t, J = 6.9 Hz, 3H); 13C NMR: 72.9, 60.2, 36.5, 31.8, 30.7, 26.1, 25.8, 24.6, 22.6, 22.2, 14.0; HRMS (TOF-MS-EI+) calcd for C13H26O: 198.1984; found: 198.1992; IR: 2962, 2925, 2862, 1447, 1081.
(E/Z)-1-Butoxyundec-2-ene (17l)
Using the general procedure for etherification described above, peroxide 16b (135 mg, 0.50 mmol) was reacted with n-BuLi (0.34 mL, 1.6 M in Hex, 0.55 mmol) to furnish ether 17l as a colorless oil (86 mg, 76%): Rf = 0.63 (10% EA/Hex); 1H NMR: 5.73–5.66 (m, 1H), 5.59–5.52 (m, 1H), 4.01 (m, 0.23 H), 3.91 (dd, J = 0.6, 6.15, 1.76 H), 3.41 (t, J = 6.6 Hz, 2H), 2.05 (m, 2H), 1.61–1.54 (m, 2H), 1.43–1.35 (m, 4H), 1.33–1.27 (m, 10H), 0.93 (t, J = 7.4 Hz, 3H), 0.89 (t, J = 7.1 Hz, 3H); 13C NMR: 134.5, 126.4, 71.6, 69.8, 32.3, 31.88, 31.87, 29.4, 29.27, 29.21, 29.0, 22.7, 19.3, 14.0, 13.9; HRMS (TOF-MS-EI+): Calcd for C15H30O: 226.2297; found: 226.2297; IR: 2954, 2923, 2852, 1465, 1105.
(E/Z)-Hexyl 2-Undecenyl Ether (17m)
Using the general procedure for etherification described above, peroxide 16b (115 mg, 0.50 mmol) was reacted with n-hexyl magnesium bromide (0.27 mL, 2 M in ether, 0.54 mmol) to furnish hexyl ether 17m as a colorless oil (104 mg, 81%): Rf = 0.67 (10% EA/Hex); 1H NMR: 5.73–5.66 (m, 1H), 5.59–5.52 (m, 1H), 4.01 (m, 0.24 H), 3.91 (dd, J = 0.6, 6.2, 1.76 H), 3.40 (t, J = 6.7, 2H), 2.05 (m, 2H), 1.58 (m, 2H), 1.43–1.27 (m, 18H), 0.90–0.87 (6H); 13C NMR: 134.5, 126.4, 71.6, 70.2, 32.3, 31.88, 31.74, 29.76, 29.46, 29.28, 29.22, 29.1, 25.9, 22.67, 22.62, 14.08, 14.03. HRMS (TOF-MS-EI+) calcd for C17H34O: 254.2610; found: 254.2610; IR: 2954, 2923, 2852, 1465, 1105.
General Procedure for sp2 C–O Bond Formation
Reactions involving commercially available reagents were conducted as described above for sp3 C–O bond formations except that reaction times were approximately 1 h. Reactions involving in situ formation of vinyl or heteroaryllithium reagents from the corresponding tributylstannanes were conducted as described below.
To a −78 °C solution of alkenyl- or aryltributyltin (0.55 mmol) in dry THF (3 mL) was added n-BuLi (0.55 mmol, 1.6 M in Hex). The reaction mixture was stirred for 10 min, after which was added a solution of the monoperoxyacetal (0.50 mmol) in THF (1 mL). The reaction mixture was stirred until no starting material could be detected on TLC (approximately 60 min) and then diluted with Hex (10 mL). The resulting solution was passed though a column of neutral alumina (1 × 2 in.) and then concentrated (an aqueous workup could also be conducted employing aq. carbonate to minimize hydrolysis). The residue was purified by silica flash chromatography using 1% ether/Hex; chromatography of enol and thienyl ethers included 0.2% triethylamine.
3,7-Dimethyl-6-octen-1-yl Phenyl Ether (18a)
[51113-53-2]: Using the general procedure described above, peroxide 8b (128 mg, 0.5 mmol) was reacted with phenyl lithium (0.30 mL, 1.8 M in dibutyl ether, 0.54 mmol) to furnish phenyl ether 18a as a colorless oil (102 mg, 87%).75 The same product was available (78 mg, 67%) by reaction of peroxide 8b (128 mg, 0.50 mmol) with phenyl magnesium bromide (0.18 mL, 3.0 M in ether, 0.54 mmol):
Rf = 0.66 (10% EA/Hex); 1H NMR: 7.32–7.30 (m, 2H), 6.97–6.93 (m, 3H), 5.15 (m, 1H), 4.03 (m, 2H), 2.08 (m, 1H), 2.02 (m, 1H), 1.88 (m, 1H), 1.75–1.71 (m, 1H), 1.73 (s, 3H), 1.65 (s, 3H), 1.64–1.61 (m, 1H), 1.46–1.41 (m, 1H), 1.30–1.24 (m, 1H), 0.98 (d, J = 6.9 Hz, 3H); 13C NMR: 151.1, 131.3, 129.4, 124.7, 120.5, 114.5, 66.1, 37.2, 36.1, 29.6, 25.8, 25.5, 19.6, 17.7.
2-Methylprop-1-en-1-yl 3-Phenylpropyl Ether (18b)
Using the general procedure described above, peroxide 9b (118 mg, 0.5 mmol) was reacted with 2-methyl-1-propenyl magnesium bromide (1.3 mL, 0.5 M in THF, 0.65 mmol) to furnish ether 18b as a colorless oil (39 mg, 41%), which decomposed during chromatography: 1H NMR: 7.34–7.30 (m, 2H), 7.26–7.21 (m, 3H), 5.83–5.82 (m, 1H), 3.72 (t, J = 6.4 Hz, 2H), 2.75 (t, J = 7.8 Hz, 2H), 2.00–1.93 (m, 2H), 1.68 (s, 3H), 1.59 (s, 3H); 13C NMR: 141.8, 140.0, 128.5, 128.4, 125.8, 110.5, 70.7, 32.0, 31.4, 19.5, 15.0. HRMS (TOF-MS-EI+) calcd for C13H18O (M)+: 190.1358; found: 190.1363; IR: 2918, 2865, 1689, 1496, 1159.
3,7-Dimethyl-6-octen-1-yl Vinyl Ether (18c)
Using the procedure described above for etherifications with in situ generated sp2 RLi, peroxyacetal 8b (128 mg, 0.5 mmol) was reacted with the reagent generated from tributyl vinylstannane (237 mg, 0.75 mmol) and n-BuLi (0.47 mL, 1.6 M in Hex, 0.75 mmol) to furnish ether 18c as a colorless oil (62 mg, 66%), which partially decomposed upon thin-layer or column chromatography: Rf = 0.7 (10% EA/Hex); 1H NMR: 6.48 (dd, J = 6.8, 14.1 Hz, 1H), 5.12 (t, J = 7.0 Hz, 1H), 4.19 (d, J = 14.1 Hz, 1H), 3.99 (d, J = 6.8 Hz, 1H), 3.78–3.68 (m, 2H), 2.08–1.93 (m, 2H), 1.77–1.69 (m, 1H), 1.70 (s, 3H), 1.65–1.58 (m, 1H), 1.62 (s, 3H), 1.54–1.44 (m, 1H), 1.42–1.33 (m, 1H), 1.25–1.16 (m, 1H), 0.93 (d, J = 6.6 Hz, 3H); 13C NMR: 152.0, 131.3, 124.7, 86.2, 66.3, 37.1, 35.9, 29.5, 25.7, 25.4, 19.5, 17.6. HRMS (TOF-MS-EI+ calcd for C12H22O (M – H)+: 181.1598; found: 181.1602; IR: 2961, 2913, 2871, 1647, 1609, 1200.
