

Structural plasticity plays a major role in determining differential binding of CXCL8 monomer and dimer to glycosaminoglycans (GAGs) and that dimer is the high-affinity GAG ligand. We propose that these properties play important roles in orchestrating in vivo chemokine-mediated neutrophil function.
Keywords: chemokine, glycosaminoglycan (GAG), heparin, nuclear magnetic resonance (NMR), structural plasticity, structure–function study
Abstract
Chemokine CXCL8/interleukin-8 (IL-8) plays a crucial role in directing neutrophils and oligodendrocytes to combat infection/injury and tumour cells in metastasis development. CXCL8 exists as monomers and dimers and interaction of both forms with glycosaminoglycans (GAGs) mediate these diverse cellular processes. However, very little is known regarding the structural basis underlying CXCL8–GAG interactions. There are conflicting reports on the affinities, geometry and whether the monomer or dimer is the high-affinity GAG ligand. To resolve these issues, we characterized the binding of a series of heparin-derived oligosaccharides [heparin disaccharide (dp2), heparin tetrasaccharide (dp4), heparin octasaccharide (dp8) and heparin 14-mer (dp14)] to the wild-type (WT) dimer and a designed monomer using solution NMR spectroscopy. The pattern and extent of binding-induced chemical shift perturbation (CSP) varied between dimer and monomer and between longer and shorter oligosaccharides. NMR-based structural models show that different interaction modes coexist and that the nature of interactions varied between monomer and dimer and oligosaccharide length. MD simulations indicate that the binding interface is structurally plastic and provided residue-specific details of the dynamic nature of the binding interface. Binding studies carried out under conditions at which WT CXCL8 exists as monomers and dimers provide unambiguous evidence that the dimer is the high-affinity GAG ligand. Together, our data indicate that a set of core residues function as the major recognition/binding site, a set of peripheral residues define the various binding geometries and that the structural plasticity of the binding interface allows multiplicity of binding interactions. We conclude that structural plasticity most probably regulates in vivo CXCL8 monomer/dimer–GAG interactions and function.
INTRODUCTION
Many classes of proteins, including chemokines, cytokines and growth factors, share the property of existing as monomers and dimers and binding glycosaminoglycans (GAGs) [1,2]. Chemokine CXCL8 [also known as interleukin-8 (IL-8)] is a well-characterized GAG-binding protein that orchestrates trafficking of various cell types such as neutrophils and oligodendrocytes during tissue injury and bacterial infection and cancer cells in metastasis [3,4]. GAG interactions have been proposed to regulate the steepness and duration of concentration gradients, which in turn regulate cellular trafficking [5,6]. CXCL8 dimerizes at micromolar concentrations and structures of the wild-type (WT) dimer and of a trapped monomer are known [7–9]. During active recruitment, local CXCL8 concentration can vary by orders of magnitude and so, in principle, could exist as monomers and/or dimers as a function of space and time. Animal model studies using trapped monomer and dimer have shown that both CXCL8 monomers and CXCL8 dimers are active in vivo and that monomer–dimer equilibrium and GAG interactions regulate neutrophil recruitment [10,11].
GAGs, such as heparan sulfate (HS), are linear sulfated polysaccharides that are ubiquitously expressed and exist as part of cell-surface and extracellular matrix proteoglycans [12–14]. HS has a modular structure with sulfated sequences (NS domain) separated by regions containing acetylated sequences (NA domain). CXCL8 and most other chemokines preferentially bind the NS domain. Heparin is commonly used for structure–function studies, as it is uniformly sulfated and size-fractionated oligosaccharides of various sizes are commercially available. CXCL8 mutants of basic residues identified as important for binding to heparin also show altered in vivo recruitment [11], indicating that heparin can function as an excellent surrogate for studying the structural basis of CXCL8–GAG interactions.
At this time, very little is known regarding the structural basis by which CXCL8 monomers and dimers bind GAG. Furthermore, there are conflicting reports in the literature regarding the binding affinities, geometry of GAG orientation and whether monomer or dimer is the high-affinity GAG ligand [15–24]. We have now characterized the binding of a series of heparin oligosaccharides to a designed monomer and WT dimer using solution NMR spectroscopy and molecular docking procedures. Our data show that basic residues mediate binding and that GAGs, independent of their length, adopt multiple geometries both in the monomer and in the dimer and that the dimer is the high-affinity GAG ligand. MD simulations on the different models in explicit solvent have provided insights into the residue-level dynamics of the binding interface and better realization of how long-range electrostatics mediates this interplay. We propose that the structural plasticity of GAG-binding residues dictates all aspects of the binding and that the roles of the individual residues are not equal: a set of core residues function as the major recognition/docking site and a second set of residues in the periphery of the core residues define the binding geometries. We further propose that the structural plasticity of the binding interface plays an important role in regulating in vivo chemokine function for homing diverse cells to their destination.
MATERIAL AND METHODS
Cloning, expression and purification of CXCL8 variants
CXCL8 WT and V27P/E29P monomer mutant (hereafter referred to as CXCL8 monomer) were expressed and purified, as described previously [25,26]. 15N- and 15N/13C-labelled proteins were prepared by growing cells in minimal medium containing 15NH4Cl and 13C glucose as the sole nitrogen and carbon sources respectively. Transformed Escherichia coli BL21 (DE3) cells were grown to an A600 of ∼0.6 and induced with 1 mM IPTG overnight at 22°C. The purity and molecular mass of the proteins were confirmed using MALDI–MS.
NMR spectroscopy
15N-labelled proteins were prepared in 50 mM sodium phosphate buffer (pH 5.5–7.5 for different experiments) containing 1 mM DSS (2,2-dimethyl-2-silapentanesulfonic acid), 1 mM sodium azide and 10% (v/v) 2H2O. NMR spectra were acquired at 30°C on a Bruker Avance III 800 MHz (equipped with a TXI cryoprobe) or 600 MHz (with QCI probe) spectrometer. Chemical shifts of the dp8 (heparin octasaccharide)-bound CXCL8 monomer were assigned using standard pulse sequences [27]. Spectra were processed with NMRPipe [28] and analysed using NMRView [29] or Bruker Topspin 3.2 software. Chemical shift indexes were calculated using the program CSI 2.0 [30].
Protein concentrations between 30 and 150 μM were used for different titration experiments. The heparin-derived oligosaccharides were purchased from Iduron or Neoparin Inc. A stock solution of heparin oligosaccharides (5–10 mM) prepared in the same buffer was added to the protein sample and a series of 1H-15N HSQC spectra were collected until essentially no changes in chemical shifts were observed. The final protein/oligosaccharide molar ratios for the CXCL8 dimer were 1:29 for dp2 (heparin disaccharide), 1:10 for dp4 (heparin tetrasaccharide), 1:6 for dp8 (heparin octasaccharide) and 1:4 for dp14 (heparin 14-mer); and for CXCL8 monomer were 1:50 for dp2, 1:22 for dp4, 1:7 for dp8 and 1:4 for dp14.
The chemical shift perturbations (CSPs; Δδobs) were calculated as a weighted average chemical shift change of 1H (ΔδH) and 15N(ΔδN),
Apparent dissociation constants (Kd) were determined by fitting binding-induced chemical shift changes for five to eight residues, as described previously [31].
Molecular docking using HADDOCK
Molecular docking of the heparin oligosaccharides to CXCL8 monomer and dimer was carried out using the High Ambiguity Driven biomolecular DOCKing (HADDOCK) approach [32,33]. For the docking simulations, the WT dimer (PDB id: 1IL8) and the monomer modelled from the L25NMe monomer (PDB id: 1IKM) were used [8,9,26]. Oligosaccharides dp2, dp4, dp8 and dp14 were generated from the NMR structure of heparin (PDBid: 1HPN) [34]. In the initial model, 1H2 ring conformation was introduced for the uronate ΔUA(2S) ring at the non-reducing end. The internal iduronates IdoA(2S) and the glucosamine GlcNS(6S) rings are in the preferred 2SO and 4C1 conformations respectively [35].
