

Abstract
GalNAc‐type O‐glycosylation is found on most proteins trafficking through the secretory pathway in metazoan cells. The O‐glycoproteome is regulated by up to 20 polypeptide GalNAc‐Ts and the contributions and biological functions of individual GalNAc‐Ts are poorly understood. Here, we used a zinc‐finger nuclease (ZFN)‐directed knockout strategy to probe the contributions of the major GalNAc‐Ts (GalNAc‐T1 and GalNAc‐T2) in liver cells and explore how the GalNAc‐T repertoire quantitatively affects the O‐glycoproteome. We demonstrate that the majority of the O‐glycoproteome is covered by redundancy, whereas distinct subsets of substrates are modified by non‐redundant functions of GalNAc‐T1 and GalNAc‐T2. The non‐redundant O‐glycoproteome subsets and specific transcriptional responses for each isoform are related to different cellular processes; for the GalNAc‐T2 isoform, these support a role in lipid metabolism. The results demonstrate that GalNAc‐Ts have different non‐redundant glycosylation functions, which may affect distinct cellular processes. The data serves as a comprehensive resource for unique GalNAc‐T substrates. Our study provides a new view of the differential regulation of the O‐glycoproteome, suggesting that the plurality of GalNAc‐Ts arose to regulate distinct protein functions and cellular processes.
Keywords: apolipoproteins, dimethylation, GALNT, mass spectrometry
Subject Categories: Methods & Resources; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Post‐translational modifications greatly expand the size and functional space of the proteome, and glycosylation is one of the most abundant and diverse modifications. GalNAc‐type O‐glycosylation is uniquely geared for differential regulation of protein functions with up to 20 polypeptide GalNAc‐transferases (GalNAc‐Ts) in humans controlling the first decisive step in glycosylation and thereby determining both which proteins and where on these proteins O‐glycans are attached 1. GalNAc‐Ts catalyze the addition of GalNAc residues to serine and threonine (and possibly tyrosine), and GalNAc‐T isoenzymes have been shown to exhibit different, albeit partly overlapping, peptide substrate specificities and kinetic properties. GALNTs are differentially expressed and the repertoire in cells changes during cellular maturation and differentiation and in cancer 1, 2, 3. It is therefore predicted that the GalNAc‐T family of enzymes furnish cells with the capacity to differentially and perhaps dynamically regulate the O‐glycoproteome and thereby modulate protein function 4, not dissimilar to the regulation of the phosphoproteome by the much larger family of kinases 5. However, currently this is a hypothesis based largely on in vitro enzyme analyses.
In vitro analyses of GalNAc‐T isoenzymes suggest that these have wide roles in glycosylation of proteins without appreciable systematic or specific roles in biological processes and pathways. However, deficiencies in GALNT genes appear to cause subtle diseases both in humans and in animal models 6. For example, loss of GALNT3 causes the rare related diseases familial tumoral calcinosis (FTC) and hyperostosis syndrome (HHS) characterized by hyperphosphatemia 7, which is due to inactivation of fibroblast growth factor 23 (FGF23) by proprotein convertase (PC) processing as a result of lack of site‐specific O‐glycosylation by GalNAc‐T3 in the PC processing site 8. Strikingly, deficiency in the FGF23 gene itself produces largely the same disease phenotype 9, indicating that GalNAc‐T3 despite its predicted broader role in protein glycosylation perhaps primarily serves to co‐regulate phosphate homeostasis. Notably, this function is conserved in rodents 10, 11, and GALNT3 has been associated with human bone mineral density and fracture risk 12. Thus, it is possible that the seemingly broad and partly overlapping roles of the many GalNAc‐Ts in protein glycosylation are deceptive and that instead the many GalNAc‐Ts serve to fine‐tune highly selective protein functions in limited cellular pathways.
A long‐standing question then is why nature evolved the large family of isoenzymes with 20 members in mammals, 9 in C. elegans and 12 in Drosophila 1, 3, and more specifically whether the evolutionary requirement for multiple GalNAc‐Ts was driven by a need to accommodate efficient glycosylation of a great diversity of acceptor peptide sequences, or—and perhaps not mutually exclusively—if this was driven by a need for differential regulation and fine‐tuning of distinct protein functions and biological pathways. Other types of protein O‐glycosylation in metazoans are catalyzed by only one or two isoenzymes 1, 13, although yeast O‐mannosylation, that in many ways resembles GalNAc‐type O‐glycosylation, is also catalyzed by a large family of isoenzymes with at least six Dol‐P‐Man O‐mannosyltransferases 14. We and others have recently identified examples of non‐redundant biological functions of site‐specific O‐GalNAc glycosylation in man and model organisms 4, 15, 16, 17, and multiple Genome‐Wide Association Studies (GWAS) point to different roles of GALNTs in human diseases 18. However, it remains a challenge to characterize the non‐redundant contributions of individual GalNAc‐Ts within the O‐glycoproteome of a cell, and within these to dissect the important glycosylation events that have biological consequences and ultimately could underlie disease. Thus, there is a need to develop quantitative strategies to probe GalNAc‐T isoform‐specific O‐glycosylation and the effects these may have on protein function, cellular processes, and ultimately whole organisms.
O‐glycoproteomics has long remained a challenge, but recent advances with the “SimpleCell” (SC) strategy 19 using genetically engineered cell lines with simplified homogenous O‐glycans have enabled proteome‐wide discovery of O‐glycosites. A first draft of the human O‐glycoproteome is available 20, and here, we present a quantitative O‐glycoproteomics strategy for sensitive mapping of the non‐redundant contributions of the major GalNAc‐Ts (GALNT1 and GALNT2) in the human hepatocellular carcinoma HepG2 liver cell. Interestingly, these non‐redundant contributions involve proteins related to cellular processes inferred from mouse knockout studies (GalNAc‐T1) or GWAS (GalNAc‐T2) 21, 22, 23. The study points to a new view of the regulation of protein O‐glycosylation, suggesting that individual GalNAc‐T isoforms actually serve more specialized roles in distinct cellular processes. The results provide a fruitful discovery platform for essential non‐redundant functions of site‐specific O‐glycosylation against the background of what might be labeled as variational noise.
Results and Discussion
A quantitative differential O‐glycoproteomics strategy to map GalNAc‐T functions
We used the so‐called SimpleCell strategy 19 to develop quantitative O‐glycoproteome analyses. The strategy relies on knockout (KO) of the COSMC gene, a private chaperone for the C1GalT1 enzyme controlling most O‐glycan extension in cells, which leaves the O‐glycoproteome with rather homogenous Tn O‐glycans that can be captured by Vicia villosa (VVA) lectin chromatography. As the GALNT2 gene has been identified as a candidate gene for dyslipidemia, we choose to use the human HepG2 cell line, which is a highly differentiated liver cell line and widely used as a model system for lipid and cholesterol metabolism 24, 25. Human liver and HepG2 cells express a limited number of GalNAc‐T isoforms, mainly the GalNAc‐T1 and GalNAc‐T2 isoforms with only low levels of expression of GalNAc‐T10 and GalNAc‐T11 (Figs 1A and EV1A). We developed two clones each of a parent HepG2 SimpleCell line (HepG2SC) and isogenic lines without GalNAc‐T1 (HepG2SCΔT1) or without GalNAc‐T2 (HepG2SCΔT2) by ZFN‐mediated knockout (KO) 4. We also introduced GalNAc‐T3 to HepG2 cells to probe the effects of expressing a dominant foreign GalNAc‐T isoform not normally expressed in liver. In contrast, to GalNAc‐T1 and GalNAc‐T2, GalNAc‐T3 is differentially expressed in cells and interestingly, markedly unregulated in cancer 26. We used a previously described site‐specific ZFN‐mediated knockin (KI) strategy 4 to introduce stable expression of GalNAc‐T3 in HepG2SC (HepG2SC+T3) and the expression level and Golgi topology estimated by immunocytology was comparable to what is found in cell lines normally expressing this isoform (Fig EV1B). All clones were validated by DNA sequencing and characterized by immunocytochemistry (ICC) for loss or gain of gene products as well as O‐glycophenotype change (Fig EV1B). The total collection of cell lines used in the study is summarized in Table EV1.