2-(3,7-Dimethyloct-6-en-1-oxy)-thiophene (18d)
Using the procedure described above for etherifications with in situ generated sp2 RLi, peroxyacetal 8b (128 mg, 0.50 mmol) was reacted with the reagent generated from 2-(tributylstannyl)thiophene (279 mg, 0.75 mmol) and n-BuLi (0.47 mL, 1.6 M in Hex, 0.75 mmol) to furnish ether 18d as a colorless oil (72 mg, 60%): Rf = 0.75 (10% EA/Hex); 1H NMR: 6.73 (dd, J = 3.7, 6.0 Hz, 1H), 6.56 (dd, J = 1.2, 6.0 Hz, 1H), 6.22 (dd, J = 1.2, 3.7 Hz, 1H), 5.13 (t, J = 7.1 Hz, 1H), 4.10–4.06 (m, 2H), 2.08–1.98 (m, 2H), 1.89–1.84 (m, 1H), 1.74–1.67 (m, 1H), 1.72 (s, 3H), 1.64 (s, 3H), 1.62–1.59 (m, 1H), 1.43–1.38 (m, 1H), 1.27–1.22 (m, 1H), 0.97 (d, J = 6.6 Hz, 3H); 13C NMR: 165.8, 131.4, 124.7, 124.6, 111.7, 104.6, 72.3, 37.0, 36.0, 29.4, 25.7, 25.4, 19.5,17.7; HRMS (TOF-MS-EI+) calcd for C14H22OS (M)+: 238.1391; found: 238.1390; IR: 2963, 2925, 2913, 2871, 1536, 1193.
2-(Decyloxy)-thiophene (18e)
Using the procedure described above, peroxide 12b (129 mg, 0.50 mmol) was reacted with the reagent generated from 2-(tributylstannyl)thiophene (279 mg, 0.75 mmol) and n-BuLi (0.47 mL, 1.6 M in Hex, 0.75 mmol) to furnish ether 18e as a colorless oil (65 mg, 54%): Rf = 0.7 (10% EA/Hex); 1H NMR: 6.74–6.72 (m, 1H), 6.56–6.54 (m, 1H), 6.23–6.21 (m, 1H), 4.04 (t, J = 6.4 Hz, 2H), 1.83–1.76 (m, 2H), 1.48–1.44 (m, 2H), 1.38–1.30 (m, 12H), 0.91 (t, J = 6.8 Hz, 3H); 13C NMR: 165.9, 124.6, 111.6, 104.5, 74.0, 31.9, 29.5, 29.3, 29.2, 25.8, 22.7, 14.1. HRMS (TOF-MS-EI+) calcd for C14H24OS (M)+: 240.1548; found: 240.1548; IR: 2920, 2853, 1536, 1456, 1193.
3,3-Difluoro-3-((10-phenyldecyl)oxy)propyl)benzene (19)
The difluoroether was prepared applying a previously reported two-step procedure (reaction of lithiated dithiane with the peroxide; fluorodesulfurization of the poorly stable difluoroether products)5b to three different precursors: 2-(3-phenylpropyl)-1,3-dithiane and monoperoxyacetal 10b (100 mg, 51% isolated yield); silyl peroxide 10d (105 mg, 54% isolated yield); t-butyl peroxide 10a (95 mg, 26%, with 35% recovered starting material). In each case, difluoroether 19 was isolated as a colorless oil with Rf = 0.5 (5% EA/Hex) and with spectra matching literature values.5b
Tributyl(1-(methoxymethoxy)-3-phenylpropyl)stannane (20)
[123294-00-8] was prepared using a modification of a literature procedure.76 A solution of diisopropylamine (0.22 g, 1.1 equiv) in 5 mL of THF was cooled to 0 °C, and n-BuLi (1.37 mL, 1.6 M in Hex, 2.2 mmol, 1.1 equiv) was added. The reaction was stirred for 10 min, after which was added Bu3SnH (0.582 g, 2.0 mmol, 1 equiv). The reaction was stirred for 15 min at 0 °C and then cooled to −78 °C, whereupon hydrocinnamaldehyde (268 mg, 2.0 mmol) was added as a solution in THF (10 mL). After stirring at −78 °C for 5 min, the reaction was quenched with sat. aq. NH4Cl (10 mL), then allowed to warm to room temperature. The separated organic layer was dried over MgSO4 and concentrated (rotary evaporator, followed by high vacuum).
The residue from the step described above was dissolved in DCM (12 mL), and the solution cooled to −10 °C. Diisopropyl ethylamine (0.335 g, 1.3 equiv) was added, followed by chloromethyl methyl ether (0.177 g, 1.1 equiv). The reaction was stirred for 7 h at rt and then washed with water (10 mL). The separated organic layer was dried (Na2SO4) and concentrated on a rotary evaporator. The residue was chromatographed on silica gel using 1% EA/Hex to afford 20 as a colorless liquid (63%, 598 mg): Rf = 0.51 (10% EA/Hex); 1H NMR: 7.33–7.27 (m, 2H), 7.23–7.19 (m, 3H), 4.65 (d, J = 6.5 Hz, 1H), 4.62 (d, J = 6.5 Hz, 1H), 4.15–4.11 (m, 1H), 3.41 (s, 3H), 2.82–2.66 (m, 2H), 2.22–2.05 (m, 2H), 1.60–1.49 (m, 6H), 1.39–1.29 (m, 6H), 0.96–0.88 (m, 12H), 0.92 (t, J = 7.3 Hz, 3H); 13C NMR: 142.4, 128.4, 128.3, 125.7, 96.6, 73.7, 55.5, 37.4, 34.5, 29.2, 27.5, 13.7, 9.2.
Benzene, 3-Methoxymethoxy-3-octyloxypropyl (21a)
To a −78 °C solution of stannane 20 (235 mg, 0.5 mmol) in dry THF (3 mL) was added a solution of n-BuLi in Hex (1.6 M, 0.5 mmol). After the reaction had stirred for 3 min, peroxide 11a (100 mg, 0.5 mmol) was added as a solution in THF (1 mL), and the reaction was stirred for an additional 30 min at −78 °C. The cold bath was removed, and the reaction was allowed to warm to rt over 30 min. The reaction was quenched with water (1 mL) and diluted with hexane (40 mL). The mixture was washed with water (10 mL), and the separated aqueous layer was extracted with Hex (10 mL). The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by silica gel chromatography using ether/Hex (1–5%) to furnish recovered 11a (10%) and then the acetal 21a as a colorless oil (88 mg, 57%): Rf = 0.36 (10% EA/hex); 1H NMR: 7.32–7.27 (m, 2H), 7.24–7.19 (m, 3H), 4.83 (d, J = 6.8 Hz, 1H), 4.70 (d, J = 6.8 Hz, 1H), 4.70–4.66 (m, 1H), 3.68–3.63 (m, 1H), 3.47–3.40 (m, 1H), 3.43 (s, 3H), 2.71–2.72 (m, 2H), 2.04–1.98 (m, 2H), 1.64–1.57 (m, 2H), 1.40–1.30 (m, 10H), 0.91 (t, J = 6.7 Hz, 3H); 13C NMR: 141.7, 128.4, 125.8, 101.4, 93.3, 66.7, 55.7, 36.4, 31.8, 30.8, 29.8, 29.4, 29.2, 26.2, 22.6, 14.1; HRMS (ESI+ TOF) calcd for C19H32O3 (M + Na)+: 331.2249; found: 331.2265; IR: 2925, 2855, 1149, 1002.
