

Abstract
Background
Collimonas is a genus belonging to the class of Betaproteobacteria and consists mostly of soil bacteria with the ability to exploit living fungi as food source (mycophagy). Collimonas strains differ in a range of activities, including swimming motility, quorum sensing, extracellular protease activity, siderophore production, and antimicrobial activities.
Results
In order to reveal ecological traits possibly related to Collimonas lifestyle and secondary metabolites production, we performed a comparative genomics analysis based on whole-genome sequencing of six strains representing 3 recognized species. The analysis revealed that the core genome represents 43.1 to 52.7 % of the genomes of the six individual strains. These include genes coding for extracellular enzymes (chitinase, peptidase, phospholipase), iron acquisition and type II secretion systems. In the variable genome, differences were found in genes coding for secondary metabolites (e.g. tripropeptin A and volatile terpenes), several unknown orphan polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS), nonribosomal peptide synthetase (NRPS) gene clusters, a new lipopeptide and type III and type VI secretion systems. Potential roles of the latter genes in the interaction with other organisms were investigated. Mutation of a gene involved in tripropeptin A biosynthesis strongly reduced the antibacterial activity against Staphylococcus aureus, while disruption of a gene involved in the biosynthesis of the new lipopeptide had a large effect on the antifungal/oomycetal activities.
Conclusions
Overall our results indicated that Collimonas genomes harbour many genes encoding for novel enzymes and secondary metabolites (including terpenes) important for interactions with other organisms and revealed genomic plasticity, which reflect the behaviour, antimicrobial activity and lifestylesof Collimonas spp.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2289-3) contains supplementary material, which is available to authorized users.
Keywords: Comparative genomics, Collimonas, Secondary metabolites, Terpenes
Background
The genus Collimonas comprises soil bacteria with the ability to grow at the expense of living fungal hyphae under nutrient-limited conditions [1–3]. Since the first description of Collimonas, more mycophagous bacteria have been detected [4], but Collimonas species are still highly interesting in view of the interactions between bacteria and fungi in soil and the associated ecosystem functions including suppression of pathogens and the production of novel bioactive compounds.
Collimonas belongs to the family Oxalobacteraceae, class Betaproteobacteria. The first Collimonas isolates were obtained within the framework of a project searching for a naturally occurring biocontrol agent of fungi pathogenic to marram grass (Ammophilia arenaria) and were determined as being dominant among the cultivable chitinolytic bacteria in the acidic Dutch dune soils [3]. Three species have been described so far: C. fungivorans, C. pratensis and C. arenae [5]. All three species display the ability to feed on fungi (mycophagy), to degrade chitin and to dissolve minerals (weathering) [2]. However, Collimonas strains differ in important ecological traits such as colony morphology, the ability to oxidize various carbon sources, in their antibacterial, antifungal and antioomycetal activities [6, 7]. A comparative genomic approach would help to reveal the genetic basis of these ecological differences. Applying a comparative genomic hybridization approach [7] showed that a gene cluster involved in the production of an antifungal polyyne was only found in the genome of a few Collimonas strains. The study of Mela et al. [7] was biased in the sense that the hybridization assay used allowed only to screen for absence/presence of genes in other strains as compared to C. fungivorans strain Ter 331, the only strain for which the complete genome was sequenced at that time. To reveal the real plasticity of Collimonas strains more genome sequences are needed. This will demonstrate constant and variable genetic elements, and hence determine the adaptations of Collimonas species and traits important for inter-specific microbial interactions in the soil. To date, only two genome sequences of Collimonas are publicly available [7, 8]. Here we report on full genome sequences of five Collimonas strains across the three recognized species and performed comparative genome analysis including the recently published C. fungivorans Ter331 genome [7]. Gene clusters with potential relevance for interactions of the Collimonas species with fungi and other microorganisms were further investigated by gene knock-out mutations and/or enzymatic characterization.
Results and discussion
Genomic features
A genome sequence analysis was performed for strains Ter6, Ter91, Ter291, Ter10 and Ter282, using a combined strategy of Illumina Hiseq and PacBio sequencing. A summary of the general genomic features (size, GC content, predicted number of coding sequences, and number of rRNAs) of each Collimonas strain is presented in Table 1. Considerable variation in genome size and differences in plasmid content was observed. The six genomes vary in size by approximately one megabase (ranging from 4.7–5.7 Mb) with the number of coding sequences (CDSs) ranging from 4436 to 5424, indicating substantial strain-to-strain variation. The genomes of C. fungivorans and C. pratensis are larger and have higher GC content than the two strains of C. arenae. Only strain C. fungivorans Ter331 has a plasmid, described in detail by Mela et al. [9]. Despite the absence of a plasmid, C. pratensis Ter91 has the largest genome size (5.7 kb) and highest number of encoding genes (5424), which is likely due to the large number of horizontally acquired genes as indicated by the number of genomic islands (see below).
Table 1.
Collimonas strains and their genomic features
| Feature | Collimonas fungivorans | Collimonas pratensis | Collimonas arenae | |||
|---|---|---|---|---|---|---|
| Ter331 | Ter6 | Ter91 | Ter291 | Ter10 | Ter282 | |
| Source | Inner coastal dune soil in Terschelling, the Netherlands | |||||
| Chromosome size (Mb) | 5.2 | 5.6 | 5.7 | 5.6 | 4.7 | 4.7 |
| Plasmid size | 40 kb | NA | NA | NA | NA | NA |
| G + C % | 59.6 % | 59 % | 58.8 % | 59 % | 56.8 % | 56.8 % |
| Protein-encoding sequences (CDSs) | 4910 | 5233 | 5424 | 5228 | 4436 | 4473 |
| CDSs on plasmid | 44 | NA | NA | NA | NA | NA |
| Hypothetical proteins | 1091 | 1209 | 1292 | 1214 | 1101 | 991 |
| Average CDS length (nt) | 927 | 923 | 904 | 917 | 834 | 899 |
| Coding (%) | 87.8 % | 86.2 % | 85.6 % | 85.8 % | 78.4 % | 85.4 % |
| rRNA | 9 | 9 | 9 | 9 | 9 | 9 |
| tRNA | 52 | 52 | 52 | 51 | 52 | 53 |
| contigs | 1 | 1 | 1 | 1 | 1 | 1 |
NA not applicable, refers to strains in which no plasmids are naturally present
Phylogenetic analysis
A phylogenetic tree based on 233 protein-coding genes (Fig. 1a) revealed that C. fungivorans and C. pratensis are more closely related to each other than to C. arenae. Similar clustering was observed based on phylogenetic trees generated with whole genome fragments (200 bp fragment sizes) (Fig. 1b) and 16S rRNA (Additional file 1: Figure S1) where each strain falls into its respective species clade.
Fig. 1.
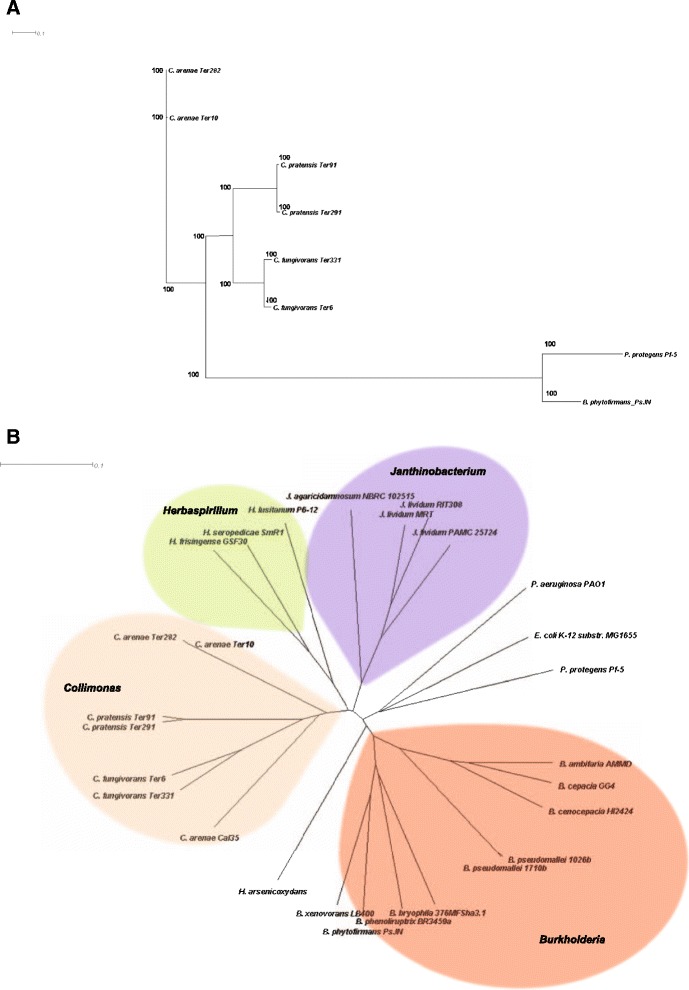
Whole genome phylogeny of the six Collimonas genomes. a Neighbor-joining tree based on concatenated sequences for 233 protein encoding genes. Pseudomonas protegens Pf-5 and Burkholderia phytofirmans PsJN were used as outgroup. Bootstrap values are shown on branches. b Phylogenetic tree based on a fragmented alignment using BLASTN made with settings 200/100. A dendrogram was produced in SplitsTree 4.13.1 (using neighbor joining method) made from a Nexus file exported from Gegenees. Burkholderia, Janthinobacterium and Herbaspirillum were set as outgroups. Bootstrap values are of all the branches are 100, for clarity reason, not shown in the figure
Core and pan-genome analysis
A core genome containing 2339 predicted orthologous groups was identified for the six Collimonas strains based on the all-vs-all BLASTp search (Fig. 2a). This core genome represents 43.1 to 52.7 % of the predicted ORF’s of each strain (Fig. 2a), illustrating a large degree of genomic diversity between these strains. Each of the six genomes includes 125 to 835 orthologous groups that are unique (Fig. 2a). Species core orthologous clusters and strain-specific unique clusters within the three Collimonas species were examined, respectively (Fig. 2b-d). In the three species, 5868, 5810 and 4546 orthologous clusters were identified and of these, 3859, 4194 and 3829 orthologs were present as the species core genome for C. fungivorans, C. pratensis, C. arenae, respectively (Fig. 2b-d). A core-pan genome evolution plot summarizing the variability in each possible combination of Collimonas species shows that the number of unique (singleton) gene clusters is stable. The variable gene clusters are increasing and the core gene clusters are decreasing (Fig. 2e). In order to determine the differences in functions encoded by the core and variable genome of each strain, the proportion of proteins in each COG (Clusters of Orthologous Groups) was plotted versus the COG function. The relative abundance of almost all the COG categories was higher in the core genome of the six strains than in the variable genome. This was not the case for COG categories N (Cell motility), U (Intracellular trafficking, secretion, and vesicular transport) and proteins that cannot be assigned in COG categories (data not shown) where the proteins were more abundant in variable genomes for C. fungivorans and C. arenae (Fig. 3a). This is most probably due to the fact that a flagellar and chemotaxis-related gene cluster is only present in these two species but not in C. pratensis (Additional file 2: Table S1). This finding is consistent with the observed reduced swimming motility of C. pratensis Ter91 and Ter291 as compared to the other four strains (Fig. 3b).