Active and passive residues were appropriately chosen based on the CSP data. The PARALLHDG force field was used for non-bonded interactions, a common force field used for NMR-based structure calculation [36]. Topology and parameter files for different oligosaccharides were generated using the PRODRG server [37].
Docking of dp2, dp4, dp8 and dp14 were performed for a 1:1 oligosaccharide:chemokine complex for the monomer and dimer. A total of 1000 structures were generated during the initial rigid body docking and the best 200 structures, based on intermolecular energies, were subjected to semi-flexible simulated annealing (SA). This step involves rigid body SA, semi-flexible SA with flexible side chain interface followed by fully flexible interface. As the last six residues (67–72) of the C-terminal helix are unstructured in the monomer, these residues were also allowed to be flexible in the monomer simulation [8]. This step was followed by explicit solvent refinement. The pair-wise ‘ligand interface RMSD matrix’ over all structures was calculated and the final structures were clustered using a RMSD cut-off value of 7.5 Å (1 Å=0.1 nm) for dp8 and dp14 and 2.0 Å for dp4 and dp2. The clusters were sorted using RMSD and HADDOCK score (weighted sum of energy terms).
MD simulations
Models of CXCL8–heparin oligosaccharide complexes from the major families obtained from HADDOCK runs were used as starting structures for unrestrained MD simulations using the NAMD 2.9 suite of programs [38] and CHARMM36 force field [39]. Modified versions of the distributed CHARMM36 carbohydrate topology and parameter files were used to incorporate GlcNS into the system by introducing a patch for 4C1 α-D-glucose [40–42]. Atomic charges for sulfamates were defined as reported by Huige and Altona [43] and scaled to be consistent with the CHARMM36 force field. The GAG segments from the HADDOCK models were read into SYBYL-X 2.1.1 (Tripos), the sulfate groups removed and the glucosamine residues converted into glucose. This structure was submitted to the Glycan Reader [44] of the CHARMM-GUI server (http://www.charmm-gui.org) to assign atom types, patched using the modified topology file via the psfgen plugin in VMD 1.9.2 [45] to generate the psf and pdb files for the heparin oligosaccharides. Neutral histidine side chains with Nε protonation were used. The system was then solvated in TIP3P water box allowing a 20 Å margin to the box boundary. Finally, Na+ counter ions were added to neutralize the system. The system was equilibrated in three stages using the constant volume NVT ensemble. Production runs were then performed using the constant pressure NPT ensemble at 300 K for ∼100 ns. The trajectories were analysed using VMD and the pyranose ring conformations were calculated using BFMP program [46], facilitated by in-house scripts. Energy calculations were performed using the NAMD energy plugin in VMD with Particle Mesh Ewald (PME) long-range electrostatics enabled. Buried surface area (BSA) calculations were performed using the ‘measure’ function in VMD. The MD runs were carried out on the shared Teal Linux cluster at VCU and the Lonestar Dell Linux cluster at the Texas Advanced Computing Center (TACC).
RESULTS
We used NMR CSP of backbone amides, HADDOCK-based docking and MD simulations to characterize the binding of heparin oligosaccharides to CXCL8 monomer and dimer. Backbone chemical shifts are exquisitely sensitive to binding and are routinely used to characterize protein–protein, protein–peptide and protein–oligonucleotide interactions, but have been challenging for GAG interactions due to aggregation, precipitation and poor quality NMR spectra. These problems are especially exacerbated for longer oligosaccharides, with some studies reporting saccharides longer than a disaccharide resulting in precipitation [47,48]. We faced similar issues, but overcame these limitations by using low protein concentrations (∼100 μM) and varying solution conditions such as pH [49]. The HSQC titration spectra of the dp8 binding to the dimer and monomer highlight the quality of the data (Figure 1).
Figure 1. NMR HSQC titrations of CXCL8 dimer and monomer binding to dp8 heparin oligosaccharide.

Sections of the 1H-15N HSQC spectra of CXCL8 dimer (A) and monomer (B) showing dp8 binding-induced chemical shift changes. Arrows indicate the direction of peak movement. The unbound peaks are in black and the final bound peaks are in red. Lys15 perturbations are shown as insets.
To characterize GAG binding to a dimer, we used WT CXCL8, which is essentially a dimer at the NMR concentrations (monomer/dimer equilibrium constant ∼12 μM) [50]. We will refer to the WT CXCL8 dimer simply as the CXCL8 dimer. We used the double proline V27P/E29P mutant for characterizing GAG binding to a monomer [26]. We previously characterized this mutant and showed that its structure is similar to that of a trapped monomer created by substituting a methyl group for the dimer-interface residue Leu25 amide proton [8]. The V27P/E29P monomer is as active as the WT monomer in cellular assays and in mouse models [26].
We characterized the binding of four oligosaccharides of increasing length from a disaccharide to a 14-mer (dp2, dp4, dp8 and dp14) by monitoring binding-induced CSP in CXCL8 dimer and monomer. A series of 1H-15N HSQC spectra were collected until essentially no chemical shift changes were detected. We observed only one set of peaks although some line broadening was observed for longer oligosaccharides indicating that binding occurs in the fast to intermediate exchange regime in the NMR time scale. The CSP profiles for the dimer and monomer are shown in Figures 2 and 3 respectively.
Figure 2. Histogram plots of CXCL8 dimer binding to heparin oligosaccharides.

CSP profiles of CXCL8 dimer binding to heparin dp14 (A), dp8 (B), dp4 (C) and dp2 (D). Basic residues lysine, arginine and histidine are shown as grey bars and buried residues (ASA < 40%) are shown in black. Residues showing CSP higher than the threshold (indicated by the dotted lines) are considered as involved in binding. Lys20 broadens out during the titration and is indicated by *.
Figure 3. Histogram plots of CXCL8 monomer binding to heparin oligosaccharides.

CSP profiles of CXCL8 monomer binding to heparin dp14 (A), dp8 (B), dp4 (C) and dp2 (D). Basic residues lysine, arginine and histidine are shown as grey bars and buried residues (ASA < 40%) are shown in black. Residues showing CSPs higher than the threshold (indicated by the dotted lines) are considered as involved in binding. Lys20 broadens out during the titration and is indicated by *. The CSP of His18 is truncated except for dp2 and actual CSPs are 0.39, 0.43 and 0.36 ppm for dp14, dp8 and dp4 respectively.
Our data show that His18, Lys20, Arg60, Lys64, Lys67 and Arg68 are perturbed both in the monomer and in the dimer and essentially on binding all oligosaccharides, whereas Lys15, Arg47, Lys23 and Lys54 showed selective perturbations. The structure shows that the first set of residues, labelled as core residues, is contiguous and forms a binding surface. The second set of residues, labelled as peripheral or secondary residues, are distributed around the core residues (Figures 4A and 4B).
Figure 4. Distribution of basic residues that mediate heparin interactions.