3-Methoxymethoxy-3-phenylpropoxy Propylbenzene (21b)
The title compound was synthesized as a colorless oil (86 mg, 55%) from reaction of 9a (104 mg, 0.50 mmol) using the general procedure described above except that 1.2 equiv each of tributylstannane and n-BuLi was used: Rf = 0.43 (10% EA/Hex); 1H NMR: 7.35–7.31 (m, 4H), 7.28–7.21 (m, 6H), 4.85 (d,, J = 6.6 Hz, 1H), 4.73 (d, J = 6.6 Hz, 1H), 4.72–4.70 (m, 1H), 3.74–3.69 (m, 1H), 3.51–3.47 (m, 1H), 3.44 (s, 3H), 2.81–2.74 (m, 4H), 2.08–2.02 (m, 2H), 2.00–1.93 (m, 2H); 13C NMR: 141.88, 141.7, 128.5, 128.49, 128.45, 128.42, 128.39, 125.9, 125.8, 101.6, 93.5, 65.8, 55.8, 36.4, 32.4, 31.4, 30.8; HRMS (TOF MS ES+) calcd for C20H26NaO3 (M + Na)+: 337.1780; found: 337.1775; IR: 2927, 1453, 1122, 1000, 697.
3-Decyloxy-3-methoxymethoxy Propyl Benzene (21c)
The title compound was synthesized as a colorless oil (89 mg, 53%) from stannane 20 (0.235 g, 0.50 mmol) and peroxyacetal 12b (0.129 g, 0.50 mmol) using the general procedure described above: Rf = 0.40 (10% EA/Hex); 1H NMR: 7.30–7.26 (m, 2H), 7.21–7.17 (m, 3H), 4.82–4.80 (m, 1H), 4.69–4.63 (m, 2H), 3.66–3.60 (m, 1H), 3.40 (s, 3H), 2.74–2.70 (m, 2H), 2.05–1.95 (m, 2H), 1.60–1.54 (m, 2H), 1.38–1.27 (m, 15H), 0.88 (t, J = 6.80 Hz, 3H); 13C NMR: 141.9, 128.4, 125.9, 101.5, 93.4, 66.9, 55.8, 36.4, 32.0, 30.9, 29.9, 29.7, 29.6, 29.5, 29.4, 26.3, 22.7, 14.2. HRMS (TOF MS ES+) calcd for C21H36NaO3 (M + Na)+: 359.2562; found: 359.2578; IR: 2925, 2856, 1149, 1000.
3-((3,7-Dimethyloct-6-en-1-yl)oxy)-3-(methoxymethoxy) Propylbenzene (21d)
Using the general procedure described above, peroxide 8c (244 mg, 0.50 mmol) was reacted with alkoxylithium generated from reaction of stannane 20 (235 mg, 0.5 mmol) and n-BuLi (0.31 mL, 1.6 M in Hex, 0.5 mmol) to furnish a mixture of mixed acetal 21d (48 mg, 28%) accompanied by 3-methoxymethyloxy-2-methyl-5-phenylpentan-2-ol 22 (23 mg, 20%). NMR analysis of the crude reaction mixture suggested the presence of 40% of mixed acetal 21d; presumably, some was lost during the separation process. The mixed acetal 16d was contaminated with a small amount of inseparable byproduct, which could be removed by subjecting a DCM solution of the crude reaction mixture to brief (∼1 min) room temperature ozonolysis with 2% O3/O2 (1 mmol O3/min) prior to chromatography. The separated byproduct was determined to be 3-methoxymethoxypropyl benzene (3 mg, ∼3%), the product of protonation of the functionalized organolithium reagent.
21d
Rf = 0.5 (5% EA/Hex); 1H NMR: 7.32–7.27 (m, 2H), 7.23–7.19 (m, 3H), 5.12 (m, 1H), 4.83 (d, J = 6.8 Hz, 1H), 4.70 (d, J = 6.8 Hz, 1H), 4.68–4.66 (m, 1H), 3.73–3.64 (m, 1H), 3.52–3.44 (m, 1H), 3.43 (s, 3H), 2.74 (m, 2H), 2.08–1.91 (m, 4H), 1.70 (s, 3H), 1.68–1.56 (m, 2H), 1.62 (s, 3H), 1.49–1.31 (m, 2H), 1.28–1.14 (m, 1H), 0.93–0.91 (m, 3H); 13C NMR: 141.7, 131.2, 128.4, 128.4, 128.39, 125.8, 124.7, 101.5, 101.4, 93.3, 64.9, 64.8, 55.7, 37.2, 37.1, 36.8, 36.78, 36.39, 30.83, 29.5, 25.7, 25.5, 19.6, 19.5, 17.6; HRMS (TOF-ESI+) calcd for C21H34O3Na (M + Na)+: 357.2406; found 357.2401; IR: 2927, 1454, 1369, 1002.
3-Methoxymethyloxy-2-methyl-5-phenylpentan-2-ol (22)
Rf = 0.3 (15% EA/Hex); 1H NMR: 7.33–7.27 (m, 2H), 7.22–7.20 (m, 3H), 4.84 (d, J = 6.7 Hz, 1H), 4.69 (d, J = 6.7 Hz, 1H), 3.58 (s, 1H), 3.49 (s, 3H), 3.29 (m, 1H), 2.94–2.87 (m, 1H), 2.66–2.58 (m, 1H), 1.88–1.81 (m, 1H), 1.78–1.70 (m, 1H), 1.19 (s, 3H), 1.15 (s, 3H); 13C NMR: 141.9, 128.5, 128.4, 125.9, 99.2, 89.9, 72.0, 56.0, 33.5, 32.7, 26.3, 23.7. HRMS (TOF-ESI+) calcd for C14H22O3Na (M + Na)+: 261.1467; found: 261.1483; IR: 3461, 2930, 2889, 1028.
3-Methoxymethoxypropylbenzene [91898-11-2] has been reported on multiple occasions without NMR characterization: Rf = 0.5 (20% EA/Hex); 1H NMR: 7.33–7.28 (m, 2H), 7.23–7.18 (m, 3H), 4.66 (s, 3H), 3.57 (t, J = 6.4 Hz, 2H), 3.39 (s, 3H), 2.73 (t, J = 7.8 Hz, 2H), 1.99–1.89 (m, 2H); 13C NMR: 141.9, 128.4, 128.3, 125.8, 96.5, 67.1, 55.2, 32.4, 31.4.
General Procedure for Synthesis of Cyclopropyl Ethers
The THP monoperoxyacetal (0.50 mmol) was dissolved in dry THF (5 mL), and the solution cooled to 0 °C. Cyclopropyl magnesium bromide (1.3 mL, 0.5 M in THF, 0.65 mmol) was then added. The cooling bath was removed, and the reaction was stirred for 45 min prior to quenching by sequential dilution with 1 M aq. hydrochloric acid (∼2 mL) and water (10 mL). The addition of HCl clarified a previously hazy solution. The combined Hex extracts (20 mL × 2) were dried over Na2SO4 and concentrated on a rotary evaporator. The residue was purified by silica gel chromatography (5 in. tall; 0.5 in. diameter) using 1–2% EA/Hex, with fraction concentrated initially at 80–120 mm. (rotary evaporator, rt) and at 0.5 mm (1 min, 0 °C).
3,7-Dimethyloct-6-en-1-yl Cyclopropyl Ether (23a)
Using the general procedure described above, THP peroxide 8b (128 mg, 0.50 mmol) was reacted with cyclopropyl magnesium bromide (1.3 mL, 0.5 M in THF, 0.65 mmol) to furnish cyclopropyl ether 23a as a colorless oil (80 mg, 80%): Rf = 0.52 (10% EA/Hex); 1H NMR: 5.13–5.09 (m, 1H), 3.58–3.48 (m, 2H), 3.28–3.24 (m, 1H), 2.05–1.91 (m, 2H), 1.70 (s, 3H), 1.62 (s, 3H), 1.58–1.49 (m, 2H), 1.42–1.31 (m, 2H), 1.23–1.09 (m, 1H), 0.91 (d, J = 6.5 Hz, 3H), 0.58–0.54 (m, 2H), 0.48–0.43 (m, 2H); 13C NMR: 131.1, 124.8, 68.9, 52.9, 37.2, 36.6, 29.5, 25.7, 25.4, 19.5, 17.6, 5.45, 5.41; HRMS (TOF-MS CI+) calcd for C13H25O (M + H)+: 197.1905; found: 197.1913; IR: 2960, 2916, 1452, 1342.
