

Abstract
Types 1 and 2 diabetes affect some 380 million people worldwide. Both result ultimately from a deficiency of functional pancreatic insulin-producing beta cells. Beta cells proliferate in humans during a brief temporal window beginning around the time of birth, with peak beta cell labeling indices achieving approximately 2% in first year of life1-4. In embryonic life and after early childhood, beta cell replication rates are very low. While beta cell expansion seems an obvious therapeutic approach to beta cell deficiency, adult human beta cells have proven recalcitrant to such efforts1-8. Hence, there remains an urgent need for diabetes therapeutic agents that can induce regeneration and expansion of adult human beta cells in vivo or ex vivo. Here, we report the results of a high-throughput small molecule screen (HTS) revealing a novel class of human beta cell mitogenic compounds, analogues of the small molecule, harmine. We also define dual specificity tyrosine-regulated kinase-1a (DYRK1A) as the likely target of harmine, and the Nuclear Factors of activated T-cells (NFAT) family of transcription factors as likely mediators of human beta cell proliferation as well as beta cell differentiation. These observations suggest that harmine analogues (“harmalogs”) may have unique therapeutic promise for human diabetes therapy. Enhancing potency and beta cell specificity are important future challenges.
Keywords: Biological Sciences, Drug Discovery, Drug Screening, High-Throughput Screening
Reasoning that the growth-mediating MYC family of proteins are essential normal drivers of cell growth for many tissues9-16, that they lie downstream of many normal, developmental and regenerative mitogenic signaling pathways9-17, that c-MYC is an essential driver of proliferation in Ins1 and RINm5F rat pancreatic beta cell lines17, that c-MYC can drive human beta cell proliferation17, and that the MYC promoter may consequently serve as a downstream “sensor” for multiple diverse upstream signals leading to proliferation, we developed a luciferase-based small molecule high-throughput screening (HTS) platform to detect molecules that directly or indirectly activate the MYC promoter (Supplementary Fig. 1, Online Methods).
We generated multiple stable cell lines expressing a luciferase reporter under control of the human MYC promoter (Supplementary Fig. 1, Online Methods). Among these, the human hepatocyte cell line HepG2 yielded most robust luciferase responses and the least variability in pilot HTS screens, and was selected for further screening using two small molecule libraries (Fig. 1): a 2300 compound FDA library and a 100,000 compound “L1” library. Of the 102,300 compounds, 4500 scored >3 for median absolute deviation (MAD)18 for luciferase activation (Fig. 1b). Among these, the 86 that generated the greatest normalized percent activation (NPA >7.5%)19 were assessed for their ability both to induce c-MYC protein expression in HepG2 cells (Supplementary Fig. 1) and to induce BrdU incorporation in dispersed rat pancreatic beta cells Fig. 1c). Only one compound, harmine, induced both mild c-MYC increments and substantial BrdU incorporation into rat beta cells. Harmine also induced notable BrdU as well as Ki67 labeling in human beta cells, with the frequent appearance of double nuclei, suggesting recent cell division (Figs. 1d−g).
Figure 1. High-throughput screening reveals harmine family members as agonists of beta cell proliferation.
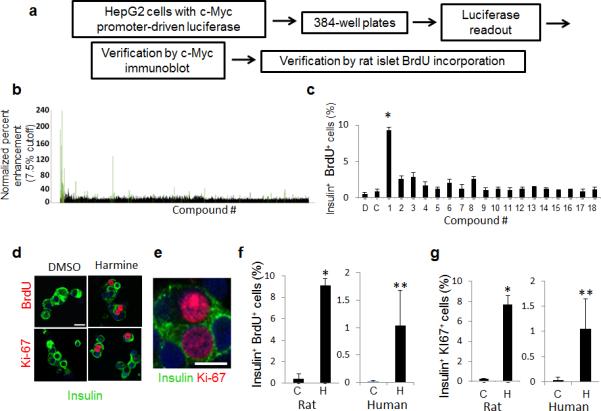
(a) Schematic outline of the screen in HepG2 cells used to identify compounds that promote beta cell replication. See text and Online Methods for details. (b) Results of the primary screen showing the 4500 initial hits (black) and the 86 compounds with a median absolute deviation (MAD) score >3 (green). (c) Examples of tertiary screening (rat beta cell BrdU incorporation) of the 86 compounds. Compound 1 is harmine. “D” is DMSO and “C” indicates rat islets treated with no vehicle. The BrdU screen was performed four times; where no error bars are seen, they are within the bar. (d) Examples of BrdU and Ki-67 labeling human beta cells treated with harmine. Note BrdU and Ki-67 nuclear “doublets” in human beta cells. (e) An enlarged view of harmine-treated human beta cells with Ki-67 nuclear “doublets” in adjacent cells. (f) Quantification of BrdU incorporation into rat (left) and human (right) beta cells. “C” indicates control (DMSO, vehicle) and “H” harmine. A minimum of 1000 beta cells was counted from multiple donors (4 rat, 6 human) for each bar. (g) Quantification of Ki67 labeling in rat and human beta cells. “C” indicates control (vehicle, DMSO) and “H” harmine. A minimum of 1000 beta cells was counted from multiple donor pairs (4 rat, 7 human) for each bar. In all relevant panels, error bars indicate s.e.m., * indicates P<0.05 and ** P<0.01 as determined by Wilcoxon Rank test, and the scale bar indicates 10 μm.
Harmine is a competitive inhibitor of ATP binding to the kinase pocket of DYRK1A, but also can inhibit other DYRK family members, monoamine oxidases (MAOs) and cdc-like kinases (CLKs). We therefore surveyed additional harmalogs (Fig. 2a)20-24. Harmaline and harmane, (inhibitors of MAO but not DYRK1A) did not induce proliferation; conversely, the Inhibitor of DYRK1A, (INDY, a DYRK1A inhibitor but not an MAO inhibitor), activated proliferation in both rat and human beta cells (Fig. 2b−d), also yielding Ki67+ and BrdU+ doublets (Fig. 2d). Harmine and INDY both also induced phosphorylation of histone-H3, a third marker of cell cycle transition (Supplementary Fig. 2). Effective harmine and INDY doses were in the 1-15 μM range, with higher doses being detrimental (Figs. 2e−h). WS6, a previously described small molecule beta cell proliferogen25, had little effect on human beta cell proliferation (Supplementary Fig. 3). Ki67 labeling induced by harmine was unaffected by glucose concentration (Supplementary Fig 2c). Collectively, these observations suggested that DYRK1A or the closely related kinases, DYRK1B, DYRK2 or CLKs 1 and 4, are the relevant targets of harmine and INDY. Harmine and INDY also activated proliferation in alpha and ductal cells, with no detectable proliferation in delta or PP cells (Supplementary Fig. 3).