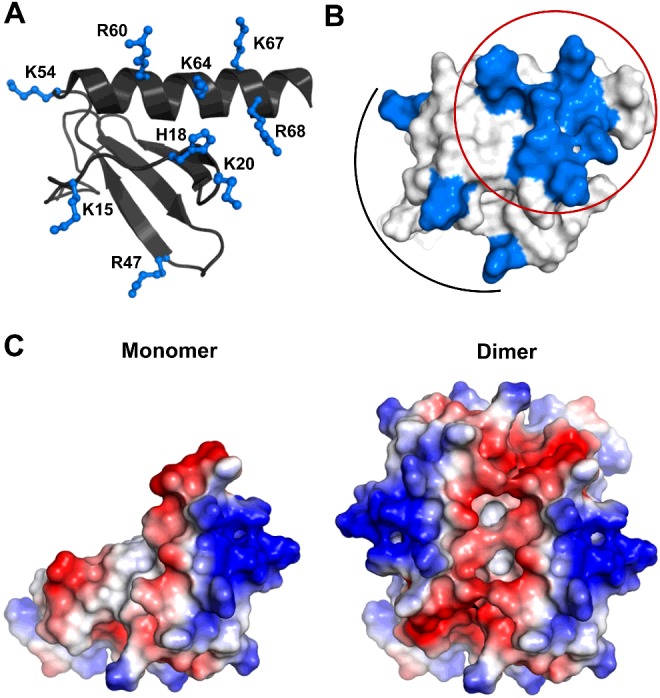
(A) A schematic of CXCL8 monomer showing the distribution of basic residues arginine, lysine and histidine in ball and stick. (B) Surface plot of CXCL8 monomer with the GAG-binding residues shown in blue. The core residues that encompass the C-helix and N-loop are circled and the peripheral residues are highlighted by an arc. (C) Electrostatic surface plots of CXCL8 monomer and dimer.
Binding of CXCL8 dimer to heparin oligosaccharides
Binding of dp8 and dp14
We first discuss the binding characteristics of the longer oligosaccharides dp8 and dp14, as their CSP were essentially identical (Figures 2A and 2B). Both solvent-exposed and buried residues showed chemical shift changes, indicating that both direct and indirect interactions mediate the binding process. Some of the largest perturbations were observed for basic residues located in the N-loop (Lys15, His18, Lys20 and Lys23) and C-helix (Arg60, Lys64 and Arg68). In addition, Arg47 in the β3-strand and Lys54 in the 50s-loop preceding the C-helix also showed perturbations that are not striking but well above the background.
Interestingly, most of the C-helix, which includes buried (Trp57, Val58, Val61 and Val62; Accessible Surface Area (ASA) < 20%) and negatively charged (Glu63 and Glu70) residues, showed perturbation, indicating that chemical shift changes in these residues are most probably due to indirect interactions such as rearrangement of the helix and not due to direct binding. On the other hand, Lys67 showed negligible perturbation, suggesting that CSP contributions from direct and indirect interactions are of opposite sign and similar magnitude and cancel out. The CSP for Lys20 could not be shown as its cross-peak broadens out early in the titration and never recovers unlike other N-loop residues, indicating binding-induced perturbation. Significant line broadening for Lys15 is surprising and unexpected considering it is relatively remote from the other GAG-binding residues and is also located in the middle of the receptor-binding site.
Binding of dp4 and dp2
The CSP for dp4, compared with dp8 and dp14, was lower, suggesting weaker interactions (Figure 2C). Nevertheless, perturbation of N-loop (Lys15, His18, Lys20 and Lys23) and C-helix (Arg60, Lys64 and Arg68 and Lys54) residues were significant compared with the background. Furthermore, similar to longer oligosaccharides, the whole C-helix showed significant perturbation indicating binding-induced rearrangement of the C-helix. Compared with dp8 and dp14, Arg47 alone was not perturbed. The CSP for dp2 was even lower when compared with dp4 and only two residues, His18 and Arg60, showed any CSP above the background (Figure 2D).
Binding of monomer to heparin oligosaccharides
We discuss the binding of dp4, dp8 and dp14 together as their CSP profiles were essentially the same (Figures 3A–3C). The binding-induced chemical shift changes were much higher in the monomer compared with the dimer. As in the dimer, chemical shift changes were observed for solvent-exposed and buried residues, indicating that direct and indirect interactions mediate the binding process. Significant perturbations were observed for N-loop (Lys15, His18, Lys20 and Lys23) and C-helix (Arg60, Lys64 and Arg68) residues. Interestingly, in contrast with the dimer, Arg47 and Lys54 showed negligible perturbation for all the three oligosaccharides.
Compared with the dimer, the perturbation profile of the C-helix in the monomer was more selective. Buried residues Trp57, Val58 and Val62 were minimally perturbed in the monomer, but significantly perturbed in the dimer, and perturbations for Phe65 and Leu66 were relatively higher, suggesting that indirect interactions vary between the monomer and the dimer. The CSP differences could also be due to structural differences in the helix between the monomer and the dimer, the last six residues (Lys67 to Ser72) are unstructured in the monomer, whereas the C-helix in the dimer is structured except for Ser72 [7–9]. In the monomer, binding resulted in amide shifts moving up-field for residues Arg68 to Glu70, whereas shift changes for the remaining C-helical residues were more or less random. The chemical shift index indicated residues Lys67 to Ala69 are now structured in the dp8-bound form (Supplementary Figure S1). Therefore, perturbations observed for the C-helix is a composite of both helix formation and GAG binding. This could also explain the lack of CSP for Lys67, although Lys67 is involved in GAG binding [11,51]. It is likely that chemical shift changes due to GAG binding is negated by changes due to structure formation in the case of monomer and helical rearrangement in the case of dimer. Furthermore, in the monomer alone, some of the N-loop residues preceding Lys15 and buried residues Ile39 from the second β-strand and Leu51 and Asp52 from the third β-strand that are proximal to the N-loop, were perturbed that can be attributed to indirect packing interactions. Considering N-loop residues are conformationally flexible, structural changes due to GAG binding, as described for the C-helix, cannot be ruled out.
The CSP profile for dp4 is similar to dp8 and dp14 (Figure 3C compared with Figures 3A and 3B), which is strikingly different compared with the lower perturbation of dp4 in the dimer (Figure 2C compared with Figures 2A and 2B). Given that the shorter dp4 cannot simultaneously interact with all of the perturbed N-loop and C-helical residues, multiple binding events must be responsible for the observed CSP. Our structural modelling studies described below have addressed this issue. The CSP due to dp2 binding was much lower compared with the longer oligosaccharides and nevertheless slightly higher perturbation was observed for Lys15, His18, Lys20, Arg60, Lys64 and Arg68 compared with the background (Figure 3D).
Structural models of CXCL8–heparin oligosaccharide complexes
Surface electrostatics of the CXCL8 monomer compared with dimer show a very different architecture of the basic residues, therefore, in principle, a given GAG could bind in more than one geometry involving different combinations of the basic residues (Figure 4C). The structures reveal that the basic residues identified from NMR studies form a binding surface running parallel to C-helix with the 310-helix of the N-loop forming the other face (Figures 4A and 4B). In the dimer, an additional surface that spans the dimer interface across the antiparallel C-helices can be envisioned (Figure 4C). In order to gain more definitive insights into the structure of the complex, we carried out HADDOCK-based calculations that use CSP data as ambiguous interaction restraints (AIRs), shape complementarity and energetics to drive the docking process.
Structural models of dimer–dp14 complex
The CXCL8 dimer structure (PDB id: 1IL8) and heparin dp14 were used as starting structures for the docking process. We carried out two different HADDOCK runs: binding of one dp14 to CXCL8 dimer with constraints given to (i) both monomers of the dimer, and (ii) only to one monomer in the dimer. The purpose of the second exercise was to ensure that all of the possible modes of binding to a monomer are captured during the docking process. Our docking results from both runs revealed two major families, dp14 binding perpendicular to the C-terminal helices and spanning the dimer interface (Model-I) and the other spanning one face of a monomer and running along the length of the C-helix at different angles about Lys64 and His18 as pivot points (Model-II).