Cyclopropyl 3-Phenylpropyl Ether (23b)
Using the general procedure described above, THP peroxide 9b (118 mg, 0.50 mmol) was reacted with cyclopropyl magnesium bromide (1.3 mL, 0.5 M in THF, 0.65 mmol) to furnish cyclopropyl ether 23b as a colorless oil (64 mg, 72%): Rf = 0.46 (10% EA/Hex); 1H NMR: 7.32–7.29 (m, 2H), 7.22–7.19 (m, 3H), 3.52 (t, J = 6.4 Hz, 2H), 3.28 (app septet, likely tt, J = 3.0, 6.0 Hz, 1H), 2.70 (t, J = 7.7 Hz, 2H), 1.94–1.87 (m, 2H), 0.61–0.57 (m, 2H), 0.49–0.45 (m, 2H); 13C NMR: 141.9, 128.4, 128.3, 125.8, 69.7, 53.0, 32.4, 31.2, 5.5; HRMS: (TOF-MS EI+) calcd for C12H16O (M+): 176.1201; found: 176.1199; IR: 3031, 2938, 2850, 1452, 1342.
Cyclopropyl Decyl Ether (23c)
Using the general procedure described above, THP peroxide 12b (129 mg, 0.50 mmol) was reacted with cyclopropyl magnesium bromide (1.3 mL, 0.5 M in THF, 0.65 mmol) to furnish cyclopropyl ether 23c as a colorless oil (79 mg, 80%): Rf = 0.56 (10% EA/Hex); 1H NMR: 3.49 (t, J = 6.7 Hz, 2H), 3.26 (app septet, likely tt, J = 3.0, 6.0 Hz, 1H), 1.58–1.54 (m, 2H), 1.34–1.27 (m, 14H), 0.89 (t, J = 7.0 Hz, 3H), 0.57–0.55 (m, 2H), 0.47–1.95 (m, 2H); 13C NMR: 70.7, 52.9, 31.9, 29.68, 29.60, 29.57, 29.5, 29.3, 26.2, 22.7, 14.1, 5.4; HRMS: (TOF-MS CI+) calcd for C13H27O (M + H)+: 199.2062; found: 199.2057; IR: 2922, 2853, 1453, 1343.
Cyclopropyl 2-Octyl Ether (23d)
Using the general procedure described above, THP peroxide 13b (115 mg, 0.50 mmol) was reacted with cyclopropyl magnesium bromide (1.3 mL, 0.5 M in THF, 0.65 mmol) to furnish cyclopropyl ether 23d as a colorless oil (60 mg, 70%): Rf = 0.6 (10% EA/Hex); 1H NMR: 3.57–3.49 (m, 1H), 3.33–3.29 (m, 1H), 1.41–1.23 (m, 10H), 1.18 (d, J = 6.1 Hz, 3H), 0.89 (t, J = 6.8 Hz, 3H), 0.61–0.56 (m, 1H), 0.54–0.51 (m, 1H), 0.49–0.47 (m, 1H), 0.46–0.40 (m, 1H); 13C NMR: 75.7, 50.7, 36.6, 31.8, 29.4, 25.4, 22.6, 19.9, 14.0, 5.96, 5.42; HRMS (TOF-MS EI+) calcd for C11H22O (M)+: 170.1671; found: 170.1678; IR: 2927, 2857, 1452, 1373.
t-Butyl 1,1-Dimethyl-2-Phenylethyl Peroxide (24a)
[133476-12-7]: 2-Bromo-2-methylpropyl benzene [23264-13-3] was prepared (3.79 g, 88%) as a colorless liquid from the corresponding alcohol (3 g, 20 mmol) using a modification of a reported procedure.40b Spectral data matched those in a previous report.77Rf = 0.8 (25% EA/Hex); 1H NMR: 7.35–7.26 (m, 5H), 3.22 (s, 2H), 1.78 (s, 6H); 13C NMR: 137.3, 130.9, 128.0, 127.0, 66.6, 53.4, 33.9.
The t-buyl peroxide was prepared from the bromide (1.89 g 8.9 mmol, 1 equiv) through reaction with t-BuOOH (1.94 mL, 5.5 M in decane 1.2 equiv, 10.6 mmol), and silver trifluoroacetate (2.34 g, 10.6 mmol 1.2 equiv) using a reported procedure.40a Following a workup which includes brief exposure to NaBH4 to destroy the trifluoroacetate ester, peroxide 24a was isolated as a colorless oil (0.79 g, 40%), which was only modestly responsive to the “peroxide” TLC stain (see the General Experimental section) even after warming: Rf = 0.7 (10% EA/Hex). Spectral properties matched those in literature reports.40a
Triethylsilyl (2-Methyl-1-phenylpropan-2-yl) Peroxide (24d)
[830345-47]: 2-Methyl-3-phenyl-1-propene (0.66 g, 5 mmol) and Et3SiH (1.16 g, 10 mmol) were sequentially added to a solution of Co(acac)2 (0.50 mmol) in ethanol (15 mL).41 The reaction mixture was stirred under an atmosphere of O2 (balloon) until no starting material was observed (TLC, ∼14 h). The residue obtained upon concentration in vacuo was purified by silica gel chromatography using 1–8% EA/Hex to furnish 24d (0.95 g, 68%) as a colorless oil. The molecule has previously been reported without NMR characterization:41bRf = 0.7 (10% EA/Hex); 1H NMR: 7.28–7.18 (m, 5H), 2.88 (s, 2H), 1.16 (s, 6H), 1.00 (m, J = 7.9 Hz, 9H), 0.70 (q, J = 7.9 Hz, 6H); 13C NMR: 138.3, 130.8, 127.8, 126.1, 82.6, 44.8, 24.3, 6.9, 4.0.
2-((2-Methyl-3-phenylpropan-1-yl)tetrahydro-2H-pyranyl Peroxide (24b)
To a 0 °C solution of silyl peroxide 24d (0.70 g, 2.5 mmol) in THF (6 mL) was added n-Bu4NF (3 mL, 1 M in THF, ∼3 mmol). After 10 min, the reaction was diluted with 50 mL of Hex and the resulting solution was washed with water (2 × 5 mL). The dried (Na2SO4) organic layer was concentrated, and the residue was purified through a short plug of silica (10% EA/hexane) to furnish 2-hydroperoxy-2-methylpropyl)benzene [1944-83-8] as a colorless oil (0.41 g, 98%), which displayed spectral data matching literature reports.78Rf = 0.35 (10% EA/hex).
The crude hydroperoxide (0.40 g, 2.4 mmol) and dihydropyran (0.2 g, 2.4 mmol) were reacted using method B to furnish monoperoxyacetal 24b as a colorless oil (0.397 g, 66%): Rf = 0.42 (10% EA/Hex); 1H NMR: 7.29–7.19 (5H), 5.12 (m, 1H), 4.06 (m, 1H), 3.63 (m, 1H), 2.97 (d, J = 13.5 Hz, 1H), 2.86 (d, J = 13.5 Hz, 1H), 1.79–175 (m, 2H), 1.67–1.59 (m, 4H), 1.25 (s, 3H), 1.18 (s, 3H); 13C NMR: 138.0, 130.7, 127.9, 126.2, 101.2, 82.9, 62.8, 45.0, 27.9, 25.3, 24.7, 24.3, 20.1. HRMS (TOF-MS-ES+) calcd for C15H22NaO3 (M + Na): 273.1461; found: 273.1461; IR: 2928, 2856, 1451.