Figure 2. Structure-activity relationship (SAR) analysis of “harmalogs” on beta cell proliferation.
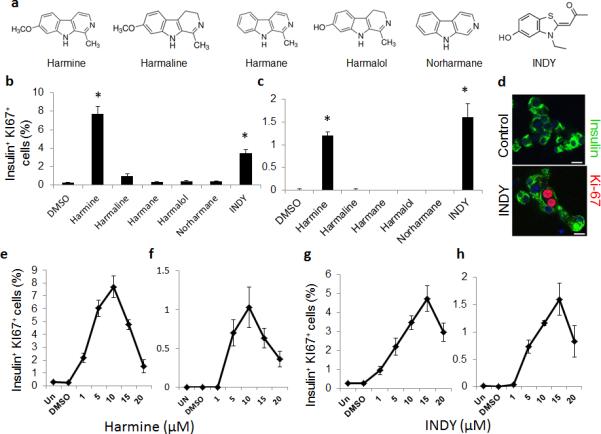
(a) Chemical structures of structural or functional harmine analogues (“harmalogs”). (b) and (c) Quantification of Ki67 labeling in rat (b) and human (c) beta cells in response to harmalogs. (d) An example of a Ki67+ “doublet” induced by INDY. The scale bar indicates 10 μm. (e−h) Dose-response curves for harmine and INDY in rat (e & g) and human (f & h) beta cell Ki67 labeling. In all relevant panels, error bars indicate s.e.m.; * indicates P<0.05 as determined by Wilcoxon Rank test. A minimum of 1000 beta cells was counted from four rat or four human donors for each graph. The scale bar indicates 10 μm.
TUNEL and p-γ-H2AX labeling revealed no evidence of beta cell death or DNA damage in response to harmine or INDY (Supplementary Fig. 2). Islet INS mRNA expression was increased; islet insulin content and glucose-stimulated insulin secretion were normal; of note, the transcription factors PDX1, NKX6.1, MAFA were increased at the mRNA, protein and beta cell immunocytochemical levels (Supplementary Fig. 4).
To determine whether calcineurin-NFAT signaling26-27 might mediate the proliferative effects of harmalogs in beta cells, we blocked the NFAT-calcineurin interaction with the NFAT inhibitor, VIVIT, and inhibited calcineurin activity with FK-506. Both inhibitors attenuated Ki67 labeling in rat and human beta cells (Fig. 3; Supplementary Fig. 5), suggesting that harmine and INDY proliferative effects may be mediated by calcineurin-NFAT signaling. We therefore surveyed NFAT family members in human beta cells. As predicted by rodent beta cells27-29 and human beta cell RNAseq30, NFATs are also detectable in the cytoplasm in quiescent human beta cells (Supplementary Fig. 6). Both harmine and INDY induce nuclear translocation of all four endogenous NFATs (Fig. 3, Supplementary Figs. 6). Concordantly, adenovirally expressed NFAT2 (gene name NFATC1) is readily apparent in the cytoplasm in vehicle-treated beta cells, but shifted to a predominantly nuclear location within harmine- and INDY-treated beta cells (Supplementary Fig. 7).
Figure 3. Calcineurin-NFAT-Dyrk1a signaling is implicated in harmine-Induced beta cell proliferation.
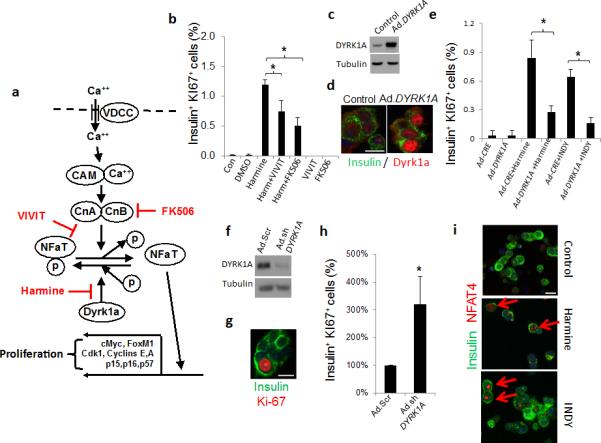
(a) A model depicting the calcium (Ca++)−calmodulin (CAM)−calcineurin (CnA and CnB subunits)−NFAT−c-MYC pathway to beta cell proliferation. The sites of action of harmine, VIVIT and FK506 are shown in red. (b) Beta cell proliferation in dispersed human islets in response to harmine, in the presence and absence of VIVIT or FK506 (n = four human preparations). (c) Adenoviral Dyrk1a overexpression in human islets, visualized by immunoblot. Representative of experiments from three islet preparations. (d) Ad.DYRK1A overexpression and immunolabeling for DYRK1A in human beta cells. Representative of experiments from three human islet preparations. (e) Effects of Ad.DYRK1A overexpression on harmine- and INDY-induced human beta cell proliferation. A minimum of 1000 beta cells was counted from four donors for each bar. (f) Effects of Ad.shDYRK1A transduction in human islets. Representative of four human islet preparations. “Ad.Scr” indicates an adenovirus expressing scrambled shRNA. (g) An example of Ki67 immunolabeling in human beta cells transduced with Ad.shDYRK1A. (h) Quantification of Ki67 in human beta cells transduced with Ad.shDYRK1A. A minimum of 1000 beta cells was counted from five donors for each bar. (i) Effects of harmine (10 μM) or INDY (15 μM) treatment on NFAT4 translocation to the nucleus of human beta cells. Examples are shown in red arrows. Translocation of other NFATs is shown in Supplementary Figure 7. In all relevant panels, the scale bar indicates 10 μm, error bars indicate s.e.m., and * indicates P<0.05 as determined by Wilcoxon Rank test.