The structures from Model-I can be further grouped into multiple subfamilies, which are related through translation by a disaccharide or tetrasaccharide unit (Figure 5). Any given binding geometry of dp14 cannot simultaneously satisfy interactions with all of the core residues (His18, Lys20, Arg60, Lys64, Lys67 and Lys68) on both monomers. A composite of the different poses, which are in fast exchange on the NMR timescale, can satisfy these interactions (Figure 5). All of the poses had relatively similar energies. In this model, interactions with Lys15, Lys23, Arg47 and Lys54 were completely missing (Figure 5).
Figure 5. Perpendicular model of the CXCL8 dimer–dp14 complex.

(A) An overlap of two poses related by a tetrasaccharide translation is shown to emphasize that the composite of these two poses are able to interact with all of the core residues on both monomers. (B) Close-up showing interactions between dp14 and the core basic residues on one of the monomer. The basic residues are coloured blue and dp14 is shown as sticks.
In Model-II, we observed three subfamilies, which span one face of the monomer, but have slightly different binding orientation with respect to the C-helix (M2A, M2B and M2C). All subfamilies showed interactions with all of the core residues and Lys15 (except in M2C), but showed differences in interactions with the peripheral residues (Figure 6). Oligosaccharide dp14 interacts with Lys54 of the second monomer across the dimer interface in M2A (Figures 6A and 6B), with Arg47 within the monomer in M2B (Figure 6C and 6D) and with Lys23 within the monomer (not shown). The network of interface interactions was relatively sparse in M2C. It is possible that Lys23 is not involved in direct binding, as the K23A mutant binds to the heparin column like the WT [51] and that the chemical shift change is due to indirect interactions. Interestingly, within the subfamilies, there was no specific preference for the directionality of the dp14 chain (the non-reducing compared with reducing end being near the C-terminal end of the helix). Both orientations in each subfamily showed very similar energies, emphasizing that the structural plasticity of the binding surface is able to accommodate different sets of sulfate and carboxylate interactions. Our modelling studies also showed that interactions involving both Arg47 and Lys54 are not possible due to their relative geometry.
Figure 6. Parallel model of the CXCL8 dimer–dp14 complex.

(A) Interactions of dp14 in subfamily M2A. Interactions of the core residues and peripheral residues Lys15 and Lys54* (of the second monomer across the dimer interface) are highlighted. (B) Surface plot showing the binding geometry of dp14 in M2A. (C) Interactions of dp14 in subfamily M2B showing interactions of the core residues and peripheral residues Lys15 and Arg47. (D) Surface plot showing the orientation of dp14 in M2B. The basic residues are coloured blue, dp14 is shown as sticks, and the second monomer of the dimer is painted in black.
Structural models of dimer–dp8 complex
We modelled the binding of dp8 to CXCL8 dimer and obtained two major families as described for dp14. Oligosaccharide dp8 can bind in a perpendicular mode (Model-I), wherein there are several translated poses in fast exchange (as observed for dp14), which are related by a disaccharide or tetrasaccharide frameshift (Supplementary Figure S2). However, this binding mode is energetically less favoured since dp8 cannot span the dimer interface and satisfy as many interactions as observed in Model-II. In Model-II, we obtained three subfamilies as in the case of dp14, except that subfamily M2A lacked interactions with Lys54 on the second monomer across the dimer interface (Supplementary Figure S3). Structures reveal that dp8 adopts most of the binding geometries observed for dp14, but some of the interactions within a given pose were missing due to its shorter length.
Structural models of monomer-bound dp8 and dp14 complexes
Our NMR studies showed that the C-terminal helix is structured in the dp8-bound form of the monomer. Therefore, we modelled binding of both dp8 and dp14 using (i) monomer from the CXCL8 dimer structure (PDB id: 1IL8), and (ii) the CXCL8 monomer structure (PDB id: 1IKM). The docking results obtained using the monomer structure of the dimer were very similar to those obtained for the dimer, as described above. In the case of the 1IKM structure, the last six residues of the helix (including Lys67 and Arg68) are unstructured. Modelling results showed that both dp8 and dp14 lie along the groove nestled between N-loop and C-helical residues (similar to pose M2A for the dimer; Supplementary Figure S4). The structures showed that both ‘forward mode’ from N- to C-terminal of the helix and ‘reverse mode’ from C- to N-terminal of the helix are equally feasible and that the core Arg60, Lys64, Lys67, Arg68, His18 and Lys20 and peripheral Lys15 residues mediate binding. Given the flexibility of the unstructured C-helix, the spatial positioning of Lys67 and Arg68 is different compared with the dimer and the interaction pattern for these residues with GAG sulfates and carboxylates were also different. As in the case of dimer, different clusters showed different sets of carboxylate, 2-O-sulfate, 6-O-sulfate and N-sulfate groups along the oligosaccharide chain can satisfy the interactions involving the CXCL8 binding surface.
Structural models of monomer and dimer bound dp4 and dp2 complexes
Oligosaccharide dp4 shows similar binding geometries both in the monomer and in the dimer and therefore our description applies to both forms. The structures reveal that dp4 can bind in different orientations and locations and engage sulfates and carboxylates in different combinations (Supplementary Figure S5). Major clusters showed that dp4 engages all of the core residues (His18, Lys20, Arg60, Lys64, Lys67 and Arg68 in a given monomer) in two partially overlapping sites: one on the N-terminal end of the helix that includes Lys15 interactions (Supplementary Figure S5A) and the other on the C-terminal end of the helix that does not include Lys15 interactions (Supplementary Figure S5B). In the case of dp2 binding to a monomer or dimer, several equal energy clusters were observed. Different clusters and the interaction patterns showed that dp2 binds to different locations, resulting in an ensemble of different populations (results not shown).
MD simulation of CXCL8–heparin oligosaccharide models
We carried out MD simulations using the NAMD suite of programs on three different CXCL8–heparin oligosaccharide models obtained from HADDOCK docking that satisfy our NMR experimental data: (i) Model-I (perpendicular mode) from dimer–dp14 models (CXCL8D–dp14, ⊥); (ii) Model-II (parallel mode) from dimer–dp8 models (CXCL8D–dp8, //), and (iii) Model-I (parallel mode) from monomer–dp8 models (CXCL8M–dp8, //). We chose dp8, and not dp14, for the parallel models because the shorter dp8 spans the entire binding surface, while minimizing any artefact arising from the dynamics of the overhanging GAG regions during the simulation. Each of the simulations was carried out for over 100 ns to sample different conformational ensembles given that the interacting interface is highly dynamic in nature. Explicit solvent and PME electrostatics facilitated characterization of the influence of long-range electrostatics at the binding interface.
Large concerted motions of lysine/arginine side chains with their interacting partners (sulfates and carboxylates) on the heparin chain were observed during the course of the simulations. The helical symmetry of the heparin chain and the fairly uniform distribution of sulfates and carboxylates around the surface allowed translational/rotational motions on the protein surface. These motions in essence result in fluent inter-conversion of sulfate/carboxylate interacting partners between adjacent basic residues. In all three simulations, the heparin chain showed appreciable flexibility and adapted to the topology and the side-chain dynamics of the CXCL8 binding surface, which either span one or both monomers. Throughout the simulation, glycosidic torsional angles (φH, ψH) fall in the allowed region [35] (Supplementary Figures S6 and S7), the glucosamine [GlcNS(6S)] pyranose rings adopt the preferred 4C1 chair conformation and the iduronate [IdoA(2S)] pyranose rings adopt the preferred 2SO(skew boat)/2,5B(boat) conformations (Supplementary Figures S8 and S9) [52].
