

SUMMARY
Tissue effector cells of the monocyte lineage can differentiate into different cell types with specific cell function depending on their environment. The phenotype, developmental requirements, and functional mechanisms of immune protective macrophages that mediate the induction of transplantation tolerance remain elusive. Here, we demonstrate that costimulatory blockade favored accumulation of DC-SIGN-expressing macrophages that inhibited CD8+ T cell immunity and promoted CD4+Foxp3+ Treg cell expansion in numbers. Mechanistically, that simultaneous DC-SIGN engagement by fucosylated ligands and TLR4 signaling was required for production of immunoregulatory IL-10 associated with prolonged allograft survival. Deletion of DC-SIGN-expressing macrophages in vivo, interfering with their CSF1-dependent development, or preventing the DC-SIGN signaling pathway abrogated tolerance. Together, the results provide new insights into the tolerogenic effects of costimulatory blockade and identify DC-SIGN+ suppressive macrophages as crucial mediators of immunological tolerance with the concomitant therapeutic implications in the clinic.
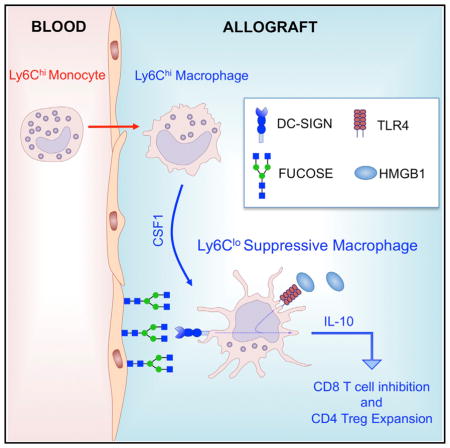
Graphical abstract
INTRODUCTION
Myeloid cells with suppressive activity inhibit graft-reactive T cell immunity and facilitate induction of regulatory T (Treg) cells, together enabling the induction of transplantation tolerance (Dugast et al., 2008; Garcia et al., 2010; Zhang et al., 2008). An emerging consensus is that myeloid cells with immune regulatory function are contained within a population of CD11b+ mononuclear cells that express the myeloid differentiation antigen Gr-1 (Bronte et al., 2000; Bronte et al., 1998). Given the wide range of myeloid cells that might be included in this category, identifying specific myeloid subsets capable of mediating suppression, understanding the molecular basis of their developmental requirements, and deciphering the mechanisms that control their immune regulatory function represents a difficult task.
In previously published work, we demonstrated that monocytic cells that co-express CD11b, Gr-1, and the macrophage colony-stimulating factor 1 receptor (CSF1R) accumulate in cardiac allografts during tolerance induction, mediate T cell suppression in vitro, and are required for long-term graft survival induced by donor-specific transfusion plus anti-CD40L mAb (Garcia et al., 2010). Building upon these published observations, and the recognition that Gr-1 comprises the distinct and independently regulated surface-expressed glycoproteins Ly6C and Ly6G (Fleming et al., 1993), we demonstrate that myeloid suppressive cells expressing CD11b+CSF1R+Ly6Clo Ly6G−CD169+ are responsible for transplantation tolerance. Transcriptome analysis revealed that graft infiltrating immune regulatory CD11b+CSF1R+Ly6CloLy6G−CD169+ monocyte-derived cells correspond to suppressive macrophages.
Blockade of the CD40L-CD40 costimulatory pathway promotes the conversion of immunogenic CD11b+CSF1R+Ly6Chi Ly6G−CD169− into suppressive CD11b+CSF1R+Ly6CloLy6G− CD169+ macrophages through partial inhibition of interferon-γ (IFN-γ) production in the transplanted allograft. The conversion process requires CSF1, and interfering with this cytokine or its receptor (CSF1R) abrogates the induction of indefinite allograft survival. Mechanistically, we demonstrate that the dendritic cell-specific ICAM-grabbing non-integrin (DC-SIGN, CD209a) is upregulated in CD11b+CSF1R+Ly6CloLy6G−CD169+-suppressive macrophages and that simultaneous DC-SIGN engagement by fucosylated ligands and TLR4 signaling is required for production of immunoregulatory interleukin-10 (IL-10) associated with immune regulation and prolonged allograft survival. In addition to delineating a unique set of phenotypic markers and offering new mechanistic insights into suppressive macrophage development and function during transplant tolerance, the data provide a foundation for developing robust protocols potentially capable of inducing immune regulatory macrophages for clinical use.
RESULTS
Suppressive Macrophages Accumulate during Tolerance Induction
To characterize myeloid cells that accumulate in allografts during tolerance induction, we transplanted BALB/c hearts (H-2d) into fully mismatched C57BL6/MaFIA (H-2b) recipient mice. These recipient animals constitutively express green fluorescent protein (GFP) under the CSF1R promoter, permitting us to identify recipient-derived graft-infiltrating myeloid cells that include monocytes, dendritic cells (DCs), macrophages, and neutrophils (Burnett et al., 2004). We treated groups of allograft recipients with anti-CD40L mAb (clone MR1) or with control anti-immuno-globulin G (IgG) mAb (Figure 1A), confirming previous work, which demonstrated that anti-CD40L mAb induced indefinite allograft survival, whereas rejection occurred by day 10 in the IgG-treated controls (Jiang et al., 2011). We harvested donor heart allografts on day 5 post-transplantation and analyzed graft-infiltrating leukocytes by flow cytometry. When we gated on live CD45+CD11b+CSF1RGFP+ recipient graft-infiltrating myeloid cells, we discerned three major populations based on differential expression patterns of Ly6C and Ly6G (Figure 1B). Quantitative analysis revealed a higher frequency of CD11b+ CSF1R+Ly6CloLy6G− cells and a lower frequency of CD11b+ CSF1R+Ly6ChiLy6G− cells in the allografts of anti-CD40L mAb-treated recipients compared to rejecting animals (p < 0.01). No differences in the frequencies of Ly6G cells were observed between groups.
Figure 1. Suppressive Macrophages Accumulate during Tolerance Induction.

(A) Graft survival of control IgG mAb (rejecting) and anti-CD40L mAb (tolerized) recipients of heterotopic cardiac allografts (n = 20 mice/group). The shaded area depicts heart allografts that were harvested at day 5 post-transplantation for subsequent analyses.
(B) Representative and quantitative flow cytometry results for Ly6C and Ly6G expression in CD45+CD11b+ CSF1RGFP myeloid cell subsets from the allografts of tolerized and rejecting recipients at day 5 post-transplantation. Results represent mean ± SEM (n = 8 mice per group).
(C) In vitro suppressive capacity of each myeloid subset for CD8+ T cells. Proliferation was measured by CSFE dilution after 96 hr by flow cytometry. Percentage of cell proliferation is presented as mean ± SEM of five independent experiments.
(D) In vitro Treg expansion of each myeloid subset. Flow cytometric analysis indicates Foxp3 expression on CD4 T cells after co-culture for 96 hr with myeloid subsets. Percentage of Treg expansion is presented as mean ± SEM of five independent experiments.
We tested the ability of each myeloid cell subset to inhibit anti-CD3 and anti-CD28 stimulated CD8+ naive T cell proliferation (Figure 1C). The CD11b+CSF1R+Ly6CloLy6G− cell subset, but not the CD11b+CSF1R+Ly6ChiLy6G− cell subset obtained from anti-CD40L mAb treated recipients, was potently suppressive. The CD11b+CSF1R+Ly6CintLy6G+ cell subset obtained from the anti-CD40L mAb-treated recipients also exhibited a modest suppressive activity. None of the myeloid cell subsets obtained from control IgG treated rejecting allografts exhibited in vitro suppression. We next tested the ability of each myeloid cell subset to induce expansion of CD4+Foxp3+ Treg cell in vitro (Figure 1D). Consistent with suppression assay results, only the CD11b+CSF1R+Ly6CloLy6G− cells obtained from anti-CD40L mAb-treated recipients, promoted the expansion of CD4+Foxp3 expressing Treg cell numbers. Thus, the graft-infiltrating CD11b+CSF1R+Ly6CloLy6G− cell subset that accumulates in anti-CD40L mAb-treated recipients possess many of the properties reported to be associated with monocytic myeloid suppressor cells, including their ability of inhibit CD8 T cell proliferation (Gallina et al., 2006) and to promote CD4+Foxp3+ Treg cell number expansion (Huang et al., 2006).