To further explore whether DYRK1A is a relevant regulator of proliferation in human beta cells, we adenovirally overexpressed DYRK1A in human islets. Notably, DYRK1A was detectable in normal quiescent human islets and could be effectively overexpressed, appearing in the nuclear compartment (Fig. 3); its overexpression competitively attenuated harmine- and INDY-induced human beta cell proliferation. Conversely, reducing endogenous DYRK1A using an adenoviral shRNA directed against human DYRK1A led to a three-fold increase in human beta proliferation (Fig 3).
Harmine and INDY occupy the ATP pocket in DYRK1A, forming two hydrogen bonds with the side chain of Lys188 and the backbone of Leu241, with hydrogen bond acceptor groups in the correct orientation to form these interactions (Supplementary Fig. 7)20-24. Conversely, harmalogs that lack DYRK1A inhibitory activity and beta cell mitogenic activity (Fig. 2) are expected to fail to bind within the ATP binding pocket of DYRK1A because they lack one of the hydrogen bond acceptor groups (harmane, norharmane), or because the hydrogen bond acceptor groups are not correctly positioned (harmaline, harmalol) due to their non-planar conformation.
Since the HTS was based on MYC activation in HepG2 cells, we next asked whether harmine-induced proliferation in human islets required c-MYC activity. Harmine induced a reproducible but mild (two-fold) increase in c-MYC protein in human islets, and the c-MYC inhibitor, 1RH31, inhibited harmine-induced proliferation (Supplementary Fig. 8a−d). Further, adenoviral-Cre-mediated excision of Myc from islets of MycloxP/loxP mice32 attenuated harmine-induced proliferation (Supplementary Fig. 8e−h). Thus, harmine-induced proliferation depends in part on c-MYC activation. Next, searching for additional downstream cell cycle mediators of harmalog action, we performed an mRNA screen of G1/S cell cycle control molecules, confirmed by immunoblot and immunocytochemistry, that revealed increases in relevant cyclins and cdks (eg., cyclins A, E, CDK1, FOXM1, and CDC25A, E2Fs1 and 2) and reductions in cell cycle inhibitors (notably p15INK4, p16INK4 and p57CIP2) in response to harmine and INDY, (Supplementary Fig. 9).
Finally, we used three different in vivo models to assess the ability of harmine to activate proliferation, augment beta cell mass and enhance glycemic control. In the first, a partial pancreatectomy (PPX) model, harmine treatment induced Ki67 labeling in mouse beta cells in both sham-operated mice and in mice subjected to PPX, with most robust proliferation in the beta cells of PPX mice (Fig. 4a−c). In the PPX model, regeneration of beta cell mass was substantially more rapid in the harmine-treated mice than in the controls, reaching near-normal values in only 14 days. In the second model, a euglycemic NOD-SCID mouse model, BrdU and Ki67 labeling increased 2-3-fold in human beta cells transplanted into the renal capsule of harmine-treated vs. control euglycemic mice, without evidence of beta cell death (Fig. 4d−h). In the third model, a marginal mass human islet transplant model in streptozotocin-diabetic NODSCID mice, harmine treatment also resulted in near normal glycemic control, as assessed both by postprandial and fasting glucose values as well as by intraperitoneal glucose tolerance challenge (Fig. 4i−j).
Figure 4. Effects of harmine in three in vivo models of beta cell replication and regeneration.
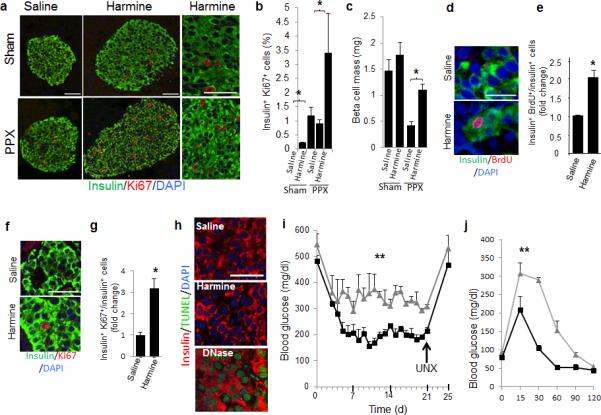
(a−c) The PPX model: (a) Examples of Ki67 immunolabeling of beta cells in mice receiving a 60% partial pancreatectomy (PPX) or a sham operation, and treated either with vehicle or harmine for seven days. (b) Quantification of Ki67 labeling of beta cells in the four treatment groups, with four mice in each group. * indicates P<0.05 as determined by Wilcoxon Rank test. (c) Beta cell mass in the four groups following two weeks of harmine or vehicle, with four mice in each group. * indicates P<0.05 as determined by Wilcoxon Rank test. (d−h) The euglycemic model: (d) examples of BrdU labeling in human beta cells following seven days of harmine or vehicle (saline) treatment of NOD-SCID mice transplanted with human islets (n=three different donors) into the renal capsule. (e) Quantification of BrdU incorporation into transplanted human beta cells in the NOD-SCID mice in (d). * indicates P<0.05 as determined by Student's unpaired t-test. (f) An example of Ki67 immunolabeling of human beta cells in the same experiment as in d. (g) Quantification of Ki67 immunolabeling in human beta cells in d. (n=4). * indicates P<0.05 as determined by Student's unpaired t-test. (h) An example of TUNEL labeling of the three human islet grafts in d; DNAse treatment is a positive control; n=4. (i) The diabetic model: streptozotocin diabetic NOD-SCID mice transplanted with human islets, treated for 14 days with saline (grey lines, n=3) or harmine (black lines, n=4). Grafts were harvested at day 21 by unilateral nephrectomy (UNX). ** indicates P<0.01 for area under the curve as determined using unpaired Student's two-tailed t-text. (j) Intrapertioneal glucose tolerance test on Day 20 in the same mice as in i. ** as in i. In all relevant panels, bars show mean ± s.e.m. The scale bars indicate 50 μm.
Harmine and INDY induce human beta cells to enter cell cycle, both in vitro and in vivo, with beta cell labeling indices that are in the range that occur physiologically in humans during in the first year of life, and a range that may be relevant to therapeutic beta cell expansion1-4. Harmine not only induces markers of proliferation in rat, mouse and human beta cells in vitro, it also increases beta cell mass and regeneration in a mouse PPX model, and enhances glycemic control and beta cell proliferation in vivo in two additional standard human islet transplant models, one euglycemic and one diabetic. Further, and unexpectedly, harmine induces production of the important beta cell transcription factors, NKX6.1, PDX1, and MAFA. It appears that harmalogs may act through the calcium-mediated, calcineurin-driven pathway, activating key cell cycle molecules such as c-MYC, CDK1, cyclins A and D, FOXM1, E2Fs, and CDC25A, as well as repressing important cell cycle inhibitors such as p15INK4, p16INK4 and p57CIP (Fig. 3a). In parallel, calcineurin-NFAT signaling has also been shown to be essential in mouse genetic models of beta cell growth and differentiation26-28.