In the CXCL8D–dp14 (⊥) model, the 14-mer runs perpendicular to the two C-helix and spans the basic surfaces on both monomers (Figure 5). Analysis of the backbone RMSDs of the CXCL8–dp14 complex showed large differences in the structure (2 ±1.5 Å) (Supplementary Figure S10). In addition, the BSA between the GAG and the protein varied by ∼200 Å2 over the course of the simulation (500±100 Å2; Supplementary Figure S11). Within the entire trajectory, we identified two ∼10 ns trajectory stretches (Traj1, 83–95 ns) and (Traj2, 98–108 ns) which differ in backbone RMSDs and BSA. Within the individual stretches, the backbone RMSDs are fairly small (1–1.5 Å) and the structures have similar BSA (results not shown). Analysis of the structures between the stretches showed large differences at the residue level hydrogen-bond and salt bridge interactions, even though the overall orientation of the GAG chain is similar with respect to the protein-binding surface (Figures 7A and 7B). The core basic residues continuously engage sulfates/carboxylates, whereas the interactions of peripheral basic residues were either transient or absent. To illustrate the dynamic nature of the interface, we show the switch in sulfate partner for the Lys64 NH3+ group between adjacent monosaccharides using snapshots and distance plots from Traj1 and Traj2 (Figures 7A and 7B). The hydrogen-bond switches from the 2-O-sulfate of IdoA(2S) to 6-O-sulfate GlcNS(6S) which corresponds to a ∼6–7 Å shift arising from translational/rotational rearrangements of the interacting partners. In a similar fashion, we also observed a single sulfate interacting with two adjacent basic residues at different time points along the trajectory (results not shown). Analysis of the residue level non-bonded interaction energies for the basic residues showed that the interactions were predominantly electrostatic in nature (Supplementary Table S1). Comparison of energetics between Traj1 and Traj2 showed that the mean residue level contribution over the entire simulation were similar, even though there is interchange of sulfate partners on the heparin chain. The large range between the minimum and the maximum energies emphasizes the highly dynamic nature of the interface.
Figure 7. Dynamic nature of the binding interface from MD simulations.

(A) Snapshots from two ∼10 ns stretches (Traj1 and Traj2) of the MD simulations of the CXCL8D dp14(⊥) showing hydrogen-bond partners of Lys64 NζH3+ switching between IdoA11–OS2 and GlcN10–OS6. (B) The distances between Lys64Nζ and the sulfates for Traj1 and Traj2 illustrate the interchange of interacting partners. (C) Snapshots from two ∼10 ns stretches (Traj1 and Traj2) of CXCL8M–dp8(//) simulations showing IdoA11(COO−) switching hydrogen-bond partners between His18 and Lys20 side chains and Lys20 exchanging between IdoA11(COO−) and GlcN12 (NS2). (D) The differences in distance of IdoA11(COO−) from His18 and Lys20 side chains during Traj1 and Traj2 illustrate the dynamic interplay. For clarity, only residues Lys64 in (A) and His18 and Lys20 in (C) are shown as sticks at the interface.
For the CXCL8D–dp8 (//) model, the binding interface was much more dynamic compared with the perpendicular model. Throughout the simulation, the interactions were confined to a single monomer, but several different poses of the GAG chain about the core basic residue as pivot were observed (results not shown). In the case CXCL8M–dp8 (//), several different angular poses of the GAG chain about the core basic residues were observed similar to the dimer (//) model. Backbone RMSD and BSA plots show large variations over the time course of the simulation (Supplementary Figures S12 and S13). The dynamic nature of the interface is illustrated for residues Lys20 and His18 (Figures 7C and 7D). Interchange of sulfate interactions for a given basic residue and as well as switching of basic residue partners for a given sulfate are shown. The residue-level energies show a large range highlighting the structural plasticity of the binding interface (Supplementary Table S2).
GAG-binding affinities of CXCL8 monomer and dimer
Binding affinities (Kd), calculated from binding-induced chemical shift changes, indicated increasing affinities with increasing oligosaccharide length for both monomer and dimer. The Kd for both monomer and dimer for various oligosaccharides were essentially similar: <10 μM for dp14, ∼30 μM for dp8, ∼200 μM for dp4 and ∼2 mM for dp2. Kd for dp14 is an upper estimate and could not be accurately determined, as binding constant measurements from NMR titrations must satisfy the requirement that the starting protein concentration is in the order of 0.5 × Kd and no more than 5 × Kd [53]. As these studies could not determine whether the monomer or the dimer is the high-affinity ligand, we adopted a different strategy for discerning the relative affinities of the monomer and dimer. We recently optimized conditions where we could observe both WT dimer and monomer peaks in the NMR spectra [54]. As our studies show that longer oligosaccharides bind with higher affinity and as the longer GAGs better reflect binding to in vivo GAGs, we simultaneously tracked the binding of dp26 (heparin 26-mer) to both WT monomer and dimer in the NMR spectra. On adding dp26, all peaks corresponding to the monomer disappeared and only the dimer peaks remained, providing unambiguous evidence that the dimer is the high-affinity ligand (Figure 8). As monomer/dimer, monomer/dp26, dimer/dp26 equilibria are coupled, the presence of only the dimer peaks indicates that the dimer binds dp26 with much higher affinity promoting dimerization and loss of monomer population.
Figure 8. NMR HSQC spectra of CXCL8–dp26 complex.
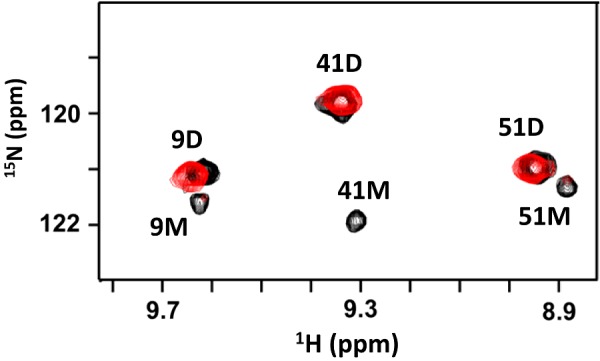
A section of the 1H-15N HSQC spectra at pH 7.5 showing the overlay of WT CXCL8 (black) and WT CXCL8–dp26 complex (red). Dimer and monomer peaks are indicated by D and M respectively. The monomer peaks disappear on dp26 binding indicating tighter binding to the dimer. At pH 7.5 and 40 μM concentration, WT CXCL8 exists as both monomers (∼10%) and dimers. The final protein/dp26 molar ratio is 1:2.
DISCUSSION
GAGs are highly negatively charged polysaccharides that bind various classes of proteins including growth factors and chemokines. Despite their fundamental roles, very little is known regarding how different residues in the context of tertiary structure bind GAGs. Mutagenesis and GAG-binding assays have shown that basic residues arginine, lysine and histidine mediate binding, but how these residues influence specificity, affinity, selectivity and activity remains largely unknown. In the present study, we show that NMR binding-induced CSP measurements are highly sensitive and can provide how basic residues in the context of tertiary and quaternary structure dictate different facets of chemokine–GAG binding properties. In particular, by characterizing the binding of oligosaccharides of various lengths to a designed monomer and WT dimer, we provide compelling evidence that conformational plasticity plays an important role in mediating the binding process. The CSP profiles and structural models reveal that binding residues can be grouped into two distinct non-overlapping clusters: a set of core residues that function as the major recognition/docking site and others around the periphery of the core residues that play a role in defining the binding geometries. Our modelling and MD exercise of both dp8 and dp14 indicates that different sets of 2-O-sulfate, 6-O-sulfate, N-sulfate and carboxylate groups along the oligosaccharide chain can satisfy the interactions involving the binding surface. The helical structure of GAG and distribution of sulfates also allows exchange between different rotational and translational poses with minimal energetic penalty.