Further gene array characterization of graft-infiltrating myeloid CD11b+CSF1R+Ly6CloLy6G− cells that accumulate in tolerized recipients revealed that suppressive CD11b+CSF1R+Ly6Clo Ly6G correspond to macrophages (Gautier et al., 2012), but not dendritic cells (Miller et al., 2012) (Figure S1A). Morphological examination of flow-sorted graft-infiltrating myeloid subsets (Figure S1B) confirmed that myeloid CD11b+CSF1R+Ly6Clo Ly6G− cells are of monocytic origin.
Suppressive Macrophages Are Required for Tolerance Induction
The transcriptional analyses of myeloid cell subsets revealed significantly higher transcript expression of CX3CR1, F4/80, CD206 (mannose macrophage receptor), CD68, CD172 (Sirp-α), CD169, and MHC-II in Ly6Clo suppressive macrophages from anti-CD40L mAb treated mice (Figure 2A). Flow cytometry confirmed higher expression of these proteins on suppressive macrophages, and we exploited their differential CD169 expression (Figure 2A and Figures S2A and S2B) along with the availability of CD169 diphtheria toxin receptor (DTR) mice (Miyake et al., 2007) to evaluate the suppressive function of Ly6CloCD169+ macrophages in vivo. We transplanted BALB/c hearts into anti-CD40L mAb-treated WT or CSF1RGFP+/ CD169DTR C57BL6 recipients and treated them with DT on the day of transplantation to deplete recipient CD169+ cells. Graft-infiltrating leukocytes by flow cytometry examined 5 days later (Figure 2B) showed specific depletion of recipient suppressive macrophages only in DT-treated animals. Depletion of Ly6CloCD169+ macrophages in the anti-CD40L mAb-treated recipients was associated with accumulation of memory or activated CD44hiCD62Llo CD8+ T cells on day 5 (Figures 2C and 2D) and a reduced percentage of graft infiltrating CD4+Foxp3+ Treg on day 21 posttransplant (Figure 2E). To verify that in vitro suppressive Ly6CloCD169+ macrophages also exhibit inhibitory function in vivo, we adoptively transferred CFSE-labeled CD8 T cells into anti-CD40L mAb-treated CD169DTR recipients and evaluated their ability to proliferate measured by CFSE dilution 5 days after DT treatment (Figure 2F). Whereas CD8 T cells transferred into tolerized recipients did not proliferate in vivo, CD8 T cells underwent proliferation in the allografts of tolerized recipients following depletion of CD169+ suppressive macrophages. Moreover, graft survival experiments showed that DT induced in vivo depletion of CD169+-suppressive macrophages resulted in graft rejection by day 30 despite tolerogenic treatment with anti-CD40L mAb (p < 0.01) (Figure 2G). Thus, Ly6CloCD169+-suppressive macrophages that accumulate in the allografts of anti-CD40L mAb-treated recipients inhibit T cell immune responses in vivo and are required for the induction of transplantation tolerance.
Figure 2. Suppressive Macrophages Are Required for Tolerance Induction.

(A) Heatmap derived from microarray data of selected myeloid markers that achieve p < 0.05 in myeloid subsets from the allografts of tolerized recipients at day 5 post-transplantation (means of n = 3 per group). Representative flow cytometry plots of the above myeloid markers on each myeloid subset. Data is representative of three independent experiments.
(B) Representative and quantitative flow cytometry results of recipient myeloid cell subsets in the allografts of tolerized CSF1RGFP (wild-type) and CSF1RGFP CD169DTR recipients at day 5 post-transplantation. Results represent mean ± SEM (n = 12 mice per group of 3 independent experiments).
(C and D) Representative and quantitative flow cytometry results depicting percentages (C) and surface memory/naive CD44/CD62L phenotypes (D) of graft infiltrating CD8 T cells after CD169+ macrophage depletion. Results represent mean ± SEM (n = 12 mice per group of 3 independent experiments).
(E) Representative flow cytometry results depicting percentages of Foxp3 expressing graft infiltrating CD4+ T cells on day 21 post-transplantation in tolerized recipients with or without CD169+ macrophage depletion. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
(F) Effects of CD169+ macrophage depletion on in vivo T cell proliferation. CFSE-labeled CD8+ T cells (5 × 106) were injected into tolerized CSF1RGFP and CSF1RGFP CD169DTR recipients. Proliferation was measured in the allograft by CSFE dilution after 120 hr by flow cytometry. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
(G) Effects of CD169+ macrophage depletion on graft survival in tolerized CSF1RGFP and CSF1RGFP CD169DTR recipients (n = 12 mice/group).
CD40L Blockade Inhibits Accumulation of Immunogenic Macrophages
Graft-infiltrating myeloid subsets express CD40, but not CD40L (Figures S3A and S3B), suggesting that tolerogenic properties of the anti-CD40L mAb treatment are not due to a direct effect on monocyte-derived cells because they do not express CD40L. To test whether anti-CD40L mAb therapy induces suppressive macrophages via inhibiting transmission of a CD40-dependent signal on the myeloid cells, we attempted to circumvent the effects of the anti-CD40L mAb blockade by co-administering an agonistic anti-CD40 antibody FGK45.5, an antibody that has been shown to transmit CD40-dependent signals to APC in the absence of CD40L (Bennett et al., 1998; Rolink et al., 1996; Schoenberger et al., 1998). Administration of the agonistic anti-CD40 mAb promoted the accumulation of immunogenic Ly6Chi macrophages in the allograft (Figure 3A). CD40-mediated accumulation of Ly6Chi macrophages might be mediated by increased IFN-γ expression in the allografts of tolerized recipients (Jutila et al., 1988). To test for a link between CD40L-CD40 ligation and IFN-γ-mediated Ly6Chi macrophage activation in our transplant model, we measured IFN-γ in the al-lografts of untreated recipients, tolerized recipients, and tolerized recipients co-treated with agonistic anti-CD40 mAb (Figure 3B). These assays showed reduction of intra-graft IFN-γ in the tolerized allografts compared to the untreated controls as previously reported (Hancock et al., 1996) but restoration of intra-graft IFN-γ observed in the anti-CD40 mAb co-treated recipient. On the contrary, agonistic CD40 ligation-mediated accumulation of immunogenic Ly6Chi macrophages and increased intra-graft IFN-γ expression was not observed in tolerized CD40-deficient (Cd40−/−) recipients (Figures 3C). Agonistic CD40 mAb abrogated the induction of tolerance despite CD40L blockade in wild-type (WT), but not in Cd40−/− recipients (Figure 3D). The data suggest that anti-CD40L mAb-induced tolerance can be abrogated by CD40 ligation, which favors the accumulation of Ly6Chi immunogenic macrophages in the allograft through IFN-γ. To confirm this hypothesis, we treated tolerized recipients with recombinant IFN-γ and observed reduced intra-graft accumulation of Ly6Clo macrophages on day 5 (Figure 3E) associated with allograft rejection (Figure 3F). Conversely, partial IFN-γ blockade restored accumulation of Ly6Clo macrophages in the allografts (Figure 3G) and reestablished indefinite allograft survival (Figure 3H) in tolerized recipients despite agonistic anti-CD40 mAb treatment. Thus, costimulation blockade with anti-CD40L mAb prevents IFN-γ production and accumulation of immunogenic Ly6Chi macrophages in the transplanted allografts.