Fig. 2.
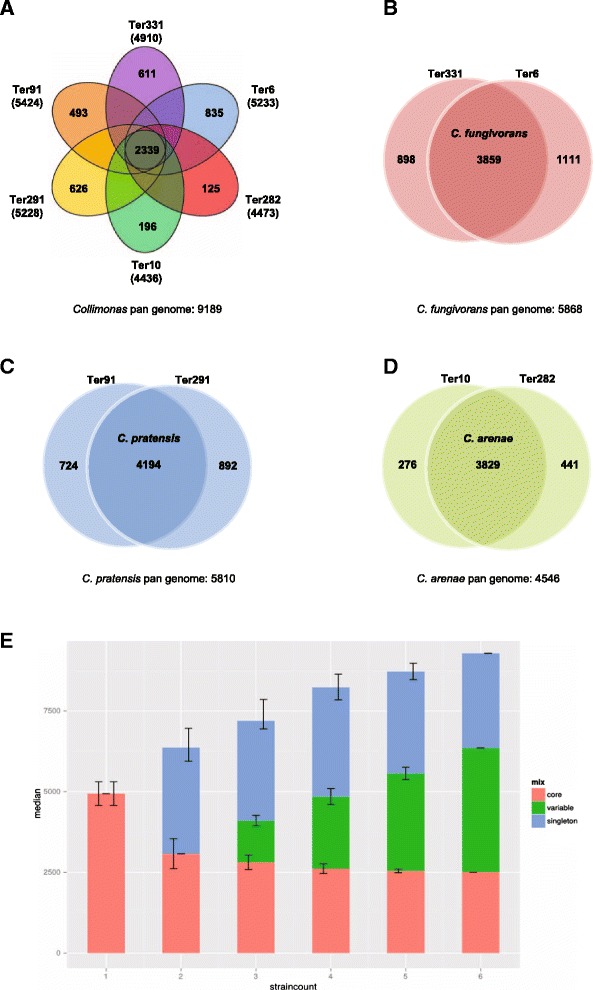
The pan-core genome of Collimonas strains. The venn diagrams illustrate the number of shared and unique genes based on clusters of orthologs. a Venn diagram showing numbers of species-specific genes commonly found in each genome of each species, (non-overlapping of each oval) and Collimonas core orthologous gene number (in the centre). The total number of protein coding genes within each genome is listed below the strain name. b Venn diagram showing numbers of unique orthologues genes in C. fungivorans strains. c Venn diagram showing numbers of unique orthologues genes in C. pratensis strains. d Venn diagram showing numbers of unique orthologues genes in C. arenae strains. e Core- and pan-genome as function of the number of genomes taken from the six Collimonas genomes in this study. The number of shared and strain specific gene clusters between strains depends on which combinations of strains (x-axis). Specific singleton gene clusters (blue bars) occur only in one strain, variable gene clusters (green bars) occur in more than one but not all strains and core gene clusters (red bars) occur in all strains of a given combination. Error bars represent the standard deviation in the core- (left error bar), variable- (middle error bar) and singleton- (right error bars) gene clusters
Fig. 3.
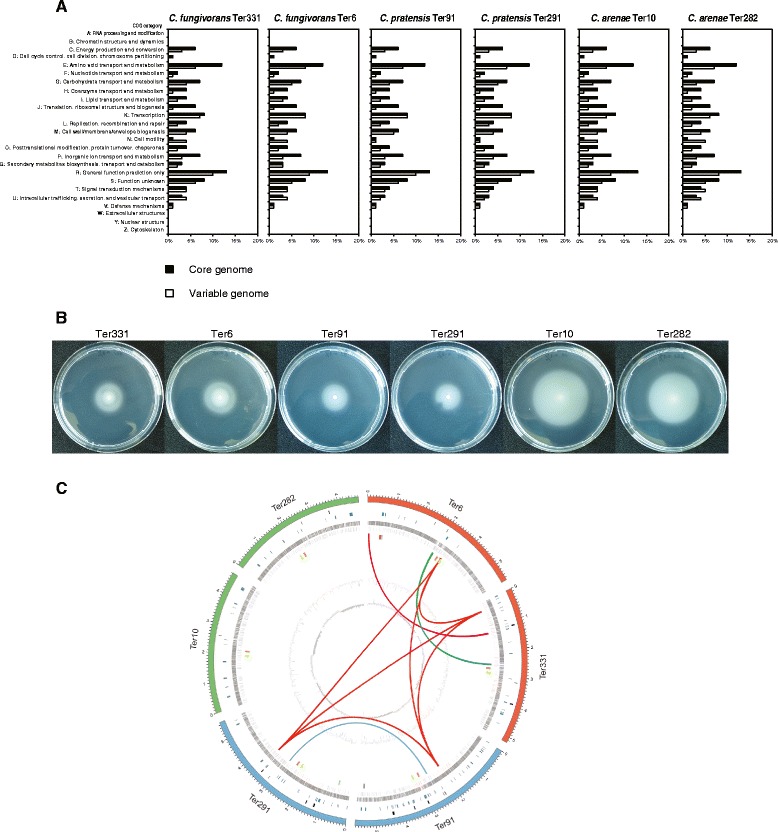
Distribution of orthologous genes based on COG category in each Collimonas strain. a The percentage of orthologous genes assigned by COG category in the core genome (black bars) and the variable genome (white bars). b Swimming motility of Collimonas strains Ter331, Ter6, Ter91, Ter291, Ter10 and Ter282 on soft (0.3 % [wt/vol]) agar plates. c Comparative genome content of the six Collimonas strains. From the outside to the inside circles: Chromosomes of all six strains (red: C. fungivorans, blue: C. pratensis, green: C. arenae). Phages/phage-like regions: black bars. Genomic islands: dark blue bars. Genes in the forward (dark grey) direction, genes in the reverse (light grey) direction, G + C content (dark grey and light grey), GC skew (dark grey: negative values, light grey: positive values). Universal and unique gene clusters are indicated with colored bars (orange: Ornibactin, yellow: Phytoene, red: T3pks-nrps (Ter6), light blue: 2-aa NRPS-1 (Ter6), purple: 2-aa NRPS-2 (Ter291), light green: Nrps-t1pks (HSAF, Ter91)). Shared secondary metabolite gene clusters are indicated with colored lines (deep pink: Collimomycin, dark red: NLP, dark green: Tripropeptin A)
Whole genome alignments of the six strains were performed to obtain information on the nucleotide level synteny (Additional file 1: Figure S2). These alignments revealed a very high level of synteny when genomes of strains from the same species were compared (Additional file 1: Figure S2A-C), but many rearrangements and inversions were observed between the genomes of all strains (Additional file 1: Figure S2D).
Genomic islands (GIs), bacteriophages and CRISPRs
Genomic islands (GIs) are mobile genetic elements acquired by horizontal transfer, which carry multiple genes that are typically involved in pathogenesis or symbiosis. The Collimonas genomes carry 7 to 47 GIs ranging from 4.0 kb to 64 kb in size (Fig. 3c). All together, the six Collimonas genomes have 139 genomic islands. The large numbers of GIs indicate a complex history of gene recombination and horizontal transfer between bacterial relatives. The genomes of all strains contain one to five possible phages, each ranging in size from 7.0 to 59.9 kb. In total, the six genomes have 18 phages with some of the phages falling into the GIs (Fig. 3c). CRISPRs (Clustered Regularly Interspaced Short Palindromic Repeats) are DNA loci that are involved in prokaryotic immunity to phage infection. Putative CRISPRs were identified using the CRISPRsFinder program [10]. Two confirmed CRISPRs were present only in C. fungivorans Ter6 genome. For the other five genomes, no or only questionable CRISPRs were found (data not shown).
Secretion systems
In Gram-negative bacteria, type II secretion systems (T2SSs) are the most ubiquitous secretion systems used by bacteria to export many extracellular enzymes. T2SSs are conserved and known as a two-step process: proteins are translocated across the inner membrane by the Sec or Tat pathway, and then transported from the periplasm to the exterior by an outer membrane secretin [11]. SecABDEFY, yajC, yidC, ftsY, and ffh and tatABC encoding genes for Sec and Tat pathways respectively were found to be present in all Collimonas genomes (Additional file 1: Figure S3A; Additional file 2: Table S2). The outer membrane secretion unit of the T2SS in the Collimonas genomes resembles the Gsp system which contains one gene cluster gspD-N, responsible for secretion of protease, lipase, and phospholipase C in Burkholderia [12] (Additional file 1: Figure S3A; Additional file 2: Table S2). A newly described subtype of T2SS, tad locus (tight adherence) [13] was identified in all six genomes. The tad locus encodes the machinery required for the assembly of adhesive Flp (fimbrial low-molecule-weight protein) pili and is necessary for bacterial adhesion to surfaces, biofilm formation, and pathogenesis as shown for Aggregatibacter actinomycetemcomitans [14], Haemophilus [15], Pseudomonas [16], Yersinia etc. [13]. For the mycophagous behavior of Collimonas, the adhesion to fungal hyphae might be of prime importance [17] and the presence of the T2SS tad locus in all strains indicates that it may be an essential trait for the mycophagy lifestyle of this species.
Type III secretion systems (T3SSs) are used by various Gram-negative bacteria to inject effector proteins into host cells, promoting either mutual benefit or pathogenesis [18, 19] and have been described as important for bacterial interaction with fungi [20]. Three Collimonas strains Ter331, Ter6 and Ter91 carry hrp-hrc1 family gene clusters of T3SS and a second T3SS (Additional file 1: Figure S3A; Additional file 2: Table S2). The T3SSs play crucial role in the virulence of plant and human pathogens [21]. However, their functions in non-pathogenic bacteria are still poorly understood; there are indications that mutation of T3SSs in a plant-growth promoting bacteria P. fluorescens SBW25 resulted in a significant reduction in the potential of the bacterium to colonize the root tips of sugar beet seedlings [22]. They might also be involved in bacterial-fungal interactions, facilitating bacteria migration along fungal hyphae [20]. For Collimonas, the T3SSs may be important to inject membrane disturbing compounds into the fungal host to get access to nutrients inside the fungal hyphae [2].