A previous NMR study of a disaccharide binding to WT dimer and affinity measurements of mutants binding to a heparin column, identified Arg60, Lys64, Lys67, Arg68, His18 and Lys20 as the GAG-binding residues [51]. MD-based and other docking studies using these data have proposed models where GAG binds perpendicular or parallel to the helix [21–24]. MS study of dp8 binding to WT dimer proposed a model where the GAG binds parallel to the helix [55]. Previous NMR and modelling studies on CXCL8–chondroitin sulfate also suggested a parallel model of binding to the helix [56]. Role of water-mediated interactions at the GAG/protein interface [57] and the importance of flexibility and explicit solvent in GAG–protein molecular docking [58] have been recently explored.
Most importantly, our studies clarify a number of ambiguities on various aspects of CXCL8 binding to GAGs that have direct impact on understanding how CXCL8–GAG interactions govern in vivo function. For the first time, we have identified Lys15, Arg47 and Lys54, located around the periphery of the core residues, as GAG-binding residues. These peripheral residues facilitate multiple binding modes and geometries that vary between monomer and dimer and between shorter and longer oligosaccharides. Our data indicate that multiple modes of parallel and perpendicular models are possible. These data have direct implications on how CXCL8 binds its receptors, CXCR1 and CXCR2. CXCL8 N-loop residues, which include Lys15, His18 and Lys20, also mediate the initial docking to both receptors [59], indicating that GAG-bound chemokine will be impaired for receptor binding.
Sequence comparison of CXCL8 and related chemokines reveal that residues corresponding to His18, Lys20, Arg60 and Lys64 are highly conserved, Lys67 and Arg68 are minimally conserved and, most interestingly, Lys15 is unique to CXCL8 (Figure 9). Considering that Lys67 and Arg68 function as core residues and Lys15 is involved in most of the binding interactions, it is likely that the role of structural plasticity will vary even among similar chemokines. We propose that the conserved core residues are essential for binding in all chemokines and that the peripheral residues promote different chemokine-specific GAG geometries. For instance, we had characterized the binding of an octasaccharide to mouse CXCL1/KC dimer using NMR spectroscopy and observed that the data are consistent with a perpendicular model [60]. Evidence for structural plasticity in GAG binding has also been observed for other chemokines. For example, it has been shown for CCL27 and CXCL12 that chemokine oligomerization properties, GAG length, protein concentration and binding-induced oligomerization and aggregation are intimately coupled [61,62].
Figure 9. Sequence alignment of CXCL8 and related chemokines.

Arginine, lysine and histidine residues of CXCL8 implicated in GAG binding and the potential GAG-binding residues in related chemokines are underlined.
A previous NMR study of CXCL8 dimer suggested that residues Glu29–Gly31 could function as a secondary GAG-binding site [63]. However, we failed to see CSP for these residues above the background. These authors characterized the binding under sub-stoichiometric conditions (1 mM CXCL8 and ∼0.15 mM dp6 heparin) and so their final molar ratio of 1:0.15 is significantly different from our final ratio of 1:6 for dp8 titration. Not surprisingly, we observed much larger CSP, considering we have 40-fold more GAG. These observations highlight the importance of characterizing binding at low protein concentrations and excess GAG to ensure that most of the chemokine is in the GAG-bound form.
Our studies highlight that differences in oligosaccharide length influence binding modes and geometries, that shorter dp4 and dp2 oligosaccharides cannot capture many of the native interactions so their results must be interpreted with caution and that structural differences between the monomer and the dimer, especially if they involve GAG-binding residues, can influence binding. Our studies also highlight the importance of interpreting CSP in the context of exposed compared with buried and direct compared with indirect interactions. Additional NMR studies that can directly detect the binding of lysine side chains and studies that can give direct insights into the GAG functional groups should result in better definition of the binding interactions.
Previous studies using WT CXCL8 and a trapped monomer had shown that the dimer is the high-affinity GAG ligand and that affinities increase with increasing GAG length [15,16]. However, a different study using fluorescence microscopy reported that the monomer binds heparin oligosaccharides (dp2–dp16) with ∼100–1000-fold higher affinity (nanomolar compared with micromolar) than the dimer [18]. These authors characterized the binding of WT CXCL8 at two different concentrations, one where the monomer dominates and the other where the dimer dominates. They reported that the monomer binds heparin dp2 with a Kd of 380 nM and dp4 with 57 nM and dimer affinities to both dp2 and dp4 of ∼100 μM. In contrast, our results show that both monomer and dimer bind dp2 with weak millimolar affinities (∼2000 μM) and dp4 with a Kd of ∼100 μM. Whereas our data show the binding affinity increase with increasing chain length, the fluorescence study showed essentially the same Kd for dimer binding to all oligosaccharides. Our ability to simultaneously track the binding of dp26 to WT monomer and dimer provide unambiguous evidence that the dimer is the high-affinity ligand, but is in disagreement with studies by Goger et al. [18].
In summary, we conclude that structural plasticity mediates GAG–CXCL8 interactions, that the dimer is the high-affinity GAG ligand, that the GAG-bound CXCL8 could be impaired for activating either receptors and that GAG-bound chemokine indirectly regulates receptor-activating function of the soluble chemokine in trafficking various cell types to their target tissue.
Acknowledgments
We thank Dr Tianzhi Wang for assistance with the NMR experiments. We are thankful for the high-performance computing resource provided by the Texas Advanced Computing Center (TACC) at University of Texas at Austin and the Center for High Performance Computing (ChiPC) at Virginia Commonwealth University.
Abbreviations
- BSA
buried surface area
- CSP
chemical shift perturbation
- dp2
heparin disaccharide
- dp4
heparin tetrasaccharide
- dp8
heparin octasaccharide
- dp14
heparin 14-mer
- dp26
heparin 26-mer
- HADDOCK
high-ambiguity-driven biomolecular docking
- HS
heparan sulfate
- IL-8
interleukin 8
- GAG
glycosaminoglycan
- IdoA
iduronic acid
- GlcN
glucosamine
- PME
Particle Mesh Ewald
- SA
simulated annealing
- WT
wild-type
AUTHOR CONTRIBUTION
Krishna Rajarathnam, Philip Mosier, Prem Joseph and Umesh Desai designed the research. Prem Joseph and Philip Mosier performed the experiments. Prem Joseph, Philip Mosier, Umesh Desai and Krishna Rajarathnam analysed the data. Prem Joseph, Philip Mosier and Krishna Rajarathnam wrote the paper.
FUNDING
This work was supported by the National Institutes of Health [grant number P01HL107152]; Sealy and Smith foundation grant to the Sealy Center for Structural Biology and Molecular Biophysics; and the National Center for Research Resources at VCU [grant number S10RR027411].