Figure 3. CD40L Blockade Inhibits Accumulation of Immunogenic Macrophages.

(A) Representative and quantitative flow cytometry results of recipient myeloid cell subsets in the allografts of tolerized WT recipients with or without co-treatment with agonistic CD40 mAb. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
(B) IFN-γ expression in cardiac allografts. Cardiac allografts were harvested 5 days after transplantation from each group. Agonistic anti-CD40 mAb was injected at 100 μg/mouse on days 0 and +1 relative to transplantation. Recombinant mouse IFN-γ was injected at 4 × 105 units/day for 10 days (n = 4 mice/group). Supernatants of single cell suspensions were analyzed for IFN-γ measured by ELISA. Bar graphs represent mean ± SEM of three independent experiments (**p < 0.01).
(C) Representative and quantitative flow cytometry results of recipient myeloid cell subsets in the allografts of tolerized CD40 deficient recipients with or without agonistic CD40 mAb treatment. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
(D) Effects of CD40 ligation on graft survival in tolerized WT and CD40 deficient recipients (n = 8 mice/group).
(E) Representative and quantitative flow cytometry results of recipient myeloid cell subsets in the allografts of tolerized WT recipients with or without recombinant IFN-γ treatment (4 × 105 units/day for 5 days). Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
(F) Effects of recombinant IFN-γ on graft survival in tolerized WT recipients (n = 8 mice/group).
(G) Representative and quantitative flow cytometry results of recipient myeloid cell subsets in the allografts of tolerized WT recipients co-treated with agonistic CD40 mAb with or without anti-IFN-γ mAb treatment. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
(H) Effects of partial IFN-γ blockade on graft survival in tolerized wild-type recipients co treated with agonistic CD40 mAb (n = 8 mice/group).
CSF1 Mediates the Development of Suppressive Macrophages
The expression of CSF1R in CD11b+CSF1R+Ly6CloLy6G− CD169+-suppressive macrophages suggests an involvement of CSF1 in the development of these cells. We quantified CSF1 transcripts in transplanted mice by RT-PCR (Figure 4A) and observed significant upregulation of CSF1 in the allografts of anti-CD40L mAb-treated recipients. To test for a mechanistic link between CSF1 and development of Ly6Clo-suppressive macrophages in anti-CD40L mAb-treated recipient mice, we transplanted BALB/c hearts into anti-CD40L mAb-treated tolerized C57BL6 recipients with or without neutralizing anti-CSF1 mAb (clone 5A1) at doses shown by others to inhibit their function in vivo (Gregory et al., 1992). Our results indicate that in vivo CSF1 blockade abrogated intra-graft accumulation of Ly6Clo suppressive macrophages (Figure 4B). CSF1 blockade also prevented the in vivo expansion of CD4+ Foxp3+ Treg cell and abrogated the induction of transplantation tolerance (Figures 4C and 4D). In vivo blockade of CSF1R receptor (clone AFS98), at doses shown by others to inhibit their function in vivo (Hashimoto et al., 2011), also abrogated tolerance, which suggests that CSF1-CSF1R signaling is necessary for the development of suppressive macrophages. Ly6Chi monocytes convert into Ly6Clo macrophages (Arnold et al., 2007), the latter being able to function as suppressive cells in tumor models (Corzo et al., 2010). To test whether analogous mechanisms apply in transplant tolerance, we isolated CD11b+CSF1R+ Ly6ChiLy6G− GFP+ bone marrow monocytes from C57BL6/ MaFIA mice and transferred them into C57BL6/WT recipients with or without anti-CD40L mAb and anti-CSF1 blocking mAb (Figure 4E). Whereas the Ly6Chi monocytic precursors converted into Ly6Clo macrophages in the allografts of anti-CD40L mAb-treated mice, Ly6Chi monocytic precursors from anti-CSF1 mAb-treated recipient mice failed to convert and maintained a Ly6Chi phenotype, similar to the untreated rejecting controls. Additional in vitro experiments confirmed that CSF1 mediates the conversion of Ly6Chi monocytic precursors into Ly6Clo myeloid cells that were functionally able to inhibit CD8+ T cell proliferation and promote Treg expansion (Figures 4F and 4G). Our in vitro human data is consistent with this hypothesis and suggests that CSF1, but not CSF2, promotes the development of CD14 monocytes into suppressive monocyte-derived cells that inhibit CD8 T cell proliferation and expand Foxp3-expressing Treg in vitro (Figure S4A) Thus, anti-CD40L mAb-induced tolerance requires prevention of IFN-γ production and upregulation of CSF1, the latter driving conversion of monocytic precursors into suppressive macrophages.
Figure 4. CSF1 Mediates the Development of Suppressive Macrophages.

(A) CSF1 expression in cardiac allografts. Cardiac allografts were harvested 5 days after transplantation from tolerized and rejecting recipients. Total single cell suspensions were analyzed for CSF1 measured by real-time PCR. Bar graphs represent mean ± SEM of three independent experiments (**p < 0.01).
(B) Representative and quantitative flow cytometry results of recipient myeloid cell subsets in the allografts of tolerized WT recipients co-treated with anti-CSF1 mAb. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
(C) Representative and quantitative flow cytometry results of Foxp3 expression on CD4 T cells in the allografts of tolerized recipients on day 21 post-transplantation following anti-CSF1 mAb treatment. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
(D) Effects of CSF1 and CSF1R blockade on graft survival in tolerized WT recipients (n = 12 mice/group).
(E) Representative and quantitative flow cytometry results of adoptively transferred CSF1R+Ly6Chi bone marrow cells into recipient mice treated with anti-CD40L mAb ± anti-CSF1 mAb 5 days after transplantation. Results represent mean ± SEM (n = 3 mice per group of 3 independent experiments).
(F) Representative and quantitative flow cytometry results of in vitro cultured Ly6Chi bone marrow cells with either recombinant CSF1 or IFNγ for 96 hr. Results represent mean ± SEM of three independent experiments.
(G) Suppressive function of Ly6Chi bone marrow cells after CSF1 or IFN-γ in vitro treatment. Results represent mean ± SEM of three independent experiments.
DC-SIGN Controls the Function of Suppressive Macrophages
CSF1 upregulates the expression of the dendritic-cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN, CD209a) (Choi et al., 2011; Domínguez-Soto et al., 2011). Our gene array, real-time PCR, flow cytometry, and immunofluorescence studies revealed higher expression of DC-SIGN in macrophages obtained from the allografts of anti-CD40L mAb-treated mice, non-rejecting human renal transplant recipients, or in vitro derived CSF1-dependent human macrophages (Figures 5A–5C, Figures S5A–S5D). To test whether DC-SIGN is required for anti-CD40L mAb-induced allograft survival, we transplanted BALB/c hearts into WT C57BL6 recipients under the cover of anti-CD40L mAb, together with either a blocking anti-DC-SIGN mAb or an iso-type IgG control. DC-SIGN blockade abrogated the induction of indefinite allograft survival in anti-CD40L mAb-treated mice (Figure 5D). When we isolated and compared graft-infiltrating leukocytes from anti-DC-SIGN mAb-treated recipients with control mice (all treated with anti-CD40L mAb), we observed similar frequencies of graft-infiltrating Ly6CloLy6G− macrophages, suggesting that anti-DC-SIGN mAb treatment does not prevent intra-graft accumulation of Ly6CloLy6G− macrophages (Figure 5E). However, whereas flow-sorted graft-infiltrating CD11b+CSF1R+Ly6CloLy6G− macrophages from tolerized recipients suppressed CD8+ T cell proliferation and expanded CD4+Foxp3+ Treg in vitro, flow-sorted CD11b+ CSF1R+Ly6CloLy6G− macrophages obtained from tolerized recipients treated with anti-DC-SIGN mAb did not exhibit either of these immune regulatory functions (Figure 5F). Finally, we flow-sorted CD11b+CSF1R+Ly6CloLy6G− -suppressive macrophages from anti-CD40L mAb-treated recipients and assessed their ability to suppress CD8+ T cell proliferation and expanded CD4+Foxp3+ Treg following in vitro blockade of DC-SIGN by adding anti-DC-SIGN mAb to the cell cultures (Figure 5G). Thus, DC-SIGN expression is required for the immune regulatory function of suppressive macrophages that mediate indefinite allograft survival. Using DC-SIGN deficient (CD209a−/−) and CD169DTR tumor bearing mice our data revealed that depletion of CD169+ macrophages or absence of DC-SIGN significantly reduces in vivo tumor growth (Figure S6A). Together with data from renal cell carcinoma patients showing increased DC-SIGN expression at the tumor site (Figure S6B), this suggests that DC-SIGN+ macrophages also participate in the immune regulatory function that controls tumor progression.