Type VI secretion systems (T6SSs) are conserved and prevalent in Gram-negative bacteria. They are known to be involved in competition, predation and inter-specific bacterial interactions [23–25]. In this study, we identified a cluster of genes encoding T6SS only in C. fungivorans and C. pratensis strains, but not in the C. arenae species. This may indicate that horizontal gene transfer events or evolutionary genes loss have occurred. Furthermore, in C. fungivorans Ter331, part of the T6SS is located on a genomic island.
Further functional studies are needed to determine the exact role of these secretion systems for Collimonas lifestyle and in particular for the attack of fungi.
Signal transduction systems
Signal transduction systems play important roles for many bacteria enabling them to detect and respond to changes and stresses in the environment [26]. Each Collimonas genome encodes 267 to 365 one-component systems (1CSs) which are the majority of signal transduction systems in prokaryotes [27] and 90 to 109 two component system (TCSs) (Additional file 2: Table S3). Additionally, 9 to 29 genes involved in chemotaxis systems were found. Extracytoplasmic function (ECF) sigma factors which comprise the largest group among the σ70 family [27] were also found in all Collimonas genomes with numbers ranging from 9 to 11. Higher numbers of signal transduction system were predicted in C. fungivorans and C. pratensis as compared to C. arenae. The overall high number of genes related to signal transduction systems (8–9 % of predicted sequences in the six Collimonas genomes) suggest that Collimonas possess the ability to sense environmental signals and cues important for their growth, survival and interactions in the heterogeneous and complex soil environment. This is in consistent with the previous report that soil bacteria contain higher number of signal transduction systems as compared to bacteria from a stable environment [28].
Our genomic analysis revealed that all six Collimonas genomes contain two QS genes: one autoinducer gene and one luxR-type transcriptional regulator. They show 40 % homology to the CepIR system from Burkholderia (Additional file 2: Table S4) which is known to regulate protease, lipases, chitinases and some other exoenzymes production [29, 30]. Quorum sensing assay performed with the indicator strain C. violaceum CV026 revealed clear short chain AHL production in C. fungivorans Te331, Ter6 and C. pratensis Ter91, Ter291 strains, but no or trace amounts in C. arenae Ter10 and Ter282 strains (Additional file 1: Figure S3B). In all strains, AHL production was detected when A. tumefaciens NT1 was used as QS bioreporter (Additional file 1: Figure S3C).
Secondary metabolome of Collimonas strains
Bacteria often produce a set of secondary metabolites with antimicrobial properties important for competition and survival in competitive environments. In Collimonas, the secondary metabolites are thought to play an important role enabling mycophagous growth, namely by disturbing the fungal membrane integrity [2]. Although Collimonas was suggested to represent a valuable resource for the discovery of novel molecules and enzymes [2], to date only two antimicrobial compounds were described for this genus, namely violacein and collimomycin [31, 32]. Violacein was identified in the Collimonas CT strain isolated from an aquatic environment and revealed antibacterial activity against Micrococcus luteus [31]. However, in the six Collimonas genomes here, no genes encoding violacein were identified.
Collimomycin is a polyacetylenic compound with alternating triple and single carbon-carbon bonds [32] which is produced by C. fungivorans Ter331 and was shown to have strong antifungal activities [32]. The corresponding biosynthesis cluster K, is only partially present in C. fungivorans Ter6, and completely absent in the other four genomes (Fig. 3c, Additional file 2: Table S5). Moreover, the part of the collimomycin gene cluster that is present only in C. fungivorans Ter331 is located in a genomic island suggesting that its presence is due to a horizontal gene transfer event.
Exoenzymes
Exoenzymes are extracellular enzymes produced inside the cell, and subsequently released outside the cell to perform extracellular digestion. Exoenzymes may be of importance for Collimonas nutrient acquisition, microbial interactions, mycophagy and weathering.
Chitinases
Chitin is a major component of fungal cell walls and is a homopolymer of N-acetyl-D-glucosamine (GlcNAc). Chitinases are able to hydrolyze the 1,4-beta-linkages of chitin [33]. Two loci A and B of chitinase biosynthesis and transport were already found in the genome of C. fungivorans Ter331 [34]. We observed that the other five Collimonas genomes also contain a complete set of genes in these two loci (Additional file 2: Table S6). This is in line with previous observations based on comparative genomic hybridization study [7] and indicates that acquisition of these genes occurred before Collimonas speciation. Phenotypic evaluation of chitinase production of these strains was confirmed by halo formation on water-agar plates containing colloidal chitin [1]. Bacterial chitinase activity has often been reported to be linked to antifungal properties [35–38]. Indeed, when adding chitinase inhibitor allosamidin, the growth of Collimonas on fungi was decreased, suggesting potential contribution to its mycophagous ability [39]. However, mutants in the chitinase loci of C. fungivorans Ter331 showed no difference during in vitro antagonism tests [34]. This suggests that chitinases might not contribute solely to the antifungal activities of Collimonas. The antifugal activities might be coupled with the production of other secondary metabolites.
Phospholipases
Phospholipases are a group of enzymes that catalyze the cleavage of phospholipids. Two major phospholipase activities can be defined by the site of cleavage, namely in the hydrophobic diacylglycerol moiety (PLA) or in the polar head group of the amphipathic phospholipid (PLC and PLD) (Schmmiel and Miller, 1999). In general, 11 to 15 phospholipases from four different groups: phospholipase A1, phospholipase C, phospholipase D and patatin phospholipase were detected in the six Collimonas genomes (Fig. 4a; Additional file 2: Table S7). Phospholipases are considered virulence factors for pathogenic bacterial species which cause tissue destruction, lung infections, hemolysis etc. [40]. Given the cleavage properties of phospholipids we speculate that they might be involved in nutrient acquisition via fungal membrane disturbing activities as well as in defense against competitors.
Fig. 4.
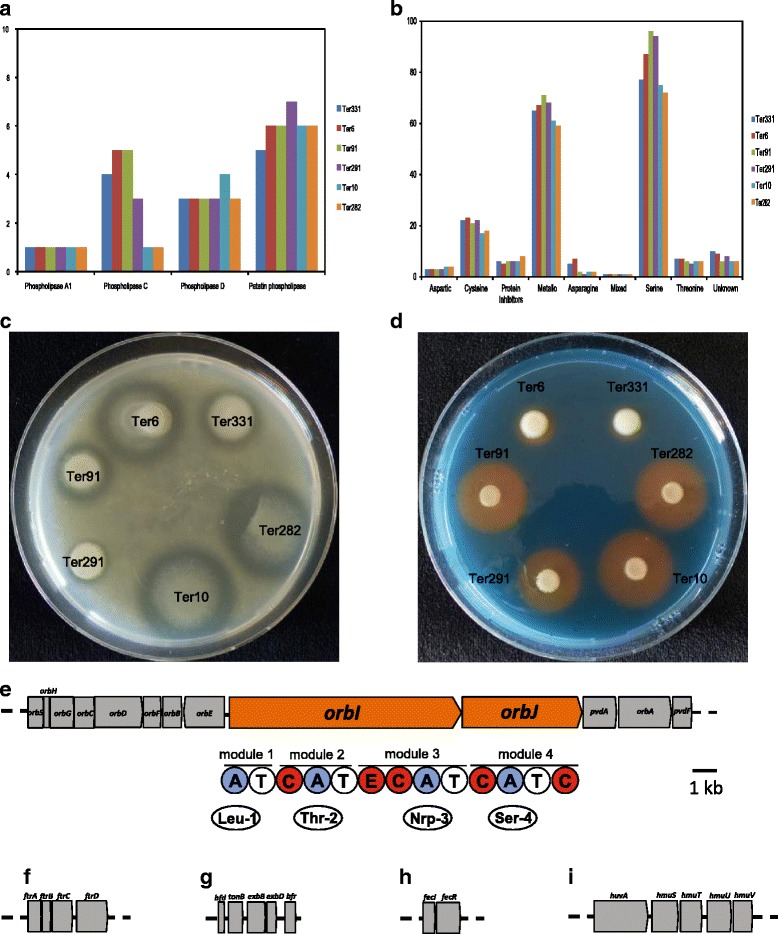
a Identification of phospholilpases in the six Collimonas genomes with PFAM signatures of the different phospholipase groups. b Identification of peptidases in the six Collimonas genomes as inferred from MEROPS 9.12 database. c Extracellular protease activity of Collimonas strains Ter331, Ter6, Ter91, Ter291, Ter10 and Ter282. A halo indicates extracellular protease production. d Siderophore production of Collimonas strains Ter331, Ter6, Ter91, Ter291, Ter10 and Ter282 plated on a CAS plate. An orange halo indicates of siderophore production. Gene clusters involved in iron acquisition in Collimonas strains. Five loci are presented: (e) Ornibactin locus. Underneath the genes are the module and domain organization of orbI and orbJ. The domains are as follows: C, condensation; A, adenylation; T, thiolation; and E, Epimerization. Underneath the domains are the amino acids that are incorporated into the peptide moiety. The number associated with the amino acid refers to the position of the amino acid in the peptide chain. f ftr bcc ABCD locus. g bfr (bacterioferritin) encoding gene and adjacent genes bfd and tonB-exbB-exbD cluster. h fecIR operon. i hmu operon (or bhu Burkholderia haem uptake operon)
Peptidases
In the six Collimonas genomes, 176 to 212 peptidases were predicted and nine families of proteolytic enzymes were identified (Fig. 4b). Among them, serine and metallo peptidases are the two dominant families. The Collimonas strains in our study were tested positive for exoprotease production (Fig. 4c). Serine proteases are one of the most abundant groups of proteolytic enzymes found in all living organisms [41] and in prokaryotes, serine proteases are involved in several biological processes associated with cell signaling, defense response and development [42–44]. Furthermore, serine protease can be involved in regulating the biosynthesis of lipopeptides, which can play a role in the suppression of other microbes [45].
Iron acquisition
Siderophores are low molecular weight, high-affinity iron chelating compounds produced by microorganisms under iron limited conditions and function in solubilization, transport and storage of iron [46, 47]. Siderophore production can act as an antagonistic mechanism by scavenging limited iron from the soil environment, thereby reducing the amount of available iron for other organisms. Our analysis revealed that all six strains encode biosynthesis clusters which resemble ornibactin (Fig. 4e), a siderophore synthesis cluster of Burkholderia cenocepacia [48]. Siderophore production was confirmed for all the six Collimonas strains, albeit with different production efficiency (Fig. 4d).