Figure 1. The isogenic HepG2 liver cell model system and schematic depiction of the workflow for quantitative identification of isoform‐specific O‐glycosylation.
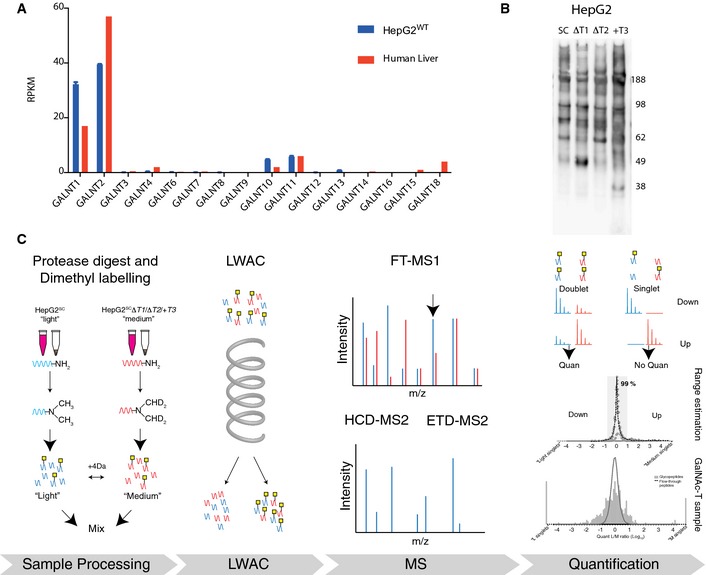
- GalNAc‐T expression levels in human liver (the body map data for human liver was kindly provided by the Gene Expression Applications research group at Illumina, http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-513/) and HepG2 wild‐type cells quantified by RNAseq (n = 2, bars representing mean ± SD).
- VVA lectin immunoblot of secretomes from HepG2 SC, SCΔT1, SCΔT2, and SC+T3.
- Digests of HepG2SC were always labeled with “light” (L) isotope and each of the three HepG2SCΔT1, HepG2SCT2, and HepG2SC+T3 were labeled with “medium” (M) isotope. After labeling, samples were mixed and glycopeptides were enriched on a long VVA lectin column. Labeled glycopeptides were analyzed using nLC‐MS/MS, the m/z of glycopeptide precursor ions in the labeled samples was measured, relative abundances calculated and glycopeptide ions selected for fragmentation were sequenced thus giving rise to total glycopeptide identification and quantitated glycopeptide identification.
Figure EV1. Characterization of GalNAc‐T expression in HepG2WT and KO model cell lines.
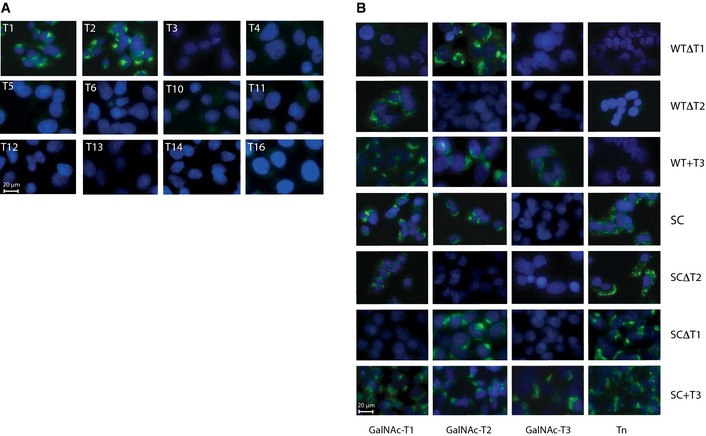
- ICC of HepG2WT cells with MAbs to a panel of human GalNAc‐Ts. Scale bar, 20 µm.
- Characterization of generated isogenic HepG2 KO cell lines by ICC with MAbs to GalNAc‐Ts and Tn (clone 5F4). The intensity of GalNAc‐T3 labeling and subcellular localization was essentially identical to what we have previously demonstrated in a variety of human cancel cell lines expressing T3 8. Scale bar, 20 µm.
We first evaluated gross effects of KO/KI of the GALNT genes in HepG2 cells by SDS–PAGE Western blotting with the anti‐Tn lectin VVA of secreted proteins from each cell line which revealed only subtle differences in labeled protein banding patterns (Fig 1B). For quantitative evaluation of changes in the O‐glycoproteome, we used stable differential isotope dimethyl labeling 27 of tryptic digests of total cell lysates and VVA lectin‐enriched culture medium (Fig 1C). Total digests from HepG2SC were labeled with light (L) reagent, and the digests from each isogenic HepG2SCΔT1/HepG2SCΔT2/HepG2SC+T3 mutant lines with medium (M) reagent. Thus, each analysis included a mixture of (L) labeled peptides from HepG2SC and (M) labeled peptides from one of the ΔT/+T mutant cell lines, which were enriched for glycopeptides by VVA Lectin Weak Affinity Chromatography (LWAC) and further analyzed by LC‐MS/MS. This approach ensured triplicate analysis of the HepG2SC glycoproteome and direct comparative and quantitative analysis of the effects of the altered cellular repertoire of GalNAc‐Ts. We analyzed total cell lysates (TCL) and secretomes (SEC) separately as this has previously resulted in substantially different O‐glycoproteomes 28, 29.
The overall O‐glycoproteome of HepG2sc
The complete datasets from the three differential O‐glycoproteomes (HepG2SCΔT1, HepG2SC ΔT2, and HepG2SC+T3) and two sources (TCL and SEC) are listed in Table EV2 and a summary shown in Fig 2 (all glycoproteome data are available at http://www.ebi.ac.uk/pride/archive/projects/PXD002770). We identified 631 O‐glycoproteins (1,742 unambiguously assigned O‐GalNAc sites) from TCL and SEC, which is a substantial expansion compared to the 74 distinct O‐glycoproteins (219 sites) identified using a non‐quantitative approach and analysis on a LTQ‐Orbitrap XL hybrid spectrometer with lower sensitivity 4 (Fig 2A). More than 50% of the O‐glycoproteins identified are novel compared to our previous analysis of 12 human SimpleCell lines from different organs 20 (Fig 2A), which may partly be attributed to use of a more sensitive mass spectrometer.
Figure 2. The overall HepG2 O‐glycoproteome.
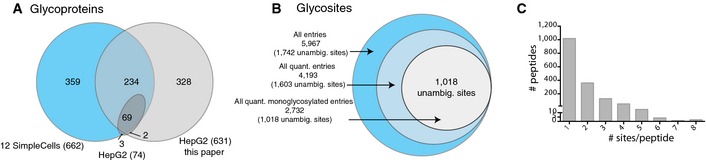
- Relative numbers of all sites, quantified sites, and quantified single sites identified in this study.
- Distribution of glycosites per protein identified in this study.
The GalNAc‐T differential O‐glycoproteomes in HepG2SC
Out of the total 1,742 unambiguously assigned glycosites, 1,603 (92%) were quantifiable (Fig 2B). Quantification takes place at the level of peptide or peptide fragment mass spectrum, and this poses a number of complications with peptides containing more than one O‐glycosite. In particular, interpretation of the contribution of individual GalNAc‐Ts and their substrate specificities is ambiguous because GalNAc‐Ts often function successively to cooperate in glycosylation of clustered glycosites through lectin‐mediated interactions with partially glycosylated substrates or through direct recognition of a substrate site with and adjacent GalNAc glycosite 1. However, most of the identified O‐glycosites were found as monoglycosylated peptides in the protease digest in agreement with our previous findings (Fig 2C) 20, and we therefore limited the analysis and exclusively considered monoglycosylated peptides in the comparative analysis of the three sample datasets (HepG2SCΔT1/HepG2SC ΔT2/HepG2SC+T3). The distribution of total quantified and monoglycosylated peptides were similar among the three samples with the same relative number of unique glycopeptide identifications (Fig EV2C and D), which provides evidence that this limitation does not bias the conclusions drawn. All identified and quantitated monoglycosylated peptides identified in the TCL and SEC from HepG2SCΔT1/HepG2SC ΔT2/HepG2SC+T3 samples are summarized in Table EV2, first sheet.