References
- 1.Raman R., Sasisekharan V., Sasisekharan R. Structural insights into biological roles of protein-glycosaminoglycan interactions. Chem. Biol. 2005;12:267–277. doi: 10.1016/j.chembiol.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 2.Imberty A., Lortat-Jacob H., Pérez S. Structural view of glycosaminoglycan-protein interactions. Carbohydr. Res. 2007;342:430–439. doi: 10.1016/j.carres.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 3.Singh S., Singh A.P., Sharma B., Owen L.B., Singh R.K. CXCL8 and its cognate receptors in melanoma progression and metastasis. Future Oncol. 2010;6:111–116. doi: 10.2217/fon.09.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelland E.E., Gilmore W., Weiner L.P., Lund B.T. The dual role of CXCL8 in human CNS stem cell function: multipotent neural stem cell death and oligodendrocyte progenitor cell chemotaxis. Glia. 2011;59:1864–1878. doi: 10.1002/glia.21230. [DOI] [PubMed] [Google Scholar]
- 5.Handel T.M., Johnson Z., Crown S.E., Lau E.K., Proudfoot A.E. Regulation of protein function by glycosaminoglycans–as exemplified by chemokines. Annu. Rev. Biochem. 2005;74:385–410. doi: 10.1146/annurev.biochem.72.121801.161747. [DOI] [PubMed] [Google Scholar]
- 6.Rot A. Chemokine patterning by glycosaminoglycans and interceptors. Front. Biosci. (Landmark Ed.) 2010;15:645–660. doi: 10.2741/3638. [DOI] [PubMed] [Google Scholar]
- 7.Baldwin E.T., Weber I.T., St Charles R., Xuan J.C., Appella E., Yamada M., Matsushima K., Edwards B.F., Clore G.M., Gronenborn A.M., Wlodawer A. Crystal structure of interleukin 8: symbiosis of NMR and crystallography. Proc. Natl. Acad. Sci. U.S.A. 1991;88:502–506. doi: 10.1073/pnas.88.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajarathnam K., Clark-Lewis I., Sykes B.D. 1H NMR solution structure of an active monomeric interleukin-8. Biochemistry. 1995;34:12983–12990. doi: 10.1021/bi00040a008. [DOI] [PubMed] [Google Scholar]
- 9.Clore G.M., Appella E., Yamada M., Matsushima K., Gronenborn A.M. Three-dimensional structure of interleukin 8 in solution. Biochemistry. 1990;29:1689–1696. doi: 10.1021/bi00459a004. [DOI] [PubMed] [Google Scholar]
- 10.Das S.T., Rajagopalan L., Guerrero-Plata A., Sai J., Richmond A., Garofalo R.P., Rajarathnam K. Monomeric and dimeric CXCL8 are both essential for in vivo neutrophil recruitment. PLoS One. 2010;5:e11754. doi: 10.1371/journal.pone.0011754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gangavarapu P., Rajagopalan L., Kolli D., Guerrero-Plata A., Garofalo R.P., Rajarathnam K. The monomer-dimer equilibrium and glycosaminoglycan interactions of chemokine CXCL8 regulate tissue-specific neutrophil recruitment. J. Leukoc. Biol. 2012;91:259–265. doi: 10.1189/jlb.0511239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hileman R.E., Fromm J.R., Weiler J.M., Linhardt R.J. Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. BioEssays. 1998;20:156–167. doi: 10.1002/(SICI)1521-1878(199802)20:2<156::AID-BIES8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 13.Li L., Ly M., Linhardt R.J. Proteoglycan sequence. Mol. Biosyst. 2012;8:1613–1625. doi: 10.1039/c2mb25021g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu D., Esko J.D. Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 2014;83:129–157. doi: 10.1146/annurev-biochem-060713-035314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoogewerf A.J., Kuschert G.S., Proudfoot A.E., Borlat F., Clark-Lewis I., Power C.A., Wells T.N. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry. 1997;36:13570–13578. doi: 10.1021/bi971125s. [DOI] [PubMed] [Google Scholar]
- 16.Frevert C.W., Kinsella M.G., Vathanaprida C., Goodman R.B., Baskin D.G., Proudfoot A., Wells T.N., Wight T.N., Martin T.R. Binding of interleukin-8 to heparan sulfate and chondroitin sulfate in lung tissue. Am. J. Respir. Cell Mol. Biol. 2003;28:464–472. doi: 10.1165/rcmb.2002-0084OC. [DOI] [PubMed] [Google Scholar]
- 17.Kuschert G.S., Coulin F., Power C.A., Proudfoot A.E., Hubbard R.E., Hoogewerf A.J., Wells T.N. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry. 1999;38:12959–12968. doi: 10.1021/bi990711d. [DOI] [PubMed] [Google Scholar]
- 18.Goger B., Halden Y., Rek A., Mosl R., Pye D., Gallagher J., Kungl A.J. Different affinities of glycosaminoglycan oligosaccharides for monomeric and dimeric interleukin-8: a model for chemokine regulation at inflammatory sites. Biochemistry. 2002;41:1640–1646. doi: 10.1021/bi011944j. [DOI] [PubMed] [Google Scholar]
- 19.Schlorke D., Thomas L., Samsonov S.A., Huster D., Arnhold J., Pichert A. The influence of glycosaminoglycans on IL-8-mediated functions of neutrophils. Carbohydr. Res. 2012;356:196–203. doi: 10.1016/j.carres.2012.02.025. [DOI] [PubMed] [Google Scholar]
- 20.Spillmann D., Witt D., Lindahl U. Defining the interleukin-8-binding domain of heparan sulfate. J. Biol. Chem. 1998;273:15487–15493. doi: 10.1074/jbc.273.25.15487. [DOI] [PubMed] [Google Scholar]
- 21.Bitomsky W., Wade R.C. Docking of glycosaminoglycans to heparin-binding proteins: validation for aFGF, bFGF, and antithrombin and application to IL-8. J. Am. Chem. Soc. 1999;121:3004–3013. doi: 10.1021/ja983319g. [DOI] [Google Scholar]
- 22.Lortat-Jacob H., Grosdidier A., Imberty A. Structural diversity of heparan sulfate binding domains in chemokines. Proc. Natl. Acad. Sci. U.S.A. 2002;99:1229–1234. doi: 10.1073/pnas.032497699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krieger E., Geretti E., Brandner B., Goger B., Wells T.N., Kungl A.J. A structural and dynamic model for the interaction of interleukin-8 and glycosaminoglycans: support from isothermal fluorescence titrations. Proteins. 2004;54:768–775. doi: 10.1002/prot.10590. [DOI] [PubMed] [Google Scholar]
- 24.Gandhi N.S., Mancera R.L. Molecular dynamics simulations of CXCL-8 and its interactions with a receptor peptide, heparin fragments, and sulfated linked cyclitols. J. Chem. Inf. Model. 2011;51:335–358. doi: 10.1021/ci1003366. [DOI] [PubMed] [Google Scholar]
- 25.Rajagopalan L., Rajarathnam K. Ligand selectivity and affinity of chemokine receptor CXCR1. role of N-terminal domain. J. Biol. Chem. 2004;279:30000–30008. doi: 10.1074/jbc.M313883200. [DOI] [PubMed] [Google Scholar]
- 26.Joseph P.R., Poluri K.M., Gangavarapu P., Rajagopalan L., Raghuwanshi S., Richardson R.M., Garofalo R.P., Rajarathnam K. Proline substitution of dimer interface β-strand residues as a strategy for the design of functional monomeric proteins. Biophys. J. 2013;105:1491–1501. doi: 10.1016/j.bpj.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Permi P., Annila A. Coherence transfer in proteins. Prog. Nucl. Mag. Reson. Spectrosc. 2004;44:97–137. doi: 10.1016/j.pnmrs.2003.12.001. [DOI] [Google Scholar]