Next to siderophore production, other mechanisms of iron acquisition were reported. For example, recently, a novel alternative siderophore-independent iron uptake system was identified in Burkholderia, named ftrbccABCD locus. This ftrABCD operon was identified in all six Collimonas genomes (Fig. 4f), indicating that there are more strategies for iron uptake besides ornibactin production. Moreover, we found genes coding for the production of bacterioferritin, a type of iron-storage protein [49]. The gene is often adjacent to genes encoding a small [2Fe-2S]-ferredoxin Bfd. Downstream of the bfd gene is the tonB-exbB-exbD cluster which encodes a system to transduce cellular energy to outer-membrane receptors for siderophores and haemin [50]. The bacterioferritin encoding gene bfr and adjacent genes bfd and tonB-exbB-exbD cluster were found in the six Collimonas genomes (Fig. 4g). Furthermore, a fecIR operon was also found in all six genomes (Fig. 4h). It is known that the transcription of genes for ferric-citrate transport in E. coli requires FecI, and FecR, a cytoplasmic membrane protein encoded by the second gene in the Fur-repressed fecIR operon which transmits an external iron signal to the cytoplasmic FecI protein [51]. A haem uptake system similar to that of Burkholderia cenocepacia J2315 was found in the genomes of C. fungivorans Ter6 and C. pratensis Ter91, encoded by the hmu operon [52]. This operon is comprised of five genes (Fig. 4i) and was suggested to be Fur-mediated. Furthermore, fungi are known to produce haem [53], and, therefore, it is plausible that Collimonas are able to use the fungal haem as source of iron.
NRPS and PKS-NRPS genes encoding metabolites
Lipopeptides
Lipopeptides (LPs) are compounds composed of a lipid tail with a linear or cyclic oligopeptide [54]. They exhibit surfactant, antimicrobial, anti-predation, and cytotoxic properties [55, 56]. LPs are synthesized in bacteria by large nonribosomal peptide synthetases (NRPSs) via a thiotemplate process. The structural diversity of the LPs is due to differences in the length, composition of the fatty acid tail, and the number, type and configuration of the amino acids in the peptide moiety. Via in silico analysis, we identified gene clusters for LP biosynthesis of tripropeptin A in the genomes of C. fungivorans Ter331 and Ter6 (Fig. 5a). Although the structure of tripropeptin A has been known for more than a decade [57, 58], genetic analysis of its biosynthesis was only recently reported [59]. Our study revealed that three NRPS genes trpA, trpB, trpC are organized in a single-operon (Fig. 5a). The trpA gene, encodes an NRPS with the first five modules, and trpB, trpC encode one and two modular NRPS, respectively. The wild type C. fungivorans Ter331 and Ter6 possess antibacterial activity against Staphylococcus aureus but the other four strains from C. pratensis and C. arenae (Fig. 5b) do not. The site-directed ∆trpA mutant of C. fungivorans Ter331 lacks this antagonism (Fig. 5b) indicating that tripropeptin A is indeed involved in antibacterial activity which is a unique trait for C. fungivorans strains.
Fig. 5.
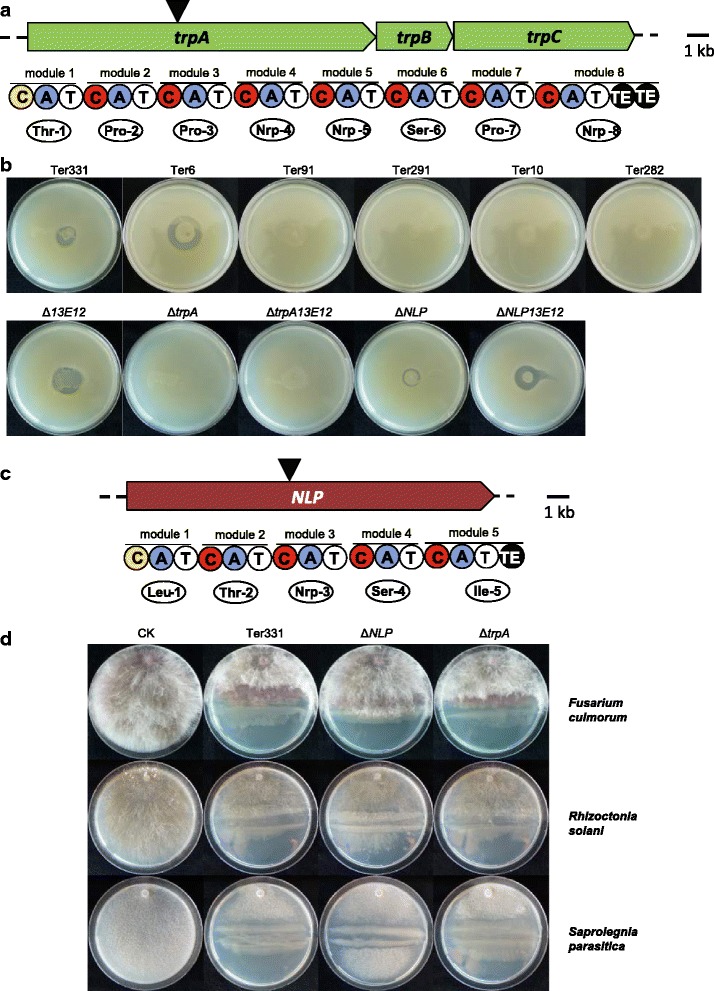
Biosynthetic gene cluster and antibacterial activity associated with tripropeptin A production by the Collimonas stains. a Organization of the gene cluster and predicted amino acid composition of the Tripropeptin A in C. fungivorans Ter331 and Ter6 genomes. Underneath the genes are the module and domain organization of trpA, trpB and trpC. The domains are as follows: C, condensation; A, adenylation; T, thiolation; and TE, thioesterification. Underneath the domains are the amino acids that are incorporated into the LP peptide moiety. The number associated with the amino acid refers to the position of the amino acid in the LP peptide chain. The black triangle indicates the position of the Gm cassette insertion in the trpA gene. b Antibacterial activity associated with tripropeptin A production. Strains Ter331, Ter6, Δ13E12 mutant (deficient in collimomycin biosynthesis), ΔNLP mutant (deficient in the new lipopeptide biosynthesis) and ΔNLP13E12 mutant (deficient in both-new lipopeptide and collimomycin biosynthesis) exhibited inhibition against Staphylococcus aureus via overlay assay. ΔtrpA mutant of C. fungivorans Ter331 (deficient in tripropeptin A biosynthesis)- loss in the inhibition activity. Biosynthetic gene cluster and antifungal/oomycetal activities associated with new lipopeptide production by the Collimonas stains. c Organization of the gene cluster and predicted amino acid composition of the new lipopeptide in C. fungivorans Ter331, Ter6 and C. pratensis Ter91, Ter291 genomes. Underneath the genes are the module and domain organization of NLP. The domains are as follows: C, condensation; A, adenylation; T, thiolation; and TE, thioesterification. Underneath the domains are the amino acids that are incorporated into the LP peptide moiety. The number associated with the amino acid refers to the position of the amino acid in the LP peptide chain. The black triangle indicates the position of the Gm cassette insertion in the nlp gene. d Antifungal/oomycetal activities associated with new lipopeptide production. Strain Ter331 which has new lipopeptide biosynthetic clusters exhibited inhibition against Fusarium culmorum, Rhizoctonia solani and Saprolegnia parasitica. Mutant ΔNLP of Ter331 deficient in new lipopeptide biosynthesis abolished the inhibition activities. CK is control grew without bacteria
Another 16.9 kb NRPS gene was found in C. fungivorans Ter331, Ter6 and C. pratensis Ter91, Ter291 strains (Figs. 3c and 5c). This gene is composed of five amino acids. There are no hits to known lipopeptides indicating a new lipopeptide. Collimonas strains are well known for their ability to suppress a range of fungi and oomycetes [3, 32, 60, 61]. However, our study revealed that the six Collimonas strains differed in the in vitro suppression against fungal and oomycetal pathogens (Fig. 5d; Additional file 2: Table S8). When the gene encoding the new lipopeptide was knocked out in C. fungivorans Ter331 by site-directed mutagenesis, reduced suppression was observed (Fig. 5d). This indicates that the new lipopeptide contributes to antimicrobial activity against both fungal and oomycetal pathogens. For the C. arenae species, no genes coding for the production of the new lipopeptide were found in the genome, but the strains showed suppressing activities against fungal and oomycetal pathogens (Additional file 2: Table S8). This indicates that apart from new lipopeptides, there may be other factor(s) involved in antimicrobial activity or that the suppression activity is due to synergistic effects rather than to a single compound. Thus, the different types of lipopeptide produced by the different Collimonas strains may partly explain the variability in the fungal inhibition behavior of these strains. The variability is also reflected in the unknown orphan gene clusters described below.
Unknown orphan NRPS and PKS-NRPS hybrid gene clusters
Within the genomes, four orphan gene clusters were identified. Two different 2-amino acids (2-aa) NRPS genes were found in Ter6 and Ter291 respectively (Additional file 2: Table S9; Additional file 1: Figure S4B, C). Next to this, a 54.5 kb unknown T3PKS-NRPS gene cluster was discovered in the genome of Ter6 (Additional file 1: Figure S4A). Another 10.6 kb NRPS-T1PKS gene cluster in Ter91 (Additional file 1: Figure S4D) resembles clusters encoding the Heat-stable antifungal factor (HSAF), also referred to as dihydromaltophilin, produced by Lysobacter species [62, 63]. HSAF exhibits inhibitory activities against a wide range of fungal species by disrupting the polarized growth or the biosynthesis of a distinct group of sphingolipids of fungi [62, 63].
Terpenes
Terpenes are a diverse family of primary and secondary metabolites which were mostly studied in plants and fungi [64]. Many terpenes of plant origin are known to be active against a wide variety of microorganisms, including Gram-positive, Gram-negative bacteria and fungi [65], but to date there are only few reports on antimicrobial activity of terpenes from microbial origin [66, 67]. In a previous study, four monoterpenes (γ-terpinene, 1S-α-pinene, β-pinene and β-myrcene) were detected in the headspace of C. pratensis strains Ter91 [68]. Here, these monoterpenes were tested individually and as a mixture for their antimicrobial activity. The β-pinene exhibited inhibition against Staphylococcus aureus and Rhizoctonia solani and the mixture of all monoterpenes revealed inhibition against these two pathogens and also to E. coli (Fig. 6a).