Figure EV2. Differential O‐glycoproteomes in HepG2SC .

-
AColumn diagram showing the relative distribution of identified glycoproteins between TCL, SEC, and overlap between TCL and SEC in all samples HepG2SCΔT1, HepG2SCΔT2, and HepG2SC+T3, illustrating that a comparable number of glycoproteins were identified in the three paired sample sets.
-
BSubcellular localization ontology of identified O‐glycoproteins from HepG2SC showing an overrepresentation of extracellular, membrane bound, Golgi‐ and ER‐resident proteins. Interestingly, we identified nuclear‐ and cytoplasmic‐resident proteins, albeit at much lower frequencies. GalNAc and GlcNAc are exact isobars, and in this type of O‐glycoproteomics studies, there is potential for including contaminating O‐GlcNAc glycopeptides 9, 10. Upon closer examination, approximately 50% of these proteins had multiple GO annotations, including terms relating to the secretory pathway. However, manual inspection of the remaining proteins revealed that these had no annotations to the secretory pathway and were predicted not to have signal peptide sequences, suggesting that these did represent contaminating glycopeptides. These proteins were excluded from the statistical analysis and listed separately (Table EV2).
-
C, DVenn diagrams showing the distribution of all quantified peptides (C) or all quantified mono‐glycosylated peptides (D) between HepG2SCΔT1, HepG2SCΔT2, and HepG2SC+T3.
To identify candidate GalNAc‐T isoform‐specific substrate glycosites, we analyzed histograms based on glycopeptide M/L ratios for each paired sample set (Fig 3A–C). Loss of function of a GalNAc‐T isoform is expected to result in loss of glycopeptides with specific acceptor sites for that isoform and appearance of L singlets (low M/L ratios), while de novo introduction of a GalNAc‐T is expected to produce M singlets (high M/L ratios). However, glycopeptides with, for example, two glycosylation sites of which only one is isoform‐specific can potentially produce a glycopeptide with only one site, which would appear as a M singlet based on the criteria of our analysis, thus explaining the non‐Gaussian distribution with both light and medium shoulders and singlets (see Fig EV3 and Appendix Supplementary Methods).
Figure 3. Differential O‐glycoproteomes in HepG2SC .

-
A–CHistograms showing the distribution of M/L quantitation ratios of monoglycosylated peptides identified in (A) HepG2SCΔT1, (B) HepG2SC ΔT2, and (C) HepG2SC+T3. Glycopeptides with M/L ratio less than −1 are colored red, and glycopeptides with a M/L ratio greater than +1 are colored green.
-
D–FVenn diagrams showing the distribution of candidates for isoform‐specific sites among HepG2SCΔT1, HepG2SCT2, and HepG2SC+T3 applying a log10 (±1) cutoff (D) (excluding sites identified in both TCL and SEC for each isoform), and (E) TCL alone and (F) SEC alone.
Figure EV3. Considerations for isoform specificity.

- Schematic depiction of how low and high M/L ratios and singlets may occur in both directions. Mono‐glycosylated peptides with isoform‐specific sites may be lost or downregulated (M/L ratio ≤ −1 (Log10)), and glycopeptides with two close adjacent O‐glycosites, of which one is isoform‐specific, may result in de novo appearance or upregulated (M/L ratio ≥1 (Log10)) of a mono‐glycosylated peptide with the remaining non‐isoform‐specific site.
- Histogram showing the proportion of mono‐glycosylated peptides that have potential glycosylation sites (sites previously identified in 12 human cell lines 2) within the peptide sequence (green). Blue, total number of quantitated monoglycosylated peptides. A slightly higher proportion of the upregulated singlet glycopeptides for GalNAc‐T1 (67% vs. 58%), GalNAc‐T2 (72% vs. 51%), and the downregulated glycopeptides for GalNAc‐T3 KI (60% vs. 41%), had previously been identified as glycopeptides with two or more O‐glycosites, indicating that at least some of these sites may appear as the result of loss of (T1/T2) or gain of (T3) an isoform‐specific glycosite.
- Distribution of isoform‐specific sites per glycoprotein.
We selected a conservative M/L cutoff ratio of 1:10 for selection of candidate isoform‐specific glycopeptides (< 1:10 for GalNAc‐T1 or GalNAc‐T2 KO and > 10:1 for GalNAc‐T3 KI) and included L or M singlets in this analysis (Fig 3 and Table EV2 sheet 2–7). Using these criteria, we identified a total of 75 glycopeptides (44 from TCL and 36 from SEC) that were essentially only found in HepG2SC and not in ΔT1 isogenic cells with this isoform (Fig 3A). The same analysis with ΔT2 isogenic cells resulted in identification of 81 glycopeptides (32 from TCL and 57 from SEC) (Fig 3B) from 67 different proteins. When GalNAc‐T3 was introduced de novo, we found 131 glycopeptides (46 from TCL and 98 from SEC) that were present only in HepG2SC+T3 (Fig 3C). The majority of identified GalNAc‐T isoform‐specific candidate sites were found on glycoproteins with only one O‐glycosite identified (Fig EV3C and Table EV2). The quantitative O‐glycoproteomics strategy thus enabled us to monitor ~1,000 O‐glycosites in isogenic cell pairs with and without a single GalNAc‐T isoform, and for the first time, we can estimate the size and nature of the non‐redundant O‐glycoproteome in a cell line expressing the two dominant GALNT1 and GALNT2 isoforms. About 10% of the identified glycosites are substantially affected (more than 10 fold) by loss or gain of individual GalNAc‐T isoforms, while the majority are unaffected or partially affected. We provided validation of a fraction of the isoform‐specific candidate O‐glycosites using in vitro enzyme analysis. We previously showed that in vitro enzyme assays with short peptides correlate well with in vivo activity and specificity in cells 20, 30, and here, we expanded the panel of peptides to include representative subsets of those identified here. For comparison, we also selected a subset of glycosites that were found to be representative of redundant functions of GalNAc‐Ts and predicted not to be isoform‐specific substrates. We tested recombinant soluble versions of the three isoforms GalNAc‐T1, GalNAc‐T2, and GalNAc‐T3, as well as two other isoforms (T4 and T11) weakly expressed in liver and HepG2 cells (Fig 1A). We also analyzed the non‐redundant O‐glycosites for prominent sequence motifs but did not identify such (Fig EV4C), although GalNAc‐T2 clearly shows preference for Pro in position −1 and −3, and GalNAc‐T3 tolerates more charged residues both N‐ and C‐terminal to glycosylation sites as previously demonstrated for synthetic random peptide libraries 31. The results confirmed that the majority of the candidate sites represent GalNAc‐T isoform‐specific acceptor substrate glycosites and thus provide further validation for the differential glycoproteomics strategy (Fig EV4A and B and Table EV3). Clearly further validation and testing in other cell systems are required for arriving at a definitive isoform‐specific dataset.
Figure EV4. In vitro GalNAc‐T enzyme assay validation of candidate isoform‐specific O‐glycosites.