Figure 5. DC-SIGN Controls the Function of Suppressive Macrophages.

(A and B) Heatmap derived from microarray data (A) and flow cytometry expression (B) of DC-SIGN in myeloid subsets from the allografts of tolerized and rejecting recipients at day 5 post-transplantation (means of n = 3 per group). Flow cytometry plots are representative of three independent experiments.
(C) Quantitative immunofluorescent analysis of tolerized and rejecting allografts at day 5 post-transplantation. Bar graphs represent frequency of DCSIGN+ cells expressed as percentage of a total of 1,000 DAPI nucleated cells from the allografts of tolerized and rejecting mice. Results represent mean ± SEM of 10 tissue sections from 4 cardiac allografts per group (**p < 0.01).
(D) Effects of DC-SIGN blockade and DC-SIGN deficiency on graft survival in tolerized WT recipients (n = 12 mice/group).
(E) Representative and quantitative flow cytometry results of recipient myeloid cell subsets in the allografts of tolerized WT recipients co-treated with anti-DC-SIGN mAb. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
(F) Representative and quantitative flow cytometry results of in vitro suppressive capacity and Treg expansion of Ly6Clo macrophages from tolerized recipients co-treated with anti-DC-SIGN. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
(G) Suppressive function of Ly6Clo macrophages from tolerized recipients after in vitro treatment with anti-DC-SIGN mAb. Results represent mean ± SEM of three independent experiments.
Fucosylated DC-SIGN Ligands Are Required for Macrophage-Mediated Suppression and Tolerance
DC-SIGN binds to carbohydrates containing mannose or fucose residues, such as LewisX (van Liempt et al., 2006). We next investigated the role of fucosylated LewisX in the induction of transplantation tolerance using the α1,3/4-fucosyltransferases (FucTs) IV-VII double-deficient (dKO) donor mice, which display impaired LewisX expression (Lowe, 2002). We next used FucT-IV and FucT-VII mice as donors to evaluate the effects of LewisX inhibition on suppressive Ly6Clo macrophages and tolerance. Figure 6A indicates that the LewisX expression was significantly reduced in tolerized dKO donor allografts, which was associated with acute rejection despite tolerogenic treatment with anti-CD40L mAb (Figure 6B). We next compared recipient graft-infiltrating leukocytes from donor dKO and WT allografts treated with anti-CD40L mAb, and we observed similar frequencies of graft-infiltrating Ly6CloLy6G− macrophages (Figure 6C). These results suggest that LewisX deficiency does not prevent intra-graft accumulation of Ly6Clo macrophages. However, whereas flow-sorted graft-infiltrating Ly6Clo macrophages from WT donors suppressed CD8+ T cell proliferation and expanded CD4+Foxp3+ Treg cell in vitro, the flow-sorted Ly6Clo macrophages obtained from dKO donor allografts did not exhibit neither of these immune regulatory functions (Figure 6D). We next investigated whether lacto-N-fucopentaose III (LNFPIII), a LewisX containing pentasaccharide that binds to DC-SIGN (Meyer et al., 2005), could overcome the fucosylated LewisX deficiency in dKO donor allograft recipients (Figure 6E). Our results indicate that unlike dextran (which does not bind to CD209a-mDC-SIGN [Takahara et al., 2004]), LNFPIII is able to restore tolerance in transplant recipients containing dKO donor allo-grafts. Flow-sorted graft-infiltrating Ly6Clo macrophages from LNFPIII-treated dKO donor allograft tolerized recipients were able to suppress CD8+ T cell proliferation and expanded CD4+ Foxp3+ (Figure 6F). Our results indicate that in vivo LNFPIII treatment restores the suppressive activity of Ly6Clo macrophages. Thus, LewisX-mediated DC-SIGN ligation is necessary for the immune regulatory function of suppressive macrophages and for the induction of indefinite allograft survival.
Figure 6. Fucosylated DC-SIGN Ligands Are Required for Macrophage Mediated Suppression.

(A) Quantitative immunofluorescent analysis of tolerized and rejecting allografts at day 5 post-transplantation. Bar graphs represent frequency of Lewis X+ cells expressed as percentage of a total of 1,000 DAPI nucleated cells from tolerized mice receiving WT and fucosyltranferase (Fut) IV and VII double-deficient donor allografts. Results represent mean ± SEM of ten tissue sections from four cardiac allografts per group (**p < 0.01).
(B) Effects of LewisX deficiency on graft survival in tolerized WT recipients (n = 12 mice/group).
(C) Representative and quantitative flow cytometry results of recipient myeloid cell subsets in tolerized mice type receiving WT and FucT IV-VII dKO donor allografts. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
(D) Representative and quantitative flow cytometry results of in vitro suppressive capacity and Treg expansion of Ly6Clo macrophages from the allografts of tolerized mice receiving WT and FucT IV-VII dKO donor allografts. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments). (E) Effects of Lacto-N-fucopentaose III (LNFPIII) on graft survival in tolerized wild-type recipients (n = 8 mice/group).