That DYRK1A inhibition can lead to beta cell expansion is surprising. Another group has reported that Dyrk1a haploinsufficiency in mice results in attenuated beta cell mass and glucose intolerance33,34; conversely transgenic Dyrk1a overexpression led to accentuated beta cell size, mass and proliferation as well as enhanced glucose tolerance35. Potential explanations for this paradox may include differences in species, age, timing of expression (developmental phenotypes in mice vs. adult treatment), duration of treatment (days in human islets, vs. permanently and constitutively in mouse genetic models), CNS targeting (the murine constructs affect all tissues), possible dominant-positive effects of heterozygous loss, and/or differences between drug-induced conformational changes in a given protein vs. ablation or overexpression of the same protein. Perhaps most importantly, Dyrk1a is an unusually complex gene, with up to 17 exons (nomenclature for Dyrk1a exons varies), and at least nine different splice variants, only one exon of which was eliminated in the gene disruption studies33,34. Unraveling the products, cell type-specific isoforms and effects of harmalogs on these various isoforms will require further study.
These studies support a role for DYRK1A as the relevant target of the harmalogs, and likely exclude closely related kinases such as DYRK1B, DYRK2, DYRK3, DYRK4, and CLKs1-4. For example, DYRKs 3 and 4, and CLKs 2 and 3 are unlikely targets, since harmalogs are weak inhibitors of these, leaving DYRK1A, DYRK1B, DYRK2, CLK1 and CLK4 as the remaining likely candidates. These cannot easily be distinguished on pharmacologic grounds, since available inhibitors largely overlap. However, the complimentary observations that overexpression DYRK1A blocks, and silencing of DYRK1A mimics, the mitogenic effects of harmine and INDY on human beta cells make DYRK1A the prime candidate for a harmalog target. Nonetheless, further specificity studies are warranted.
Since DYRK and CLK family members are widely distributed, existing harmalogs likely will have off-target effects20-24. For example, harmine derived from plant extracts is a CNS stimulant36, and activates proliferation in some non-beta islet cell types (Supplementary Fig. 4). Also, harmine is a PPAR-γ activator and mediator of adipogenesis in mice, and leads to enhanced glucose sensitivity and disposal in obese, diabetic db/db mice37. Beta cell proliferation, mass and function were not examined in that study. Thus, as is the case for all potential beta cell therapeutics25,37-40, there is an urgent need to develop strategies to target harmalogs specifically to the beta cell.
In addition, understanding and optimizing the duration and dosing of future potential harmalogs will be important: excessive (~50-150x) Myc expression in mice has adverse consequences, leading to beta cell transformation, death and diabetes12-17. Cell death serves as an evolutionary fail-safe mechanism that prevents accidental unrestrained production of c-MYC and tumor growth in all tissues9-14. The observations that harmine causes only modest, two-fold, increases in c-MYC, that proliferation induced by harmine in human beta cells (~1-1.5%) is similar to that observed in neonatal life1-4, and that the duration of physiologic proliferation in humans is confined to the first year or few years of life1-4 provide room for optimism. The absence of evidence of beta cell death, DNA damage or de-differentiation in response to harmalogs is also encouraging.
In summary, harmalogs are able to induce adult human beta cell cycle entry at rates that are in the physiologic and potentially therapeutic range. Further approaches to optimizing the potency of the harmalog backbone, of unequivocally defining its molecular target(s), and of developing methods to direct it specifically to the beta cell are important future challenges.
ONLINE METHODS
General experimental approaches
No samples, mice or data points were excluded from the reported analyses. For islet studies, equivalent aliquots of every islet batch were randomly assigned to culture wells or microscope slides. For mouse studies, mice were randomly selected to groups to receive harmine or vehicle control, r to receive partial pancreatectomy or sham pancreatectomy. Analyses were not performed in a blinded fashion.
Reagents
Reagents were as follows: INDY (4997, Tocris Biosciences), BrdU (RPN20, GE Healthcare), harmaline (51330, Sigma), harmane (103276, Sigma), harmalol (H125, Sigma), harmine (286044, Sigma, for in vitro studies), harmine hydrochloride (CAS 343-27-1, Santa Cruz, for in vivo studies), VIVIT (502306392, Fisher scientific), FK506 (tlrl-fk5, Invivogen), 1RH (10058, 475956, Calbiochem), etoposide (E1383, Sigma), recombinant human IL1-β (201-lb-005, R&D Systems), recombinant human TNF-α (210-TA-010, R&D Systems), WS6 (M60097-2s, Xcess Biosciences).
Cell lines
Four cell lines were initially assessed for the HTS: rat insulinoma cells (Ins1 823/13), mouse insulinoma cells (βTC3), human hepatoma cells (HepG2) and human colon cancer cells (HCT116). The human hepatoma cell line used for these studies, HepG2 (ATCC), was cultured in EMEM medium supplemented with 10% FCS, 1% penicillin-streptomycin. The adenovirus packaging cell line, HEK-293A (Life Technologies) was cultured in DMEM medium supplemented with 10% FCS, and 1% penicillin-streptomycin, and 1x MEM containing non-essential amino acids. Cell lines were not validated by genomic testing, and we not tested for mycoplasma.
Generation of stable cell lines
HepG2 cells, βTC3 cells, Ins1 823/13 cells and HCT116 cells were transduced in T25 flasks with plasmid DNA encoding the MYC promoter-driven luciferase reporter construct described above. After transfection for 48 hr, the cells were trypsinized and re-seeded using serial dilution into 96-well plates containing 1 μg ml−1 puromycin for selection. After three weeks, individual clones were picked and reconsolidated into 24 well plates. Clones were verified as containing the plasmid by luciferase activity.