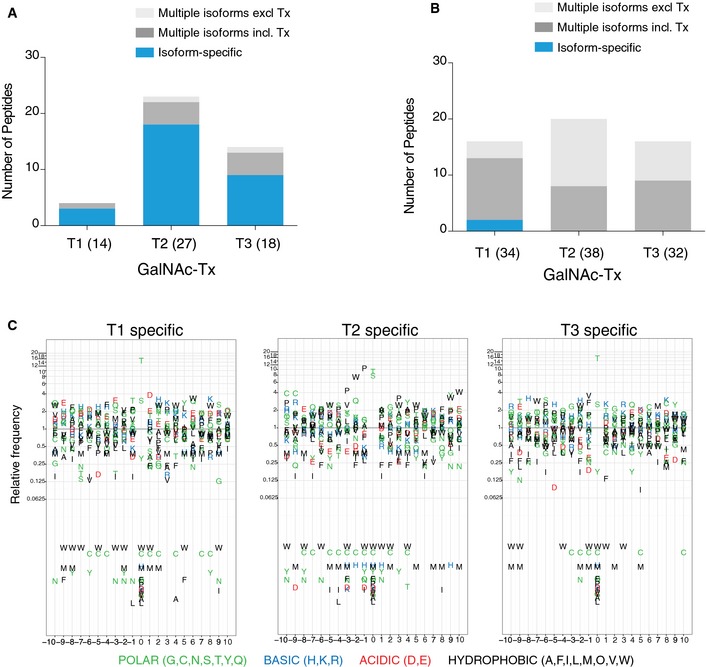
- 20‐mer peptides designed to cover identified candidate isoform‐specific O‐glycosites as acceptor substrates for recombinant human GalNAc‐T1, GalNAc‐T2, and GalNAc‐T3 enzymes (see Table EV3 for a summary of data). Total number of peptides tested for each isoform is shown in parenthesis. Since GalNAc‐T3 is not endogenously expressed in HepG2 cells, we disregarded overlapping activity of this enzyme with a few substrates in qualifying isoform specificities of GalNAc‐T1 and GalNAc‐T2 in the HepG2 cellular context. Blue color marks isoform‐specific peptides, dark gray marks peptides glycosylated by multiple isoforms, including the specific isoform (Tx), and light gray color the peptides glycosylated by multiple isoforms excluding the specific isoform (Tx). In testing GalNAc‐T1 candidate isoform‐specific sites, only 4/14 peptide designs served as substrates for any of the enzyme isoforms tested, and of these, 3/4 were found to be specific substrates for T1. For GalNAc‐T2, 23/27 peptides were used by the enzymes tested and of these, 19/23 were specific for T2. For GalNAc‐T3, 14/18 peptides served as substrates for the tested enzymes and of these, 9/14 were specifically glycosylated by T3.
- For comparative testing of the glycosites that were not predicted to be isoform‐specific in the HepG2SCΔT1 analysis, we found that 2/35 peptides were weak substrates for T1, 14/35 was glycosylated by several isoforms, and 19/35 were not glycosylated by any isoform tested. For the isoform non‐specific sites found in the HepG2SCΔT2, 0/38 peptides were specific for T2, 20/38 were glycosylated by several isoforms and 18/38 did not serve as substrates for any isoforms. For HepG2SCΔT3, 0/32 peptides were specific for T3, 16/32 were glycosylated by several isoforms and 16/32 were not glycosylated in vitro.
- Substantial efforts have been devoted to searching for consensus motifs for O‐GalNAc glycosylation 11. We therefore aligned the candidate isoform‐specific glycosite sequences and analyzed these by frequency plots showing the relative frequency of amino acids ± 10 sites from GalNAc‐T1, GalNAc‐T2, or GalNAc‐T3 isoform‐specific glycosylation sites. For GalNAc‐T1‐specific sites, we demonstrated a preference for acidic amino acids in position +1 and −3 and for basic His in position +2 and −4. For GalNAc‐T2‐specific sites, we found a more clear preference (4‐ to 12‐fold enrichment) for Pro in positions −1 and −3 and Trp in position −2. Interestingly, we also observed a diminishment of certain charged residues in position −3 to +1, as His and Asp residues are never found in these positions. For GalNAc‐T3‐specific sites, we found a subtle enrichment for basic and hydrophobic residues in positions −1, −2, +1, and +3. Interestingly, in general methionine, cysteine and the large bulky tryptophan residues are under‐represented in sequences surrounding isoform‐specific sites of all three isoforms. Overall, these results are well in line with previous in vitro glycosylation studies 11, and support the idea that isoform discriminating amino acid preference exists at least to some extent.
Comparison of the identified candidate glycosites for the three GalNAc‐T isoforms (Fig 3D–F) revealed that these were largely different for each GalNAc‐T isoform, which provides strong support for the validity of the strategy and the identified candidates. Among proteins with T1‐specific sites (Table EV2), we found several proteins involved in basement membrane (BM)/extracellular matrix (ECM) organization (e.g., HSPG2, FN1, VCAN, DAG1, SCD4, FRAS1, FGA, MIA3, VWA1, MATN3), which is in agreement with and confirms the proposed association of murine Galnt1 and BM/ECM formation 32, 33. Furthermore, we found a T1 isoform‐specific glycosite on Osteopontin (Ser280), which correlates well with and confirms a previous report demonstrating reduced O‐glycosylation of Osteopontin in Galnt1 −/− KO mice 34. In agreement with our previous studies 4, we found ApoC‐III (Thr94) among the T2‐specific glycosites. Moreover, among the T2‐specific sites identified in our previous non‐quantitative study 4 for which quantitative data were obtained in this study, > 80% were identified as T2‐specific candidates. Interestingly, a total of 13 of the 67 proteins with identified T2 candidate‐specific O‐glycosites (Table EV2) are involved in lipid or cholesterol metabolism (Apo‐CII, Apo‐CIII, Apo‐AIV, Apo‐AV, Apo‐E and Apo‐H, MIA2, NMB, NUCB2, PTGFRN, DLK1, LIPG and LSR). The remaining proteins constitute a diverse set of proteins with no interpretable connection.
De novo introduction of GalNAc‐T3 resulted in a large expansion of the O‐glycoproteome of HepG2 comparable to the loss observed with knockout of GalNAc‐T1 and GalNAc‐T2 (Fig 3). GalNAc‐T3 has unique substrate specificity and accepts charged residues in near proximity of acceptor sites (Table EV2 and Fig EV4). Pro‐proteins rely on limited proteolytic activity upon activation and such cleavage sites are often composed of basic residues. Interestingly, several of the T3‐specific sites were located in close proximity to such activation sites either on the active protein or in the propeptide (PROS1, FV, ASHG, LOX, F2RL1, and RARRES2). A number of the identified candidates for T3‐specific glycosylation have previously been found in human cell lines endogenously expressing GalNAc‐T3, for example, Pro‐SAAS (Thr53) and PAR‐2 (Thr49) 20. The analysis shows that de novo introduction of a GalNAc‐T can have dramatic effects on the O‐glycoproteome of a cell and may serve to help understand the functional consequences of the aberrant expression of GalNAc‐T3 found in many cancers 35, 36, 37.
HepG2 and liver express minor levels of other less well‐characterized GalNAc‐T isoforms (GalNAc‐T4, GalNAc‐T10, GalNAc‐T11) (Figs 1 and EV1A), and their contribution was not addressed in the present study. GalNAc‐T4 and GalNAc‐T10 are mainly functioning as so‐called follow‐up enzymes using partially GalNAc‐glycosylated substrates to complete glycosylation 1, and the analysis of these functions in a quantitative way is not simple with the current strategy where we have limited the analysis to single O‐glycosites. GalNAc‐T11 is unique in controlling O‐glycosylation of the low‐density‐lipoprotein (LDL) receptor in the LDL binding region 38, and here, we indentified one of these O‐glycosites (T108), and the glycosylation of this site was as predicted not affected by loss or gain of the three other GALNT genes studied (Table EV2). We recently found that GalNAc‐T11‐mediated O‐glycosylation is required for LDL binding and uptake (unpublished observations), and it is our hypothesis that this isoform serves a selective role in these functions. However, at this stage, the finding that loss or gain of GalNAc‐T1, GalNAc‐T2, and GalNAc‐T3 isoforms in HepG2 does not affect LDLR O‐glycosylation provides additional validation for the strategy.