(F) Representative and quantitative flow cytometry results of in vitro suppressive capacity and Treg expansion of Ly6Clo macrophages from the allografts of tolerized mice receiving WT and FucT IV-VII dKO donor allografts following administration of LNFPIII. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
IL-10 Is Essential for DC-SIGN-Mediated Suppression
Fucose-specific DC-SIGN signaling results in production of IL-10 (Caparrós et al., 2006; Gringhuis et al., 2014). We observed a significant IL-10 upregulation in the allografts of anti-CD40L mAb-treated WT recipients compared to untreated rejecting controls (Figure 7A). In contrast, IL-10 was essentially absent in anti-CD40L mAb-treated DC-SIGN-deficient (CD209a−/−) recipient mice. Among graft-infiltrating leukocytes, we detected the highest IL-10 expression in Ly6Clo macrophages obtained from anti-CD40L mAb treated recipients, but the same Ly6Clo macrophages obtained from the allografts of CD209a−/− recipients exhibited significant less IL-10 expression despite anti-CD40L mAb treatment (Figure 7A; Figure S7A). To specifically test whether IL-10 is required for regulatory macrophage function, we sorted intra-graft Ly6Clo macrophages from anti-CD40L mAb treated IL-10-deficient (Il10−/−) recipient mice and tested their ability to suppress CD8+ T cell proliferation and to expand CD4+Foxp3+ Treg cell in vitro (Figure 7B). In the absence of IL-10, Ly6Clo macrophages did not exhibit either of these immune regulatory functions despite tolerogenic treatment with anti-CD40L mAb. Using CD209a−/−recipient mice, we next investigated whether recombinant IL-10 could restore the suppressive function of Ly6Clo graft-infiltrating macrophages (Figure 7C). Although intra-graft CD209a−/−Ly6Clo macrophages were unable to suppress CD8+ T cell proliferation and to expand CD4+Foxp3+ Treg in vitro, IL-10 addition rescued the immune regulatory function of CD209a−/− Ly6Clo macrophages. Thus, graft-infiltrating DC-SIGN+Ly6Clo macrophages exert their immune regulatory function in part through an IL-10-dependent mechanism. Because crosstalk between DC-SIGN and TLR4 signaling is required for fucose binding-meditated production of IL-10 (Gringhuis et al., 2007; Gringhuis et al., 2014), we explored the effects of TLR4 deficiency in suppressive macrophages using tolerized TRL4 recipients, and showed that in the absence of TLR4 stimulation, IL-10 production was reduced in Ly6Clo macrophages (Figure 7D), and their in vitro suppressive function was defective (Figure 7E). To demonstrate that synergistic DC-SIGN and TRL4 signaling was necessary for IL-10 production, we cultured bone marrow cells from WT, CD209a−/−, and TRL4-deficient (Tlr4−/−) mice and stimulated them with the DC-SIGN ligand LewisX and the TLR4 ligand high mobility group box 1(HMGB1) (Figure 7F). Simultaneous DC-SIGN and TRL4 signaling was necessary for optimal IL-10 production, and interfering with one of the signals resulted in impaired IL-10 production. We investigated whether ligation of DC-SIGN and/or TLR4 resulted in an increased inhibitory function (Figure 7G). Addition of both DC-SIGN and TLR4 agonists resulted in the highest suppressive function observed in monocyte-derived human cells in comparison with each of the ligands alone. Thus, DC-SIGN+ macrophages stimulated though DC-SIGN and TLR4 are negative regulators of the immune response and that their manipulation will open new avenues for therapeutic intervention either by inhibiting their function (i.e., in cancer patients) or by enhancing their suppressive effects and promoting their expansion (i.e., in transplant recipients).
Figure 7. IL-10 Is Essential for DC-SIGN Mediated Suppression.

(A) IL-10 expression in cardiac allografts and Ly6Clo macrophages. Cardiac allografts from WT untreated, WT tolerized, and DC-SIGN KO tolerized recipients were harvested 5 days after transplantation. Total single cell suspensions and Ly6Clo cells were analyzed for IL-10 measured by real-time PCR. Bar graphs represent mean ± SEM of three independent experiments (**p < 0.01).
(B) Representative and quantitative flow cytometry results of in vitro suppressive capacity and Treg expansion of Ly6Clo macrophages from the allografts of tolerized IL-10 deficient recipient mice. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
(C) Representative and quantitative flow cytometry results of in vitro suppressive capacity and Treg expansion of Ly6Clo macrophages from the allografts of tolerized DC-SIGN deficient recipient mice receiving in vitro IL-10 stimulation for 72 hr at 10 ng/ml. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
(D) IL-10 expression in cardiac allografts. Cardiac allografts from tolerized WT and TLR4 KO recipients were harvested 5 days after transplantation. Total single cell suspensions were analyzed for IL-10 measured by real-time PCR. Bar graphs represent mean ± SEM of three independent experiments (**p < 0.01). (E) Representative and quantitative flow cytometry results of in vitro suppressive capacity and Treg expansion of Ly6Clo macrophages from the allografts of tolerized TLR4 deficient recipient mice. Results represent mean ± SEM (n = 4 mice per group of 3 independent experiments).
(F) IL-10 expression in stimulated bone marrow cells from WT, DC-SIGN-deficient, and TLR4-deficient mice. Bone marrow cells were stimulated with LewisX (10 μg/ml) and recombinant HMGB1 (10 μg/ml) for 72 hr in vitro stimulation (control group non-stimulated). Supernatants of single cell suspensions were analyzed for IL-10 measured by ELISA. Results represent mean ± SEM of three independent experiments (**p < 0.01).
(G) Representative and quantitative flow cytometry results for in vitro suppressive capacity of human monocytes cultured with CSF1 plus IL-4. Results represent mean ± SEM of three independent experiments (*p < 0.05, **p < 0.01).
DISCUSSION
We demonstrate here that DC-SIGN-expressing macrophages are required for the induction of transplantation tolerance. DC-SIGN is a type II transmembrane C-type lectin with a carbohydrate recognition domain, which is expressed in human DCs and macrophages (Geijtenbeek et al., 2000; Soilleux et al., 2002), and is involved in multiple aspects of the immunological response (van Kooyk and Geijtenbeek, 2003). Broxmeyer and colleagues reported that in vitro differentiation of monocytes in the presence of M-CSF and IL-4, which induces DC-SIGN expression (Martinez et al., 2006), are less efficient inductors of allogeneic mixed lymphocyte reactions (Li et al., 2004; Li et al., 2005). Here, we extend these findings to newly demonstrate that DC-SIGN+ macrophages inhibit T cell proliferation in vitro and in vivo in an experimental mouse model of solid organ transplantation. Additionally, we demonstrate that human DC-SIGN expressing macrophages stimulated with M-CSF and IL-4 (Figure S5D) induced the expansion of Foxp3-expressing Treg from allogeneic naive CD4+ T cell precursors in vitro, whereas macrophages treated with GM-CSF and IL-4 did not drive Treg expansion (Figure S4A).
The ability of murine DC-SIGN+ macrophages to promote IL-10-mediated transplantation tolerance requires two synergistic signals: DC-SIGN engagement by fucosylated ligands and TLR4 signaling. The CDR domain of human DC-SIGN recognizes fucosylated Lewis glycans (van Liempt et al., 2006) expressed by self and non-self antigens (Geijtenbeek et al., 2004). In humans, DC-SIGN ligation potentiates the secretion of IL-10 (Geijtenbeek et al., 2003). Because DC-SIGN macrophages secrete IL-10 upon fucose ligand engagement (Gringhuis et al., 2014) and participate in the generation of regulatory T cells (Cai et al., 2013; Smits et al., 2005), DC-SIGN could actively contribute to the maintenance of an immunosuppressive tissue environment, as proposed by Yvette van Kooyk’s laboratory (van Gisbergen et al., 2005). Indeed, DC-SIGN ligation by non-immune cells, such as pathogens and tumor tissue results in immune escape (Geijtenbeek and Gringhuis, 2009), suggesting that both tumor and pathogens have ways to escape immune activation by targeting DC-SIGN. Consistent with this hypothesis our data reveals that depletion of CD169+ macrophages or absence of DC-SIGN significantly reduces in vivo tumor growth (Figure S6A), suggesting that DC-SIGN+ macrophages might participate in the immune regulatory function that controls tumor progression (Figure S6B). Our transplant results indicate that fucosylated glycans are present in the donor allografts of tolerized recipients that serve as ligands of DC-SIGN expressing macrophages. Using fucosyltransferase-deficient donor heart allografts inhibits the expression of LewisX glycoproteins and prevents the induction of indefinite allograft survival despite tolerogenic treatment with anti-CD40L mAb treatment. This suggests common mechanisms of immune regulation following engagement of DC-SIGN by tumor and transplant microenvironment via LewisX recognition that lead to the production of IL-10 producing macrophages (Domínguez-Soto et al., 2011; Nonaka et al., 2008; van Gisbergen et al., 2005).