Reaction of Monoperoxyacetal24bwith Organometallic Reagents.
2-Butoxy-2-methylpropylbenzene (25a)
Using the procedure described for reaction of n-BuLi with THP monoperoxyacetals, reaction of 24b (125 mg, 0.50 mmol) with n-BuLi (0.34 mL, 1.6 M in Hex, 0.55 mmol) at −78 C for 15 min furnished, after workup and chromatography, recovered 24b (9 mg, 7%), alcohol 26 (15 mg, 10%), and butyl ether 25a (56 mg, 55%).
25a
Colorless oil; Rf = 0.75 (10% EA/Hex); 1H NMR: 7.29–7.26 (m, 2H), 7.23–7.19 (m, 3H), 3.42 (t, J = 6.6 Hz, 2H), 2.79 (s, 2H), 1.59–1.52 (m, 2H), 1.44–1.35 (m, 2H), 1.14 (s, 6H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR: 138.8, 130.7, 127.8, 126.0, 74.9, 61.2, 47.2, 32.8, 25.3, 19.6, 14.1; HRMS (TOF MS EI+) calcd for C14H22O (M)+: 206.1671; found: 206.1677; IR: 2956, 2927, 1453, 1362, 1080.
(2-Hexyloxy-2-methylpropyl)benzene (25b)
By the general procedure described for reaction of Grignard reagents with monoperoxyacetals, a solution of 24c (125 mg, 0.5 mmol) in THF (3 mL) was reacted with hexylMgBr (0.60 mL, 2 M in ether, 2.4 equiv) to furnish, after workup and chromatography, hexyl ether 25b as a colorless oil (88 mg, 75%): Rf = 0.75 (10% EA/Hex); 1H NMR: 7.29–7.25 (m, 2H), 7.22–7.19 (m, 3H), 3.41 (t, J = 6.7 Hz, 2H), 2.78 (s, 2H), 1.60–1.52 (m, 2H), 1.39–1.30 (m, 6H), 1.14 (s, 6H), 0.91 (t, J = 6.8 Hz, 3H); 13C NMR: 138.8, 130.7, 127.8, 126.0, 74.9, 61.5, 47.1, 31.9, 30.7, 26.1, 25.3, 22.7, 14.2. HRMS Calcd for C16H26ONa (M + Na)+: 257.1879; found: 257.1881; IR: 2956, 2927, 1453, 1362, 1080.
Reaction of t-Butyl Peroxide Probe 24a with n-BuLi
Using the general procedure described for reaction of n-BuLi with THP peroxides as above, peroxide 24a (222 mg, 1.0 mmol) was reacted with n-BuLi (0.62 mL, 1.6 M in Hex, 1.1 mmol) at room temperature for 5 h to furnish, following workup and chromatography, the following products: unreacted starting material (101 mg, 45%); alcohol 26 (48 mg, 32%); and pentylbenzene (5 mg, 3%): 2-Methyl-1-phenylpropan-2-ol (26) [100-86-7]: colorless oil; Rf = 0.35 (10% EA/Hex); 1H NMR: 7.33–7.21 (m, 5H), 2.77 (s, 2H), 1.23 (s, 6H); 13C NMR: 137.9, 130.5, 128.3, 126.6, 70.8, 49.8, 29.2. Pentyl benzene [538-68-1]: colorless oil; Rf = 0.8 (10% EA/Hex); 1H NMR: 7.31–7.29 (m, 2H), 7.23–7.19 (m, 3H), 2.63 (t, J = 7.7 Hz, 2H), 1.67–1.62 (m, 2H), 1.40–1.33 (m, 4H), 0.93 (t, J = 7.0 Hz, 3H); 13C NMR: 142.9, 128.4, 128.2, 125.5, 35.9, 31.5, 31.2, 22.5, 14.0.
Reaction of Silyl Peroxide Probe 24d with n-BuLi
Peroxide 24d (280 mg, 1.0 mmol) was reacted with n-BuLi (0.62 mL, 1.6 M in Hex, 1 mmol) at room temperature for 6 h, to furnish a mixture of products which were separated by chromatography: recovered 24d (115 mg, 41%); Et3SiOH (15 mg, 24%); pentylbenzene (16 mg, 11%) and alcohol 26 (15 mg, 10%).
Triethylsilanol [597-52-4]
Colorless oil; Rf = 0.5 (10% EA/Hex); 1H NMR: 1.49–1.44 (bs, 1H), 0.99 (t, J = 8.0 Hz, 9 H), 0.618 (q, J = 8.0 Hz, 6H); 13C NMR: 6.58, 5.79.
Competition for n-BuLi: Monoperoxyacetal vs Peroxide
To a −78 °C solution containing peroxyacetal 12b (0.50 mmol, 129 mg) and peroxide 9a (0.50 mmol, 104 mg) in THF (3 mL) was added n-BuLi (0.31 mL, 1.6 M in Hex, 0.50 mmol). The reaction was quenched after 3 min with an excess of water to furnish, as a colorless oil, n-butyl decyl ether (27, 70 mg, 65%) accompanied by recovered 9a (90 mg, 86%). Butyl decyl ether (27) [111082-32-7]: Rf = 0.8 (10% EA/Hex); 1H NMR: 3.41–3.37 (m, 4H), 1.61–1.51 (m, 4H), 1.41–1.26 (m, 16H), 0.91 (t, J = 7.4 Hz, 3H); 0.87 (t, J = 6.8 Hz, 3H); 13C NMR: 71.1. 70.7, 32.02. 32.00, 29.9, 29.73, 29.69, 29.62, 29.4, 26.3, 22.8, 19.5, 14.2, 14.0.
Competition for Monoperoxyacetal: n-BuLi vs PhLi
Solutions of n-BuLi (0.47 mL, 0.762 mmol, 1.6 M in Hex) and PhLi (0.42 mL, 0.762 mmol, 1.8 M in dibutyl ether, 0.42 mL) were sequentially drawn into the same 1 mL syringe, and the contents were rapidly added to a rapidly stirring −78 °C solution of 9b (0.762 mmol, 180 mg) in dry THF (5 mL). The reaction was quenched at −78 C after 5 min to furnish dialkyl ether 28 (78 mg, 53%), phenyl alkyl ether 29 (30 mg, 19%), and biphenyl (5 mg, 4%): 3-Butoxypropyl benzene (28): Spectral data were nearly identical to those previously reported.79Rf = 0.7 (10% EA/Hex); 1H NMR: 7.32–7.28 (m, 2H), 7.23–7.20 (m, 3H), 3.46–3.43 (m, 4H), 2.72 (t, J = 7.6 Hz, 2H), 1.95–1.91 (m, 2H), 1.63–1.57 (m, 2H)1.44–1.40 (m, 2H), 0.96 (t, J = 7.4 Hz, 3H); 13C NMR: 142.1, 128.5, 128.3, 125.7, 70.7, 69.9, 32.4, 31.9, 31.3, 19.4, 14.0. 3-Phenoxy propyl benzene [64806-63-9] (29): Colorless oil; Rf = 0.6 (10% EA/Hex); 1H NMR: 7.34–7.21 (m, 7H); 6.99–6.92 (m, 3H); 4.00 (t, J = 6.3 Hz, 2H), 2.85 (t, J = 7.5 Hz, 2H), 2.18 (m, 2H); 13C NMR: 159.1, 141.7, 129.5, 128.6, 128.5, 126.0, 120.7, 114.7, 66.9, 32.3, 30.9.