Small molecule screening
A 2.5 kb human MYC promoter plasmid was obtained from Addgene41. The MYC promoter fragment was excised with SacI and HindIII and ligated into the PGL4.20 luciferase vector (Promega). This was stably introduced into HepG, βTC3, Ins1 823/13 and HCT116 cell lines. HepG2 cell clone 13 (Supplementary Fig. 1) was ultimately selected for the screen because of its stability and robust response (Z’ of 0.75)18,19,42 to the positive controls, IL1-β and TNF-α, in a preliminary screen. These cells were maintained in complete DMEM medium with 1μg ml-1 puromycin. As a positive control, a combination of IL-1β and TNFα was selected because of the NFkB sites in the MYC promoter.
Screening was carried out in 384-well format using two commercially-sourced libraries. The “FDA” library (2300 compounds, Microsource Discovery Systems) composed of compounds approved for use in humans or animals, was used in a pilot screen, and to evaluate the robustness of the assay format. The “L1” library (100,000 compounds; Chembridge) is comprised of a structurally diverse set of small molecules selected based on their adherence to Lipinski's rule of 542, an indication of drug-like properties based on small molecule structure. All library compounds are stored as 10 mM stocks (DMSO).
For screening, 384-well assay plates (Perkin Elmer ProxiPlate 6008230) were pre-filled with HepG2 cells (5000 cells/well in 10 μl) expressing the luciferase reporter construct. Twenty-four hours after cell addition, compounds from library plates were transferred by pin tool (V&P Scientific) into assay plates at a final concentration of 7.5 uM. As positive control, 1 μl IL-1β was added at a final assay concentration of 5 ng ml−1 (Multidrop Combi; Thermo Scientific). Following 24 hr of incubation, luciferase expression was evaluated by the addition of 5 ul luciferase substrate reagent (Neolite Perkin Elmer); luminescence was detected after 10 minutes using an EnVision plate reader (Perkin Elmer).
Assay plates were validated using Z-factor (0.5 cutoff)42 comparing positive control wells to blank wells. Data were normalized against plate controls using normalized percent activation (NPA)19. A subset of 86 positive compounds showing an NPA >15% was selected for secondary screens and IC50 determination in primary beta cells based on proliferation. Additionally, the entire data set was renormalized using Robust Z-score (a method developed for the analysis of RNAi screens)18 and compounds showing a score >3 median absolute deviation (MAD) above the background signal were used for ranked scoring in subsequent structure-activity relationship (SAR) analysis. (Compounds with a Robust Z-score <3 MAD were considered to be “negative”).
Human, rat and mouse islets
Human islets from 63 donors were obtained through the NIH-supported Integrated Islet Distribution Program (IIDP). Donors ranged in age from 18 to 69 y.o. (mean 43.5 y.o.); 28 were female, 35 were male. There was no relationship between age and the ability of harmine to activate proliferation within these age constraints. Mean BMI was 29.4 (range 15-44), and cold ischemia time was 635 min (range 121-1150). Purity ranged from 50– 95%. Rat islets were isolated from 8- to 10-week-old male Wistar rats (Charles River Laboratories, Wilmington, MA) as described previously44,45. Mouse islets were isolated from 8- to 10-week-old MycloxP/loxP mice32. Isolated rat and mouse islets were cultured in RPMI 1640 medium (Life Technologies) containing 10% FCS, 5.5 mM glucose, and 1% penicillin-streptomycin for 24 hr. Islets were dispersed by trypsinization. All animal studies were performed in compliance with, and with approval of, the Icahn School of Medicine at Mount Sinai Institutional Animal Care and Use Committee.
Islet dispersion
Islets were dispersed as described46-49, centrifuged at 1500 rpm for 10 min, washed twice in PBS, re-suspended in 1 ml of 1 mg ml−1 trypsin, and incubated for 10 min at 37°C. During this digestion, the islets were dispersed by gentle pipetting up and down every 5 min for 10 s. Complete RPMI medium containing 5.5 mM glucose, 1% penicillin/streptomycin with 10% FBS was then added to stop the digestion. The cells were then centrifuged for 5 min at 1500 rpm, the supernatants removed, the pellet re-suspended in complete medium, and cells then plated on coverslips with 50 μl cell suspension per coverslip. For rat islets, Poly-DLysine/Laminin-treated cover slides were used. Cells were then allowed to attach for 2 hr at 37°C or were transduced with adenovirus for 2 hr. For MycLoxP/LoxP mouse islet studies, islets were isolated and dispersed on Poly-D-Lysine/Laminin-treated cover slips and transduced with Ad.Lacz or Ad.Cre. After 2 hr, 500 μl complete RPMI was added in each well to terminate the adenoviral transduction. Cells were cultured for 48–96 hr as described in the Figure legends.
Compound treatments
For compound treatment, after dispersed islets were allowed to recover from dispersal on coverslip 24 hr, complete medium was replaced with the medium containing compounds for 3-96 hr. Specifically, for Ki67 staining and BrdU staining, the cells were treated with compounds for 72 hr, and BrdU labeling was for 72 hr. For phospho-histone-3 staining, the cells were treated with compounds for 96 hr. For Ad.NFATC1-GFP nuclear translocation, the cells were transduced with adenovirus for 48 hours, after which cells were treated with harmine or INDY for 3 hr. For inhibitor experiments with VIVIT or FK506, cells were pretreated with inhibitor for 2 hr before addition of harmine or INDY.