Induction of transplantation mediated by DC-SIGN+-suppressive macrophages depends on simultaneous TLR4 signaling. DC-SIGN signaling crosstalk with TLR4 has been demonstrated to mediate IL-10 production (Geijtenbeek et al., 2003; Gringhuis et al., 2007; Gringhuis et al., 2014). Here we report that DC-SIGN+ macrophages from TLR4-deficient heart recipients produce significantly less IL-10 and do not exhibit suppressive function. Consistent with these results, a recent study indicates that during peripheral tolerance, DC-SIGN and TLR4 are required for IL-10 secretion and decreased T cell proliferation in mixed leukocyte reactions possibly caused by an increased frequency of Treg cell, which is associated with a high fucosyltransferase expression (García-Vallejo et al., 2014). In transplantation, while absence of absence of innate MyD88 signaling prevents acute allograft rejection and promotes inducible allograft acceptance (Goldstein et al., 2003; Walker et al., 2006), it is possible that specific signaling molecules of the MyD88 pathway, such as TRL4, might have a critical role in the induction of tolerance mediated by suppressive myeloid cells. In this respect, the TLR4 agonist HMGB1, which is upregulated during tissue damage associated with ischemia reperfusion (Wu et al., 2007) and surgical transplantation (Huang et al., 2007), has been recently demonstrated to enhance the immune-suppressive capacity of myeloid-derived suppressor cells through the production of IL-10 (Parker et al., 2014).
In conclusion, we demonstrate that graft-infiltrating DC-SIGN+-suppressive macrophages mediate the induction of transplantation tolerance, revealing a previously unknown function of mouse DC-SIGN. Our delineation of a specific cell-surface phenotype for immunoregulatory, graft-protective suppressive macrophages in transplantation, as well as the mechanistic insights underlying the requirements for their differentiation in vivo, have important implications for understanding and potentially manipulating pathogenic immune responses. The C-type lectin DC-SIGN (CD209a) has a critical function in the induction of transplantation tolerance as demonstrated by its absence or in vivo blockade and might be used as phenotypic marker to define immune regulatory macrophages. The data provide a framework for developing CSF1-based in vitro protocols to induce therapeutic macrophages for clinical use to prevent transplant rejection and suggest that depleting or blocking suppressive macrophage development by targeting CSF1, which upregulates the expression of DC-SIGN, could be exploited to enhance anti-tumor immunity.
EXPERIMENTAL PROCEDURES
Mice
BALB/c, C57BL/6, C57BL/6-Foxp3tm1Flv/J, B6.129P2-Cd40tm1Kik/J, and B6.B10ScN-Tlr4lps-del/JthJ mice 8 weeks of age were purchased from The Jackson Laboratory. DC-SIGN-deficient mice (DC-Sign-KO, B6 [FVB]-Cd209atm1.1Cfg/Mmcd) were from the Mutant Mouse Regional Resource Centers, Consortium for Functional Glycomics (Scripps Res. Institute). The alpha(1,3)fucosyltransferases FucT-IV and FucT-VII double-deficient mice were from John Lowe (University of Michigan). The C57BL/6-Tg (Csf1r-EGFP-NGFR/FKBP1A/TNFRSF6) 2Bck/J MaFIA mice from D. Cohen (University of Kentucky) (Burnett et al., 2004). The C57BL/6 CD169DTR mice have been previously described (Miyake et al., 2007). All experiments were performed with age- and sex-matched mice in accordance with Institutional Animal Care and Utilization Committee-approved protocols.
Vascularized Heart Transplantation
BALB/c hearts were transplanted as fully vascularized heterotopic grafts into C57BL/6 mice as previously described (Corry et al., 1973). Recipient mice were treated with 250 μg anti-CD40L mAb (clone MR1, BioXcell) for tolerance induction on days 0, 2, and 4 as previously described (Jiang et al., 2011). Graft function was monitored every other day by abdominal palpation. Untreated control mice received hamster IgG in PBS. Rejection was defined as complete cessation of a palpable beat and confirmed by direct visualization at laparotomy.
In Vivo Cell Depletion
For depletion of CD169 expressing CD11b+CSF1R+Ly6CloLy6G− regulatory macrophages heterozygous CD169-DTR recipients were injected intraperitoneally (i.p.) with 10 ng/g body weight of DT (Sigma-Aldrich) 24, 48, and 72 hr after transplantation (Miyake et al., 2007). Ly6G+ cell depletion was induced with anti-Ly6G mAb clone 1A8 (BioXcell) injected at 0.5 mg i.p. on days –3, –2, and –1 relative to transplantation as previously described (Daley et al., 2008; Garcia et al., 2010).
Antibody-Mediated In Vivo Treatment
Agonistic anti-CD40 mAb (clone FGK4.5 mAb) was produced by BioXcell. CD40-mediated priming independent of CD40L was achieved by intravenous (i.v.) injection of 100 μg of agonistic anti-CD40 mAb on days 0 and +1 relative to transplantation (Gorbachev and Fairchild, 2004). Blocking antibody to IFN-γ (Clone R4-6A2) was produced by BioXcell. Anti-IFN-γ mAb was injected at 500 μg on days 0 and +1 relative to transplantation. Blocking antibody to CSF1 (clone 5A1) (Lokeshwar and Lin, 1988) and CSF1R (clone AFS98) (Sudo et al., 1995) were produced by BioXcell. Anti-CSF1 mAb was injected at 150 μg i.p. on days −1, +1, +2, +3, and +4 relative to transplantation, which is known to neutralize the biological functions of CSF1 in vivo (Gregory et al., 1992). Anti-CSF1R mAb was injected at 2 mg/mouse on day −5 and 0.5 mg/mouse on days −4 and −3, which is known to neutralize the biological functions of CSF1R in vivo (Hashimoto et al., 2011). Blocking antibody to DC-SIGN (CD209a) (Cheong et al., 2010) was mAb was purified from culture supernatant, grown in a CELLine Flask (BD) in serum-free medium (PFHM-II; Invitrogen) and injected at 250 μg i.p. on days +1, +2, +3, and +4 relative to transplantation.
Mouse Suppression Assay
Spleens of C57BL/6 or C57BL/6-Foxp3tm1Flv/J (H-2b) mice were gently dissociated into single-cell suspensions, and red blood cells were removed using hypotonic ACK lysis buffer. Splenocytes were either stained with anti-CD4 mAb, or labeled with CFSE at 5 μM concentration (Molecular probes - In-vitrogen) followed by staining with anti-CD8 mAb for 30 min on ice. Responder FoxP3+CD4+ and CFSE+CD8+ T cells were sorted using FACS Aria II (BD Biosciences) with a purity > 98%. Spleens of BALB/c (H-2d) mice were gently dissociated into single-cell suspensions and were enriched for CD11c+ cells using the EasySep Mouse CD11c Positive Selection Kit (StemCell). Enriched CD11c+ splenocytes were stained with anti-mouse CD11c mAb and sorted using FACS Aria II (BD Biosciences) and were used together with anti-CD3 plus CD28 mAb (1 μg/ml) as stimulators. Stimulated FoxP3+CD4+ or CFSE+CD8+ T cells were cultures with graft infiltrating CD11b+CSF1R+Ly6ChiLy6G−, CD11b+CSF1R+Ly6CloLy6G−, and CD11b+CSF1R+Ly6CintLy6G+ myeloid cells for 4 days at 37°C in a 5% CO2 incubator. T cell proliferation was measured by flow cytometric analysis of CFSE dilution on CD8+ T cells. Treg expansion was measured by flow cytometric analysis of Foxp3-RFP on CD4+ T cells.
Supplementary Material
Highlights.
DC-SIGN+ macrophages inhibit CD8+ T cell proliferation and expand CD4+Foxp3+ Treg
In vivo development of DC-SIGN+ macrophages is regulated by IFN-γ and CSF1
IL-10 is essential for DC-SIGN+ macrophage-mediated suppression
Simultaneous Fucose-DC-SIGN and HMGB1-TLR4 signaling is required for IL-10 production
Acknowledgments
We thank Andres Hidalgo (Centro Nacional de Investigaciones Cardiovasculares, ISCIII) and Emmanuel Gautier (Department of Pathology & Immunology, Washington University) for critical review of the manuscript. We acknowledge the technical contributions of the Flow Cytometry, Microsurgery, and the Bio-repository/Pathology Centers of Research Excellence at Mount Sinai, and the Alberta Glycomics Centre for their help on the LNFPIII synthesis. Acknowledgments to Marcy Kuentzel and Sridar Chittur at the University of Albany Center for Functional Genomics Microarray Core facility for their assistance in generating the microarray data. This work was supported by the COST Action BM1305: Action to Focus and Accelerate Cell Tolerogenic Therapies (A FACTT), the Mount Sinai Recanati/Miller Transplantation Institute developmental funds, AST/Pfizer Basic Science Faculty Development Grant, Ministerio de Educacióny Ciencia SAF2010-15062, SAF2013-48834-R, and Fundación Mutua Madrileñ a grants to J.O. A portion of this work appears as part of the doctoral thesis of P.C.