Reactivity of Methoxyethoxy Alkyl Peroxide
Using the general procedure for etherification described above, methoxyethoxy peroxide 9e (105 mg, 0.5 mmol) was reacted with n-BuLi (0.34 mL, 1.6 M in Hex, 0.55 mmol) to furnish 3-butoxypropyl benzene (28, 25 mg, 26%) as a colorless oil, accompanied by a small amount (4 mg, 6%) of 3-phenyl-1-propanol.
Note on Safety
Although no safety issues were encountered in the course of this work, any preparative work with peroxides should be conducted with an awareness of the potential for spontaneous and exothermic decomposition reactions. The reader is directed to a web-published collection of peroxide-related safety information.80
Acknowledgments
We thank the NSF (CHE-1057982 and CHE-1464914) for support and Profs. James Takacs, Andrzej Rajca, and Stephen DiMagno for useful discussions. Portions of this research were conducted in facilities renovated with support from the NIH (RR0116544-01).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.5b02043.
The authors declare no competing financial interest.
Supplementary Material
References
- a Forsyth C. J., Ed. Science of Synthesis, v. 27: Ethers; George Thieme Verlag: Stuttgart, 2008; p 992. [Google Scholar]; b Shade R. E.; Hyde A. M.; Olsen J.-C.; Merlic C. A. J. Am. Chem. Soc. 2010, 132, 1202. 10.1021/ja907982w. [DOI] [PubMed] [Google Scholar]
- Leading references to metal-catalyzed etherification:; a Hartwig J. F. Acc. Chem. Res. 1998, 31, 852. 10.1021/ar970282g. [DOI] [Google Scholar]; b Gowrisankar S.; Sergeev A. G.; Anbarasan P.; Spannenberg A.; Neumann H.; Beller M. J. Am. Chem. Soc. 2010, 132, 11592. 10.1021/ja103248d. [DOI] [PubMed] [Google Scholar]; c Wu X.; Fors B. P.; Buchwald S. L. Angew. Chem., Int. Ed. 2011, 50, 9943. 10.1002/anie.201104361. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lin H.; Sun D. Org. Prep. Proced. Int. 2013, 45, 341. 10.1080/00304948.2013.816208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Example of electrophile-promoted etherification:Larrosa I.; Romea P.; Urpí F. Tetrahedron 2008, 64, 2683. 10.1016/j.tet.2007.11.092. [DOI] [Google Scholar]
- For a leading reference to syntheses based upon metal-catalyzed decomposition of peroxides or peresters in the presence of hydrocarbons, see:Gephart R. T.; McMullin C. L.; Sapiezynski N. G.; Jang E. S.; Aguila M. J. B.; Cundari T. R.; Warren T. H. J. Am. Chem. Soc. 2012, 134, 17350. 10.1021/ja3053688. [DOI] [PubMed] [Google Scholar]
- a Willand-Charnley R. W.; Puffer B. W.; Dussault P. H. J. Am. Chem. Soc. 2014, 136, 5821. 10.1021/ja5026276. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kyasa S. K.; Dussault P. H. Org. Lett. 2014, 16, 5235. 10.1021/ol502726p. [DOI] [PubMed] [Google Scholar]
- Gilman H.; Adams C. E. J. Am. Chem. Soc. 1925, 47, 2816. 10.1021/ja01688a027. [DOI] [Google Scholar]
- Baramki G. A.; Chang H. S.; Edward J. T. Can. J. Chem. 1962, 40, 441. 10.1139/v62-070. [DOI] [Google Scholar]
- Hou Y.; Meyers C. Y.; Akomeah M. J. Org. Chem. 2009, 74, 6362. 10.1021/jo901086s. [DOI] [PubMed] [Google Scholar]
- a Campbell T. W.; Burney W.; Jacobs T. L. J. Am. Chem. Soc. 1950, 72, 2735. 10.1021/ja01162a108. [DOI] [Google Scholar]; b Jarvie A. W. P.; Skelton D. J. Organomet. Chem. 1971, 30, 145. 10.1016/S0022-328X(00)90194-3. [DOI] [Google Scholar]
- Nugent W. A.; Bertini F.; Kochi J. K. J. Am. Chem. Soc. 1974, 96, 4945. 10.1021/ja00822a036. [DOI] [Google Scholar]
- a Camici L.; Dembech P.; Ricci A.; Seconi G.; Taddei M. Tetrahedron 1988, 44, 4197. 10.1016/S0040-4020(01)86666-7. [DOI] [Google Scholar]; b Dembech P.; Ricci A.; Seconi G.; Taddei M. A. Org. Synth 1997, 74, 84. [Google Scholar]; c Neumann H.; Seebach D. Chem. Ber. 1978, 111, 2785. 10.1002/cber.19781110807. [DOI] [Google Scholar]; d Camici L.; Ricci A.; Taddei M. Tetrahedron Lett. 1986, 27, 5155. 10.1016/S0040-4039(00)85158-8. [DOI] [Google Scholar]
- Brandes D.; Blaschette A. J. Organomet. Chem. 1975, 99, C33. 10.1016/S0022-328X(00)88467-3. [DOI] [Google Scholar]
- a Panek E. J.; Kaiser L. R.; Whitesides G. M. J. Am. Chem. Soc. 1977, 99, 3708. 10.1021/ja00453a032. [DOI] [Google Scholar]; b Boche G.; Bosold F.; Lohrenz J. C. W. Angew. Chem., Int. Ed. Engl. 1994, 33, 1161. 10.1002/anie.199411611. [DOI] [Google Scholar]; c Minko Y.; Marek I. Org. Biomol. Chem. 2014, 12, 1535. 10.1039/c3ob42349b. [DOI] [PubMed] [Google Scholar]
- Razuvaev G. A.; Brilkina T. G. Usp. Khim. 1976, 45122196. [Google Scholar]
- a Chen B.-C.; Zhou P.; Davis F. A.; Ciganek E. Org. React. 2003, 62, 1. 10.1002/0471264180.or062.01. [DOI] [Google Scholar]; b Wang D.; Xu C.; Zhang L.; Luo S. Org. Lett. 2015, 17, 576.and references within 10.1021/ol503592n. [DOI] [PubMed] [Google Scholar]
- a Schwaebe M.; Little R. D. Tetrahedron Lett. 1996, 37, 6635. 10.1016/S0040-4039(96)01494-3. [DOI] [Google Scholar]; b Ziegert E.; Bräse S. Synlett 2006, 2006, 2119. 10.1055/s-2006-948201. [DOI] [Google Scholar]
- Lawesson S.-O.; Yang N. C. J. Am. Chem. Soc. 1959, 81, 4230. 10.1021/ja01525a028. [DOI] [Google Scholar]
- a Goodman R. M.; Kishi Y. J. Am. Chem. Soc. 1998, 120, 9392. 10.1021/ja982188g. [DOI] [Google Scholar]; b Criegee R. Liebigs Ann. Chemie 1948, 560, 127. 10.1002/jlac.19485600106. [DOI] [Google Scholar]; c Kropf H. In Methoden der Organischen Chemie, 4th ed.; Kropf H., Ed.; Thieme: Stuttgart, 1988; Vol. E13, pp 1095.–. [Google Scholar]