Fig. 6.
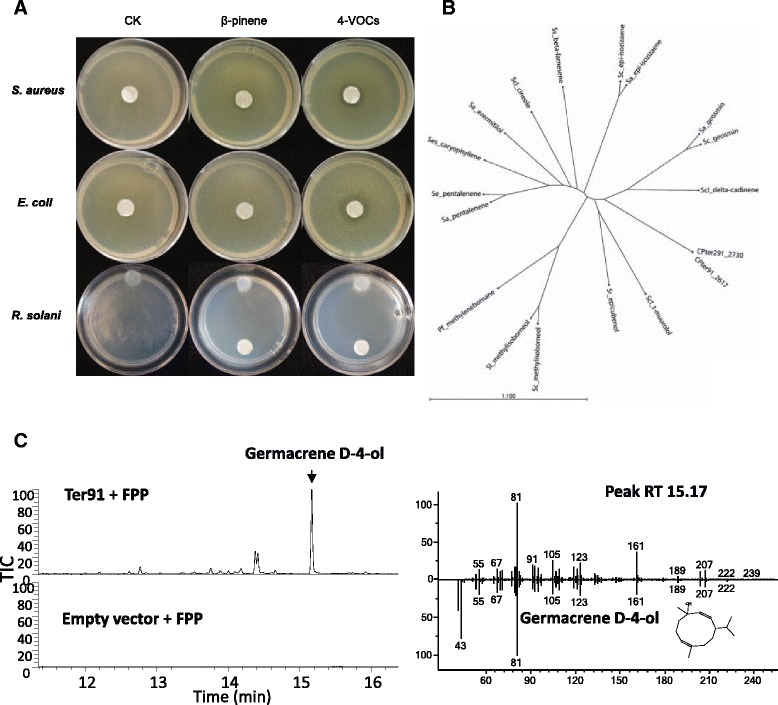
a Antimicrobial activities of pure β-pinene and 4-VOCs (mix of β-pinene, α-pinene, myrcene and terpinene in 1:1:1:1 ratio) against Staphylococcus aureus, Escherichia coli and Rhizoctonia solani. CK is control without terpenes. 5 mm sterilized white filter papers were placed in the centre or on the bottom of the petri dishes for antibacterial and antifungal assays respectively. 2 μl of each VOC was added accordingly to the white filter papers. b Phylogenetic tree of characterized bacterial terpene cyclase proteins. The protein name indicates the bacterial species and the major terpene produced by these terpene cyclases. Sequences included are listed in Additional file 2: Table S13. c Ter91 terpene synthase with FPP. TIC 100 % = 1.76E5. Major sesquiterpene product at RT 15.17, identified as Germacrene D-4-ol by comparison of mass spectra to NIST14 spectral library
Biosynthesis of terpenes is mediated by terpene cyclases, and starts from polyprenyl pyrophosphate precursors. The precursor accepted by the cyclase determines which class of terpenes it produces: the C10 precursor geranyl pyrophosphate (GPP) will lead to formation of monoterpenes, while C15 precursorfarnesyl pyrophosphate (FPP) will lead to sesquiterpenes, and the C20 precursor geranylgeranyl pyrophosphate (GGPP) will lead to di-terpenes, or to phytoene (C40). In particular mono- and sesquiterpene synthases can occur in many cyclization patterns, leading to a huge diversity of molecules.
The genomes of the six Collimonas strains were screened for gene clusters possibly involved in terpene biosynthesis. All strains carry genes related to phytoene biosynthesis (Additional file 2: Table S9). Only C. pratensis strains Ter91 and Ter291 harbored an additional cluster comprising terpene synthases genes (Fig. 3c; CPter91_2617 and CPter291_2730). Terpene cyclases are widely distributed in bacteria, but mostly characterised in Streptomyces species [69, 70]. To date only one terpene cyclase from Proteobacteria has been functionally characterised, the 2-methylenebornane synthase from Pseudomonas fluorescens Pf0-1 [71]. CPter91_2617 and CPter291_2730 both encode a 330-amino acid protein that differs only in 2 amino acid residues (Additional file 1: Figure S5). When compared to other functionally characterized terpene cyclases, the Collimonas protein sequences showed maximally 23 % aa-identity to any previously characterized bacterial terpene cyclase (Fig. 6b).
The product specificity of mono- and sesquiterpene cyclases cannot be predicted from their primary sequence. For biochemical characterization, CPter91_2617 and CPter291-2730 genes were expressed in E. coli, partially purified and tested in vitro using FPP, GPP or GGPP as substrates. When the produced terpenes were analyzed by GC-MS, both Collimonas enzymes converted FPP to a mix of sesquiterpenes and sesquiterpene alcohols. The major peak was putatively identified as germacrene D-4-ol by comparison of the mass spectrum to the NIST 2014 spectral library, and several minor sesquiterpene peaks, including δ-cadinene (Fig. 6c). When GPP was applied as a substrate, the production of two monoterpenes identified as β-pinene and β-linalool was observed (Additional file 1: Figure S6A). A small amount of product could be observed upon the incubation of GGPP as substrate, which was putatively identified as 13-epimanool (Additional file 1: Figure S6B). Thus we characterized CPter91_2617 and CPter291_2730 as mixed mono-, sesqui- and diterpene cyclases, with major product germacrene D-4-ol. The sesquiterpene products suggest that they are functionally related to plant and fungal cadinene/cadinol and germacrene D-4-ol synthases, although sequence homology to these enzymes is low [72, 73]. One of the monoterpene products of CPter91_2617 and CPter291_2730, β-pinene, was also observed in the headspace of C. pratensis and showed antibacterial activity, suggesting a role of the Collimonas terpene cyclases in antimicrobial activity. Volatiles terpenes may have synergistic antimicrobial effects in combination with antibiotics. For example a synergistic effect of terpenes and penicillin on multiresistant strains S. aureus and E. coli was reported [74]. Beside antimicrobial activity the production of terpenes by Collimonas may point to another important ecological role, namely chemical communication. Since terpenes volatilize easily and can be produced by all kingdoms of life including plant, fungi and bacteria, we assume that terpenes may play a significant role for Collimonas long-distance inter-kingdom interactions and communication.
Conclusions
The comparative analysis of six completely sequenced Collimonas genomes representing three species revealed a high degree of genomic diversity between strains with a core genome representing 43.1 to 52.7 % of the genome of all strains (Summarized in Fig. 3c). Although the genomes were largely syntenic, genome rearrangements were observed both between and within the species indicating high genomic plasticity. All Collimonas genomes carry large numbers of Genomic Islands pointing to a complex history of gene recombination and horizontal transfer between bacterial relatives. Type two secretion systems were present in all genomes suggesting that it may be important trait for the mycophagous lifestyle of Collimonas.
Genomic analysis of secondary metabolism of Collimonas revealed that genes encoding for exoenzymes such as chitinase, peptidase, phospholipase are well conserved but that there is a high variability of genes encoding for the production of other secondary metabolites such as collimomycin, lipopeptides, (PKS-)NRPS, and terpenes. Genes encoding the production of the polyacetylenic compound collimomycin and lipopeptide tripropeptin A are present only in C. fungivorans while the gene clusters encoding the production of a putative new lipopeptide is present in both C. fungivorans and C. pratensis, but not C. arenae. Moreover, several unknown orphan (PKS-)NRPS gene clusters are present in the genome of C. fungivorans and C. pratensis.
Mutational and phenotypical analyses indicated that tripropeptin A and the designated new lipopeptide have antibacterial and antifungal (oomycetal) activities, respectively. The biochemical characterization of the terpene synthases genes revealed that Collimonas are able to produce a set of sesquiterpenes and/or monoterpens that are considered to be mainly of plant origin. The in vitro assay of pure terpene compounds indicated their contributions to both antibacterial and antifungal activities.
Overall, our exploration of Collimonas genomes revealed that this bacterial group represents a valuable resource for the discovery of novel secondary metabolites and enzymes. The results gained here will be certainly helpful for designing future experimental studies that will lead to comprehensive understanding of the unique ecology of Collimonas species.
Methods
Strains and growth conditions
All bacterial strains used in this study are listed in Table 1 and Additional file 2: Table S10. Collimonas strains were cultured in 0.1 Tryptic Soy Broth (0.1 TSB) (5 g/L NaCl, 1 g/L KH2PO4, 3 g/L TSB, 20 gL − 1 CMN-Boom Agar, pH = 6.7) or King’s B medium (20 g/L proteose peptone, 1.5 g/L MgSO4, 1.2 g/L KH2PO4, 10 g/L glycerol, 15 g/L agar). Escherichia coli strain DH5α was used as a host for the plasmids used for site-directed mutagenesis. E. coli strains were grown on Luria-Bertani (LB) plates (10 g/L NaCl, 10 g/L Bacto™ Tryptone, 5 g/L Bacto™ Yeast extract, 20 g/L Merck Agar) or in LB broth amended with the appropriate antibiotics.
Genomic DNA isolation
Genomic DNA from each Collimonas strain was extracted from overnight grown cells using QIAamp® DNA Mini Kit and Qiagen® MagAttract® HMW kit and used for Illumina and PacBio RS II sequencing respectively.
Genome sequencing
Illumina paired-end sequences were obtained for C. arenae Ter10, C. arenae Ter282, C. pratensis Ter91, C. pratensis Ter291 and C. fungivorans Ter6 from BaseClear B.V. on the Illumina HiSeq2000 platform (2 x 51 bp paired-end reads, except strain Ter91 which was sequenced at 2 x 100 bp paired-end reads) and assembled using the Ray [75] assembler version 2.3.1 (Additional file 2: Table S11).
PacBio RS II sequences were obtained from 5 SMRT cells, one for each strain. Sequences were filtered using SMRT Analysis server v2.2.0 with default settings. The RS_HGAP Assembly.3 (HGAP3) [76] protocol was used to assemble the filtered reads, the RS_AH_Scaffolding protocol was used if the initial assembly yielded more than one contig, followed by a final Quiver correction using the RS_Resequencing protocol (See Additional file 2: Table S12).
The singular contigs were checked using a custom script and overlapping ends trimmed. The final circular contig for each chromosome was rearranged to start at the dnaA gene in the forward direction. There was no evidence of plasmids in the sequence data.
All Collimonas sequences including the previously published genome of Ter331 were annotated using the IGS annotation pipeline [77]. The IGS annotation of Ter331 is attached as a Additional file 3: The accession numbers of the other five genomes after submission to NCBI are Ter6 (CP013232); Ter91 (CP013234); Ter291 (CP013236); Ter10 (CP013233) and Ter282 (CP013235).