Footnotes
Accession Number
ACCESSION NUMBER
The GEO accession number for the microarray data reported in this paper is GSE68648.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2015.05.009.
References
- Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med. 2007;204:1057–1069. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- Bronte V, Wang M, Overwijk WW, Surman DR, Pericle F, Rosenberg SA, Restifo NP. Apoptotic death of CD8+ T lymphocytes after immunization: induction of a suppressive population of Mac-1+/Gr-1+ cells. J Immunol. 1998;161:5313–5320. [PMC free article] [PubMed] [Google Scholar]
- Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, Restifo NP, Zanovello P. Identification of a CD11b(+)/Gr-1(+)/ CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood. 2000;96:3838–3846. [PMC free article] [PubMed] [Google Scholar]
- Burnett SH, Kershen EJ, Zhang J, Zeng L, Straley SC, Kaplan AM, Cohen DA. Conditional macrophage ablation in transgenic mice expressing a Fas-based suicide gene. J Leukoc Biol. 2004;75:612–623. doi: 10.1189/jlb.0903442. [DOI] [PubMed] [Google Scholar]
- Cai M, Wu J, Mao C, Ren J, Li P, Li X, Zhong J, Xu C, Zhou T. A Lectin-EGF antibody promotes regulatory T cells and attenuates nephrotoxic nephritis via DC-SIGN on dendritic cells. J Transl Med. 2013;11:103. doi: 10.1186/1479-5876-11-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caparrós E, Munoz P, Sierra-Filardi E, Serrano-Gómez D, Puig-Kröger A, Rodríguez-Fernández JL, Mellado M, Sancho J, Zubiaur M, Corbí AL. DC-SIGN ligation on dendritic cells results in ERK and PI3K activation and modulates cytokine production. Blood. 2006;107:3950–3958. doi: 10.1182/blood-2005-03-1252. [DOI] [PubMed] [Google Scholar]
- Cheong C, Matos I, Choi JH, Schauer JD, Dandamudi DB, Shrestha E, Makeyeva JA, Li X, Li P, Steinman RM, Park CG. New monoclonal anti-mouse DC-SIGN antibodies reactive with acetone-fixed cells. J Immunol Methods. 2010;360:66–75. doi: 10.1016/j.jim.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Cheong C, Dandamudi DB, Park CG, Rodriguez A, Mehandru S, Velinzon K, Jung IH, Yoo JY, Oh GT, Steinman RM. Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity. 2011;35:819–831. doi: 10.1016/j.immuni.2011.09.014. [DOI] [PubMed] [Google Scholar]
- Corry RJ, Winn HJ, Russell PS. Primarily vascularized allo-grafts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation. 1973;16:343–350. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
- Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- Domínguez-Soto A, Sierra-Filardi E, Puig-Kröger A, Peérez-Maceda B, Gómez-Aguado F, Corcuera MT, Sánchez-Mateos P, Corbí AL. Dendritic cell-specific ICAM-3-grabbing nonintegrin expression on M2-polarized and tumor-associated macrophages is macrophage-CSF dependent and enhanced by tumor-derived IL-6 and IL-10. J Immunol. 2011;186:2192–2200. doi: 10.4049/jimmunol.1000475. [DOI] [PubMed] [Google Scholar]
- Dugast AS, Haudebourg T, Coulon F, Heslan M, Haspot F, Poirier N, Vuillefroy de Silly R, Usal C, Smit H, Martinet B, et al. Myeloid-derived suppressor cells accumulate in kidney allograft tolerance and specifically suppress effector T cell expansion. J Immunol. 2008;180:7898–7906. doi: 10.4049/jimmunol.180.12.7898. [DOI] [PubMed] [Google Scholar]
- Fleming TJ, Fleming ML, Malek TR. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J Immunol. 1993;151:2399–2408. [PubMed] [Google Scholar]
- Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, Basso G, Brombacher F, Borrello I, Zanovello P, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. 2006;116:2777–2790. doi: 10.1172/JCI28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MR, Ledgerwood L, Yang Y, Xu J, Lal G, Burrell B, Ma G, Hashimoto D, Li Y, Boros P, et al. Monocytic suppressive cells mediate cardiovascular transplantation tolerance in mice. J Clin Invest. 2010;120:2486–2496. doi: 10.1172/JCI41628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Vallejo JJ, Ilarregui JM, Kalay H, Chamorro S, Koning N, Unger WW, Ambrosini M, Montserrat V, Fernandes RJ, Bruijns SC, et al. CNS myelin induces regulatory functions of DC-SIGN-expressing, antigen-presenting cells via cognate interaction with MOG. J Exp Med. 2014;211:1465–1483. doi: 10.1084/jem.20122192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. 2009;9:465–479. doi: 10.1038/nri2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijtenbeek TB, Torensma R, van Vliet SJ, van Duijnhoven GC, Adema GJ, van Kooyk Y, Figdor CG. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell. 2000;100:575–585. doi: 10.1016/s0092-8674(00)80693-5. [DOI] [PubMed] [Google Scholar]
- Geijtenbeek TB, Van Vliet SJ, Koppel EA, Sanchez-Hernandez M, Vandenbroucke-Grauls CM, Appelmelk B, Van Kooyk Y. Mycobacteria target DC-SIGN to suppress dendritic cell function. J Exp Med. 2003;197:7–17. doi: 10.1084/jem.20021229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijtenbeek TB, van Vliet SJ, Engering A, ’t Hart BA, van Kooyk Y. Self- and nonself-recognition by C-type lectins on dendritic cells. Annu Rev Immunol. 2004;22:33–54. doi: 10.1146/annurev.immunol.22.012703.104558. [DOI] [PubMed] [Google Scholar]
- Goldstein DR, Tesar BM, Akira S, Lakkis FG. Critical role of the Toll-like receptor signal adaptor protein MyD88 in acute allograft rejection. J Clin Invest. 2003;111:1571–1578. doi: 10.1172/JCI17573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbachev AV, Fairchild RL. CD40 engagement enhances antigen-presenting langerhans cell priming of IFN-gamma-producing CD4+ and CD8+ T cells independently of IL-12. J Immunol. 2004;173:2443–2452. doi: 10.4049/jimmunol.173.4.2443. [DOI] [PubMed] [Google Scholar]
- Gregory SH, Wing EJ, Tweardy DJ, Shadduck RK, Lin HS. Primary listerial infections are exacerbated in mice administered neutralizing antibody to macrophage colony-stimulating factor. J Immunol. 1992;149:188–193. [PubMed] [Google Scholar]
- Gringhuis SI, den Dunnen J, Litjens M, van Het Hof B, van Kooyk Y, Geijtenbeek TB. C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity. 2007;26:605–616. doi: 10.1016/j.immuni.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Gringhuis SI, Kaptein TM, Wevers BA, Mesman AW, Geijtenbeek TB. Fucose-specific DC-SIGN signalling directs T helper cell type-2 responses via IKKε- and CYLD-dependent Bcl3 activation. Nat Commun. 2014;5:3898. doi: 10.1038/ncomms4898. [DOI] [PubMed] [Google Scholar]