- For leading references, see:Fisher T. J.; Dussault P. H. Tetrahedron Lett. 2010, 51, 5615. 10.1016/j.tetlet.2010.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo M.; Komatsu T.; Tsuruta K.; Sonoda Y. Bull. Chem. Soc. Jpn. 1980, 53, 829. 10.1246/bcsj.53.829. [DOI] [Google Scholar]
- Okubo M.; Saito H.; Tomiyoshi T. Bull. Chem. Soc. Jpn. 1974, 47, 1289. 10.1246/bcsj.47.1289. [DOI] [Google Scholar]
- Kyasa S.; Puffer B. W.; Dussault P. H. J. Org. Chem. 2013, 78, 3452. 10.1021/jo4001564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dussault P. H.; Sahli A. J. Org. Chem. 1992, 57, 1009. 10.1021/jo00029a043. [DOI] [Google Scholar]
- Milas N. A.; Peeler R. L. Jr.; Mageli O. L. J. Am. Chem. Soc. 1954, 76, 2322. 10.1021/ja01638a012. [DOI] [Google Scholar]
- Rigaudy J.; Izoret G. Compt. Rend. 1953, 236, 2086. [Google Scholar]
- a Doskotch R. W.; El-Feraly F. S.; Fairchild E. H.; Huang C.-T. J. Org. Chem. 1977, 42, 3614. 10.1021/jo00442a037. [DOI] [Google Scholar]; b Kim H.-S.; Nagai Y.; Ono K.; Begum K.; Wataya Y.; Hamada Y.; Tsuchiya K.; Masuyama A.; Nojima M.; McCullough K. J. J. Med. Chem. 2001, 44, 2357. 10.1021/jm010026g. [DOI] [PubMed] [Google Scholar]; c Tokuyasu T.; Masuyama A.; Nojima M.; Kim H.-S.; Wataya Y. Tetrahedron Lett. 2000, 41, 3145. 10.1016/S0040-4039(00)00372-5. [DOI] [Google Scholar]; d Cookson P. G.; Davies A. G.; Roberts B. P. J. Chem. Soc., Chem. Commun. 1976, 1022. 10.1039/c39760001022. [DOI] [Google Scholar]
- a Clark G. R.; Nikaido M. M.; Fair C. K.; Lin J. J. Org. Chem. 1985, 50, 1994. 10.1021/jo00211a047. [DOI] [Google Scholar]; b Adam W.; Catalani L. H.; Saha-Möller C. R.; Will B. Synthesis 1989, 1989, 121. 10.1055/s-1989-27167. [DOI] [Google Scholar]
- a Tietz J. I.; Seed A. J.; Sampson P. Org. Lett. 2012, 14, 5058.and references within 10.1021/ol3022897. [DOI] [PubMed] [Google Scholar]; b Sonpatki V. M.; Herbert M. R.; Sandvoss L. M.; Seed A. J. J. Org. Chem. 2001, 66, 7283. 10.1021/jo015644j. [DOI] [PubMed] [Google Scholar]; c Kiryanov A. A.; Sampson P.; Seed A. J. J. Org. Chem. 2001, 66, 7925. 10.1021/jo016063x. [DOI] [PubMed] [Google Scholar]; d Pedersen E. B.; Lawesson S. O. Tetrahedron 1971, 27, 3861. 10.1016/S0040-4020(01)98247-X. [DOI] [Google Scholar]
- Kornblum N.; DeLaMare H. E. J. Am. Chem. Soc. 1951, 73, 880. 10.1021/ja01146a542. [DOI] [Google Scholar]
- a Seebach D.; Corey E. J. J. Org. Chem. 1975, 40, 231. 10.1021/jo00890a018. [DOI] [Google Scholar]; b Smith A. B. III; Adams C. M. Acc. Chem. Res. 2004, 37, 365. 10.1021/ar030245r. [DOI] [PubMed] [Google Scholar]
- a Landelle G.; Panossian A.; Leroux F. R. Curr. Top. Med. Chem. 2014, 14, 941. 10.2174/1568026614666140202210016. [DOI] [PubMed] [Google Scholar]; b Leroux F. R.; Manteau B.; Vors J.-P.; Pazenok S. Beilstein J. Org. Chem. 2008, 4, 13. 10.3762/bjoc.4.13. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jeschke P. Pest Manage. Sci. 2010, 66, 10. 10.1002/ps.1829. [DOI] [PubMed] [Google Scholar]
- Liang T.; Neumann C. N.; Ritter T. Angew. Chem., Int. Ed. 2013, 52, 8214. 10.1002/anie.201206566. [DOI] [PubMed] [Google Scholar]
- a Ma J.-A.; Cahard D. Chem. Rev. 2008, 108, PR1. 10.1021/cr800221v. [DOI] [PubMed] [Google Scholar]; b Liu X.; Xu C.; Wang M.; Liu Q. Chem. Rev. 2015, 115, 683. 10.1021/cr400473a. [DOI] [PubMed] [Google Scholar]
- a Rubiales G.; Alonso C.; Martinez de Marigorta E.; Palacios F. ARKIVOC 2014, 362. [Google Scholar]; b Prakash G. K. S.; Zhang Z.; Wang F.; Munoz S.; Olah G. A. J. Org. Chem. 2013, 78, 3300. 10.1021/jo400202w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Prakash G. K. S.; Jog P. V.; Batamack P. T. D.; Olah G. A. Science 2012, 338, 1324. 10.1126/science.1227859. [DOI] [PubMed] [Google Scholar]; b Kyasa S. Synlett 2015, 26, 1911. 10.1055/s-0034-1380924. [DOI] [Google Scholar]
- a Still W. C. J. Am. Chem. Soc. 1978, 100, 1481. 10.1021/ja00473a025. [DOI] [Google Scholar]; b Perry M. A.; Rychnovsky S. D. Nat. Prod. Rep. 2015, 32, 517–533 10.1039/C4NP00125G. [DOI] [PubMed] [Google Scholar]; c Chinchilla R.; Nájera C.; Yus M. Tetrahedron 2005, 61, 3139. 10.1016/j.tet.2004.08.011. [DOI] [Google Scholar]
- For leading references, see:; a Schöllkopf U. Angew. Chem., Int. Ed. Engl. 1968, 7, 588. 10.1002/anie.196805881. [DOI] [Google Scholar]; b Olofson R. A.; Lotts K. D.; Barber G. N. Tetrahedron Lett. 1976, 17, 3779. 10.1016/S0040-4039(00)93108-3. [DOI] [Google Scholar]; c Simmons H. E.; Cairns T. L.; Vladchik S. A.; Hoiness C. M. Org. React. 1973, 20, 1. [Google Scholar]; d Hollingworth G. J.; Dinnell K.; Dickinson L. C.; Elliott J. M.; Kulagowski J. J.; Swain C. J.; Thomson C. G. Tetrahedron Lett. 1999, 40, 2633. 10.1016/S0040-4039(99)00235-X. [DOI] [Google Scholar]; e Alnasleh B. K.; Sherrill W. M.; Rubina M.; Banning J.; Rubin M. J. Am. Chem. Soc. 2009, 131, 6906. 10.1021/ja900634m. [DOI] [PubMed] [Google Scholar]
- Longone D. T.; Miller A. H. Tetrahedron Lett. 1967, 8, 4941. 10.1016/S0040-4039(01)89942-1. [DOI] [Google Scholar]
- Arends I. W. C. E.; Ingold K. U.; Wayner D. D. M. J. Am. Chem. Soc. 1995, 117, 4710. 10.1021/ja00121a031. [DOI] [Google Scholar]
- a Magri D. C.; Workentin M. S. Org. Biomol. Chem. 2003, 1, 3418. 10.1039/b305348b. [DOI] [PubMed] [Google Scholar]; b Precursor bromide: Masada H.; Murotani Y. Bull. Chem. Soc. Jpn. 1980, 53, 1181. 10.1246/bcsj.53.1181. [DOI] [Google Scholar]