Immunocytochemistry
Islet cells on coverslips were fixed in fresh 4% paraformaldehyde for 15 min at 25°C, washed with PBS and incubated in blocking buffer (1.0% BSA, 0.5% Triton, and 5% normal goat serum in PBS) for 1 hr at 25°C. Cells on coverslips were incubated with primary antisera overnight at 4°C in blocking buffer. Secondary antisera were added for 1 h at 25°C in secondary buffer (1% BSA, 0.5% Triton in PBS, 5% NGS). For phosphohistone-3 and phospho-γ-H2AX, primary antibody was exposed 2 h at 25°C. TUNEL labeling was performed according to Promega instructions (G3250, Promega). BrdU immunocytochemistry was performed using 1N HCL antigen-retrieval for 30 min at 37°C after fixation. Primary antisera were as follows: BrdU (ab6326, Abcam), Ki67 (RM-9106-s1, Thermo Scientific), p-Histone-3 (06-570, Millipore), insulin (A0564, DAKO), c-MYC (9402, Cell Signaling), NFAT1 (ab2722, Abcam), NFAT2 (556602, BD Pharmingen), NFAT3 (Sc-13036, Santa Cruz), NFAT4 (Sc-8321, Santa Cruz), p-γH2AX (MA1-2022 Thermo Scientific), DYRK1A (D-1694, Sigma), FoxM1 (Sc-500 Santa Cruz), p57 (2557s, Cell Signaling), E2F2 (Sc-632, Santa Cruz), Nkx6.1 (F55A10-c, University of Iowa), Pdx1 (07-696, Millipore), MafA (Ab26405, Abcam), glucagon (2760s Cell Signaling), somatostatin (Sc-20999, Santa Cruz), pancreatic polypeptide (A0619, DAKO), CK19 (Ab52625, Abcam). Labeled cells were then visualized using laser confocal microscopy (Leica SP5 DM). Results shown are representative of three to six separate human or rat islet preparations. TUNEL labeling was preformed using the DeadEnd Fluormetric TUNEL System (Cat#G3250, Promega)
Immunoblotting
Islet extracts (20–50 μg) were resolved using 10 or 12% SDS-PAGE, and transferred to Immobilon-P membranes (Millipore). Primary antisera included c-MYC (9402, Cell Signaling 1:2000), α-tubulin (CP06, Calbiochem 1:1000), DYRK1A (D-1694, Sigma 1:000), Cdk1 (Sc-747, Santa Cruz), cyclin A (Sc-239,Santa Cruz), cyclinE (Sc-481, Santa Cruz), FoxM1(Sc-500 Santa Cruz), p16INK4a (Sc-468, Santa Cruz), p57CIP2 (2557s, Cell Signaling), E2F1 (Sc-193, Santa Cruz), E2F2 (Sc-632, Santa Cruz), Nkx6.1 (F55A10-c, University of Iowa), Pdx-1 (07-696, Millipore), MafA (Ab26405, Abcam), GAPDH (Sc-25778,Santa Cruz). Quantitative densitometry was performed using Image J software (NIH).
Gene expression
RNA was isolated and quantitative RT-PCR was performed as described previously46. Gene expression in dispersed islets or cell lines was analyzed by real-time PCR performed on an ABI 7500 System. Primer sequences are in Supplementary Table 1.
Plasmids and adenoviruses
Ad.GFP, Ad.NFATC1 were provided by Dr. Djamel Lebeche (Icahn School of Medicine at Mount Sinai, N.Y., N.Y.). Ad.DYRK1A was made using the pAd/CMV/V5-DEST Gateway recombination system (Life Technologies) after cloning the human full-length DYRK1A into the pENTR vector. An Ad.shRNA directed against human DYRK1A was prepared using the Block-It RNAi kit (Life Technologies) targeting GGAACTTAAAGAAGACCAAAG using the U6 promoter). Adenoviruses were packaged and produced in 293A cells. Titers were determined by plaque assay (pfu). Dispersed rat or human islets on coverslips were transduced with either experimental or control adenoviruses (Ad.Cre, Ad.Scrambled, or Ad.LacZ) at 200 moi in serum-free medium for 2 hr. Transduction was stopped by adding complete medium containing 10% FCS and cultured for 48 to 96 h as described in the Figures.
Glucose-stimulated insulin secretion and insulin content
Insulin release was measured in triplicate from human islets treated either with vehicle (DMSO), harmine or INDY treated for 72 hr46-49. Briefly, islets were preincubated in Krebs-Ringer bicarbonate buffer supplemented with 10 mmol/l HEPES, 1% BSA, and 2.8 mmol/l glucose for 1 h at 37°C in a 5% CO2 incubator, then treated with harmine or INDY for 24 hr. After washing once with the same solution, groups of 15 islet equivalents (IEQs) per condition were incubated in 1 ml fresh Krebs-Ringer bicarbonate buffer plus 1% BSA and either 2.8 or 16.7 mmol/l glucose for 30 min. Buffer was removed and frozen at −20°C for insulin measurement by insulin ELISA kit (EZHI-14K, Millipore). Islets were then digested overnight in NaOH at 37°C, and protein was measured by the Bradford method after neutralization with HCl. Insulin values are normalized to protein content.
Mouse partial pancreatectomy model (PPX)
This model has been described in detail50,51. Briefly, two- to three-month old male C57B6 mice were randomized into two groups (PPX or sham PPX), and a 60% PPX (splenic portion) (n=16) or sham PPX (laparotomy only) (n=16) was performed. Following PPX, mice were allowed to recover for 24 hours, and then further randomized to receive vehicle (saline) or 10 mg kg−1 harmine HCl by intraperitoneal injection daily for 7 or 14 days. They were sacrificed on day 7 for Ki67 studies (n=8) or on day 14 (n=8) for beta cell mass determination. Beta cell mass and islet number were measured in four insulin-stained pancreas sections per mouse using ImageJ software (National Institutes of Health, Bethesda, MD) (12,13). Sections were also stained for Ki67 and insulin. A minimum of 2,000 beta cells/pancreas were counted. Investigators were blinded as to group assignments.
Euglycemic human islet transplantation model
1000 human islet equivalents (IEQ) from three different human cadaveric donors were transplanted under the renal capsule of six euglycemic three month old NOD-SCID mice as detailed previously46-48. Animals were allowed to recover for seven days, and were then randomly selected to be given 10 mg kg−1 harmine HCl intraperitoneally (n=3) or vehicle (saline) (n=3) every 12 hours for seven days as described by Waki37. On the evening of day 13, animals were given BrdU intraperitoneally, and on the AM of day 14, animals were sacrificed, kidneys harvested, fixed, embedded, sectioned and immunostained for insulin and BrdU as detailed previously47-49. BrdU incorporation in the controls was 0.12% ± 0.08%; beta cell Ki67 labeling in the controls was 0.22% ± 0.07%. Investigators were blinded as to group assignments.
Diabetic marginal mass human islet transplantation model
500 IEQ from four different donors were transplanted under the renal capsule of seven male NOD-SCID mice rendered diabetic by 200 mg/kg streptozotocin as detailed previously47-49. Mice were randomly selected for treatment with 10 mg kg−1 harmine HCl (n=4) or saline (n=3) as above. Blood glucose was at least 400 mg/dl prior to islet transplant and was measured daily following transplant. An intraperitoneal glucose tolerance test (2 mg/kg)46-48,50 was performed on day 21, and islet grafts were excised by unilateral nephrectomy (UNX) as described in detail47-49,51. Investigators were blinded as to group assignments.