Our study provides the first comprehensive and quantitative cell‐based evidence that the O‐glycoproteome of a cell is differentially regulated by its repertoire of GalNAc‐T isoenzymes expressed. The expression of individual GalNAc‐T isoenzymes thus affects a limited subset of the O‐glycoproteome, while the majority of the O‐glycoproteome is covered by redundancy among the GalNAc‐Ts expressed in a cell. This provides strong evidence that cells have the potential for differential and dynamic regulation of O‐glycosylation of distinct subsets of proteins by the non‐redundant functions of individual GalNAc‐Ts. Moreover, the results suggest that the non‐redundant contributions to the O‐glycoproteome of the three GalNAc‐T isoenzymes studied can be categorized to different biological processes. In particular, we found strong support for the putative roles in lipid metabolism for GalNAc‐T2 and in basement membrane/extracellular matrix composition for GalNAc‐T1, where each isoenzyme controlled O‐glycosites in multiple proteins with pivotal roles in these pathways 22, 23, 32.
The non‐redundant functions identified for each GalNAc‐T are based on complete loss or gain of function, and hence display the extreme effects. It is therefore still unknown how quantitative changes in expression of individual GalNAc‐Ts will affect the O‐glycoproteome, and we are currently approaching this with reintroduction of inducible GalNAc‐Ts in cell line systems. Within the non‐redundant subsets, there will be a broad range of acceptor substrates for which the kinetic properties of each particular GalNAc‐T isoform will clearly be very different as suggested by extensive in vitro enzyme analyzes (Fig EV4) 30, and it may be expected that only a fraction of the non‐redundant O‐glycosites identified, in fact, will be substantially affected by minor quantitative changes in expression of individual GalNAc‐Ts. Thus, physiological regulation of individual GalNAc‐Ts in cells may have even more subtle and specific consequences for the O‐glycoproteome and a major challenge will be to identify O‐glycosites that are truly regulatable by quantitative changes in GalNAc‐T expression. More broadly, the challenge is to identify the few non‐redundant O‐glycosites that are essential for health, and that when not glycosylated, due to deficiencies in or dysregulation of GALNT genes, cause disease or susceptibility to disease.
Transcriptional analysis of isogeneic HepG2WT cell lines with different GalNAc‐T repertoires
To evaluate potential transcriptional compensation of GalNAc‐T genes in response to loss‐of‐function mutations, we generated a panel of isogenic HepG2WT cell lines with the same KO and KI of GALNTs as analyzed for the O‐glycoproteomics studies (Fig EV1B and Table EV1). We used wild‐type HepG2 cells for these experiments such that potential effects in the transcriptomes could be related solely to loss or gain of the GalNAc‐T isoforms. First, we used immunocytology to compare expression of 12 GalNAc‐T isoforms for which we have developed monoclonal antibodies to, and this demonstrated that the protein expression of these was essentially unaltered by loss or gain of GalNAc‐T1, GalNAc‐T2, and GalNAc‐T3 in HepG2 SC and WT cells (Fig EV1 and data not shown). The transcriptomes of two independent clones of each HepG2WTΔT1/HepG2WTΔT2/HepG2WT+T3 KOs were analyzed by RNA‐sequencing (RNAseq, data are available at www.ebi.ac.uk/arrayexpress with accession number E‐MTAB‐3844), and although we saw minor differences in GALNT1 and GALNT2 transcript levels, we found no evidence of compensatory changes in transcript levels in any members of the large GALNT family (Fig 4) or other known glycosyltransferase genes (for an updated list of glycosyltransferase genes, see 39), suggesting that they serve independent functions without cross talk.
Figure 4. RNAseq transcriptome analysis of isogenic HepG2 cells.
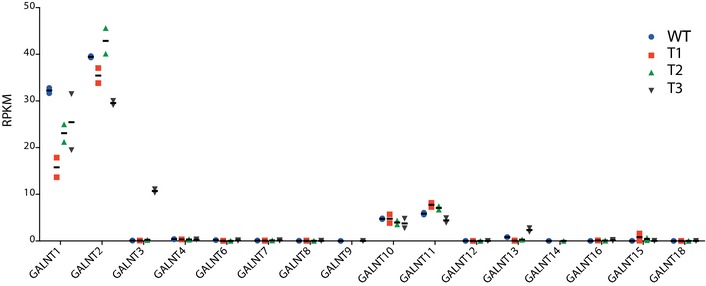
Transcript levels in RPKM (reads per kilobase transcriptome per million mapped reads) of GalNAc‐Ts in two individual clones of isogenic HepG2 lines as indicated. Apart from minor changes in the GALNT1 and GALNT2 transcript levels, all transcript levels were similar. Bars represent mean values, and error bars represent SD.
To further explore the transcriptome data, we used a stringent and conservative statistical analysis for differential RNAseq transcriptomic data 40, and comparison of the transcriptomes of HepG2WT and HepG2WTΔT1 showed that 908 genes were significantly upregulated and 791 genes downregulated (Fig EV5 and Table EV4). Considerably fewer transcripts were affected in HepG2WTΔT2 with 254 genes upregulated and 210 downregulated (Fig EV5 and Table EV5). Remarkably, the de novo introduction of the GalNAc‐T3 isoform in HepG2 that resulted in a marked expansion of the O‐glycoproteome (Table EV2) had little effect on the transcriptome (Fig EV5 and Table EV6) as only 14 genes were significantly upregulated and 53 genes downregulated in HepG2WT+T3. This demonstrates for the first time that loss of endogenous GalNAc‐T isoforms has major impact on cells, while in contrast aberrant expression of GalNAc‐T3 in the context of a liver cell only appears to have little effect.
Figure EV5. RNAseq transcriptome analysis of isogenic HepG2 cells.
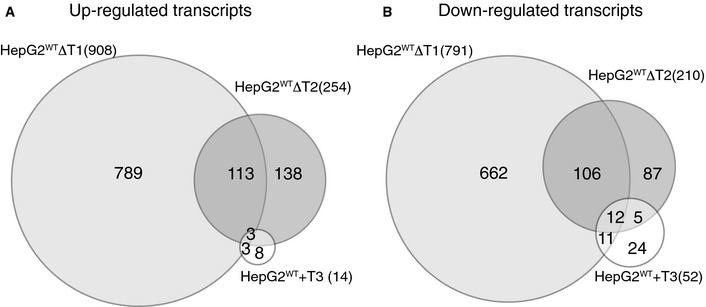
-
A, BVenn diagram showing distribution of up‐ (A) and downregulated (B) genes between HepG2WTΔT1, HepG2WTΔT2, and HepG2WT+T3 compared to HepG2WT.
Despite a limited number of transcripts being affected by de novo introduction of GalNAc‐T3 (Fig EV5 and Table EV6), it may be interesting to note that matrix metalloproteinase 14 (MMP14 also known as MT1‐MMP) was downregulated more than 50‐fold by expression of GalNAc‐T3 (HepG2WT+T3). MMP14 promotes cellular migration and invasion in vivo, and a number of specific substrates have been reported 41. Specifically, we identified a non‐redundant glycosite (T263) in close proximity to the MMP14 cleavage site in matrix metalloproteinase 11 (MMP11 also known as Stromelysin‐3 42) (253VQHL↓YGQPWPTVTSRT268, cleavage site arrow and glycosite underlined). MMP11 is inactivated by the MMP14 processing in this site 42. MMP11 is upregulated in cancer and suggested to play a role in invasive growth 43, 44, 45, and it is possible that the upregulation of GalNAc‐T3 (and perhaps its paralog GalNAc‐T6 with similar specificity 46 often found in cancer 47, 48 may help block MMP14‐mediated inactivation of MMP11 and thus promote invasive growth. Clearly, this is speculation and further studies are needed to address the functions of these GalNAc‐T isoforms, but the example demonstrates the potential connectivity of site‐specific O‐glycosylation and relevant cellular processes.