Bioinformatic analysis
Core, pan and variable genome analysis
Protein coding genes from the six Collimonas strains were clustered together with the protein coding genes of Burkholderia phytofirmans PsJN (NC_010681.1, NC_010676.1) and Pseudomonas protegens Pf-5 (NC_004129.6) using cd-hit [78] with word length 3 (−n 3), global identity (−G 1) and a minimal alignment coverage of 60 % for the shortest protein (−aS 0.6). Cd-hit clusters were parsed into an absence-presence matrix from which the core, pan and variable genomes were parsed using custom scripts. COG annotations were determined using kognitor [79]. Core and pan evolution plot is generated based on [80]. At each number of strains (n) out of strain set (s), s!/n!*(s-n)! combinations are possible. The median number of specific, variable and core genes for all combinations are plotted as a function of n. Clusters containing one gene per strain were selected from the core cluster set of the Collimonas, Burkholderia and Pseudomonas strains were aligned with MAFFT and combined in one pseudoalignment. Redundant colums were removed and maximum likelihood phylogenetic trees were calculated with RAxML [81].
The annotation of the previously-published genome of Ter331 was updated and manually curated as part of this study. Synteny analyses were performed using Progressive MAUVE [82]. Phylogenetic analyses on the whole genome was performed using Gegenees [83], and 16S rDNA analysis was performed using MEGA6 [84–87]. Whole genome peptidases prediction was conducted by MEROPS [88]. Phospholipases were predicted by searching on the basis of the profile HMM using PFAM domains of phospholipase A1 (PF02253), phospholipase A2 (PF09056), phospholipase C (PF05506) and phospholipase D (PF00614). Secondary metabolite production clusters were examined using the antiSMASH program [89, 90]. The amino acid composition of products from NRPS sequences were predicted using NRPSpredictor 2 [91]. Genomic islands were identified using IslandViewer [92, 93] and phages elements and features were identified using PHAST [94]. CRISPRs were identified based on CRISPRfinder [10]. Whole genome analysis for type VI secretion system was conducted with SecRet6 [95] and circular genome diagrams were visualized using Circos [96].
Availability of supporting data
Further methodological details are provided in the Additional file 4: Materials and Methods.
Acknowledgements
This research was supported in part by funds from Ecolink (260–45140) and by The Netherlands Organization for Scientific Research (NWO) VIDI personal grant to P.G (864.11.015). This publication is No.5983 of the Netherlands Institute of Ecology (NIOO-KNAW).
Abbreviations
- PKS
Polyketide synthase
- NRPS
Nonribosomal peptide synthetase
- COG
Clusters of Orthologous Groups
- GIs
Genomic islands
- CRISPRs
Clustered Regularly Interspaced Short Palindromic Repeats
Additional files
Phylogenetic tree based on 16S rDNA depicting the relationships of sequenced strains of Collimonas. Underneath the genes are the module and domain organization of PKS or NRPS genes. The domains are as follows: C, condensation; A, adenylation; T, thiolation; E, Epimerization; TE, thioesterification; CAL, Co-enzyme A ligase domain; KS, Ketosynthase domain; AT, Acyltransferase domain; ER, Enoylreductase domain; KR, Ketoreductase domain; DH, Dehydratase domain; and ACP, Acyl-carrier protein domain. Underneath the domains are the amino acids that are incorporated into the peptide moiety. The number associated with the amino acid refers to the position of the amino acid in the peptide chain. Figure S2. Synteny of the six Collimonas genomes. Pairwise alignments of genomes were generated using Mauve (A) C. fungivorans (B) C. pratensis (C) C. arenae and (D) The three species together. Colored outlined blocks surround the regions of the genomic sequence that aligned to another genome. The colored bars inside the blocks are related to the level of sequence similarities. The analysis showed that the highest number of rearrangements was evident between all the three species. Figure S3. (A) Conserved gene clusters for type II (T2SSa/T2SSb), III (T3SSa/T3SSb) and VI (T6SS) secretion systems identified in Collimonas strains. Quorum sensing assays of the Collimonas strains. Quorum sensing activity of Collimonas strains Ter331, Ter6, Ter91, Ter291, Ter10 and Ter282 with indicator strain (B) C. violaceum CV026 and (C) A. tumefaciens NT1 (outer colonies). A purple (C. violaceum CV026) or blue (A. tumefaciens NT1) pigment produced by the indicator strains is indicative of quorum sensing activity of the tested strains. Figure S4. Organization of the orphan gene clusters and predicted amino acid compositions. Figure S5. Amino acid alignment of Collimonas terpene synthases CPter91_2617 and CPter291_2730 with previously characterised bacterial terpene synthases. The Collimonas terpene synthases were aligned with the Streptomyces exfoliatus pentalenene synthase (Se_pentalenene), S. coelicolor geosmin synthase (Sc_geosmin, 336 amino acids of the N-terminus), Streptosporangium roseum epi-cubenol synthase (Sr_epicubenol), S. avermitilis avermitilol synthase (Sa_avermitilol), S. clavuligerus 1,8-cineole synthase (Scl_cineole) and Pseudomonas fluorescens 2-methylenebornane synthase (Pf_methylenebornane). The characteristic terpene synthase divalent metal-binding motifs, namely the acidic amino acid-rich motif and the NSE triad, are boxed. Figure S6. GC-MS chromatograms of Ter91 terpene cyclase incubated with different substrates, and mass spectra of major products. The GC-MS chromatograms of Ter291 were identical to Ter91 (data not shown). Empty vector chromatograms shows products from a control enzyme extract (pACYC-duet-1). (A) Ter91 terpene synthase with GPP. TIC 100 % = 1.19E4. Major monoterpene product at RT 6.79, identified as Beta-pinene by authentic standard. (B) Ter91 terpene synthase with GGPP. TIC 100 % = 2.12E4. Major diterpene product at RT 20.24, identified as 13-epimanool comparison of mass spectra to the NIST8 library. (PDF 570 kb)
Gene locus of the flagellar biosynthetic gene cluster of the Collimonas strains. Table S2. Gene locus of the secretion systems of the Collimonas strains. Table S3. Genomic distribution of signal transduction proteins in six Collimonas genomes. Table S4. Presence of quorum-sensing proteins in six Collimonas genomes. Table S5. Gene locus of the collimomycin biosynthetic gene cluster of the Collimonas strains. Table S6. Gene locus of the chitinase biosynthetic gene clusters of the Collimonas strains. Table S7. Gene locus of the phopholipase genes of the Collimonas strains. Table S8. Antimicrobial activities of the Collimonas strains. Table S9. Genes encoding secondary metabolites and secretion systems in Collimonas strains. Table S10. Strains and mutants used in this study. Table S11. Summary of Illumina paired-end sequences assembly data. Table S12. Summary of PacBio sequences assembly data. Table S13. Sequences of characterized bacterial terpene synthases used in the phylogenetic analysis. (XLSX 56 kb)
The IGS annotation of Collimonas fungivorans Ter331 genome. (GBF 10511 kb)
Materials and Methods. Additional detailed materials and methods. (DOCX 45 kb)
Footnotes
Competing interests
The authors declare no conflict of interest.
Authors’ contributions
CS and PG designed the experiments. CS, RS and VJ performed the bioinformatics analysis. CS, DK, EJ, KC, AV conducted the experiments. CS drafted the manuscript. PG, JB, KC, WB and JV revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Chunxu Song, Email: c.song@nioo.knaw.nl.
Ruth Schmidt, Email: r.schmidt@nioo.knaw.nl.
Victor de Jager, Email: v.dejager@nioo.knaw.nl.
Dorota Krzyzanowska, Email: dorota.krzyzanowska@biotech.ug.edu.pl.
Esmer Jongedijk, Email: esmer.jongedijk@wur.nl.
Katarina Cankar, Email: katarina.cankar@wur.nl.
Jules Beekwilder, Email: jules.beekwilder@wur.nl.
Anouk van Veen, Email: anouk_v_veen@hotmail.com.
Wietse de Boer, Email: w.deboer@nioo.knaw.nl.
Johannes A. van Veen, Email: h.vanveen@nioo.knaw.nl
Paolina Garbeva, Email: p.garbeva@nioo.knaw.nl.
References
- 1.de Boer W, Leveau JH, Kowalchuk GA, Klein Gunnewiek PJ, Abeln EC, Figge MJ, et al. Collimonas fungivorans gen. nov., sp. nov., a chitinolytic soil bacterium with the ability to grow on living fungal hyphae. Int J Syst Evol Microbiol. 2004;54(Pt 3):857–864. doi: 10.1099/ijs.0.02920-0. [DOI] [PubMed] [Google Scholar]
- 2.Leveau JH, Uroz S, de Boer W. The bacterial genus Collimonas: Mycophagy, weathering and other adaptive solutions to life in oligotrophic soil environments. Environ Microbiol. 2010;12(2):281–292. doi: 10.1111/j.1462-2920.2009.02010.x. [DOI] [PubMed] [Google Scholar]
- 3.De Boer W, Gunnewiek PJAK, Lafeber P, Janse JD, Spit BE, Woldendorp JW. Anti-fungal properties of chitinolytic dune soil bacteria. Soil Biol Biochem. 1998;30(2):193–203. doi: 10.1016/S0038-0717(97)00100-4. [DOI] [Google Scholar]
- 4.Rudnick M-B: Mycophagous soil bacteria. PhD thesis. 2015:87–124.