Statistical analysis
All mouse and in vitro experiments were repeated multiple times in multiple batches of mouse, rat and human islets as indicated in the Figure legends, and were analyzed blindly at the conclusion of the experiment. Key analyses from one author were repeated in a blind manner by another author. All data are expressed as the mean ± SE. Results were accepted as statistically significant at P<0.05, as determined using two tailed Wilcoxon Rank Test or Students unpaired t-test as indicated in the figures and legends. Sample size was based on prior studies in which 3-7 sets of human or rat islets were used45-49,51. A minimum of 1000 beta cells were counted for each graph shown.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Drs. R. Vasavada, N. Fiaschi-Taesch, H. Chen, K. Takane, M. Ohlmeyer, R. DeVita, E. Schadt, C.Argmann, B. Losic, D. Lebeche, S. Kim, and B. Wagner for their many helpful discussions during this study. We thank the NIDDK-supported Integrated Islet Distribution Program (IIDP), Dr. T. Kin at the University of Alberta in Edmonton CA, and Dr. P. Witkowski at the University of Chicago for providing human islets. This work was supported by grants from the National Institutes of Health {R-01 DK55023 (A.F.S.), U-01 DK089538 (A.F.S.), R-01 DK065149 (D.K.S.), R-01 DK067351 (A.G.-O.) and R-01 DK077096 (A.G.-O.)}, the JDRF {17-2011-598 and 1-2011-603 (A.F.S.), and the American Diabetes Association (1-14-BS-059) (A.G.-O.)}.
Footnotes
AUTHOR CONTRIBUTIONS
P.W., J.C.A-P, D.P.F., H.L., S.S., A.B., A.K. R.S., D.K.S., A.G-O and A.F.S. designed and performed experiments. P.W., A.G-O. and A.F.S wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B. Beta cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes. 2000;49:1325–1333. doi: 10.2337/diabetes.49.8.1325. [DOI] [PubMed] [Google Scholar]
- 2.Meier JJ, et al. Beta cell replication is the primary mechanism subserving the postnatal expansion of beta cell mass in humans. Diabetes. 2008;57:1584–94. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohler CU, Olewinski M, Tannapfel A, Schmidt WE, Fritsch H, Meier JJ. Cell cycle control of beta cell replication in the prenatal and postnatal human pancreas. Am J Physiol Endocrinol Metab. 2011;300:E221–230. doi: 10.1152/ajpendo.00496.2010. [DOI] [PubMed] [Google Scholar]
- 4.Gregg BE, et al. Formation of a human beta cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab. 2012;97:3197–3206. doi: 10.1210/jc.2012-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta cell deficit and increased beta cell apoptosis in humans with diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 6.Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. Beta cell mass and turnover in humans: effects of obesity and aging. Diabetes Care. 2013;36:111–7. doi: 10.2337/dc12-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kulkarni RN, Bernal-Mizrachi E, Garcia-Ocaña A, Stewart AF. Human β-Cell Proliferation and Intracellular Signaling: Driving in the Dark Without a Roadmap. Diabetes. 2012;61:2205–2213. doi: 10.2337/db12-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernal-Mizrachi E, Kulkarni RN, Stewart AF, Garcia-Ocaña A. Human β-Cell Proliferation and Intracellular Signaling Part 2: Still Driving in the Dark without a Roadmap. Diabetes. 2014;63:819–31. doi: 10.2337/db13-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soucek L, Evan GI. The ups and downs of Myc biology. Curr Opin Genetic Dev. 2009;20:91–5. doi: 10.1016/j.gde.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wierstra I, Alves J. The c-myc Promoter: Still MysterY and Challenge. Adv Cancer Res. 2008;99:113–333. doi: 10.1016/S0065-230X(07)99004-1. [DOI] [PubMed] [Google Scholar]
- 11.Bretones G, Delgado MD, Leon J. Myc and cell cycle control. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbagrm.2014.03.013. http://doi.org/10.1016/j.bbagrm2014.03.013. [DOI] [PubMed]
- 12.Pelengaris S, Kahn M, Evan GI. Suppression of myc-induced apoptosis in beta cells exposes multiple oncogenic properties of myc and triggers carcinogenic progression. CELL. 2002;3:321–34. doi: 10.1016/s0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- 13.Pelengaris S, Khan M. Oncogenic co-operation in β-cell tumorigenesis. Endocr Relat Cancer. 2001;8:307–314. doi: 10.1677/erc.0.0080307. [DOI] [PubMed] [Google Scholar]
- 14.Finch A, et al. Bcl-X gain of function and p19ARF L loss of function cooperate oncogenically with Myc in vivo by distinct mechanisms. Cancer Cell. 2006;10:113–120. doi: 10.1016/j.ccr.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 15.Laybutt DR, et al. Overexpression of c-myc in beta cells of transgenic mice causes proliferation and apoptosis, downregulation of insulin gene expression and diabetes. Diabetes. 2002;51:1703–804. doi: 10.2337/diabetes.51.6.1793. [DOI] [PubMed] [Google Scholar]
- 16.Cano DA, et al. Regulated beta-cell regeneration in the adult mouse pancreas. Diabetes. 2008;57:958–66. doi: 10.2337/db07-0913. [DOI] [PubMed] [Google Scholar]
- 17.Karslioglu E, et al. cMyc is the principal upstream driver of beta cell proliferation in rat insulinoma cell lines and Is an effective mediator of human beta cell replication. Mol Endocrinol. 2011;25:1760–72. doi: 10.1210/me.2011-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chung N, et al. Median absolute deviation to improve hit selection for genome-scale RNAi screens. J Biomol Screen. 2008;13:149–158. doi: 10.1177/1087057107312035. [DOI] [PubMed] [Google Scholar]
- 19.Goktug AN, Chai SCC, Chen T. Data analysis approaches in high throughput screening. Drug Discovery (Chapter 7) 2013 doi:10.5772/52508. [Google Scholar]
- 20.Becker W, Sippl W. Activation, Regulation and Inhibition of Dyrk1a. FEBS J. 2010;278:246–56. doi: 10.1111/j.1742-4658.2010.07956.x. [DOI] [PubMed] [Google Scholar]
- 21.Ogawa Y, et al. Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1a. Nat Comm. 2010;1:86. doi: 10.1038/ncomms1090. doi:10.1038/ncomms1090. [DOI] [PubMed] [Google Scholar]