Finally, to further explore the marked differential gene expression observed in cells without GALNT1 or GALNT2, we analyzed the differential transcriptomes for HepG2WTΔT1 and ΔT2 by GO enrichment analysis to evaluate potential global effects. We found significant (hypergeometric test, P‐value < 0.05) enrichment of distinct combinations of biological process terms for each isoform (Fig EV6), suggesting that individual GalNAc‐Ts affect the transcriptome of cells in isoform‐specific manner and that complete loss of function of a GalNAc‐T gene induces a selective transcriptional response. Clearly, further studies are needed to validate these initial findings and probe into cause and effects. Interestingly, however, GO terms significantly enriched for among T2‐specific upregulated transcripts were mainly related to lipid metabolism biological processes, which is in line with the isoform‐specific glycosylation functions identified here and the genetic studies associating GALNT2 polymorphisms with dyslipidemia 22, 23.
Figure EV6. Differential transcriptomics and Gene Ontology Enrichment analysis.

-
A–CRNAseq expression data plotted for HepG2WT/HepG2WTΔT1 (A), HepG2WT/HepG2WTΔT2 (B), and HepG2WT/HepG2WT+T3 (C) with expression level as a function of fold change. Top 10 biological process ontology terms among up‐ or downregulated transcripts are shown in table format. The biological coefficients of variations between HepG2WT clones with the same gene KO/KI were 19, 25, and 29% for GalNAc‐T1, GalNAc‐T2, and GalNAc‐T3, respectively (not shown). These variations are within the boundaries of what is expected when two individual clonal cell lines are compared. In contrast, the biological coefficients of variations between the two individual datasets for pools of HepG2WT cells were as expected much lower (2–3%).
An increasing number of GALNT genes have been identified as candidate genes for human diseases (for review see 18). Apart from the strong evidence for GALNT2 and dyslipidemia, studies have pointed to associations of GALNT4 with coronary artery disease 49, GALNT7 with Alzheimer's disease 50, GALNT10 with obesity and high body mass index 51, GALNT13 with elevated tricuspid regurgitation jet in sickle cell disease patients 52, and GALNT14 with hepatocellular carcinoma 53. The strategy presented here produced a list of candidate glycoproteins and glycosites specifically controlled by the GalNAc‐T2 isoform in a liver cell line that may aid in dissection of molecular mechanisms underlying the association with dyslipidemia. More generally, the quantitative O‐glycoproteomics strategy should be applicable to any GalNAc‐T isoform in discovery and dissection of non‐redundant biological functions in appropriate cell line systems and eventually in vivo in model organisms or patients. Although perhaps premature, the quantitative transcriptomics of appropriate isogenic cell models may also support discovery of disease‐causing functions.
In conclusion, the repertoire of GalNAc‐Ts of a cell direct its O‐glycoproteome, and individual GalNAc‐Ts serve limited non‐redundant functions that may affect distinct subsets of metabolic processes. The developed isogenic cell models open for wider discovery of functions of the large GalNAc‐T family and deciphering of molecular mechanisms causing disease.
Materials and Methods
ZFN gene targeting
HepG2ΔT1 and HepG2ΔT2 in WT and SC background were generated as previously described 4. HepG2+T3 cells were generated by stable integration of GalNAc‐T3 as previously described 4 (see Appendix Supplementary Methods). Cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 1% glutamine and 1% NEAA.
For ICC characterization, cells were grown on sterile teflon‐coated slides (Clearcell, Histolab) and briefly fixed with ice‐cold acetone. Cells were then sequentially incubated with MAbs to GalNAc‐T1 (4D8), GalNAc‐T2 (4C4), GalNAc‐T3 (2D10), or Tn O‐glycans (5F4) overnight and FITC‐conjugated rabbit anti‐mouse immunoglobulin (Dako) for 45 min and mounted with ProLong Gold antifade reagent (DAPI) (Invitrogen). Fluorescence microscopy was performed using a Zeiss Axioskop 2 plus with an AxioCam MR3.
Dimethyl labeling and lectin weak affinity chromatography
Samples of 100 ml cell culture supernatant or 0.5 ml packed cells were processed 4 (see Appendix Supplementary Methods). For quantitative differential glycoproteomics, tryptic digests of isogenic pairs of SCs were labeled with light (L) and medium (M) isotopomeric dimethyl labels 27. Cleared acidified digests were loaded in equal amounts (peptide concentration) onto equilibrated SepPak C18 cartridges (Waters) followed by 3 × CV 0.1% TFA wash. Digests were labeled on‐column by adding 5 CV 30 mM NaBH3CN and 0.2% formaldehyde in 50 mM sodium phosphate buffer pH 7.5 (L‐labeling), or 30 mM NaBH3CN and 0.2% D‐formaldehyde in 50 mM sodium phosphate buffer pH 7.5 (M‐labeling). Columns were washed using 3 CV 0.1% FA and eluted with 0.5 ml 50% MeOH in 0.1% FA. Labeled GalNAc glycopeptides were separated from non‐glycosylated peptides using a long VVA‐agarose LWAC 19.
Mass spectrometry
LWAC elution fractions were fractionated by isoelectric focusing according to 28 and analyzed by EASY‐nLC 1000 UHPLC (Thermo Scientific) interfaced via nanoSpray Flex ion source to an LTQ‐Orbitrap Velos Pro spectrometer (Thermo Scientific) as previously described 29. Data were processed using Proteome Discoverer 1.4 software (Thermo Scientific) (see Appendix Supplementary Methods).
In vitro glycosylation
Peptides were designed and synthesized with acceptor residues in center position of a 20‐mer peptide (NeoBioScience). GalNAc‐transferase assays, monitored by MALDI‐TOF mass spectrometry, were performed as previously describes 30.
RNA transcriptomic analysis
Total RNA was extracted from exponentially growing cells using RNeasy® kit (Qiagen). RNA integrity and quality were determined using Bioanalyzer instrumentation (Agilent Technologies). RNAseq was performed by Beijing Genetics Institute (BGI). Briefly, library construction was performed using Illumina Truseq RNA Sample Preparation Kit and subjected to next‐generation sequencing using Illumina HiSeq 2000 System (Illumina, USA). See Appendix Supplementary Methods for more details.
Bioinformatic analysis
The aligned reads from the BGI analysis were analyzed combining DESeq 54 and EdgeR 55 methods to determine the differentially expressed transcripts (see Appendix Supplementary Methods). GO enrichment analyses of glycoproteomic and transcriptomic data were performed in R using GO and GOStats 56.
Data availability
The mass spectrometry glycoproteomics data have been deposited to the ProteomeXchange Consortium 57 via the PRIDE partner repository with the dataset identifier PXD002770 and RNAseq data can be found at www.ebi.ac.uk/arrayexpress with accession number E‐MTAB‐3844.
Author contributions
KTS, HJJ, SYV, and HC designed and performed experiments, analyzed data, and wrote the paper. HHW and EPB analyzed data. YK, CKG, and SLK performed experiments.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Review Process File
Acknowledgements
We thank James Paulson, Ramon Hurtado Guerrero, and Frederic Bard for critical reading of the manuscript. This work was supported by The Danish Research Councils (Sapere Aude Research Talent Grant to KTS), The Mizutani Foundation, Kirsten og Freddy Johansen Fonden, A.P. Møller og Hustru Chastine Mc‐Kinney Møllers Fond til Almene Formaal, The Novo Nordisk Foundation, a program of excellence from the University of Copenhagen (CDO2016), and The Danish National Research Foundation (DNRF107).