- a Isayama S.; Mukaiyama T. Chem. Lett. 1989, 573. 10.1246/cl.1989.573. [DOI] [Google Scholar]; b Tokuyasu T.; Kunikawa S.; McCullough K. J.; Masuyama A.; Nojima M. J. Org. Chem. 2005, 70, 251. 10.1021/jo048359j. [DOI] [PubMed] [Google Scholar]; c O'Neill P. M.; Hindley S.; Pugh M. D.; Davies J.; Bray P. G.; Park B. K.; Kapu D. S.; Ward S. A.; Stocks P. A. Tetrahedron Lett. 2003, 44, 8135. 10.1016/j.tetlet.2003.09.033. [DOI] [Google Scholar]
- Houk K. N.; Rondan N. G.; Schleyer P. v. R.; Kaufmann E.; Clark T. J. Am. Chem. Soc. 1985, 107, 2821. 10.1021/ja00295a053. [DOI] [Google Scholar]
- Kaufmann E.; Schleyer P. V.; Houk K. N.; Wu Y. D. J. Am. Chem. Soc. 1985, 107, 5560. 10.1021/ja00305a058. [DOI] [Google Scholar]
- dos Passos Gomes G.; Vil V.; Terent'ev A.; Alabugin I. V. Chem. Sci. 2015, 6, 6783. 10.1039/C5SC02402A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julia M.; Pfeuty-Saint Jaimes V.; Plé K.; Verpeaux J.-N.; Hollingworth G. Bull. Soc. Chim. Fr. 1996, 133, 15. [Google Scholar]
- Kurskii Y. A.; Baryshnikov N.; Vesnovskaya G. I.; Kaloshina N. N.; Yu A. Dokl. Akad. Nauk SSSR 1981, 258, 936.(CAN 95: 114414). [Google Scholar]
- a Dussault P. H.; Lee I. Q.; Lee H.-J.; Lee R. J.; Niu J.; Schultz J. A.; Zope U. R. J. Org. Chem. 2000, 65, 8407. 10.1021/jo991714z. [DOI] [PubMed] [Google Scholar]; b Dussault P. H.; Lee H.-J.; Liu X. J. Chem. Soc., Perkins Trans. 1 2000, 3006. 10.1039/b001391i. [DOI] [Google Scholar]
- Smith L. L.; Hill F. L. J. Chromatogr. 1972, 66, 101. 10.1016/S0021-9673(01)82933-2. [DOI] [PubMed] [Google Scholar]
- a Becke A. D. J. Chem. Phys. 1993, 98, 5648. 10.1063/1.464913. [DOI] [Google Scholar]; b Lee C.; Yang W.; Parr R. G. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Hehre W. J.; Ditchfield R.; Pople J. A. J. Chem. Phys. 1972, 56, 2257. 10.1063/1.1677527. [DOI] [Google Scholar]
- Clark T.; Chandrasekhar J.; Spitznagel G. W.; Schleyer P. v. R. J. Comput. Chem. 1983, 4, 294. 10.1002/jcc.540040303. [DOI] [Google Scholar]
- Frisch M. J.; Pople J. A.; Binkley J. S. J. Chem. Phys. 1984, 80, 3265. 10.1063/1.447079. [DOI] [Google Scholar]
- Merrick J. P.; Moran D.; Radom L. J. Phys. Chem. A 2007, 111, 11683. 10.1021/jp073974n. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. J. Phys. Chem. B 2009, 113, 6378. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Dussault P.Chem. Eng. News 1993, 71 (32), 2, http://pubs.acs.org/cen/safety/19930809.html. [Google Scholar]
- Hoffman R. V.; Tao J. J. Org. Chem. 1998, 63, 3979. 10.1021/jo980003i. [DOI] [Google Scholar]
- Ghorai P.; Dussault P. H. Org. Lett. 2008, 10, 4577. 10.1021/ol801859c. [DOI] [PubMed] [Google Scholar]
- Hocquaux M.; Semeria D.; Saint-Leger D.; Vingler P. Ger. Offen. DE 3825536 A1 19890209, 1989.
- Oh C. H.; Kang J. H. Tetrahedron Lett. 1998, 39, 2771. 10.1016/S0040-4039(98)00335-9. [DOI] [Google Scholar]
- Harris J. R.; Haynes M. T.; Thomas A. M.; Woerpel K. A. J. Org. Chem. 2010, 75, 5083. 10.1021/jo1008367. [DOI] [PubMed] [Google Scholar]
- Klement I.; Lütjens H.; Knochel P. Tetrahedron 1997, 53, 9135. 10.1016/S0040-4020(97)00603-0. [DOI] [Google Scholar]
- Camuzat-Dedenis B.; Provot O.; Cointeaux L.; Perroux V.; Berrien J.-F.; Bories C.; Loiseau P. M.; Mayrargue J. Eur. J. Med. Chem. 2001, 36, 837. 10.1016/S0223-5234(01)01278-8. [DOI] [PubMed] [Google Scholar]
- Duttwyler S.; Chen S.; Lu C.; Mercado B. Q.; Bergman R. G.; Ellman J. A. Angew. Chem., Int. Ed. 2014, 53, 3877. 10.1002/anie.201310517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert C.; Bégue J.-P. Synthesis 1985, 1985, 759. 10.1055/s-1985-31336. [DOI] [Google Scholar]
- Silbert L. S. Analyst 1992, 117, 745. 10.1039/an9921700745. [DOI] [Google Scholar]
- Beard C. D.; Baum K. J. Org. Chem. 1974, 39, 3875. 10.1021/jo00940a016. [DOI] [Google Scholar]
- Dai P.; Dussault P. H.; Trullinger T. K. J. Org. Chem. 2004, 69, 2851. 10.1021/jo035191d. [DOI] [PubMed] [Google Scholar]
- Morales-Serna J. A.; Llaveria J.; Diaz Y.; Matheu M. M.; Castillon S. Org. Biomol. Chem. 2008, 6, 4502. 10.1039/b814882a. [DOI] [PubMed] [Google Scholar]
- Camps F.; Gasol V.; Guerrero A. Synthesis 1987, 1987, 511. 10.1055/s-1987-27988. [DOI] [Google Scholar]
- Dahlen A.; Sundgren A.; Lahmann M.; Oscarson S.; Hilmersson G. Org. Lett. 2003, 5, 4085. 10.1021/ol0354831. [DOI] [PubMed] [Google Scholar]
- Burger B. V.; Petersen W. G. B. J. Chem. Ecol. 2002, 28, 501. 10.1023/A:1014583826875. [DOI] [PubMed] [Google Scholar]
- Livneh M.; Sutter J. K.; Sukenik C. N. J. Org. Chem. 1987, 52, 5039. 10.1021/jo00231a042. [DOI] [Google Scholar]
- Bodipati N.; Palla S. R.; Komera V.; Peddinti R. K. Tetrahedron Lett. 2014, 55, 6878. 10.1016/j.tetlet.2014.10.093. [DOI] [Google Scholar]
- Huo C.; Chan T. H. Adv. Synth. Catal. 2009, 351, 1933. 10.1002/adsc.200900102. [DOI] [Google Scholar]
- a Odinokov V. N.; Kukovinets O. S.; Zainullin R. A.; Kasradze V. G.; Dolidze A. V.; Tolstikov G. A. Zh. Org. Khim. 1992, 28, 1178. [Google Scholar]
- Pratt A. J.; Thomas E. J. J. Chem. Soc., Perkin Trans. 1 1989, 1521. 10.1039/p19890001521. [DOI] [Google Scholar]
- Moerdyk J. P.; Bielawski C. W. Chem. - Eur. J. 2014, 20, 13487. 10.1002/chem.201403407. [DOI] [PubMed] [Google Scholar]
- Watanabe E.; Kaiho A.; Kusama H.; Iwasawa N. J. Am. Chem. Soc. 2013, 135, 11744. 10.1021/ja406028c. [DOI] [PubMed] [Google Scholar]
- Dinnocenzo J. P.; Zuilhof H.; Lieberman D. R.; Simpson T. R.; McKechney M. W. J. Am. Chem. Soc. 1997, 119, 994. 10.1021/ja963112s. [DOI] [Google Scholar]
- http://digitalcommons.unl.edu/chemistryperoxides/1/ (accessed August 2015).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.