- 5.Hoppener-Ogawa S, de Boer W, Leveau JH, van Veen JA, de Brandt E, Vanlaere E, et al. Collimonas arenae sp. nov. and Collimonas pratensis sp. nov., isolated from (semi-)natural grassland soils. Int J Syst Evol Microbiol. 2008;58(Pt 2):414–419. doi: 10.1099/ijs.0.65375-0. [DOI] [PubMed] [Google Scholar]
- 6.Mela F, Fritsche K, de Boer W, van Veen JA, de Graaff LH, van den Berg M, et al. Dual transcriptional profiling of a bacterial/fungal confrontation: Collimonas fungivorans versus Aspergillus niger. ISME J. 2011;5(9):1494–1504. doi: 10.1038/ismej.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mela F, Fritsche K, de Boer W, van den Berg M, van Veen JA, Maharaj NN, et al. Comparative genomics of bacteria from the genus Collimonas: Linking (dis)similarities in gene content to phenotypic variation and conservation. Environ Microbiol Rep. 2012;4(4):424–432. doi: 10.1111/j.1758-2229.2012.00336.x. [DOI] [PubMed] [Google Scholar]
- 8.Wu JJ, de Jager VC, Deng WL, Leveau JH: Finished genome sequence of Collimonas arenae Cal35. Genome announcements 2015, 3(1) doi: 10.1128/genomeA.01408-14. [DOI] [PMC free article] [PubMed]
- 9.Mela F, Fritsche K, Boersma H, van Elsas JD, Bartels D, Meyer F, et al. Comparative genomics of the pIPO2/pSB102 family of environmental plasmids: Sequence, evolution, and ecology of pTer331 isolated from Collimonas fungivorans Ter331. FEMS Microbiol Ecol. 2008;66(1):45–62. doi: 10.1111/j.1574-6941.2008.00472.x. [DOI] [PubMed] [Google Scholar]
- 10.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cianciotto NP. Many substrates and functions of type II secretion: Lessons learned from Legionella pneumophila. Future Microbiol. 2009;4(7):797–805. doi: 10.2217/fmb.09.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeShazer D, Brett PJ, Burtnick MN, Woods DE. Molecular characterization of genetic loci required for secretion of exoproducts in Burkholderia pseudomallei. J Bacteriol. 1999;181(15):4661–4664. doi: 10.1128/jb.181.15.4661-4664.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomich M, Planet PJ, Figurski DH. The tad locus: Postcards from the widespread colonization island. Nat Rev Microbiol. 2007;5(5):363–375. doi: 10.1038/nrmicro1636. [DOI] [PubMed] [Google Scholar]
- 14.Planet PJ, Kachlany SC, Fine DH, DeSalle R, Figurski DH. The widespread colonization island of Actinobacillus actinomycetemcomitans. Nat Genet. 2003;34(2):193–198. doi: 10.1038/ng1154. [DOI] [PubMed] [Google Scholar]
- 15.Nika JR, Latimer JL, Ward CK, Blick RJ, Wagner NJ, Cope LD, et al. Haemophilus ducreyi requires the flp gene cluster for microcolony formation in vitro. Infect Immun. 2002;70(6):2965–2975. doi: 10.1128/IAI.70.6.2965-2975.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durand E, Bernadac A, Ball G, Lazdunski A, Sturgis JN, Filloux A. Type II protein secretion in Pseudomonas aeruginosa: the pseudopilus is a multifibrillar and adhesive structure. J Bacteriol. 2003;185(9):2749–2758. doi: 10.1128/JB.185.9.2749-2758.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoppener-Ogawa S, Leveau JH, Hundscheid MP, van Veen JA, de Boer W. Impact of Collimonas bacteria on community composition of soil fungi. Environ Microbiol. 2009;11(6):1444–1452. doi: 10.1111/j.1462-2920.2009.01872.x. [DOI] [PubMed] [Google Scholar]
- 18.Cornelis GR, Wolf-Watz H. The Yersinia Yop virulon: A bacterial system for subverting eukaryotic cells. Mol Microbiol. 1997;23(5):861–867. doi: 10.1046/j.1365-2958.1997.2731623.x. [DOI] [PubMed] [Google Scholar]
- 19.Galan JE, Collmer A. Type III secretion machines: Bacterial devices for protein delivery into host cells. Science. 1999;284(5418):1322–1328. doi: 10.1126/science.284.5418.1322. [DOI] [PubMed] [Google Scholar]
- 20.Warmink JA, van Elsas JD. Selection of bacterial populations in the mycosphere of Laccaria proxima: Is type III secretion involved? ISME J. 2008;2(8):887–900. doi: 10.1038/ismej.2008.41. [DOI] [PubMed] [Google Scholar]
- 21.Tseng TT, Tyler BM, Setubal JC. Protein secretion systems in bacterial-host associations, and their description in the gene ontology. BMC Microbiol. 2009;9(Suppl 1):S2. doi: 10.1186/1471-2180-9-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson RW, Preston GM, Rainey PB. Genetic characterization of Pseudomonas fluorescens SBW25 rsp gene expression in the phytosphere and in vitro. J Bacteriol. 2005;187(24):8477–8488. doi: 10.1128/JB.187.24.8477-8488.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwarz S, Hood RD, Mougous JD. What is type VI secretion doing in all those bugs? Trends Microbiol. 2010;18(12):531–537. doi: 10.1016/j.tim.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russell AB, Hood RD, Bui NK, LeRoux M, Vollmer W, Mougous JD. Type VI secretion delivers bacteriolytic effectors to target cells. Nature. 2011;475(7356):343–U392. doi: 10.1038/nature10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Records AR. The type VI secretion system: A multipurpose delivery system with a phage-like machinery. Mol Plant Microbe In. 2011;24(7):751–757. doi: 10.1094/MPMI-11-10-0262. [DOI] [PubMed] [Google Scholar]
- 26.Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 27.Ulrich LE, Koonin EV, Zhulin IB. One-component systems dominate signal transduction in prokaryotes. Trends Microbiol. 2005;13(2):52–56. doi: 10.1016/j.tim.2004.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krell T, Busch A, Lacal J, Silva-Jimenez H, Ramos JL. The enigma of cytosolic two-component systems: A hypothesis. Environ Microbiol Rep. 2009;1(3):171–176. doi: 10.1111/j.1758-2229.2009.00020.x. [DOI] [PubMed] [Google Scholar]
- 29.Lewenza S, Conway B, Greenberg EP, Sokol PA. Quorum sensing in Burkholderia cepacia: identification of the LuxRI homologs CepRI. J Bacteriol. 1999;181(3):748–756. doi: 10.1128/jb.181.3.748-756.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aguilar C, Friscina A, Devescovi G, Kojic M, Venturi V. Identification of quorum-sensing-regulated genes of Burkholderia cepacia. J Bacteriol. 2003;185(21):6456–6462. doi: 10.1128/JB.185.21.6456-6462.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hakvag S, Fjaervik E, Klinkenberg G, Borgos SE, Josefsen KD, Ellingsen TE, et al. Violacein-producing Collimonas sp. from the sea surface microlayer of costal waters in Trondelag, Norway. Mar Drugs. 2009;7(4):576–588. doi: 10.3390/md7040576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fritsche K, van den Berg M, de Boer W, van Beek TA, Raaijmakers JM, van Veen JA, et al. Biosynthetic genes and activity spectrum of antifungal polyynes from Collimonas fungivorans Ter331. Environ Microbiol. 2014;16(5):1334–1345. doi: 10.1111/1462-2920.12440. [DOI] [PubMed] [Google Scholar]
- 33.Rabea EI, Badawy MET, Stevens CV, Smagghe G, Steurbaut W. Chitosan as antimicrobial agent: Applications and mode of action. Biomacromolecules. 2003;4(6):1457–1465. doi: 10.1021/bm034130m. [DOI] [PubMed] [Google Scholar]
- 34.Fritsche K, de Boer W, Gerards S, van den Berg M, van Veen JA, Leveau JH. Identification and characterization of genes underlying chitinolysis in Collimonas fungivorans Ter331. FEMS Microbiol Ecol. 2008;66(1):123–135. doi: 10.1111/j.1574-6941.2008.00547.x. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi DY, Reedy RM, Bick J, Oudemans PV. Characterization of a chitinase gene from Stenotrophomonas maltophilia strain 34S1 and its involvement in biological control. Appl Environ Microb. 2002;68(3):1047–1054. doi: 10.1128/AEM.68.3.1047-1054.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang WT, Chen CS, Wang SL. An antifungal chitinase produced by Bacillus cereus with shrimp and crab shell powder as a carbon source. Curr Microbiol. 2003;47(2):102–108. doi: 10.1007/s00284-002-3955-7. [DOI] [PubMed] [Google Scholar]
- 37.Chernin L, Ismailov Z, Haran S, Chet I. Chitinolytic Enterobacter agglomerans antagonistic to fungal plant-pathogens. Appl Environ Microb. 1995;61(5):1720–1726. doi: 10.1128/aem.61.5.1720-1726.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dahiya N, Tewari R, Tiwari R, Hoondal G. Production of an antifungal chitinase from Enterobacter sp NRG4 and its application in protoplast production. World J Microb Biot. 2005;21(8–9):1611–1616. doi: 10.1007/s11274-005-8343-6. [DOI] [Google Scholar]
- 39.De Boer W, Gunnewiek PJAK, Kowalchuk GA, Van Veen JA. Growth of chitinolytic dune soil beta-subclass Proteobacteria in response to invading fungal hyphae. Appl Environ Microb. 2001;67(8):3358–3362. doi: 10.1128/AEM.67.8.3358-3362.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmiel DH, Miller VL. Bacterial phospholipases and pathogenesis. Microbes Infect. 1999;1(13):1103–1112. doi: 10.1016/S1286-4579(99)00205-1. [DOI] [PubMed] [Google Scholar]
- 41.Page MJ, Di Cera E. Serine peptidases: classification, structure and function. Cell Mol Life Sci. 2008;65(7–8):1220–1236. doi: 10.1007/s00018-008-7565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gottesman S. Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Bi. 2003;19:565–587. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]
- 43.Hengge R, Bukau B. Proteolysis in prokaryotes: Protein quality control and regulatory principles. Mol Microbiol. 2003;49(6):1451–1462. doi: 10.1046/j.1365-2958.2003.03693.x. [DOI] [PubMed] [Google Scholar]
- 44.Jenal U, Hengge-Aronis R. Regulation by proteolysis in bacterial cells. Curr Opin Microbiol. 2003;6(2):163–172. doi: 10.1016/S1369-5274(03)00029-8. [DOI] [PubMed] [Google Scholar]
- 45.de Bruijn I, Raaijmakers JM. Regulation of cyclic lipopeptide biosynthesis in Pseudomonas fluorescens by the ClpP protease. J Bacteriol. 2009;191(6):1910–1923. doi: 10.1128/JB.01558-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chu BC, Garcia-Herrero A, Johanson TH, Krewulak KD, Lau CK, Peacock RS, et al. Siderophore uptake in bacteria and the battle for iron with the host; a bird’s eye view. Biometals: Intl J Role Met Ions Biol Biochem Med. 2010;23(4):601–611. doi: 10.1007/s10534-010-9361-x. [DOI] [PubMed] [Google Scholar]