- Hancock WW, Sayegh MH, Zheng XG, Peach R, Linsley PS, Turka LA. Costimulatory function and expression of CD40 ligand, CD80, and CD86 in vascularized murine cardiac allograft rejection. Proc Natl Acad Sci USA. 1996;93:13967–13972. doi: 10.1073/pnas.93.24.13967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto D, Chow A, Greter M, Saenger Y, Kwan WH, Leboeuf M, Ginhoux F, Ochando JC, Kunisaki Y, van Rooijen N, et al. Pretransplant CSF-1 therapy expands recipient macrophages and ameliorates GVHD after allogeneic hematopoietic cell transplantation. J Exp Med. 2011;208:1069–1082. doi: 10.1084/jem.20101709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- Huang Y, Yin H, Han J, Huang B, Xu J, Zheng F, Tan Z, Fang M, Rui L, Chen D, et al. Extracellular hmgb1 functions as an innate immune-mediator implicated in murine cardiac allograft acute rejection. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2007;7:799–808. doi: 10.1111/j.1600-6143.2007.01734.x. [DOI] [PubMed] [Google Scholar]
- Jiang X, Sun W, Guo D, Cui Z, Zhu L, Lin L, Tang Y, Wang X, Liang J. Cardiac allograft acceptance induced by blockade of CD40-CD40L costimulation is dependent on CD4+CD25+ regulatory T cells. Surgery. 2011;149:336–346. doi: 10.1016/j.surg.2010.08.012. [DOI] [PubMed] [Google Scholar]
- Jutila MA, Kroese FG, Jutila KL, Stall AM, Fiering S, Herzenberg LA, Berg EL, Butcher EC. Ly-6C is a monocyte/macrophage and endothelial cell differentiation antigen regulated by interferon-gamma. Eur J Immunol. 1988;18:1819–1826. doi: 10.1002/eji.1830181125. [DOI] [PubMed] [Google Scholar]
- Li G, Hangoc G, Broxmeyer HE. Interleukin-10 in combination with M-CSF and IL-4 contributes to development of the rare population of CD14+CD16++ cells derived from human monocytes. Biochem Biophys Res Commun. 2004;322:637–643. doi: 10.1016/j.bbrc.2004.07.172. [DOI] [PubMed] [Google Scholar]
- Li G, Kim YJ, Broxmeyer HE. Macrophage colony-stimulating factor drives cord blood monocyte differentiation into IL-10(high)IL-12absent dendritic cells with tolerogenic potential. J Immunol. 2005;174:4706–4717. doi: 10.4049/jimmunol.174.8.4706. [DOI] [PubMed] [Google Scholar]
- Lokeshwar BL, Lin HS. Development and characterization of monoclonal antibodies to murine macrophage colony-stimulating factor. J Immunol. 1988;141:483–488. [PubMed] [Google Scholar]
- Lowe JB. Glycosylation in the control of selectin counter-receptor structure and function. Immunol Rev. 2002;186:19–36. doi: 10.1034/j.1600-065x.2002.18603.x. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177:7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- Meyer S, van Liempt E, Imberty A, van Kooyk Y, Geyer H, Geyer R, van Die I. DC-SIGN mediates binding of dendritic cells to authentic pseudo-LewisY glycolipids of Schistosoma mansoni cercariae, the first parasite-specific ligand of DC-SIGN. J Biol Chem. 2005;280:37349–37359. doi: 10.1074/jbc.M507100200. [DOI] [PubMed] [Google Scholar]
- Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, Pandey G, Leboeuf M, Elpek KG, Helft J, et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol. 2012;13:888–899. doi: 10.1038/ni.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, Tanaka M. Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. J Clin Invest. 2007;117:2268–2278. doi: 10.1172/JCI31990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka M, Ma BY, Murai R, Nakamura N, Baba M, Kawasaki N, Hodohara K, Asano S, Kawasaki T. Glycosylation-dependent interactions of C-type lectin DC-SIGN with colorectal tumor-associated Lewis glycans impair the function and differentiation of monocyte-derived dendritic cells. J Immunol. 2008;180:3347–3356. doi: 10.4049/jimmunol.180.5.3347. [DOI] [PubMed] [Google Scholar]
- Parker KH, Sinha P, Horn LA, Clements VK, Yang H, Li J, Tracey KJ, Ostrand-Rosenberg S. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res. 2014;74:5723–5733. doi: 10.1158/0008-5472.CAN-13-2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolink A, Melchers F, Andersson J. The SCID but not the RAG-2 gene product is required for S mu-S epsilon heavy chain class switching. Immunity. 1996;5:319–330. doi: 10.1016/s1074-7613(00)80258-7. [DOI] [PubMed] [Google Scholar]
- Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- Smits HH, Engering A, van der Kleij D, de Jong EC, Schipper K, van Capel TM, Zaat BA, Yazdanbakhsh M, Wierenga EA, van Kooyk Y, Kapsenberg ML. Selective probiotic bacteria induce IL-10-producing regulatory T cells in vitro by modulating dendritic cell function through dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin. J Allergy Clin Immunol. 2005;115:1260–1267. doi: 10.1016/j.jaci.2005.03.036. [DOI] [PubMed] [Google Scholar]
- Soilleux EJ, Morris LS, Leslie G, Chehimi J, Luo Q, Levroney E, Trowsdale J, Montaner LJ, Doms RW, Weissman D, et al. Constitutive and induced expression of DC-SIGN on dendritic cell and macrophage subpopulations in situ and in vitro. J Leukoc Biol. 2002;71:445–457. [PubMed] [Google Scholar]
- Sudo T, Nishikawa S, Ogawa M, Kataoka H, Ohno N, Izawa A, Hayashi S, Nishikawa S. Functional hierarchy of c-kit and c-fms in intra-marrow production of CFU-M. Oncogene. 1995;11:2469–2476. [PubMed] [Google Scholar]
- Takahara K, Yashima Y, Omatsu Y, Yoshida H, Kimura Y, Kang YS, Steinman RM, Park CG, Inaba K. Functional comparison of the mouse DC-SIGN, SIGNR1, SIGNR3 and Langerin, C-type lectins. Int Immunol. 2004;16:819–829. doi: 10.1093/intimm/dxh084. [DOI] [PubMed] [Google Scholar]
- van Gisbergen KP, Aarnoudse CA, Meijer GA, Geijtenbeek TB, van Kooyk Y. Dendritic cells recognize tumor-specific glycosylation of carcinoembryonic antigen on colorectal cancer cells through dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin. Cancer Res. 2005;65:5935–5944. doi: 10.1158/0008-5472.CAN-04-4140. [DOI] [PubMed] [Google Scholar]
- van Kooyk Y, Geijtenbeek TB. DC-SIGN: escape mechanism for pathogens. Nat Rev Immunol. 2003;3:697–709. doi: 10.1038/nri1182. [DOI] [PubMed] [Google Scholar]
- van Liempt E, Bank CM, Mehta P, Garciá-Vallejo JJ, Kawar ZS, Geyer R, Alvarez RA, Cummings RD, Kooyk Yv, van Die I. Specificity of DC-SIGN for mannose- and fucose-containing glycans. FEBS Lett. 2006;580:6123–6131. doi: 10.1016/j.febslet.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Walker WE, Nasr IW, Camirand G, Tesar BM, Booth CJ, Goldstein DR. Absence of innate MyD88 signaling promotes inducible allograft acceptance. J Immunol. 2006;177:5307–5316. doi: 10.4049/jimmunol.177.8.5307. [DOI] [PubMed] [Google Scholar]
- Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Liang S, Wu J, Horuzsko A. Human inhibitory receptor immunoglobulin-like transcript 2 amplifies CD11b+Gr1+ myeloid-derived suppressor cells that promote long-term survival of allografts. Transplantation. 2008;86:1125–1134. doi: 10.1097/TP.0b013e318186fccd. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.