- 28.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 29.Johnson B.A., Blevins R.A. NMR View: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 30.Wishart D.S., Sykes B.D. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 31.Ravindran A., Joseph P.R., Rajarathnam K. Structural basis for differential binding of the interleukin-8 monomer and dimer to the CXCR1 N-domain: role of coupled interactions and dynamics. Biochemistry. 2009;48:8795–8805. doi: 10.1021/bi901194p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dominguez C., Boelens R., Bonvin A.M. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 33.de Vries S.J., van Dijk A.D., Krzeminski M., van Dijk M., Thureau A., Hsu V., Wassenaar T., Bonvin A.M. HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 2007;69:726–733. doi: 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- 34.Mulloy B., Forster M.J., Jones C., Davies D.B. N.M.R. and molecular-modelling studies of the solution conformation of heparin. Biochem. J. 1993;293:849–858. doi: 10.1042/bj2930849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z., McCallum S.A., Xie J., Nieto L., Corzana F., Jimenez-Barbero J., Chen M., Liu J., Linhardt R.J. Solution structures of chemoenzymatically synthesized heparin and its precursors. J. Am. Chem. Soc. 2008;130:12998–13007. doi: 10.1021/ja8026345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linge J.P., Nilges M. Influence of non-bonded parameters on the quality of NMR structures: a new force field for NMR structure calculation. J. Biomol. NMR. 1999;13:51–59. doi: 10.1023/A:1008365802830. [DOI] [PubMed] [Google Scholar]
- 37.Schüttelkopf A.W., van Aalten D.M. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta. Crystallogr. D Biol. Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 38.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., MacKerell A.D., Jr CHARMM General Force Field (CGenFF): a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guvench O., Hatcher E., Venable R.M., Pastor R.W., MacKerell A.D., Jr CHARMM additive all-atom force field for glycosidic linkages between hexopyranoses. J. Chem. Theory Comput. 2009;5:2353–2370. doi: 10.1021/ct900242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guvench O., Mallajosyula S.S., Raman E.P., Hatcher E., Vanommeslaeghe K., Foster T.J., Jamison F.W., II, MacKerell A.D., Jr CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate–protein modeling. J. Chem. Theory Comput. 2011;7:3162–3180. doi: 10.1021/ct200328p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mallajosyula S.S., Guvench O., Hatcher E., MacKerell A.D., Jr CHARMM additive all-atom force field for phosphate and sulfate linked to carbohydrates. J. Chem. Theory Comput. 2012;8:759–776. doi: 10.1021/ct200792v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huige C.J.M., Altona C. Force field parameters for sulfates and sulfamates based on ab initio calculations: extensions of AMBER and CHARMm fields. J. Comput. Chem. 1995;16:56–79. doi: 10.1002/jcc.540160106. [DOI] [Google Scholar]
- 44.Jo S., Song K.C., Desaire H., MacKerell A.D., Jr, Im W. Glycan reader: automated sugar identification and simulation preparation for carbohydrates and glycoproteins. J. Comput. Chem. 2011;31:3135–3141. doi: 10.1002/jcc.21886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 46.Makeneni S., Foley B.L., Woods R.J. BFMP: a method for discretizing and visualizing pyranose conformations. J. Chem. Inf. Model. 2014;54:2744–2750. doi: 10.1021/ci500325b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao B., Liwang P.J. Characterization of the interactions of vMIP-II, and a dimeric variant of vMIP-II, with glycosaminoglycans. Biochemistry. 2010;49:7012–7022. doi: 10.1021/bi100549y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rajasekaran D., Keeler C., Syed M.A., Jones M.C., Harrison J.K., Wu D., Bhandari V., Hodsdon M.E., Lolis E.J. A model of GAG/MIP-2/CXCR2 interfaces and its functional effects. Biochemistry. 2012;51:5642–5654. doi: 10.1021/bi3001566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joseph P., Poluri K., Sepuru K., Rajarathnam K. Characterizing protein–glycosaminoglycan interactions using solution NMR spectroscopy. Methods Mol. Biol. 2015;1229:325–333. doi: 10.1007/978-1-4939-1714-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rajagopalan L., Chin C.C., Rajarathnam K. Role of intramolecular disulfides in stability and structure of a noncovalent homodimer. Biophys. J. 2007;93:2129–2134. doi: 10.1529/biophysj.107.108761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuschert G.S., Hoogewerf A.J., Proudfoot A.E., Chung C.W., Cooke R.M., Hubbard R.E., Wells T.N., Sanderson P.N. Identification of a glycosaminoglycan binding surface on human interleukin-8. Biochemistry. 1998;37:11193–11201. doi: 10.1021/bi972867o. [DOI] [PubMed] [Google Scholar]
- 52.Mulloy B., Forster M.J. Conformation and dynamics of heparin and heparan sulfate. Glycobiology. 2000;10:1147–1156. doi: 10.1093/glycob/10.11.1147. [DOI] [PubMed] [Google Scholar]
- 53.Williamson M.P. Using chemical shift perturbation to characterize ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 54.Joseph P.R., Rajarathnam K. Solution NMR characterization of WT CXCL8 monomer and dimer binding to CXCR1 N-terminal domain. Protein Sci. 2015;24:81–92. doi: 10.1002/pro.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seo Y., Andaya A., Bleiholder C., Leary J.A. Differentiation of CC vs CXC chemokine dimers with GAG octasaccharide binding partners: an ion mobility mass spectrometry approach. J. Am. Chem. Soc. 2013;135:4325–4332. doi: 10.1021/ja310915m. [DOI] [PubMed] [Google Scholar]
- 56.Pichert A., Samsonov S.A., Theisgen S., Thomas L., Baumann L., Schiller J., Beck-Sickinger A.G., Huster D., Pisabarro M.T. Characterization of the interaction of interleukin-8 with hyaluronan, chondroitin sulfate, dermatan sulfate and their sulfated derivatives by spectroscopy and molecular modeling. Glycobiology. 2012;22:134–145. doi: 10.1093/glycob/cwr120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Samsonov S.A., Teyra J., Pisabarro M.T. Docking glycosaminoglycans to proteins: analysis of solvent inclusion. J. Comput. Aided Mol. Des. 2011;25:477–489. doi: 10.1007/s10822-011-9433-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Samsonov S.A., Gehrcke J.P., Pisabarro M.T. Flexibility and explicit solvent in molecular-dynamics-based docking of protein-glycosaminoglycan systems. J. Chem. Inf. Model. 2014;54:582–592. doi: 10.1021/ci4006047. [DOI] [PubMed] [Google Scholar]
- 59.Rajagopalan L., Rajarathnam K. Structural basis of chemokine receptor function–a model for binding affinity and ligand selectivity. Biosci. Rep. 2006;26:325–339. doi: 10.1007/s10540-006-9025-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poluri K.M., Joseph P.R., Sawant K.V., Rajarathnam K. Molecular basis of glycosaminoglycan heparin binding to the chemokine CXCL1 dimer. J. Biol. Chem. 2013;288:25143–25153. doi: 10.1074/jbc.M113.492579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jansma A.L., Kirkpatrick J.P., Hsu A.R., Handel T.M., Nietlispach D. NMR analysis of the structure, dynamics, and unique oligomerization properties of the chemokine CCL27. J. Biol. Chem. 2010;285:14424–14437. doi: 10.1074/jbc.M109.091108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ziarek J.J., Veldkamp C.T., Zhang F., Murray N.J., Kartz G.A., Liang X., Su J., Baker J.E., Linhardt R.J., Volkman B.F. Heparin oligosaccharides inhibit chemokine (CXC motif) ligand 12 (CXCL12) cardioprotection by binding orthogonal to the dimerization interface, promoting oligomerization, and competing with the chemokine (CXC motif) receptor 4 (CXCR4) N terminus. J. Biol. Chem. 2013;288:737–746. doi: 10.1074/jbc.M112.394064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nordsieck K., Pichert A., Samsonov S.A., Thomas L., Berger C., Pisabarro M.T., Huster D., Beck-Sickinger A.G. Residue 75 of interleukin-8 is crucial for its interactions with glycosaminoglycans. ChemBioChem. 2012;13:2558–2566. doi: 10.1002/cbic.201200467. [DOI] [PubMed] [Google Scholar]