- 47.Hider RC, Kong XL. Chemistry and biology of siderophores. Nat Prod Rep. 2010;27(5):637–657. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- 48.Agnoli K, Lowe CA, Farmer KL, Husnain SI, Thomas MS. The ornibactin biosynthesis and transport genes of Burkholderia cenocepacia are regulated by an extracytoplasmic function sigma factor which is a part of the Fur regulon. J Bacteriol. 2006;188(10):3631–3644. doi: 10.1128/JB.188.10.3631-3644.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andrews SC. Iron storage in bacteria. Adv Microb Physiol. 1998;40:281–351. doi: 10.1016/S0065-2911(08)60134-4. [DOI] [PubMed] [Google Scholar]
- 50.Schalk IJ, Yue WW, Buchanan SK. Recognition of iron-free siderophores by TonB-dependent iron transporters. Mol Microbiol. 2004;54(1):14–22. doi: 10.1111/j.1365-2958.2004.04241.x. [DOI] [PubMed] [Google Scholar]
- 51.Braun V, Mahren S, Ogierman M. Regulation of the FecI-type ECF sigma factor by transmembrane signalling. Curr Opin Microbiol. 2003;6(2):173–180. doi: 10.1016/S1369-5274(03)00022-5. [DOI] [PubMed] [Google Scholar]
- 52.Holden MTG, Seth-Smith HMB, Crossman LC, Sebaihia M, Bentley SD, Cerdeno-Tarraga AM, et al. The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients (vol 191, pg 261, 2009). J Bacteriol. 2009;191(8):2907–7. [DOI] [PMC free article] [PubMed]
- 53.Franken ACW, Lokman BC, Ram AFJ, van den Hondel CAMJJ, de Weert S, Punt PJ. Analysis of the role of the Aspergillus niger aminolevulinic acid synthase (hemA) gene illustrates the difference between regulation of yeast and fungal haem- and sirohaem-dependent pathways. Fems Microbiol Lett. 2012;335(2):104–112. doi: 10.1111/j.1574-6968.2012.02655.x. [DOI] [PubMed] [Google Scholar]
- 54.Raaijmakers JM, de Bruijn I, Nybroe O, Ongena M. Natural functions of lipopeptides from Bacillus and Pseudomonas: more than surfactants and antibiotics. Fems Microbiol Rev. 2010;34(6):1037–1062. doi: 10.1111/j.1574-6976.2010.00221.x. [DOI] [PubMed] [Google Scholar]
- 55.Raaijmakers JM, de Bruijn I, de Kock MJD. Cyclic lipopeptide production by plant-associated Pseudomonas spp.: Diversity, activity, biosynthesis, and regulation. Mol Plant Microbe In. 2006;19(7):699–710. doi: 10.1094/MPMI-19-0699. [DOI] [PubMed] [Google Scholar]
- 56.Raaijmakers JM, Mazzola M. Diversity and natural functions of antibiotics produced by beneficial and plant pathogenic bacteria. Annu Rev Phytopathol. 2012;50:403–424. doi: 10.1146/annurev-phyto-081211-172908. [DOI] [PubMed] [Google Scholar]
- 57.Hashizume H, Igarashi M, Hattori S, Hori M, Hamada M, Takeuchi T. Tripropeptins, novel antimicrobial agents produced by Lysobacter sp. I. Taxonomy, isolation and biological activities. J Antibiot. 2001;54(12):1054–1059. doi: 10.7164/antibiotics.54.1054. [DOI] [PubMed] [Google Scholar]
- 58.Hashizume H, Hirosawa S, Sawa R, Muraoka Y, Ikeda D, Naganawa H, et al. Tripropeptins, novel antimicrobial agents produced by Lysobacter sp - II. Struct Elucidation J Antibiot. 2004;57(1):52–58. doi: 10.7164/antibiotics.57.52. [DOI] [PubMed] [Google Scholar]
- 59.Medema MH, Paalvast Y, Nguyen DD, Melnik A, Dorrestein PC, Takano E, et al. Pep2Path: Automated mass spectrometry-guided genome mining of peptidic natural products. Plos Comput Biol. 2014;10(9):e1003822. doi: 10.1371/journal.pcbi.1003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adesina MF, Lembke A, Costa R, Speksnijder A, Smalla K. Screening of bacterial isolates from various European soils for in vitro antagonistic activity towards Rhizoctonia solani and Fusarium oxysporum: Site-dependent composition and diversity revealed. Soil Biol Biochem. 2007;39(11):2818–2828. doi: 10.1016/j.soilbio.2007.06.004. [DOI] [Google Scholar]
- 61.Opelt K, Berg G. Diversity and antagonistic potential of bacteria associated with bryophytes from nutrient-poor habitats of the Baltic Sea coast. Appl Environ Microb. 2004;70(11):6569–6579. doi: 10.1128/AEM.70.11.6569-6579.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu FG, Zaleta-Rivera K, Zhu XC, Huffman J, Millet JC, Harris SD, et al. Structure and biosynthesis of heat-stable antifungal factor (HSAF), a broad-spectrum antimycotic with a novel mode of action. Antimicrob Agents Ch. 2007;51(1):64–72. doi: 10.1128/AAC.00931-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lou LL, Qian GL, Xie YX, Hang JL, Chen HT, Zaleta-Riyera K, et al. Biosynthesis of HSAF, a tetramic acid-containing macrolactam from Lysobacter enzymogenes. J Am Chem Soc. 2011;133(4):643–645. doi: 10.1021/ja105732c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trapp SC, Croteau RB. Genomic organization of plant terpene synthases and molecular evolutionary implications. Genetics. 2001;158(2):811–832. doi: 10.1093/genetics/158.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tholl D. Terpene synthases and the regulation, diversity and biological roles of terpene metabolism. Curr Opin Plant Biol. 2006;9(3):297–304. doi: 10.1016/j.pbi.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 66.Gurtler H, Pedersen R, Anthoni U, Christophersen C, Nielsen PH, Wellington EM, et al. Albaflavenone, a sesquiterpene ketone with a zizaene skeleton produced by a streptomycete with a new rope morphology. J Antibiot. 1994;47(4):434–439. doi: 10.7164/antibiotics.47.434. [DOI] [PubMed] [Google Scholar]
- 67.Zhao B, Lin X, Lei L, Lamb DC, Kelly SL, Waterman MR, et al. Biosynthesis of the sesquiterpene antibiotic albaflavenone in Streptomyces coelicolor A3(2) J Biol Chem. 2008;283(13):8183–8189. doi: 10.1074/jbc.M710421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garbeva P, Hordijk C, Gerards S, de Boer W. Volatile-mediated interactions between phylogenetically different soil bacteria. Front Microbiol. 2014;5:289. doi: 10.3389/fmicb.2014.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cane DE, Ikeda H. Exploration and mining of the bacterial terpenome. Acc Chem Res. 2012;45(3):463–472. doi: 10.1021/ar200198d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamada Y, Kuzuyama T, Komatsu M, Shin-Ya K, Omura S, Cane DE, et al. Terpene synthases are widely distributed in bacteria. Proc Natl Acad Sci U S A. 2015;112(3):857–862. doi: 10.1073/pnas.1422108112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chou WKW, Ikeda H, Cane DE. Cloning and characterization of Pfl_1841, a 2-methylenebornane synthase in Pseudomonas fluorescens PfO-1. Tetrahedron. 2011;67(35):6627–6632. doi: 10.1016/j.tet.2011.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoshikuni Y, Martin VJ, Ferrin TE, Keasling JD. Engineering cotton (+)-delta-cadinene synthase to an altered function: Germacrene D-4-ol synthase. Chem Biol. 2006;13(1):91–98. doi: 10.1016/j.chembiol.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 73.Lauchli R, Pitzer J, Kitto RZ, Kalbarczyk KZ, Rabe KS. Improved selectivity of an engineered multi-product terpene synthase. Org Biomol Chem. 2014;12(23):4013–4020. doi: 10.1039/C4OB00479E. [DOI] [PubMed] [Google Scholar]
- 74.Gallucci MN, Oliva M, Casero C, Dambolena J, Luna A, Zygadlo J, et al. Antimicrobial combined action of terpenes against the food-borne microorganisms Escherichia coli, Staphylococcus aureus and Bacillus cereus. Flavour Frag J. 2009;24(6):348–354. doi: 10.1002/ffj.1948. [DOI] [Google Scholar]
- 75.Boisvert S, Laviolette F, Corbeil J. Ray: Simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J Comput Biol. 2010;17(11):1519–1533. doi: 10.1089/cmb.2009.0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10(6):563. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 77.Galens K, Orvis J, Daugherty S, Creasy HH, Angiuoli S, White O, et al. The IGS standard operating procedure for automated prokaryotic annotation. Stand Genomic Sci. 2011;4(2):244–251. doi: 10.4056/sigs.1223234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang Y, Niu BF, Gao Y, Fu LM, Li WZ. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics. 2010;26(5):680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Snel B, Bork P, Huynen MA. The identification of functional modules from the genomic association of genes. Proc Natl Acad Sci U S A. 2002;99(9):5890–5895. doi: 10.1073/pnas.092632599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A. 2005;102(39):13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Agren J, Sundstrom A, Hafstrom T, Segerman B. Gegenees: Fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. Plos One. 2012;7(6):e39107. doi: 10.1371/journal.pone.0039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Saitou N, Nei M. The neighbor-joining method - a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 86.Felsenstein J. Phylogenies and the comparative method. Am Nat. 1985;125(1):1–15. doi: 10.1086/284325. [DOI] [PubMed] [Google Scholar]
- 87.Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci U S A. 2004;101(30):11030–11035. doi: 10.1073/pnas.0404206101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rawlings ND, Waller M, Barrett AJ, Bateman A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014;42(Database issue):D503–509. doi: 10.1093/nar/gkt953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, et al. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39:W339–W346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, et al. antiSMASH 2.0-a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41(W1):W204–W212. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rottig M, Medema MH, Blin K, Weber T, Rausch C, Kohlbacher O. NRPSpredictor2-a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011;39:W362–W367. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Langille MGI, Brinkman FSL. IslandViewer: An integrated interface for computational identification and visualization of genomic islands. Bioinformatics. 2009;25(5):664–665. doi: 10.1093/bioinformatics/btp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dhillon BK, Chiu TA, Laird MR, Langille MGI, Brinkman FSL. IslandViewer update: Improved genomic island discovery and visualization. Nucleic Acids Res. 2013;41(W1):W129–W132. doi: 10.1093/nar/gkt394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou Y, Liang YJ, Lynch KH, Dennis JJ, Wishart DS. PHAST: A fast phage search tool. Nucleic Acids Res. 2011;39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li J, Yao Y, Xu HH, Hao L, Deng Z, Rajakumar K, et al. SecReT6: A web-based resource for type VI secretion systems found in bacteria. Environ Microbiol. 2015;17(7):2196–202. doi: 10.1111/1462-2920.12794. [DOI] [PubMed] [Google Scholar]
- 96.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: An information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]