EMBO Reports (2015) 16: 1713–1722
References
- 1. Bennett EP, Mandel U, Clausen H, Gerken TA, Fritz TA, Tabak LA (2012) Control of mucin‐type O‐glycosylation: a classification of the polypeptide GalNAc‐transferase gene family. Glycobiology 22: 736–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gill DJ, Clausen H, Bard F (2011) Location, location, location: new insights into O‐GalNAc protein glycosylation. Trends Cell Biol 21: 149–158 [DOI] [PubMed] [Google Scholar]
- 3. Tran DT, Ten Hagen KG (2013) Mucin‐type O‐glycosylation during development. J Biol Chem 288: 6921–6929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schjoldager KT, Vakhrushev SY, Kong Y, Steentoft C, Nudelman AS, Pedersen NB, Wandall HH, Mandel U, Bennett EP, Levery SB et al (2012) Probing isoform‐specific functions of polypeptide GalNAc‐transferases using zinc finger nuclease glycoengineered SimpleCells. Proc Natl Acad Sci USA 109: 9893–9898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298: 1912–1934 [DOI] [PubMed] [Google Scholar]
- 6. Tabak LA (2010) The role of mucin‐type O‐glycans in eukaryotic development. Sem Cell Dev Biol 21: 616–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, Khamaysi Z, Behar D, Petronius D, Friedman V et al (2004) Mutations in GALNT3, encoding a protein involved in O‐linked glycosylation, cause familial tumoral calcinosis. Nat Genet 36: 579–581 [DOI] [PubMed] [Google Scholar]
- 8. Kato K, Jeanneau C, Tarp MA, Benet‐Pages A, Lorenz‐Depiereux B, Bennett EP, Mandel U, Strom TM, Clausen H (2006) Polypeptide GalNAc‐transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O‐glycosylation. J Biol Chem 281: 18370–18377 [DOI] [PubMed] [Google Scholar]
- 9. Benet‐Pages A, Orlik P, Strom TM, Lorenz‐Depiereux B (2005) An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet 14: 385–390 [DOI] [PubMed] [Google Scholar]
- 10. Esapa CT, Head RA, Jeyabalan J, Evans H, Hough TA, Cheeseman MT, McNally EG, Carr AJ, Thomas GP, Brown MA et al (2012) A mouse with an N‐Ethyl‐N‐nitrosourea (ENU) Induced Trp589Arg Galnt3 mutation represents a model for hyperphosphataemic familial tumoural calcinosis. PLoS ONE 7: e43205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ichikawa S, Sorenson AH, Austin AM, Mackenzie DS, Fritz TA, Moh A, Hui SL, Econs MJ (2009) Ablation of the Galnt3 gene leads to low‐circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression. Endocrinology 150: 2543–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Duncan EL, Danoy P, Kemp JP, Leo PJ, McCloskey E, Nicholson GC, Eastell R, Prince RL, Eisman JA, Jones G et al (2011) Genome‐wide association study using extreme truncate selection identifies novel genes affecting bone mineral density and fracture risk. PLoS Genet 7: e1001372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leonhard‐Melief C, Haltiwanger RS (2010) O‐fucosylation‘ of thrombospondin type 1 repeats. Methods Enzymol 480: 401–416 [DOI] [PubMed] [Google Scholar]
- 14. Loibl M, Strahl S (2013) Protein O‐mannosylation: what we have learned from baker's yeast. Biochim Biophys Acta 1833: 2438–2446 [DOI] [PubMed] [Google Scholar]
- 15. Boskovski MT, Yuan S, Pedersen NB, Goth CK, Makova S, Clausen H, Brueckner M, Khokha MK (2013) The heterotaxy gene GALNT11 glycosylates Notch to orchestrate cilia type and laterality. Nature 504: 456–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van der Post S, Subramani DB, Backstrom M, Johansson ME, Vester‐Christensen MB, Mandel U, Bennett EP, Clausen H, Dahlen G, Sroka A et al (2013) Site‐specific O‐glycosylation on the MUC2 mucin protein inhibits cleavage by the Porphyromonas gingivalis secreted cysteine protease (RgpB). J Biol Chem 288: 14636–14646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang L, Syed ZA, van Dijk Hard I, Lim JM, Wells L, Ten Hagen KG (2014) O‐glycosylation regulates polarized secretion by modulating Tango1 stability. Proc Natl Acad Sci USA 111: 7296–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schjoldager KT, Clausen H (2012) Site‐specific protein O‐glycosylation modulates proprotein processing ‐ deciphering specific functions of the large polypeptide GalNAc‐transferase gene family. Biochim Biophys Acta 1820: 2079–2094 [DOI] [PubMed] [Google Scholar]
- 19. Steentoft C, Vakhrushev SY, Vester‐Christensen MB, Schjoldager KT, Kong Y, Bennett EP, Mandel U, Wandall H, Levery SB, Clausen H (2011) Mining the O‐glycoproteome using zinc‐finger nuclease‐glycoengineered SimpleCell lines. Nat Methods 8: 977–982 [DOI] [PubMed] [Google Scholar]
- 20. Steentoft C, Vakhrushev SY, Joshi HJ, Kong Y, Vester‐Christensen MB, Schjoldager KT, Lavrsen K, Dabelsteen S, Pedersen NB, Marcos‐Silva L et al (2013) Precision mapping of the human O‐GalNAc glycoproteome through SimpleCell technology. EMBO J 32: 1478–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tenno M, Ohtsubo K, Hagen FK, Ditto D, Zarbock A, Schaerli P, von Andrian UH, Ley K, Le D, Tabak LA et al (2007) Initiation of protein O glycosylation by the polypeptide GalNAcT‐1 in vascular biology and humoral immunity. Mol Cell Biol 27: 8783–8796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R, Heath SC, Timpson NJ, Najjar SS, Stringham HM et al (2008) Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet 40: 161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kathiresan S, Melander O, Guiducci C, Surti A, Burtt NP, Rieder MJ, Cooper GM, Roos C, Voight BF, Havulinna AS et al (2008) Six new loci associated with blood low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol or triglycerides in humans. Nat Genet 40: 189–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hozumi Y, Kawano M, Jordan VC (2000) In vitro study of the effect of raloxifene on lipid metabolism compared with tamoxifen. Eur J Endocrinol 143: 427–430 [DOI] [PubMed] [Google Scholar]
- 25. Vallianou I, Peroulis N, Pantazis P, Hadzopoulou‐Cladaras M (2011) Camphene, a plant‐derived monoterpene, reduces plasma cholesterol and triglycerides in hyperlipidemic rats independently of HMG‐CoA reductase activity. PLoS ONE 6: e20516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sutherlin ME, Nishimori I, Caffrey T, Bennett EP, Hassan H, Mandel U, Mack D, Iwamura T, Clausen H, Hollingsworth MA (1997) Expression of three UDP‐N‐acetyl‐alpha‐D‐galactosamine:polypeptide GalNAc N‐acetylgalactosaminyltransferases in adenocarcinoma cell lines. Cancer Res 57: 4744–4748 [PubMed] [Google Scholar]
- 27. Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJ (2009) Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc 4: 484–494 [DOI] [PubMed] [Google Scholar]
- 28. Vakhrushev SY, Steentoft C, Vester‐Christensen MB, Bennett EP, Clausen H, Levery SB (2013) Enhanced mass spectrometric mapping of the human GalNAc‐type O‐glycoproteome with SimpleCells. Mol Cell Proteomics 12: 932–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang Z, Halim A, Narimatsu Y, Joshi HJ, Steentoft C, Schjoldager KT, Schulz MA, Sealover NR, Kayser KJ, Bennett EP et al (2014) The GalNAc‐type O‐glycoproteome of CHO cells characterized by the SimpleCell strategy. Mol Cell Proteomics 12: 3224–3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kong Y, Joshi HJ, Schjoldager KT, Madsen TD, Gerken TA, Vester‐Christensen MB, Wandall HH, Bennett EP, Levery SB, Vakhrushev SY et al (2015) Probing Polypeptide GalNAc‐Transferase Isoform Substrate Specificities by In Vitro Analysis. Glycobiology 25: 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gerken TA, Raman J, Fritz TA, Jamison O (2006) Identification of common and unique peptide substrate preferences for the UDP‐GalNAc:polypeptide alpha‐N‐acetylgalactosaminyltransferases T1 and T2 derived from oriented random peptide substrates. J Biol Chem 281: 32403–32416 [DOI] [PubMed] [Google Scholar]