- 22.Tahtouh T, et al. Selectivity, co-crystal structures and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J Med Chem. 2012;55:9312–30. doi: 10.1021/jm301034u. [DOI] [PubMed] [Google Scholar]
- 23.Waite A, et al. Mechanism of dual specificity kinase activity of Dyrk1a. FEBS J. 2013;280:4495–4511. doi: 10.1111/febs.12411. [DOI] [PubMed] [Google Scholar]
- 24.Jain P, Karthikeyan C, Moorthy NS, Walker DK, Jain AK, Trivedu P. Human cdc2-like kinase 1 (CLK1): a novel target for Alzheimer’s disease. Curr Drug Targets. 2014;15:539–50. doi: 10.2174/1389450115666140226112321. [DOI] [PubMed] [Google Scholar]
- 25.Shen W, et al. Small molecule inducer of beta cell proliferation identified by high-throughput screening. J Am Chem Soc. 2013;135:1669–72. doi: 10.1021/ja309304m. [DOI] [PubMed] [Google Scholar]
- 26.Gallo EM, Cante-Barrett K, Crabtree GR. Lymphocyte calcium signaling from membrane to nucleus. Nat Immunol. 2006;7:25–32. doi: 10.1038/ni1295. [DOI] [PubMed] [Google Scholar]
- 27.Heit JJ, et al. Calcineurin/NFAT signaling regulates pancreatic β-cell growth and function. Nature. 2006;443:345–349. doi: 10.1038/nature05097. [DOI] [PubMed] [Google Scholar]
- 28.Goodyer WR, Gu X, Liu Y, Bottino R, Crabtree GR, Kim SK. Neonatal beta cell development in mice and humans is regulated by calcineurin/NFaT. Dev Cell. 2012;23:21–34. doi: 10.1016/j.devcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demozay D, Tsunekawa S, Briaud I, Shah R, Rhodes CJ. Specific glucose-induced control of insulin receptor-supstrate-2 expression is mediated by Ca2+-dependent calcineurin-NFAT signaling in primary pancreatic islet ß-cells. Diabetes. 2011;60:2892–2902. doi: 10.2337/db11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nica AC, et al. Cell-type, allelic and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res. 2013;23:1554–62. doi: 10.1101/gr.150706.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Hammoudeh DI, Follis AV, Reese BE, Lazo JS, Prochownik EV. Improved low molecular weight Myc-Max inhibitors. Mol Cancer Ther. 2007;6:2399–2408. doi: 10.1158/1535-7163.MCT-07-0005. [DOI] [PubMed] [Google Scholar]
- 32.de Alboran IM, et al. Analysis of cMYC function in normal cells via conditional gene-targeted mutation. Immunity. 2001;14:45–55. doi: 10.1016/s1074-7613(01)00088-7. [DOI] [PubMed] [Google Scholar]
- 33.Fotaki V, et al. Dyrk1a haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol Cell Biol. 2002;22:6636–47. doi: 10.1128/MCB.22.18.6636-6647.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rachdi L, et al. Dyrk1a haploinsufficiency induces diabetes in mice through decreased pancreatic beta cell mass. Diabetologia. 2014;57:960–969. doi: 10.1007/s00125-014-3174-3. [DOI] [PubMed] [Google Scholar]
- 35.Rachdi L, et al. Dyrk1a induces pancreatic beta cell mass expansion and improves glucose tolerance. Cell Cycle. 2014;13:2221–9. doi: 10.4161/cc.29250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brierley DI, Davidson C. Developments in harmine pharmacology – implications for ayahuasca use and drug-dependence treatment. Prog Neuro-Psychopharmacol Biol Psychiatry. 2012;39:263–72. doi: 10.1016/j.pnpbp.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 37.Waki H, et al. The small molecule harmine is an antidiabetic cell-type specific regulator of PPARγ expression. Cell Metab. 2007;5:357–70. doi: 10.1016/j.cmet.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 38.Purwana I, et al. GABA promotes human beta-cell proliferation and modulates glucose homeostasis. Diabetes. 2014;63:4197–4205. doi: 10.2337/db14-0153. [DOI] [PubMed] [Google Scholar]
- 39.Wang W, et al. Identification of small molecule inducers of pancreatic beta cell proliferation. Proc Nat Acad Sci. 2009;106:1427–32. doi: 10.1073/pnas.0811848106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chamberlain CE, et al. Menin determines K-Ras proliferative outputs in endocrine cells. J Clin Invest. 2014;124:4093–4101. doi: 10.1172/JCI69004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He TC, et al. Identification of cMyc as a target of the APC pathway. Science. 1998;281:1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, Chung T, Oldenburg K. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 43.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 44.Ricordi C, Rastellini C. Methods in pancreatic islet separation. In: Ricordi C, editor. Methods in Cell Transplantation. R.G. Landes Co; Austin, TX: 2000. pp. 433–438. [Google Scholar]
- 45.Cozar-Castellano I, et al. Lessons from the first comprehensive molecular characterization of cell cycle control in rodent insulinoma cell lines. Diabetes. 2008;57:3056–68. doi: 10.2337/db08-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Metukuri MR, et al. ChREBP mediates glucose-stimulated pancreatic beta cell proliferation. Diabetes. 2012;61:2004–15. doi: 10.2337/db11-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fiaschi-Taesch NM, et al. Hepatocyte growth factor (HGF) enhances engraftment and function of non-human primate islets. Diabetes. 2008;57:2745–54. doi: 10.2337/db08-1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fiaschi-Taesch NM, et al. A survey of the human pancreatic beta cell G1/S proteome reveals a potential therapeutic role for cdk-6 and cyclin D1 in enhancing human beta cell replication and function in vivo. Diabetes. 2009;58:882–93. doi: 10.2337/db08-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fiaschi-Taesch NM, et al. Induction of human beta cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes. 2010;59:1926–36. doi: 10.2337/db09-1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peshavaria M, et al. Regulation of pancreatic beta cell regeneration in the normoglycemic 60% pancreatectomy mouse. Diabetes. 2006;55:3289–98. doi: 10.2337/db06-0017. [DOI] [PubMed] [Google Scholar]
- 51.Alvarez-Perez JC, et al. Hepatocyte growth factor/c-Met signaling is required for β-cell regeneration. Diabetes. 2014;63:216–223. doi: 10.2337/db13-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.