- 32. Tian E, Hoffman MP, Ten Hagen KG (2012) O‐glycosylation modulates integrin and FGF signalling by influencing the secretion of basement membrane components. Nat Commun 3: 869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tian E, Stevens SR, Guan Y, Springer DA, Anderson SA, Starost MF, Patel V, Ten Hagen KG, Tabak LA (2015) Galnt1 is required for normal heart valve development and cardiac function. PLoS ONE 10: e0115861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miwa HE, Gerken TA, Jamison O, Tabak LA (2010) Isoform‐specific O‐glycosylation of osteopontin and bone sialoprotein by polypeptide N‐acetylgalactosaminyltransferase‐1. J Biol Chem 285: 1208–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang ZQ, Bachvarova M, Morin C, Plante M, Gregoire J, Renaud MC, Sebastianelli A, Bachvarov D (2014) Role of the polypeptide N‐acetylgalactosaminyltransferase 3 in ovarian cancer progression: possible implications in abnormal mucin O‐glycosylation. Oncotarget 5: 544–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taniuchi K, Cerny RL, Tanouchi A, Kohno K, Kotani N, Honke K, Saibara T, Hollingsworth MA (2011) Overexpression of GalNAc‐transferase GalNAc‐T3 promotes pancreatic cancer cell growth. Oncogene 30: 4843–4854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kato K, Takeuchi H, Kanoh A, Miyahara N, Nemoto‐Sasaki Y, Morimoto‐Tomita M, Matsubara A, Ohashi Y, Waki M, Usami K et al (2010) Loss of UDP‐GalNAc:polypeptide N‐acetylgalactosaminyltransferase 3 and reduced O‐glycosylation in colon carcinoma cells selected for hepatic metastasis. Glycoconj J 27: 267–276 [DOI] [PubMed] [Google Scholar]
- 38. Pedersen NB, Wang S, Narimatsu Y, Yang Z, Halim A, Schjoldager KT, Madsen TD, Seidah NG, Bennett EP, Levery SB et al (2014) Low density lipoprotein receptor class a repeats are o‐glycosylated in linker regions. J Biol Chem 289: 17312–17324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hansen L, Lind‐Thomsen A, Joshi HJ, Pedersen NB, Have CT, Kong Y, Wang S, Sparso T, Grarup N, Vester‐Christensen MB et al (2015) A glycogene mutation map for discovery of diseases of glycosylation. Glycobiology 25: 211–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD (2013) Count‐based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 8: 1765–1786 [DOI] [PubMed] [Google Scholar]
- 41. Itoh Y (2015) Membrane‐type matrix metalloproteinases: their functions and regulations. Matrix Biol 44‐46C: 207–223 [DOI] [PubMed] [Google Scholar]
- 42. Buache E, Thai R, Wendling C, Alpy F, Page A, Chenard MP, Dive V, Ruff M, Dejaegere A, Tomasetto C et al (2014) Functional relationship between matrix metalloproteinase‐11 and matrix metalloproteinase‐14. Cancer Med 3: 1197–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rouyer N, Wolf C, Chenard MP, Rio MC, Chambon P, Bellocq JP, Basset P (1994) Stromelysin‐3 gene expression in human cancer: an overview. Invasion Metastasis 14: 269–275 [PubMed] [Google Scholar]
- 44. Anderson IC, Sugarbaker DJ, Ganju RK, Tsarwhas DG, Richards WG, Sunday M, Kobzik L, Shipp MA (1995) Stromelysin‐3 is overexpressed by stromal elements in primary non‐small cell lung cancers and regulated by retinoic acid in pulmonary fibroblasts. Cancer Res 55: 4120–4126 [PubMed] [Google Scholar]
- 45. Porte H, Chastre E, Prevot S, Nordlinger B, Empereur S, Basset P, Chambon P, Gespach C (1995) Neoplastic progression of human colorectal cancer is associated with overexpression of the stromelysin‐3 and BM‐40/SPARC genes. Int J Cancer 64: 70–75 [DOI] [PubMed] [Google Scholar]
- 46. Bennett EP, Hassan H, Mandel U, Hollingsworth MA, Akisawa N, Ikematsu Y, Merkx G, van Kessel AG, Olofsson S, Clausen H (1999) Cloning and characterization of a close homologue of human UDP‐N‐acetyl‐alpha‐D‐galactosamine: Polypeptide N‐acetylgalactosaminyltransferase‐T3, designated GalNAc‐T6. Evidence for genetic but not functional redundancy. J Biol Chem 274: 25362–25370 [DOI] [PubMed] [Google Scholar]
- 47. Li Z, Yamada S, Inenaga S, Imamura T, Wu Y, Wang KY, Shimajiri S, Nakano R, Izumi H, Kohno K et al (2011) Polypeptide N‐acetylgalactosaminyltransferase 6 expression in pancreatic cancer is an independent prognostic factor indicating better overall survival. Br J Cancer 104: 1882–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Freire‐de‐Lima L, Gelfenbeyn K, Ding Y, Mandel U, Clausen H, Handa K, Hakomori SI (2011) Involvement of O‐glycosylation defining oncofetal fibronectin in epithelial‐mesenchymal transition process. Proc Natl Acad Sci USA 108: 17690–17695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Erbilgin A, Civelek M, Romanoski CE, Pan C, Hagopian R, Berliner JA, Lusis AJ (2013) Identification of CAD candidate genes in GWAS loci and their expression in vascular cells. J Lipid Res 54: 1894–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, Myers AJ, Corneveaux JJ, Hardy J, Vonsattel JP, Younkin SG et al (2014) Genome‐wide association meta‐analysis of neuropathologic features of Alzheimer's disease and related dementias. PLoS Genet 10: e1004606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ng MC, Hester JM, Wing MR, Li J, Xu J, Hicks PJ, Roh BH, Lu L, Divers J, Langefeld CD et al (2012) Genome‐wide association of BMI in African Americans. Obesity 20: 622–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Desai AA, Zhou T, Ahmad H, Zhang W, Mu W, Trevino S, Wade MS, Raghavachari N, Kato GJ, Peters‐Lawrence MH et al (2012) A novel molecular signature for elevated tricuspid regurgitation velocity in sickle cell disease. Am J Respir Crit Care Med 186: 359–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yeh CT, Liang KH, Lin CC, Chang ML, Hsu CL, Hung CF (2014) A single nucleotide polymorphism on the GALNT14 gene as an effective predictor of response to chemotherapy in advanced hepatocellular carcinoma. Int J Cancer 134: 1214–1224 [DOI] [PubMed] [Google Scholar]
- 54. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. McCarthy DJ, Chen Y, Smyth GK (2012) Differential expression analysis of multifactor RNA‐Seq experiments with respect to biological variation. Nucleic Acids Res 40: 4288–4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Falcon S, Gentleman R (2007) Using GOstats to test gene lists for GO term association. Bioinformatics 23: 257–258 [DOI] [PubMed] [Google Scholar]
- 57. Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N et al (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol 32: 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Review Process File
Data Availability Statement
The mass spectrometry glycoproteomics data have been deposited to the ProteomeXchange Consortium 57 via the PRIDE partner repository with the dataset identifier PXD002770 and RNAseq data can be found at www.ebi.ac.uk/arrayexpress with accession number E‐MTAB‐3844.